

ABSTRACT
It has been demonstrated that some commonly used Extracellular Vesicle (EV) isolation techniques can lead to substantial contamination with non-EV factors. Whilst it has been established that this impacts the identification of biomarkers, the impact on apparent EV bioactivity has not been explored. Extracellular vesicles have been implicated as critical mediators of therapeutic human mesenchymal stem cell (hMSC) paracrine signalling. Isolated hMSC-EVs have been used to treat multiple in vitro and in vivo models of tissue damage. However, the relative contributions of EVs and non-EV factors have not been directly compared. The dependence of hMSC paracrine signalling on EVs was first established by ultrafiltration of hMSC-conditioned medium to deplete EVs, which led to a loss of signalling activity. Here, we show that this method also causes depletion of non-EV factors, and that when this is prevented proangiogenic signalling activity is fully restored in vitro. Subsequently, we used size-exclusion chromatography (SEC) to separate EVs and soluble proteins to directly and quantitatively compare their relative contributions to signalling. Non-EV factors were found to be necessary and sufficient for the stimulation of angiogenesis and wound healing in vitro. EVs in isolation were found to be capable of potentiating signalling only when isolated by a low-purity method, or when used at comparatively high concentrations. These results indicate a potential for contaminating soluble factors to artefactually increase the apparent bioactivity of EV isolates and could have implications for future studies on the biological roles of EVs.
KEYWORDS: Extracellular vesicles, exosomes, microvesicles, purification, mesenchymal stem cell, paracrine effect, contamination, angiogenesis, size exclusion, polymer precipitation
Introduction
Many organ systems in adult humans cannot fully regenerate after injury. In many tissues, damaged tissue is replaced with a fibrous scar with little or no functional capacity. This insufficiency underlies some of the most common causes of death and disability in the developed world, including myocardial infarction [1], stroke [2], kidney damage [3] and more. Efforts to develop therapies that allow functional regeneration often make use of multipotent stem cells. In particular, human mesenchymal stem cells (hMSCs) have been used as a source of autologous cells that can be readily obtained from adult patients, overcoming many of the immunological, safety and ethical barriers associated with induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs). It was initially thought that after administration to the site of injury, hMSCs would engraft, expand, differentiate, and eventually replace the damaged tissue. Preclinical models in which hMSCs were injected into infarcted hearts and damaged kidneys have shown encouraging results, with some treatments advancing to clinical trials [4,5]. However, it soon became apparent that the observed beneficial effects could not be attributed to expansion and differentiation of the injected hMSCs. In a rat model of myocardial infarction, limitation of infarct size and improvements in heart function were observed within 72 h of administration, too soon to be due to the development of new tissue [6]. HMSCs were found to engraft at low frequency in both hearts and kidneys, with little long-term persistence in tissue [7,8]. It is now generally thought that any benefits from hMSC transplantation are due to paracrine signalling from hMSCs to tissue near or at the site of injury [9,10]. In 2005, it was demonstrated for the first time that hMSC-conditioned culture medium could recapitulate the cardio-protective effects of hMSC transplantation in infarcted hearts [11]. Since then, considerable work has been undertaken to establish the key paracrine factors underlying this signalling [6,12–14], as they could have potential as cell-free therapies for a variety of conditions and injuries.
Paracrine signalling from hMSCs has been proposed to act by several mechanisms. In the case of myocardial infarction, multiple studies have found that hMSCs upregulate several key soluble growth factors in response to hypoxia, including vascular endothelial growth factor (VEGF), Insulin-like growth factor 1 (IGF-1), basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), Thymosin β4 (Tβ4), and stromal cell-derived factor (SDF1) [6,12–14]. These factors appear to function by reducing apoptosis and encouraging neoangiogenesis within the host tissue. In many cases, these factors have been investigated as potential therapies to improve cardiac remodelling [15,16]. Reno- and neuro-protective signalling from hMSCs has also been attributed to these and other soluble factors [17–23]. These factors have been identified by consensus from multiple studies. In addition, there is also evidence that this paracrine signalling occurs via extracellular vesicles. Extracellular vesicles (EVs) are nanoscale cell-derived lipid vesicles [24]. They are subcategorised by their cellular origin; microvesicles are formed by budding from the plasma membrane, whereas exosomes are released when internal multivesicular bodies fuse with the plasma membrane, releasing their internal vesicles. Although exosomes tend to be smaller than microvesicles and their typical protein populations are not identical, there is substantial overlap in their physical and biochemical properties and as such they are thought to be co-isolated by most if not all isolation methods. Both are known to contain cytosolic and membrane proteins, mRNA, and miRNA, and can transfer these biomolecules to recipient cells either by direct fusion with the plasma membrane or through endocytosis. EV membrane proteins may also interact with cells via ligand/receptor interactions, without uptake or fusion. By these mechanisms, EVs can act as signalling factors [25].
The first indication that EVs might have a role in the paracrine effect was the observation that hMSC-conditioned medium passed through an ultrafiltration membrane to exclude EVs was no longer capable of reducing infarct size in a porcine model of myocardial infarction [26]. These results also implied little or no contribution by soluble factors. Subsequently, EVs were purified and found to be capable of mediating a reduction in infarct size [27]. Since then, EVs from hMSCs have been explored for the treatment of a wide variety of conditions [28–39]. However, few subsequent studies have explicitly re-demonstrated that depleting EVs from hMSC-conditioned medium removes the therapeutic effect; rather, the majority of studies have focused on the use of EVs after their isolation from conditioned medium. There have been recent concerns over the reproducibility of the reported effects of hMSC-EVs, difficulties in establishing a consensus on their mechanism of action, and the quality of characterisation of hMSC-EV preparations [40]. Within the wider EV field, there is an increasing awareness that many of the techniques commonly used for EV isolation are prone to substantial contamination with soluble factors [41–46]. This has so far mostly been discussed in the context of the use of EVs as diagnostic biomarkers [46], and the implications of these observations for the apparent therapeutic capacity of EVs have not yet been explored.
Here, we show for the first time that these and other methodological issues can lead to misidentification of EVs as critical factors in hMSC-mediated signalling. Ultrafiltration of hMSC-conditioned medium removed EVs and reduced proangiogenic signalling, but unexpectedly also removed soluble factors below the membrane pore size. Preventing depletion of soluble factors while maintaining exclusion of EVs fully restored proangiogenic activity in vitro. Chromatographical separation of EVs and soluble protein revealed that proangiogenic and pro-wound healing activity was exclusively associated with soluble protein and not EVs. EVs isolated by a commercial polymer precipitation method stimulated both angiogenesis and wound healing; however, this isolation method was found to co-isolate biologically active contaminants to which this activity could be attributed. More effectively purified EV samples were able to weakly stimulate angiogenesis, but only at concentrations far higher than those at which they were present in conditioned medium, consistent with a minimal role in hMSC paracrine signalling, at least in vitro.
Materials and methods
Cell culture
NIH-3T3, MDA-MB-231 and A549 cells were obtained from the ATCC (Manassas, VA, USA). NIH-3T3 cells were cultured in 4.5 g/L high glucose DMEM with 10% (v/v) FBS and antibiotic/antimycotic (A/A) (Gibco 15240062). MDA-MB-231 and A549 cells were maintained in 4.5 g/L DMEM, supplemented with 10% FBS, 1x non-essential amino acids, 25 mM HEPES and 1x penicillin/streptomycin. Pooled HUVECs (Gibco C0155C) were cultured in M200 media (Gibco M200500) with A/A and Low-Serum Growth Supplement (LSGS) (Gibco S00310). Human Mesenchymal Stem Cells were obtained from Lonza (Basel, Switzerland). Donor 1 was used throughout the paper; donors 2 and 3 were used to confirm the results from analytical SEC. Donor lot numbers, age, sex, and ethnicity are shown in Table 1. HMSCs were cultured in DMEM with 1 g/L glucose with A/A and 20% (v/v) hMSC-qualified FBS. All cells were grown in Corning (NY, U.S.A) cell culture flasks at 37°C, 5% CO2. All cell lines were tested and found to be free of mycoplasma.
Table 1.
Bone marrow-derived human mesenchymal stem cell donor details.
| Donor | Lot number | Age | Sex | Ethnicity |
|---|---|---|---|---|
| 1 | 372262 | 39 | M | Black |
| 2 | 539540 | 18 | F | Black |
| 3 | 588695 | 22 | M | Hispanic |
Conditioned medium production
To prepare hMSC-conditioned medium, hMSCs were expanded from p4 stocks to p6, splitting from 1x T175 to 8x T225, and then to 72x T225 flasks. Near-confluent cells were washed once with sterile PBS before addition of 35 mL per flask DMEM (1 g/L glucose) with A/A, without FBS, for 40 h prior to harvest. 2.5 L of conditioned medium was harvested, and each 500 mL of conditioned medium was passed through a 0.45 μm bottle-top filter (Jet Bio-filtration, Guangzhou, China) and aliquoted prior to immediate storage at −80°C. To obtain conditioned medium from MDA-MB-231 and A549 cells, cells were grown to 80% confluence prior a 72 h incubation in basal DMEM, before 0.45 μm filtration as before.
Ultrafiltration
Either 3 kDa or 100 kDa Amicon Ultra-15 centrifugal filter devices (Merck Millipore) were used (see text). 12 mL of media was added to the top compartment prior to centrifugation in a fixed-angle rotor at 5000 x g, for 3 min at 4°C. The flowthrough was removed and media topped up to 12 mL. This was repeated, increasing the time of each spin as necessary, until processing was complete.
To block concentrator membranes, 100 kDa centrifugal filter devices were washed once with 18.2 MΩ water by adding 12 mL to the upper compartment prior to centrifugation (5000 x g, 3 min). Subsequently, 12 mL of 1% (w/v) bovine serum albumin (BSA) in PBS was added to the top compartment and left undisturbed for 2 ½ h. The upper compartment was then washed out 4 times with PBS, without centrifugation. The filter was then washed once with PBS by centrifugation before immediate use.
Size exclusion chromatography
A 30 × 1 cm Bio-Rad Econo-column was packed to a depth of 28 cm (volume ≈ 22 cm3) with Sepharose CL-2B (Sigma). A flow adaptor (Bio-Rad #7380015) was then fitted. Columns were stored in 20% (v/v) de-gassed EtOH and washed with > 60 mL particle-free cell culture PBS (Gibco #10010015) prior to use. A valve switching system was used to apply 500 μL of sample before immediately switching to a PBS reservoir. 30 × 1 mL fractions were collected from the point of the switch. Columns were then washed with > 60 mL PBS and 60 mL of 20% EtOH for storage.
Total exosome isolation
From 100 mL of pre-processed hMSC-conditioned medium, 750 μL was removed for later analysis, and the remaining media was concentrated to a final volume of 1 mL using 4 × 3 kDa Amicon centrifugal concentrators. From the concentrated media, 165 μL was removed for later analysis. 417 μL of TEI reagent was added to the remaining 835 μL of concentrated medium in a 1.5 mL Eppendorf tube before vortexing to mix. The TEI/concentrated media mix was incubated for 16 h at 4°C before centrifugation (10 min, 10,000 g, 4°C) to pellet precipitated material. Supernatant was thoroughly aspirated, and the pellet was resuspended in 100 μL particle-free PBS by thorough pipetting before immediate aliquoting and storage at −80°C.
Nanoparticle tracking analysis
Particle concentration was quantified using a Nanosight NS300 (532 nm laser, SCMOS camera) (Malvern Instruments, Salisbury, UK). EVs were diluted in particle-free water from a Select Fusion Milli-Q water purifier (Suez Water UK, Thame, U.K.) at the point of measurement to a concentration between 108–109 particles/mL. Using NTA V3.0 software, three 60-s videos were recorded and analysed per sample, with software parameters Camera Level 15 and Detection Threshold 5 used, and FTLA smoothing disabled.
Dot blots
A 0.45 µm nitrocellulose membrane (Bio-Rad 162–0117) was soaked in Tris-buffered Saline (TBS, Bio-Rad 170–6435) for 10 min, before assembly into the Bio-Dot cassette (Bio-Rad 170–6545). Wells were filled with TBS and drained under vacuum prior to addition of sample to each well. 100 μL of conditioned medium or 50 μL of SEC fraction samples was added per well and allowed to drain without vacuum. Wells were then washed 3x with TBS, with vacuum, prior to retrieving the membrane and blotting with 5% (w/v) skim milk (Bio-Rad 170–6404) in TBS-T on a rocking platform (1 h, r.t). The membrane was washed 3x in TBS-T (5 min each) prior to addition of primary antibodies and overnight incubation. Primaries were used at 1:1000 dilution in 5% (w/v) BSA in TBS-T (CD63: 10628D, CD81: 10630D, CD9: 10626D, Thermo Fisher). The membrane was washed as before prior to addition of secondary antibody (1:10,000 in 5% (w/v) BSA/TBS-T, goat anti-mouse 800 nm, Li-COR 926–32210) and incubated in the dark for 1 h. After washing as before, membranes were imaged on a Li-COR Odyssey infrared fluorescence imager. Dot intensity quantification was performed using Image Studio software, generating a relative intensity (R.I) value for each dot that was then normalised to the average value within each experimental replicate (N.R.I).
Western blots
Protein was isolated from both cell (crude lysate) and EV (peak fraction) samples. Lysates were prepared in RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) supplemented with phosphatase and protease inhibitors (Roche, Basel, Switzerland), and sonicated on ice for 20 seconds at 20% amplitude using a VibraCell VCX500 sonicator (Sonics & Materials Inc., Newtown, CT, USA). Lysates were gently mixed for 1 h at 4°C, after which they were centrifuged for 10 min at 20,000 x g at 4°C. Supernatants were transferred to clean tubes and pellets discarded. Protein concentrations were determined using the BCA assay (Pierce, Thermo Fisher Scientific, Inc.). 15 μg of protein, mixed with Laemmli sample buffer (Bio-Rad Laboratories, Inc., Hercules, CA, USA) without reducing agent, was loaded unto Mini-protean TGX precast 4–20% gradient gels (Bio-Rad) and separated by SDS-PAGE. Next, protein was blotted onto PVDF membranes (EMD Millipore) prior to antibody incubation and fluorescent imaging as performed with dot blots.
Cryo-TEM
SEC-EV peak fractions were pooled and concentrated as previously described. TEI-EV samples were used without further concentration. Holey Carbon 200 mesh Grids (Electron Microscopy Supplies) were glow-discharged (15 sec O2/H2 1:1) with a Solaris plasma cleaner (Gatan, Pleasanton, CA, U.S.). Samples were prepared with a Leica EM GP plunge-freezer. Sample grids were prepared by adding 4 µL of sample within an environmental chamber (20°C, 90% relative humidity). Excess sample was blotted away on filter paper prior to plunging into liquid ethane. Samples were stored and transferred in liquid nitrogen in a Gatan model 914 cryo transfer holder. Samples were imaged at −170°C in a JEOL 2100 Plus Transmission Electron Microscope (JEOL, Tokyo, Japan) at 200 kV, using a Minimum Dose System software. Images were acquired with an Orius SC 100 camera (Gatan) at 30 k magnification over 5-sec exposure time.
ELISA
The VEGF Human ELISA kit (Thermo Fisher KHG0112) was used according to manufacturer instructions.
Nano-orange
The NanoOrange assay kit (Thermo Fisher N6666) was used according to manufacturer instructions.
Tubule formation assay
HUVECs were thawed from p2 stocks and grown for 6 days prior to the assay. 6 h prior to use, HUVECs were transferred to basal M200 media. 1 h prior to use, GFR Geltrex (Gibco A1413202) was mixed at a 6:1 ratio with ice-cold PBS, before addition (on ice) of 10 μL of diluted Geltrex to each well of an Ibidi 96-well angiogenesis plate (Ibidi, 89646). The plate was spun in a plate centrifuge at 50 g, 60 s, before incubation at 37°C for gelation. HUVECs were detached with Trypsin, centrifuged and resuspended in basal M200 medium before measurement of viable cell concentration with a Countess II FL cell counter (ThermoFisher). Viable cell concentration was adjusted to 106/mL. Cells, samples and buffers (DMEM or PBS) were combined and added gently to wells. Sample volumes are shown in Table 2.
Table 2.
Sample volumes for tubule formation assay.
| For 12 replicates | M200 (basal) | M200 + LSGS (10x) | Sample/buffer | Cells at 1 x 106/mL |
|---|---|---|---|---|
| Positive Ctrl | 120 µL | 60 µL | 300 µL (buffer) | 120 µL |
| Negative Ctrl | 180 µL | 0 µL | 300 µL (buffer) | 120 µL |
| Samples | 180 µL | 0 µL | 300 µL (sample) | 120 µL |
After 16 h incubation at 37°C, 5% CO2, 20 mL of 2 µM Calcein-AM (Invitrogen C3099) in basal M200 media was gently added to the top compartment of the plate before returning to the incubator, protected from light, for 20 min. The plate was then washed once and imaged using an Axio Observer live cell imaging microscope with an HXP 120 light source (Zeiss) at 37°C, 5% CO2. A 2.5X objective was used and the entirety of each well was imaged.
Images were imported, cropped circularly to remove well edges, and converted to a binary image using a manually set threshold using FIJI [47]. The threshold was set to highlight all tubule structures without including background. The binary images were then analysed using the “Angiogenesis Analyzer” plugin [48]. Total tubule length was used to compare the pro-angiogenic activity of samples.
Transfection
Cytolight Green Lentivirus (Incucyte 4481, Sartorius, Göttingen, Germany) was used according to the manufacturer’s instructions to generate a GFP-positive NIH-3T3 cell line (GFP-3T3).
Scratch assay
24-well plates were coated with Collagen-I (100 μL/well, 50 μg/mL, in 0.02 N acetic acid, Corning 354236) for 2 h before aspirating fluid and allowing to dry fully for at least 3 h. Coated plates were stored at 4°C and used within 2 weeks. GFP-3T3 cells were seeded at 50 K cells/well 24 h prior to the assay. After 24 h, a monolayer had formed. A scratch was made across the centre of each well using a p200 pipette tip steadied with a ruler. The media was replaced with fresh basal DMEM, and each scratch visually inspected. Scratches that were ragged, wider than the field of view or less than 1/3rd of the width of the field of view were excluded. The basal DMEM was removed before adding samples and controls, all diluted 1:1 in fresh basal DMEM (C+: DMEM + 10% (v/v) FBS, C-: sample buffer (DMEM or PBS)). Each well was imaged for GFP fluorescence at 3 manually selected positions along the scratch, every hour for 15 h (16 timepoints) using an Axio Observer live cell imaging microscope (Zeiss). Images were analysed with FIJI [47], using the “MRI Wound Healing Tool” [49]. The area of the gap in each image was quantified, and the rate of area reduction until closure determined by linear regression for each individual image set. Image sets with improper gap/cell segmentation (due to reflections, uneven illumination or floating cells) were excluded from the final calculation of each well’s closure rate. Three wells were used for each condition, forming three technical replicates.
Statistics
Calculations were performed using Graphpad Prism 8. Experiments were designed such that paired or within-subjects statistical tests could be used. For comparison of two groups with paired replicates, 2-tailed paired Student’s t-tests were used. For comparison of three of more groups with paired replicates, one-way within-subjects ANOVAs (Analysis of Variance) were used, with a post-hoc Tukey’s HSD test (for comparison of all means) or Dunnett’s test (for comparison to a single control value). Numerical data within body of text is expressed as mean ± standard deviation.
EV-TRACK
We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase [50] (EV-TRACK ID: EV190075).
Results
We initially sought to recapitulate the pioneering observation that passage through an ultrafiltration membrane depletes the bioactivity of hMSC conditioned medium (hMSC-CM) [26,27]. HMSC-conditioned medium was prepared and passed through a 100 kDa Molecular Weight Cut-Off (MWCO) centrifugal spin filter (Figure 1(a)), collecting five sequential ~20 mL samples (Flowthrough 1–5, or FT1-5). It was expected that EVs would be retained while soluble proteins smaller than 100 kDa would pass through unimpeded. To establish the presence and concentration of EVs in the hMSC-conditioned medium and the flowthrough, the particle concentration was quantified by nanoparticle tracking analysis (NTA) (Figure 1(b)) and the availability of CD63 epitopes investigated by dot blotting (Figure 1(c)). Nanoparticles and CD63 were detected in the hMSC-conditioned medium but not in any of the flowthrough fractions, confirming that EVs were present in conditioned medium but reduced to undetectable levels after passage through the spin filter.
Figure 1.
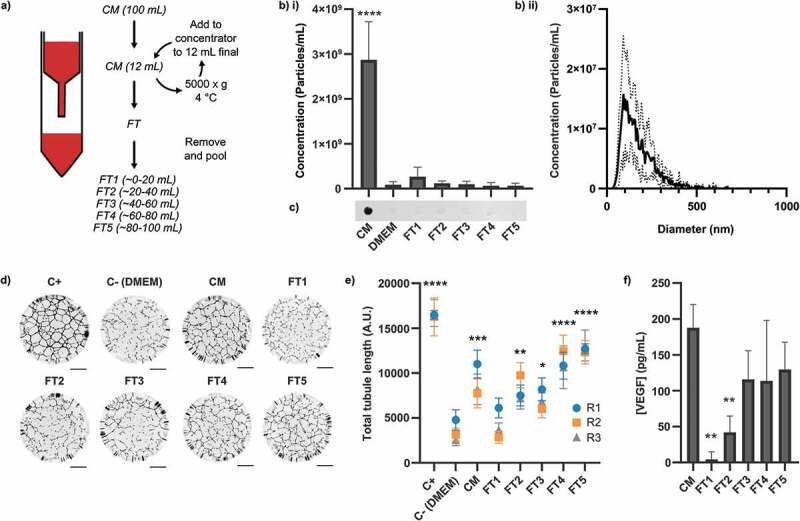
Contents and signalling properties of hMSC-conditioned medium after 100 kDa ultrafiltration. Media (CM) was passed through a 100 kDa membrane and collected in sequential ~20 mL batches (FT1-5), as described in the main text. (a) Schematic of experimental workflow. CM = Conditioned medium, FT = Flowthrough. (b) NTA measurements of i) concentration and ii) size distribution (of CM sample), N = 3, n = 3 x 60s videos, mean ± SD shown. Particles were only detected in CM (1-way within-subjects ANOVA and post-hoc Dunnett’s test vs DMEM control). (c) Dot blots against CD63. CD63 was only detected in CM. Independently prepared replicate samples were blotted onto the same membrane and analysed simultaneously (N = 3, representative blot shown). (d) Representative whole-well images of tubule structures. Images inverted for clarity. Scale bar = 1 mm. (e) Quantification of total tubule length in tubule formation assay. All samples besides FT1 had a greater proangiogenic activity than DMEM (N = 3, n = 12, mean ± SD of n shown for each N (replicates 1–3, R1-3), 1-way within-subjects ANOVA and post-hoc Dunnett’s test vs DMEM control). (f) ELISA against VEGF. VEGF was reduced relative to the level in CM in FT1 and FT2 only (N = 3, n = 2, mean ± SD shown, 1-way within-subjects ANOVA and post-hoc Dunnett’s test vs CM). *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
We next evaluated the pro-angiogenic activity of the hMSC-conditioned medium and flowthrough samples using an in vitro human umbilical vein endothelial cell (HUVEC) tubule formation assay, in which HUVECs were incubated for 16 h on a growth factor-reduced basement membrane mimic in the presence of 1:1 v/v fresh basal medium and sample. Cells were visualised by Calcein-AM fluorescence and the total length of the tubules formed was quantified for each well using FIJI, as described in the methods. We observed that the in vitro pro-angiogenic activity of the conditioned medium was completely removed in the first ~20 mL of flowthrough (Figure 1(d,e)), consistent with results reported elsewhere [26,27]. However, the subsequent fractions exhibited increasing pro-angiogenic activity, which rose to a level similar to that of the unfiltered hMSC-conditioned medium. As we did not detect EVs in these fractions, we hypothesised that non-EV factors might contribute to signalling. Many small protein signalling factors secreted by hMSCs have been implicated in their signalling [6,12–14,17,18]. Of these, we selected VEGF to serve as a representative indicator of the presence and abundance of these factors as a group and evaluated its concentration in the conditioned medium and flowthrough. Sample analysis with an Enzyme-Linked Immunosorbent Assay (ELISA) found that VEGF was present in the unfiltered conditioned medium, significantly depleted in the first two flowthrough fractions, but progressively increased in subsequent flowthrough fractions to a level similar to that of the unfiltered conditioned medium (Figure 1(f)). This matched the pattern of bioactivity observed in the tubule formation assay, indicating contribution by soluble factors.
VEGF exists in solution as a 45 kDa homodimer [51] and so the observation that it did not pass through a 100 kDa pore size membrane unimpeded was unexpected. We attributed this observation to attractive electrostatic or hydrophobic interactions between the soluble factors and the ultrafiltration membrane. Under this model, the membrane surface becomes passivated as progressively more protein is adsorbed and removed from the flowthrough. Eventually, a passivating layer of protein builds up on the surface, blocking subsequent protein/surface interactions and allowing unimpeded passage of proteins below the membrane pore size. While these effects may not be significant when filtering a large quantity of protein, it could have a significant impact on dilute solutions containing low-abundance soluble factors such as VEGF. We tested this hypothesis by passing hMSC-conditioned medium through membranes that had been pre-passivated with 1% (w/v) Bovine Serum Albumin (BSA) (Figure 2).
Figure 2.
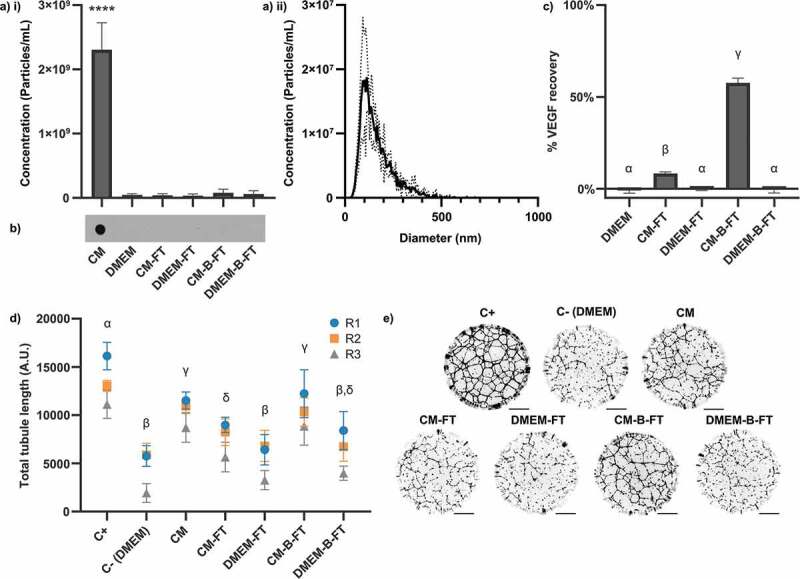
Effects of membrane surface BSA-blocking on 100 kDa ultrafiltration of hMSC-CM. Membranes were either BSA-blocked (-B-FT) or left unblocked (-FT) prior to filtration of 10 mL of CM or DMEM. (a) NTA measurements (3 x 60 s videos/sample, N = 3, mean ± SD shown) of i) particle concentration and ii) size distribution (of CM sample). Particles were only detected in CM (1-way within-subjects ANOVA and post-hoc Dunnett’s test vs DMEM control). (b) Dot blots against CD63. CD63 was only detected in CM. Independently prepared replicate samples were blotted onto the same membrane and analysed simultaneously (N = 3, representative blot shown). (c) ELISA against VEGF. % recovery of VEGF in CM for each sample is shown (N = 3, n = 2, mean ± SD shown). (d) Quantification of total tubule length in tubule formation assay (N = 3, n = 12, mean ± SD of n shown for each N (replicates 1–3, R1-3)). (e) Representative whole-well images of tubule structures formed in (d). Images inverted for clarity. Scale bar = 1 mm. Samples are significantly different unless they share a letter (p < 0.05, one-way within-subjects ANOVA and post-hoc Tukey’s HSD test).
No particles (Figure 2(a)) or CD63 epitopes (Figure 2(b)) were detected in the flowthrough of pre-passivated or untreated membranes. However, the loss of VEGF was substantially mitigated by pre-passivation, with 57.6 ± 2.6% recovery compared to 8.3 ± 0.9% for the un-passivated control (Figure 2(c)). The same pattern was seen in stimulation of in vitro angiogenesis (Figure 2(d,e)). Flowthrough from non-passivated filters had a reduced capacity to stimulate angiogenesis compared to the unfiltered conditioned medium, whereas the flowthrough from pre-passivated filters retained its original pro-angiogenic capacity. Non-conditioned medium passed through the membrane did not stimulate angiogenesis compared to the negative control, with or without membrane pre-blocking. We concluded that in our experimental system the reduction of pro-angiogenic signalling by ultrafiltration was artefactual rather than due to the depletion of a critical factor larger than 100 kDa.
For subsequent experiments, we chose to also investigate the ability of hMSC-conditioned medium-derived material to stimulate fibroblast migration in a scratch-wound model of wound healing. The inclusion of this model allowed us to test whether our observed results were specific to our tubule formation assay. As it was necessary to concentrate hMSC-conditioned medium in order to observe significant stimulation of wound healing, it was also necessary to investigate whether this concentration step could cause a bias in component recovery or a loss of stimulatory activity.
To investigate this, 100 mL of hMSC-conditioned medium was concentrated against an ultrafiltration spin filter. A 3 kDa MWCO membrane was used in order to retain both EVs and soluble protein (Figure 3). Conditioned medium (1x) was concentrated 5-fold (5x) and 25-fold (25x). The 25x concentrated conditioned medium was then diluted 5-fold (5x-d) and 25-fold (1x-d) with fresh basal DMEM. Any loss due to the concentration process was anticipated to manifest as differences between the unconcentrated and the concentrated/re-diluted samples (i.e. 5x vs 5x-d, 1x vs 1x-d). It was found that concentration led to a significant decrease in the total number of particles (Figure 3(a)) but no significant loss of VEGF (Figure 3(b)). CD63 epitopes were detected in all samples (Figure 3(c)). Importantly, the ability of hMSC-conditioned medium to stimulate tubule formation (Figure 3(d)) and wound healing (Figure 3(e)) was unaffected by concentration/dilution. It therefore appeared that although sample recovery after concentration was biased in favour of non-EV factors, the factors responsible for stimulation were recovered without significant loss. This was consistent with our previous observations that EVs were dispensable for stimulation of our assays.
Figure 3.
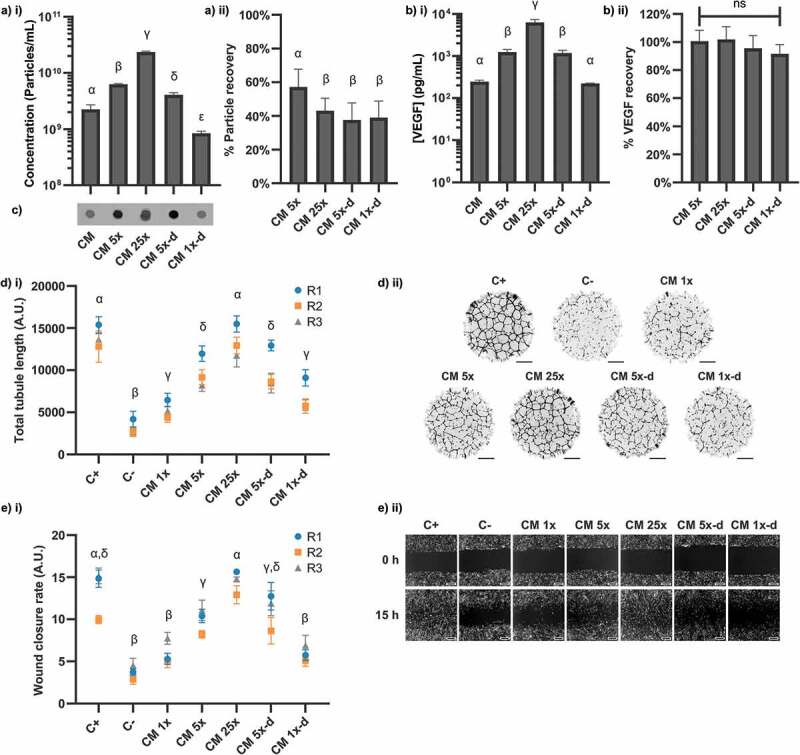
Effects of concentration on contents and signalling properties of hMSC-CM. CM was concentrated 5x against a 3 kDa membrane, then 25x, before subsequent dilution to 5x (5x-d) and 1x (1x-d) concentrations. (a) NTA concentration measurements of samples, expressed as i) absolute concentration values and ii) % recovered from input CM. (N = 3, n = 3 x 60s videos, mean ± SD shown). (b) ELISA against VEGF, expressed as i) absolute concentration values and ii) % recovered from input CM. (N = 3, n = 2, mean ± SD shown). (c) Anti-CD63 dot blots. Independently prepared replicate samples were blotted onto the same membrane and analysed simultaneously. (d) i) Quantification of total tubule length in tubule formation assay (N = 3, n = 12, mean ± SD of n shown for each N (replicates 1–3, R1-3)). (d) ii) Representative whole-well images of tubule structures formed in i). Images inverted for clarity. Scale bar = 1 mm. (e) i) Quantification of wound closure rate for each condition (N = 3, n = 3, mean ± SD shown for each N (replicates 1–3, R1-3)). (e) ii) Representative images of scratch wounds for each condition are shown for t = 0 h and t = 15 h. Scale bar = 100 μm. Samples are significantly different unless they share a letter (p < 0.05, one-way within-subjects ANOVA and post-hoc Tukey’s HSD test). Where plotted on a logarithmic axis, data were log10-transformed prior to statistical analysis.
Our results so far had indicated that EVs were not necessary for stimulation of angiogenesis, and their loss during concentration of conditioned medium did not impact wound healing in vitro. However, the possibility remained that EVs could stimulate angiogenesis and wound healing independently of soluble factors. We used analytical Size Exclusion Chromatography (aSEC) to separate the components of pre-concentrated (3 kDa, 100 mL -> 0.5 mL) hMSC-conditioned medium by size. 30 × 1 mL fractions were collected. The nanoparticle concentration was quantified in F6-25, and the presence of EV marker proteins (CD63, CD81 and CD9) was determined as before by dot blots on each fraction (Figure 4(a)). Antibodies had been previously validated by Western blot (Figure S1). The majority of EVs appeared to elute in a major peak at F9-10, as defined by particle concentration and the intensity of dot blots against EV markers. However, one or more EV markers were detectable up to F20, with an apparent minor secondary peak detected by all markers and NTA at F18. EVs were not detectable beyond F20.
Figure 4.
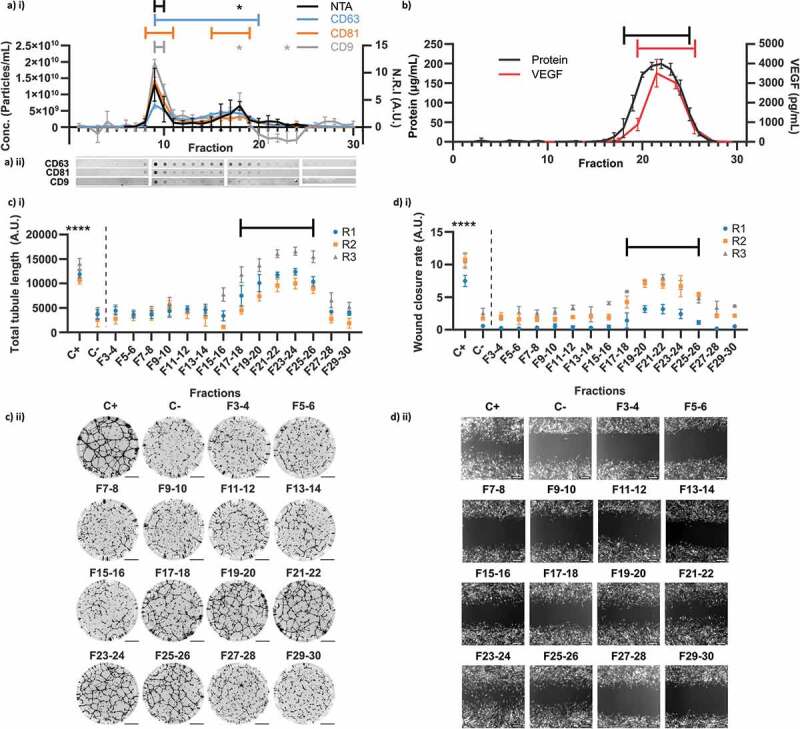
Analytical Size Exclusion Chromatography of hMSC-CM. (a) EV content of fractions evaluated by particle concentration measured by NTA i) (n = 3 x 60s videos, N = 3) and dot blots against CD63, CD81 and CD9 (N = 3, quantified by Normalised Relative Intensity, N.R.I, as in methods). (a) ii) Representative dot blot images as used for quantification in a) i) (images re-cut to align with graph). (b) Total protein and VEGF concentrations of each fraction (VEGF measured on pooled adjacent pairs and plotted at mid-point). (c) i) Quantification of total tubule length in tubule formation assay for pooled adjacent fraction pairs (N = 3, n = 6, mean ± SD of n shown for each N (replicates 1–3, R1-3)). (c) ii) Representative whole-well images of tubule structures formed in C i). Images inverted for clarity. Scale bar = 1 mm. (d) i) Quantification of wound closure rate for pooled adjacent fraction pairs (N = 3, n = 3, mean ± SD shown for each N (replicates 1–3, R1-3)). (d) ii) Representative images of scratch wounds for each fraction pair are shown for t = 15 h (endpoint). Scale bar = 100 μm. * and brackets: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, (one-way within-subjects ANOVA and post-hoc Dunnett’s test vs earliest measured fraction/s or negative control (NTA: F6, DB: F1, Total protein: F1, VEGF: F7-8, Tubule length: C-, Wound Closure Rate: C-)).
The protein concentration in each fraction was quantified (Figure 4(b)). Protein was observed between F18-F25, with a peak between F21-22, but was not detected in association with the EV peak. However, as the peak EV concentration was measured to be ~2 x1010 particles/mL, pure EVs are reported to have a particle to protein ratio of approximately ~3 x1010 particles/μg [42] and the limit of detection for the protein quantification assay was ~1 μg/mL, this observation was consistent with the presence of high-purity EVs. An ELISA against VEGF was performed on pooled adjacent fractions (Figure 4(b)). This analysis revealed the presence of VEGF between F19/20 – 25/26, with a peak at F21/22, consistent with the elution profile of soluble proteins. We concluded that our aSEC setup was capable of separating the majority of EVs from the majority of soluble factors, with a small zone of overlap between F18-20.
We subsequently performed our tubule formation and scratch-wound assays on pooled adjacent fractions (excluding F1-2) (Figure 4(c,d)). For both assays, a significant increase in activity was observed only between F17/18 – F25/26, the fractions containing the highest levels of soluble protein and VEGF. Contrastingly, there was no detectable stimulation of angiogenesis or wound healing in the fractions containing the highest concentrations of EVs. The aSEC experiments were repeated with two additional independent hMSC donors (Figures S2-3) and similar results were observed, with no EV-associated stimulation and robust soluble protein-associated stimulation. The average modal EV diameter at F9 across all 3 donors was 141 ± 17.3 nm (125 ± 11.3 nm when a single outlier was excluded [Table S1]). As before, the concentrators were found to preferentially recover VEGF, but there was no difference detected between EV and VEGF recovery from aSEC, either overall or in the peak fractions (Figure S4).
Our evidence so far indicated that the majority of pro-angiogenic and pro-migratory signalling from hMSC-conditioned medium was mediated by conventional soluble factors and not by EVs. However, EVs are widely reported to potentiate wound healing and angiogenesis. We hypothesised that this discrepancy could be partially due to differences in EV purity, given that there is increasing concern that many EV purification protocols do not effectively remove contaminants. In order to investigate whether less stringent purification procedures could lead to artefactual results by co-isolation of biologically active soluble factors, we chose to purify EVs using a commercial kit for polymer precipitation-based EV isolation (Total Exosome Isolation Reagent, or TEI). Polymer precipitation remains in common use as a primary isolation method; however, it has been reported that the resulting EV samples may be impure [44]. 100 mL of conditioned medium was concentrated against a 3 kDa membrane to a final volume of 1 mL as before. EVs were isolated from this 100x concentrated medium according to manufacturer instructions, resuspending EVs in 0.1 mL of particle-free PBS.
EVs isolated by TEI were able to significantly stimulate both angiogenesis (Figure 5(a)) and wound healing (Figure 5(b)) at minimum in-assay concentrations of 2 × 109 and 5 × 109 particles/mL respectively. This is in contrast to the lack of any stimulation by higher concentrations of particles from the F9-10 EV peak after analytical SEC (Figure 4, S1, S2). The average combined particle concentrations of F9 and F10 across all 3 donors was 1.11 × 1010 particles/mL – consequently, the average in-assay concentration would have been 5.55 × 109 particles/mL due to dilution. This is a higher concentration than the minimum threshold for stimulation by TEI-isolated EVs for both the tubule formation and wound healing assays. We therefore concluded that there was a difference in the content of these EV isolates besides the number of particles. We assessed the particle (Figure 5(c)) and VEGF concentrations (Figure 5(d)) of the conditioned medium, concentrated conditioned medium, and final TEI-isolated EV samples to determine whether TEI might have co-isolated VEGF or other contaminants. We also confirmed the presence of the EV marker CD63 in all samples by dot blots as before (Figure 5(e)), and the size distribution of the TEI-EVs (modal diameter = 115.8 ± 10.6 nm) (Figure 5(f)). The protein concentration of the TEI-isolated EV sample was 589.2 ± 20.9 μg/mL, an estimated yield of 0.59 μg per mL of the initial conditioned medium. TEI isolation removed 98.7 ± 0.2% (by mass) of VEGF from the concentrated conditioned medium; however, due to the high initial concentration and low final volume, the final concentration of VEGF was over an order of magnitude higher than that of conditioned medium: 2.52 ± 0.67 μg/mL. The particle-to-protein ratio in the final isolate was calculated to be 7.84 ± 0.46 x 108 particles/μg. Values below 1.5 × 109 are typically considered impure [42]. The ratio of particles-to-VEGF was 1.98 ± 0.79 x 108 particles/pg. The average F9 particle concentration across all 3 donors was 1.44 × 1010 particles/mL, and the F9-10 value was 1.11 x 1010, as previously mentioned. At these ratios, the expected associated mean protein concentration at F9 would have been 18.3 μg/mL, and the VEGF concentration at F9-10 would have been 56.1 pg/mL. These values are well within the detection limits of each assay; however, no protein was detected in F9 or VEGF in F9-10 (Figure 4). It was therefore concluded that they were likely present not as EV-associated material but as contaminants that had been removed by SEC but not TEI. This was consistent with Cryo-TEM images that showed that although both SEC-isolated EV samples and TEI-isolated EV samples contained vesicular structures of the expected diameter (Figure S5), TEI-isolated EV samples also contained dark amorphous clusters of material that may have been non-vesicular protein aggregates. These results indicated that the activity of our TEI-isolated EV samples was driven by the presence of contaminating soluble factors and not by EVs.
Figure 5.
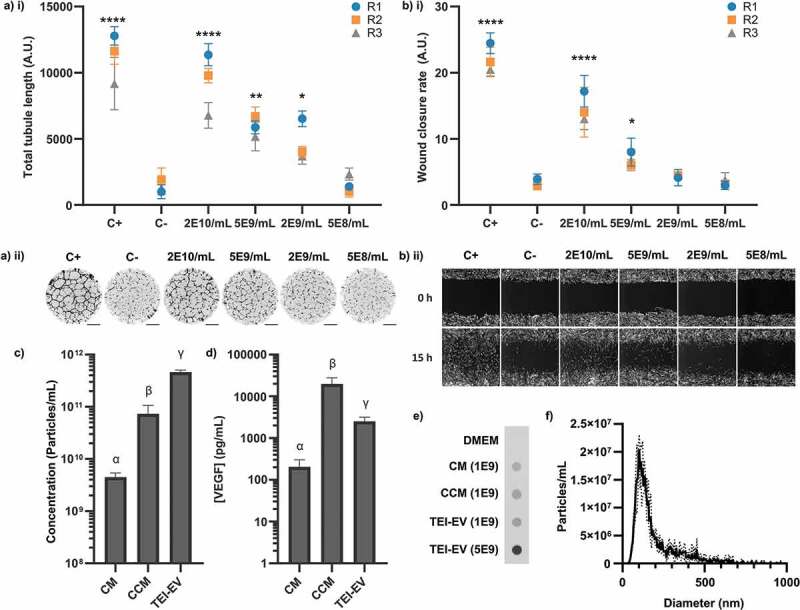
Biological activity of TEI-isolated hMSC-EVs. (a) i) Quantification of total tubule length in tubule formation assay stimulated by different TEI-EV concentrations. (N = 3, n = 12, mean ± SD of n shown for each N (replicates 1–3, R1-3). (a) ii) Representative whole-well images of tubule structures formed in (a) i). Images inverted for clarity. Scale bar = 1 mm. (b) i) Quantification of wound closure rate stimulated by different TEI-EV concentrations. (N = 3, n = 3, mean ± SD shown for each N (replicates 1–3, R1-3)). (b) ii) Representative images of scratch wounds are shown for t = 0 and t = 15 h. Scale bar = 100 μm. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, (one-way within-subjects ANOVA and post-hoc Dunnett’s test vs C-). (c) Particle concentrations (as measured by NTA) in each sample (N = 3, n = 3 x 60s videos, mean ± SD shown). (d) VEGF concentrations (as measured by ELISA) in each sample (N = 3, n = 2, mean ± SD shown). Samples in (c) and (d) are significantly different unless they share a letter (p < 0.05, one-way within-subjects ANOVA and post-hoc Tukey’s HSD test). Where plotted on a logarithmic axis, data were log10-transformed prior to statistical analysis. (e) Dot blot against CD63 for CM, concentrated CM (CCM) and TEI-EV samples Number denotes NTA-quantified particles loaded. (N = 3, one representative image shown). CD63 is present in all samples. (f) Size distribution of TEI-EV as measured by NTA (N = 3, n = 3 x 60s videos, mean ± SD shown) (same dataset as (c)).
In previously reported research, the quantity of EVs that were used to stimulate angiogenesis or wound healing has not typically been explicitly compared to the quantity of EVs in the original conditioned medium. Doses used are often given in terms of protein concentrations rather than particle concentrations; this metric is susceptible to influence from non-EV protein contaminants. Additionally, EV-associated protein alone cannot readily be measured in conditioned medium prior to isolation. Hundreds of mL of conditioned media are often required to purify sufficient EVs for downstream use, despite the stimulatory activity originally having been apparent in unconcentrated, unprocessed medium. We hypothesised that EVs were not a major signalling component at the concentrations at which they were present in our conditioned medium, but that it might be possible to stimulate our assays using SEC-purified EVs if they were used at a much higher concentration. EVs were purified in batches from 1.4 L of hMSC-conditioned medium, concentrating conditioned medium to 1.5 mL with centrifugal ultrafiltration (100 kDa MWCO) prior to separation of 3 × 0.5 mL batches using SEC as in Figure 4. Fractions previously established to contain the major EV peak (F8-14) were pooled and further concentrated by ultrafiltration. To maximise the amount of EV material available for stimulation, only particle and protein concentration measurements were performed on the final samples. The mean particle-to-protein ratio was 5.23 ± 0.31 x 109 particles/μg. This indicated moderate but not exceptional purity, suggesting that the increased concentration of the conditioned medium prior to column loading might have slightly affected particle/protein separation. Modal particle diameter was 116 ± 15.8 nm.
It was found that SEC-purified EVs at in-assay concentrations of 2 × 1010 particles/mL and 1 × 1011 particles/mL were weakly stimulatory compared to the negative control (Figure 6(a)) in a tubule formation assay. To allow direct comparison to conditioned medium, SEC-purified EVs were transferred to DMEM by ultrafiltration. This sample and 100-fold concentrated conditioned medium were both measured by NTA and diluted to matched particle concentrations. Modal pure EV particle diameter was 110 ± 9.4 nm. No significant stimulation of tubule formation by SEC-purified EVs was observed at any concentration (Figure 6(b)); contrastingly, conditioned medium was stimulatory at all particle concentrations, the lowest of which was 125x lower than the highest SEC-EV concentration.
Figure 6.
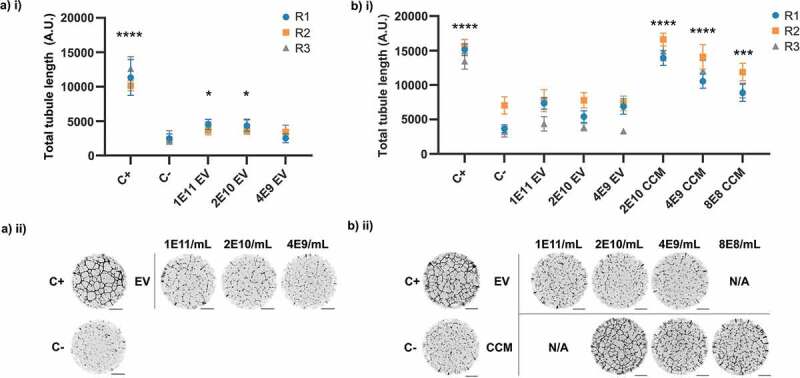
Pro-angiogenic activity of SEC-isolated EVs. (a) Assay with EVs in PBS. (a) i) Quantification of total tubule length. (N = 3, n = 8–10, mean ± SD of individual replicates shown (replicates 1–3, R1-3)). (a) ii) Representative whole-well images of tubule structures formed in Ai. (b) Assay with EVs in DMEM and CCM. (b) i) Quantification of total tubule length. (N = 3, n = 10, mean ± SD of individual replicates shown (replicates 1–3, R1-3)). (b) ii) Representative whole-well images of tubule structures formed in b) i). *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 (one-way within-subjects ANOVA and post-hoc Dunnett’s test against C-). Scale bar = 1 mm.
Discussion
In the work presented here, it was found that the presence or absence of EVs did not affect the ability of hMSC-conditioned medium to stimulate in vitro models of angiogenesis and wound healing. Rather, smaller soluble factors such as VEGF appeared to be responsible. Ultrafiltration of conditioned medium to deplete EVs, which was previously used to implicate the criticality of EVs to therapeutic hMSC paracrine signalling in in vivo models of cardiac ischaemia, was found to generate misleading results due to off-target depletion of soluble factors. HMSC-EVs were only found to stimulate angiogenesis and wound healing in vitro when either insufficiently purified away from other factors or when used at concentrations far higher than those at which they were present in conditioned medium. The extent to which these methodological risks may have affected previously-reported hMSC-EV studies is unclear, as many do not report EV-depleted conditioned medium controls, sufficiently detailed data describing EV sample purity, or the EV doses used in relation to the quantity of EVs in the source material.
However, it is important to note that this study does not exclude the possibility that hMSC-EVs are genuinely capable of stimulating regenerative processes under other experimental conditions in vitro or in vivo. HMSC phenotype is known to be affected by culture condition. Our protocol for EV collection involved the culture of hMSCs in basal DMEM, with no supplementation by EV-depleted FBS or exogenous growth factors. This was necessary in order to allow a direct comparison between hMSC-secreted EVs and soluble factors. EVs isolated from hMSCs cultured in basal medium in normoxic 2D culture (as in this study) have been previously reported to have beneficial effects in multiple models of disease [52–57], including in vitro models of wound healing and angiogenesis similar to those presented here [55–57]. However, it may be the case that some culture conditions produce viable, potent hMSC-EVs in sufficient quantities and that the basal conditions required in this study do not. HMSC-EVs have been widely reported to have various beneficial therapeutic effects and assessing the whole spectrum of culture conditions, cell sources, isolation methods and therapeutic applications is beyond the scope of this study. We suggest that our results indicate a “caution signal” – in effect, a requirement for greater awareness of the potential for contamination by non-EV factors to lead to apparently positive results.
The methodological hazards identified in this study are relevant to EV bioactivity research outside of hMSC-EV signalling. To the best of our knowledge, co-depletion of soluble factors when removing EVs from conditioned medium by ultrafiltration has not been previously reported. In our study, this led to artefactual attribution of pro-angiogenic signalling to EVs. Polymer precipitation-based methods for EV isolation remain in common use despite concerns over their ability to remove contaminants. We are not aware of any previous demonstration that co-isolated contaminants are active in signalling, nor that their activity can be misattributed to EVs.
We also identified the quantity of EVs used as a potential methodological point of concern. The quantity of EVs in the final isolate used for functional testing is not typically compared to the quantity of EVs in the initial conditioned medium or biofluid. Additionally, cells simultaneously secrete EVs and soluble factors, but cell-derived soluble factors are normally discarded during the purification process and may not be distinguishable from soluble factors added to support cell growth. This means that the respective contributions to signalling of EVs and soluble factors cannot typically be simultaneously evaluated or compared. Under these circumstances, EVs that are only weakly stimulatory compared to other cell-secreted factors could be used at a far higher concentration than that at which they were originally present and then found to be able to stimulate the effect originally stimulated by conditioned medium. The context of the higher potency of other factors would be absent, leading to an erroneous impression that the EVs under investigation have a more critical role in signalling than may be the case.
Conclusions
We unexpectedly found that extracellular vesicles were dispensable for pro-angiogenic and pro-migratory paracrine signalling from hMSC-conditioned basal media in vitro. Previously undescribed experimental artefacts relating to below-cut-off protein depletion by ultrafiltration membranes, co-isolation of soluble protein by less-stringent purification, and experimental usage of comparatively high EV concentrations were all able to obscure this result and led to apparently positive results incorrectly indicating EV function. HMSC-EVs produced and assessed in a variety of experimental conditions have been widely reported to stimulate angiogenesis and wound healing, and EVs may nevertheless genuinely contribute to signalling under other conditions not evaluated here. We interpret our results to be a “caution signal”, indicating a strong requirement to be aware of these experimental hazards when designing and conducting studies on EV bioactivity, including those unrelated to hMSCs.
Supplementary Material
Acknowledgements
We thank Akemi Nogiwa-Valdez for help in manuscript preparation. We thank James P.K. Armstrong, Isaac Pence, and Andrea Serio for their valuable advice and expertise. T.E.W. and M.M.S. acknowledge support from the Rosetrees Trust. A.N. and M.M.S. acknowledge support from GlaxoSmithKline Engineered Medicines Laboratory. M.M.S and U.K. acknowledge support from the Deutsche Forschungsgemeinschaft [KA 4370/1-1]. V.N. acknowledges support from the Ermenegildo Zegna Founder’s Scholarship program and the Rosetrees Trust. We also acknowledge use of microscopy facilities within the Harvey Flower Electron Microscopy Suite, Department of Materials, Imperial College London.
Data access statement
Raw data are available upon request from rdm-enquiries@imperial.ac.uk.
Disclosure statement
No funding agency was involved in the study design, data collection, data management, analysis, interpretation or preparation of the manuscript. No conflicts of interest are declared.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018). Eur Heart J. 2018;39(42):3757–3758. [DOI] [PubMed] [Google Scholar]
- [2].George PM, Steinberg GK.. Novel stroke therapeutics: unraveling stroke pathophysiology and its impact on clinical treatments. Neuron. 2015;87(2):297–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hopkins C, Li J, Rae F, et al. Stem cell options for kidney disease. J Pathol. 2009;217(2):265–281. [DOI] [PubMed] [Google Scholar]
- [4].Morigi M. Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J Am Soc Nephrol. 2004;15(7):1794–1804. [DOI] [PubMed] [Google Scholar]
- [5].Choi YH, Kurtz A, Stamm C. Mesenchymal stem cells for cardiac cell therapy. Hum Gene Ther. 2011;22(1):3–17. [DOI] [PubMed] [Google Scholar]
- [6].Gnecchi M, He H, Noiseux N, et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. Faseb J. 2006;20(6):661–669. [DOI] [PubMed] [Google Scholar]
- [7].Mummery CL, Davis RP, Krieger JE. Challenges in using stem cells for cardiac repair. Sci Transl Med. 2010;2(27):27ps17. [DOI] [PubMed] [Google Scholar]
- [8].Humphreys BD, Bonventre JV. Mesenchymal stem cells in acute kidney injury. Annu Rev Med. 2008;59:311–325. [DOI] [PubMed] [Google Scholar]
- [9].Wollert KC, Drexler H. Cell therapy for the treatment of coronary heart disease: a critical appraisal. Nat Rev Cardiol. 2010;7(4):204–215. [DOI] [PubMed] [Google Scholar]
- [10].Iso Y, Spees JL, Serrano C, et al. Multipotent human stromal cells improve cardiac function after myocardial infarction in mice without long-term engraftment. Biochem Biophys Res Commun. 2007;354(3):700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gnecchi M, He H, Liang OD, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005;11(4):367–368. [DOI] [PubMed] [Google Scholar]
- [12].Uemura R, Xu M, Ahmad N, et al. Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Circ Res. 2006;98(11):1414–1421. [DOI] [PubMed] [Google Scholar]
- [13].Xu M, Uemura R, Dai Y, et al. In vitro and in vivo effects of bone marrow stem cells on cardiac structure and function. J Mol Cell Cardiol. 2007;42(2):441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Korf-Klingebiel M, Kempf T, Sauer T, et al. Bone marrow cells are a rich source of growth factors and cytokines: implications for cell therapy trials after myocardial infarction. Eur Heart J. 2008;29(23):2851–2858. [DOI] [PubMed] [Google Scholar]
- [15].Thiagarajan H, Thiyagamoorthy U, Shanmugham I, et al. Angiogenic growth factors in myocardial infarction: a critical appraisal. Heart Fail Rev. 2017;22(6):665–683. [DOI] [PubMed] [Google Scholar]
- [16].Yanagisawa-Miwa A, Uchida Y, Nakamura F, et al. Salvage of infarcted myocardium by angiogenic action of basic fibroblast growth factor. Science. 1992;257(5075):1401–1403. [DOI] [PubMed] [Google Scholar]
- [17].Togel F, Weiss K, Yang Y, et al. Vasculotropic, paracrine actions of infused mesenchymal stem cells are important to the recovery from acute kidney injury. Am J Physiol Renal Physiol. 2007;292(5):F1626–35. [DOI] [PubMed] [Google Scholar]
- [18].Yuan L, Wu M-J, Sun H-Y, et al. VEGF-modified human embryonic mesenchymal stem cell implantation enhances protection against cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol. 2011;300(1):F207–18. [DOI] [PubMed] [Google Scholar]
- [19].Honmou O, Onodera R, Sasaki M, et al. Mesenchymal stem cells: therapeutic outlook for stroke. Trends Mol Med. 2012;18(5):292–297. [DOI] [PubMed] [Google Scholar]
- [20].Nomura T, Honmou O, Harada K, et al. I.V. infusion of brain-derived neurotrophic factor gene-modified human mesenchymal stem cells protects against injury in a cerebral ischemia model in adult rat. Neuroscience. 2005;136(1):161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen Q, Long Y, Yuan X, et al. Protective effects of bone marrow stromal cell transplantation in injured rodent brain: synthesis of neurotrophic factors. J Neurosci Res. 2005;80(5):611–619. [DOI] [PubMed] [Google Scholar]
- [22].Onda T, Honmou O, Harada K, et al. Therapeutic benefits by human mesenchymal stem cells (hMSCs) and Ang-1 gene-modified hMSCs after cerebral ischemia. J Cereb Blood Flow Metab. 2008;28(2):329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Miki Y, Nonoguchi N, Ikeda N, et al. Vascular endothelial growth factor gene-transferred bone marrow stromal cells engineered with a herpes simplex virus type 1 vector can improve neurological deficits and reduce infarction volume in rat brain ischemia. Neurosurgery. 2007;61(3):586–94; discussion 594–5. [DOI] [PubMed] [Google Scholar]
- [24].Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].EL Andaloussi S, Mäger I, Breakefield XO. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12(5):347–357. [DOI] [PubMed] [Google Scholar]
- [26].Timmers L, Lim SK, Arslan F, et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 2007;1(2):129–137. [DOI] [PubMed] [Google Scholar]
- [27].Lai RC, Arslan F, Lee MM, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4(3):214–222. [DOI] [PubMed] [Google Scholar]
- [28].Bian X, Ma K, Zhang C, et al. Therapeutic angiogenesis using stem cell-derived extracellular vesicles: an emerging approach for treatment of ischemic diseases. Stem Cell Res Ther. 2019;10(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yao Y, Chen R, Wang G, et al. Exosomes derived from mesenchymal stem cells reverse EMT via TGF-β1/Smad pathway and promote repair of damaged endometrium. Stem Cell Res Ther. 2019;10(1):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McBride JD, Rodriguez-Menocal L, Guzman W, et al. Bone marrow mesenchymal stem cell-derived CD63(+) exosomes transport Wnt3a exteriorly and enhance dermal fibroblast proliferation, migration, and angiogenesis in vitro. Stem Cells Dev. 2017;26(19):1384–1398. [DOI] [PubMed] [Google Scholar]
- [31].Bian S, Zhang L, Duan L, et al. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J Mol Med (Berl). 2014;92(4):387–397. [DOI] [PubMed] [Google Scholar]
- [32].Feng Y, Huang W, Wani M, et al. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS One. 2014;9(2):e88685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yu B, Kim HW, Gong M, et al. Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int J Cardiol. 2015;182:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao Y, Sun X, Cao W, et al. Exosomes derived from human umbilical cord mesenchymal stem cells relieve acute myocardial ischemic injury. Stem Cells Int. 2015;2015:761643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bruno S, Grange C, Deregibus MC, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20(5):1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bruno S, Grange C, Collino F, et al. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS One. 2012;7(3):e33115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gatti S, Bruno S, Deregibus MC, et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant. 2011;26(5):1474–1483. [DOI] [PubMed] [Google Scholar]
- [38].Xin H, Li Y, Buller B, et al. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells. 2012;30(7):1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xin H, Li Y, Cui Y, et al. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J Cereb Blood Flow Metab. 2013;33(11):1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Witwer KW, Van Balkom BW, Bruno S, et al. Defining mesenchymal stromal cell (MSC)-derived small extracellular vesicles for therapeutic applications. J Extracell Vesicles. 2019;8(1):1609206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Auber M, Fröhlich D, Drechsel O, et al. Serum-free media supplements carry miRNAs that co-purify with extracellular vesicles. J Extracell Vesicles. 2019;8(1):1656042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Webber J, Clayton A. How pure are your vesicles? J Extracell Vesicles. 2013;2(1):19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Witwer KW, Buzás EI, Bemis LT, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2:20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Van Deun J, Mestdagh P, Sormunen R, et al. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J Extracell Vesicles. 2014;3 DOI: 10.3402/jev.v3.24384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gamez-Valero A, Monguió-Tortajada M, Carreras-Planella L, et al. Size-exclusion chromatography-based isolation minimally alters extracellular vesicles’ characteristics compared to precipitating agents. Sci Rep. 2016;6:33641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Popović, M and de Marco, A. Canonical and selective approaches in exosome purification and their implications for diagnostic accuracy. Translational Cancer Research. 2017;7:S209–S225. [Google Scholar]
- [47].Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Carpentier G, Contribution: angiogenesis Analyzer. ImageJ News, 2012.
- [49].Baecker V. ImageJ macro tool sets for biological image analysis. ImageJ User and Developer Conference 2012, Luxembourg; 2012. [Google Scholar]
- [50].Van Deun J, Mestdagh P, Agostinis P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14(3):228. [DOI] [PubMed] [Google Scholar]
- [51].Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161(2):851–858. [DOI] [PubMed] [Google Scholar]
- [52].Reis LA, Borges FT, Simoes MJ, et al. Bone marrow-derived mesenchymal stem cells repaired but did not prevent gentamicin-induced acute kidney injury through paracrine effects in rats. PLoS One. 2012;7(9):e44092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhou Y, Xu H, Xu W, et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res Ther. 2013;4(2):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rager TM, Olson JK, Zhou Y, et al. Exosomes secreted from bone marrow-derived mesenchymal stem cells protect the intestines from experimental necrotizing enterocolitis. J Pediatr Surg. 2016;51(6):942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang B, Wang M, Gong A, et al. HucMSC-exosome mediated-Wnt4 signaling is required for cutaneous wound healing. Stem Cells. 2015;33(7):2158–2168. [DOI] [PubMed] [Google Scholar]
- [56].Nakamura Y, Miyaki S, Ishitobi H, et al. Mesenchymal-stem-cell-derived exosomes accelerate skeletal muscle regeneration. FEBS Lett. 2015;589(11):1257–1265. [DOI] [PubMed] [Google Scholar]
- [57].Hu L, Wang J, Zhou X, et al. Exosomes derived from human adipose mensenchymal stem cells accelerates cutaneous wound healing via optimizing the characteristics of fibroblasts. Sci Rep. 2016;6(1):32993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kowal EJK, Ter-Ovanesyan D, Regev A, et al. Extracellular vesicle isolation and analysis by Western blotting. Methods Mol Biol. 2017;1660:143–152. [DOI] [PubMed] [Google Scholar]
- [59].Ageberg M, Lindmark A. Characterisation of the biosynthesis and processing of the neutrophil granule membrane protein CD63 in myeloid cells. Clin Lab Haematol. 2003;25(5):297–306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.