

ABSTRACT
Tissue-resident stem cell senescence leads to stem cell exhaustion, which is a major cause of physiological and pathological ageing. Stem cell-derived extracellular vesicles (SC-EVs) have been reported in preclinical studies to possess therapeutic potential for diverse diseases. However, whether SC-EVs can rejuvenate senescent tissue stem cells to prevent age-related disorders still remains unknown. Here, we show that chronic application of human embryonic stem cell-derived small extracellular vesicles (hESC-sEVs) rescues the function of senescent bone marrow mesenchymal stem cells (BM-MSCs) and prevents age-related bone loss in ageing mice. Transcriptome analysis revealed that hESC-sEVs treatment upregulated the expression of genes involved in antiaging, stem cell proliferation and osteogenic differentiation in BM-MSCs. Furthermore, liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis identified 4122 proteins encapsulated in hESC-sEVs. Bioinformatics analysis predicted that the protein components in the hESCs-sEVs function in a synergistic way to induce the activation of several canonical signalling pathways, including Wnt, Sirtuin, AMPK, PTEN signalling, which results in the upregulation of antiaging genes in BM-MSCs and then the recovery of senescent BM-MSCs function. Collectively, our findings reveal the effect of hESC-sEVs in reversing BM-MSCs senescence and age-related osteogenic dysfunction, thereby preventing age-related bone loss. Because hESC-sEVs could alleviate senescence of tissue-resident stem cells, they might be promising therapeutic candidates for age-related diseases.
KEYWORDS: Extracellular vesicle, embryonic stem cells, cellular senescence, bone loss, bone marrow MSCs
Introduction
Ageing is an inevitable biological process characterized by the progressive loss of physiological function, leading to a variety of age-associated disorders [1]. In mammals, senescence of the tissue-resident stem cells is one of the primary risk factors for organismal ageing [1,2]. Senescent tissue-resident stem cells exhibit properties of reduced self-renewal and multilineage differentiation, which leads to a decline in tissue repair and regeneration [2,3]. Bone marrow-derived mesenchymal stem cells (BM-MSCs), the common progenitors from which osteoblasts arise, play a critical role in maintaining the balance of bone metabolism [4]. Recently, a study conducted by Zhou et.al. reported that BM-MSCs in aged individuals exhibited decreased proliferation and osteogenic differentiation potential, as well as a variety of senescent phenotypes, such as the expression of senescence-associated β-galactosidase (SA-β-gal), the cell cycle inhibitors P16 and P21, and the senescence-associated secretory phenotype (SASP) [5]. Similarly, BM-MSCs extracted from patients with osteoporosis also acquired several abnormalities indicative of ageing, including reduced proliferation and mineralization capacity [6]. Thus, BM-MSC senescence is regarded as a major driver in the pathogenesis of age-related bone loss [7,8]. Accordingly, several studies have reported that transplantation of young BM-MSCs into aged osteoporotic mice significantly ameliorates BM-MSCs senescence, slows down the reduction in bone mass, and increases the lifespan in aged mice [9,10]. Therefore, therapeutic strategies aimed at rejuvenating senescent BM-MSCs to restore their function hold great potential in preventing age-related bone loss.
Extracellular vesicles (EVs), such as microvesicles and exosome, are nanosized vesicles that are secreted by most cells [11,12]. EVs contain proteins, lipids, and various nucleic acid species from the source cell [13,14]. EVs can mediate intercellular communication and modulate the cellular behaviour of recipient cells via the transfer of their encapsulated bioactive components, and thus are increasingly being explored as potential therapeutic agents [14]. Recently, adult stem cell-derived EVs have been reported to achieve similar effects as those of their parental cells in various disease models, such as myocardial infarction and reperfusion injury [15], liver and kidney injury [16–18], hind limb ischaemia [19], inflammatory disease [20], and degenerative diseases [21–23]. In addition to adult stem cells, embryonic stem cells (ESCs) have also been shown to be a promising source of therapeutic EVs due to their unique ability to proliferate indefinitely and pluripotent capacity [24]. In addition, ESC-derived EVs have no risk of tumorigenesis and a low possibility of immune rejection [25]. Moreover, mounting evidence has indicated that ESC-derived EVs possess pro-regenerative and antiaging effects. For instance, intramyocardial injection of exosomes from mouse ESCs enhances the healing of infarcted hearts by increasing cardiomyocyte survival and neovascularization [26]. Our previous study demonstrated that local application of human ESC-derived small EVs (hESC-sEVs) accelerated the wound healing process by alleviating endothelial senescence [27]. However, the effect of ESC-sEVs in rejuvenating senescent tissue-resident stem cells to prevent age-related disorders still remains unknown.
In this study, we investigated whether hESC-sEVs could improve the senescent phenotypes of BM-MSCs in an age-induced bone loss animal model, and explored the underlying mechanisms. We show that hESC-sEVs rejuvenate senescent BM-MSCs both in vitro and in vivo, and thereby prevent age-related bone loss. By high-throughput transcriptome analysis, we found that hESC-sEVs treatment up-regulates the expression of antiaging-related genes and those that regulate stem cell proliferation and osteogenic differentiation. Furthermore, 4122 proteins were identified in hESC-sEVs by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). Bioinformatics analysis predicted that multiple proteins function synergistically to activate several canonical pathways, including Wnt, Sirtuin, AMPK, and PTEN signalling, that are involved in ameliorating BM-MSCs senescence and promoting osteogenic differentiation. These data show for the first time that hESC-sEVs are effective in counteracting BM-MSCs ageing and thereby prevent age-related bone loss.
Materials and methods
Cell culture and characterization
The hESC line H9 was kindly provided by the Stem Cell Bank, Chinese Academy of Science (Shanghai, China). The cells were expanded under serum- and feeder-free conditions using ncEpic medium (Nuwacell Biotechnologies Co., Ltd, China, Cat#RP01001), and cultured on vitronectin (VTN; Nuwacell Biotechnologies Co., Ltd, China, Cat#RP01002) coated plate in a humidified incubator at 37°C with 5% CO2. The culture medium was changed daily. Upon reaching ~85% confluency, the cells were passaged as clumps using Dissociation Buffer (Nuwacell Biotechnologies Co., Ltd, China, China; Cat#RP01007), and re-plated in ncEpic medium on VTN coated plates. Pluripotency-related markers (NANOG, OCT4, SSEA4, TRA-1-60 and TRA-1-81) were detected by immunofluorescence (IF) staining. The antibodies used for IF staining were listed as follows: NANOG (1:500; 4903s, Cell Signaling Technology), OCT4 (1:500; 2840s, Cell Signaling Technology), SSEA4 (1:1000; 4755, Cell Signaling Technology), TRA-1-60 (1:500; 4746, Cell Signaling Technology), TRA-1-81 (1:500; 4745, Cell Signaling Technology). Cells were tested for mycoplasma and found to be negative.
Isolation of hESC-sEVs
hESC-sEVs were isolated by serial centrifugation with ultracentrifugation as previously described [28,29]. Conditioned medium (CM) was collected during the subculture process, typically 24 h after changing the medium of 2-day-old hESC cultures. All centrifugation steps were performed at 4°C. Firstly, 500 mL CM was collected and centrifuged at 300 × g for 5 min to pellet and remove dead cells. Next, the supernatant was spun at 2,000 × g for 20 min to remove debris and apoptotic bodies. Then, the supernatant was centrifuged at 10,000 × g for 30 min to pellet large EVs (IEVs). Afterwards, the media supernatant was passed through a 0.22 μm pore PES filter (Millipore, Cat# SCGPU05RE) to further remove any remaining IEVs. This supernatant was next subjected to ultracentrifugation at 100,000 × g for 114 min in an SW 32 Ti Rotor Swinging Bucket rotor (k factor of 256.8, 28536 rpm, Beckman Coulter, Fullerton, CA) to pellet hESC-sEVs. The hESC-sEVs pellet was re-suspended in a large volume of PBS followed by ultracentrifugation at 100,000 × g for 114 min in the SW 32 Ti Rotor Swinging Bucket rotor to wash the sample. After PBS washing, the sEVs-enriched fraction pellet was re-suspended in 200 μL PBS and stored at −80°C. We used this type of small EVs preparation in all studies unless indicated. We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV190059) [30]. The EV-metric score is 56%.
Transmission electron microscopy (TEM)
A total of 10 μL hESC-sEVs-enriched solution were placed on a formvar-carbon-coated grid (400 meshes) and left to dry at room temperature for 20 min. After washing with PBS, the sEVs enriched fraction was fixed in 1% glutaraldehyde for 5 min, washed in water and stained with saturated aqueous uranyl oxalate for 5 min. The excess liquid was blotted on a whatman paper. The grid was then dried at room temperature for 10 min and imaged in Transmission Electron Microscope (TF20, FEI, USA).
Size distribution and particle concentration
The size distribution and particle concentration of hESC-sEVs were measured using qNano platform (iZON® Science, UK) as described previously [31]. In brief, the NP100 nanopores of the measuring system are calibrated using particles of known size (CPC100 standard solutions, iZON® Science), then washed 3 times with PBS. The sEVs were diluted with 1000-fold PBS and then added to the nanopores for measurement. Particle size measurement and data analysis were performed with a particle analyser (qNano platform, iZON® Science) and Control Suite software v2.2 (iZON® Science).
Protein concentration
The protein concentration of hESC-sEVs enriched fraction was quantified by Pierce BCA Protein Assay Kit (Thermo Scientific, Cat#23225) according to the product manual. A total of 10 μL samples were loaded into each well of a 96-well plate and 200 μL of the WR solution was added. The plate was incubated at 37°C for 30 min and the absorbance was detected at 562 nm. A standard curve was used to determine the protein concentration of each sEVs enriched fraction sample.
Western blot analysis
Cells or hESC-sEVs were lysed with RIPA buffer containing a complete protease inhibitor tablet (Roche). Lysates were cleared by centrifugation at 12,000 g for 20 min. The supernatant fractions were used for western blot analysis. Protein extracts were resolved by 10% SDS-PAGE (10 μg protein/lane) and probed with the indicated antibodies. The antibodies against the following proteins were used for western blot analysis: CD63 (1:1000; ab134045, Abcam), CD9 (1: 2000; ab92726, Abcam), TSG101 (1: 1000; sc-7964, Santa cruz), GM130 (1: 1000; ab52649, Abcam), PRKAA1 (1:1000; 5831 T, Cell Signaling Technology), CTNNB1(1:1000; 8480 T, Cell Signaling Technology), GSK3B (1:1000; 12,456 T, Cell Signaling Technology), STK11(1:1000; ab199970, Abcam), LRP1(1: 20,000; ab92544, Abcam), NAMPT (1:2000; AG-20A-0034, Adipogen), AKT1 (1:1000; 9272, Cell Signaling Technology), TGFBR1 (1:1000; ab31013, Abcam), POU5F1 (1:1000; 2750s, Cell Signaling Technology), and SLC2A1 (1:1000; ab652, Abcam). Anti-rabbit IgG or anti-mouse IgG, HRP-linked antibody (1: 2,000; Cell Signaling Technology) was used as secondary antibody.
Animal experiments
Animal care and experimental procedures were approved by the Animal Research Committee of the Shanghai Jiao Tong University Affiliated Sixth People’s Hospital (approval code: DWSY2018-118), and were conformed to the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (NIH publication, 8th edition, 2011). Male senescence-accelerated mouse prone eight (SAMP8) mice were purchased from the First Teaching Hospital of Tianjin University of Traditional Chinese Medicine (Tianjin, China). All mice were housed under pathogen-free conditions and provided a standard diet and water. At the end of the experiment, the mice were euthanized with an overdose of pentobarbital sodium (150 mg/kg, i.p). For the in vivo study, 6-month-old male SAMP8 mice was gavage treated every other day for 6 months with 100 μl vehicle control (PBS, n = 8 mice) or sEVs (1 × 1010 particles per mouse, dissolved in PBS, n = 8 mice). Mice were sacrificed for further analysis after 6 months of treatment.
Labelling of hESC-sEVs
hESC-sEVs were obtained as described in the “Isolation of hESC-sEVs” section and were labelled with the DiR (Thermo Scientific, Cat#D12731) according to the manufacture’s protocol and as described previously with modifications in the removal of unbound dyes [32]. In brief, the labelled sEVs were loaded at the bottom of an iodixanol cushion (0%, 20%, 30% and 50%) and centrifuged at 100,000 × g for 114 min (SW 32-Ti Rotor) to separate them from the free unbound dye. The DiR-labelled sEVs were collected from the interphase between 20% and 30%, then washed in PBS and centrifuged at 100,000 × g for 114 min in the SW 32 Ti Rotor Swinging Bucket rotor.
Analysis of DiR-sEVs distribution
Purified DiR-labelled hESC-sEVs (1 × 1010 particles/ml,100 μl) were applied into SAMP8 mice, followed by euthanasia and ex vivo imaging of dissected organs (gastrointestine (GI) tract, liver, spleen, kidney, heart, lung, brain, femur and tibia) at 6, 12, 24, and 48 hours after application. Bio-distribution of DiR-labelled sEVs was examined by using IVIS Spectrum (Perkin Elmer). The harvested organs were imaged for 1–2 seconds (excitation 710 nm, emission 760 nm). The data were analysed with the IVIS software (Living Image Software for IVIS).
Micro-computed tomography (micro-CT) analysis
Micro-CT images were obtained to evaluate trabecular bone structure of the femur. The dissected, formalin-fixed samples were scanned with the femur axis perpendicular to the plane at 9 μm resolution. The 3D representation of the regenerated bone was reconstructed with NRecon software (Version 1.5.1.4, Skyscan). The region of interest (ROI) was defined as the cancellous bone of the distal femur. The values of trabecular bone volume fraction (BV/TV; %), trabecular number (Tb. N; 1/mm), trabecular thickness (Tb. Th; mm), trabecular separation (Tb. Sp; mm) were selected to evaluate the bone structure. For cortical bone, we selected the region of mid-diaphysis on the 10% femoral length to analyse the cortical thickness (mm), cortical area (mm2), and bone total area (mm2). For bone mineral density (BMD; mg/cm3) measurement, we selected bone area under the first mandibular molar to analyse cancellous bone. All imaging was performed and analysed in a blinded fashion.
Isolation, culture and characterization of mouse BM-MSCs
The proximal and distal metaphysis of the femurs and tibias was cut and the bone marrow was flushed from the diaphysis with 1% FBS (Gibco, USA) in PBS. Single-cell suspension of all nuclear cells was seeded at a density of 2 × 105 cells per square centimetre on 10-cm culture dish (Corning, NY, USA) at 37°C in 5% CO2. Non-adherent cells were removed after 24 hours, and the attached cells were maintained BM-MSCs. Surface antigens of BM-MSCs were analysed using flow cytometry. The monoclonal antibodies used (BD Biosciences) were CD44-FITC, CD29-PE, Sca-1-PE, CD34-APC, CD45-FITC and CD11b-PE. The cells were incubated with 1% bovine serum albumin (BSA; Gibco) for 30 minutes to block non-specific antigens. Following two washes in PBS, the cells were resuspended and analysed using the Guava easyCyte™ system (Millipore, Billerica, MA, USA). Non-Specific fluorescence was determined with isotype-matched mouse monoclonal antibodies (BD Biosciences).
Senescence-associated β-galactosidase (SA-β-gal) assay
SA-β-gal staining of BM-MSCs was performed using an SA-β-gal staining kit (Beyotime Biotechnology, China; Cat#C0602) according to the manufacturer’s protocol. In brief, cells were fixed in 4% paraformaldehyde for 5 min. After washing with PBS three times, samples were incubated in SA-β-gal solution at 37°C for 16–18 h. Ice-cold PBS was then used to stop the enzymatic reaction. In blinded analyses, for each sample, 10 images were taken from random fields using microscope. The ratio of positive cells was determined by counting the blue cells and dividing by the total number of observed cells.
Colony-forming unit (CFU) assay
BM-MSCs were suspended and seeded at a density of 200 cells into 6-well plates. The cells were cultured in complete medium for 10 days. Cell colonies were washed with PBS, fixed with 4% PFA for 30 min at RT, and were stained with 0.5% crystal violet solution for 30 min at RT on a rocking shaker. Afterwards, excess stain was washed off and plates were air-dried. Colonies were counted by investigators blinded to the experimental conditions.
Osteogenic differentiation assay
For ex vivo experiment, BM-MSCs were isolated, cultured to 80% confluence, and then incubated with osteogenic differentiation medium (Cyagen Biosciences, USA; Cat# MUBMX-90021). Half of the induction medium was replaced every other day. For in vitro experiment, BM-MSCs were isolated from 6-month-old SAMP8 mice, cultured to 80% confluence, and then incubated with osteogenic differentiation medium containing 1 × 1010 particles/mL hESC-sEVs or vehicle (PBS). Half of the induction medium was replaced by an equivalent volume of fresh osteogenic medium supplemented with 1 × 1010 particles/mL hESC-sEVs or with PBS every other day. The expression levels of osteogenic-related genes were assayed by RT-qPCR on day 7 after the osteogenic induction. To assess osteogenic differentiation, the cells were stained with 1% Alizarin Red (Cyagen Biosciences) on day 21 after the osteogenic differentiation. Calcified nodules were eluted with 10% cetylpyridinium chloride (CPC), and the absorbance was calculated at 562 nm.
Alkaline phosphatase (ALP) staining
For ALP staining, BM-MSCs were fixed with 4% paraformaldehyde after osteogenic induction for 10 days in vitro and incubated with a solution of 0.25% naphthalol AS-BI phosphate and 0.75% Fast Blue BB dissolved in 0.1 M Tris buffer. ALP activity was quantified by using a commercial kit according to the manufacturer’s protocol (Cell Biolab, CA, USA).
Immunofluorescence staining
BM-MSCs were fixed with 4% paraformaldehyde, permeabilized in PBS containing 0.5% Triton X-100 for 15 min at room temperature and pre-incubated with 5% BSA for 1 h at room temperature to block non-specific staining. Fixed cells were then incubated overnight at 4°C with primary antibodies against Ki-67 (1:200; 9129s, Cell Signaling Technology), p21 (1:500; ab188224, Abcam), γ-H2AX (1:400; 9718s, Cell Signaling Technology), followed by secondary antibody conjugated to Alexa Fluor 594 (1:1000; Thermo Scientific). Nuclei were labelled with DAPI (1:1000; D9542, Sigma) at room temperature for 5 min. Fluorescence images were captured under fluorescence microscope (Leica Microsystems). Quantification of the number of positively stained cells was performed by using the ImageJ software.
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Targeted gene expression analyses were performed by RT-qPCR. Briefly, total RNA was isolated using QIAzol Lysis Reagent and RNeasy Mini Columns (Qiagen, Valencia, CA). RNA quantity and purity were confirmed with a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE). Reverse transcriptase was performed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, CA; Cat#K1622). PCR reactions were run using the ABI Prism 7900HT Real Time System (Applied Biosystems, Carlsbad, CA) with SYBR green (Roche Applied Science; Cat# 04913850001). The primer sequences used in this study are listed in Supplementary Table 1.
Histological analysis
The samples were fixed in 4% formaldehyde, decalcified in 10% EDTA solution and then embedded in paraffin. The embedded samples were cut into 5 μm sections with a microtome (Leica Microsystems, Germany). Sections were stained with haematoxylin and eosin (H&E) and visualized under a light microscope (Leica Microsystems, Germany).
RNA-sequencing analysis
RNA-sequencing (RNA-seq) analysis was performed by Shanghai Biotechnology Corporation (Shanghai, China). In brief, total RNA was isolated using RNeasy mini kit (Qiagen, Germany). Paired-end libraries were synthesized by using the TruSeq™ RNA Sample Preparation Kit (Illumina, USA) following TruSeq™ RNA Sample Preparation Guide. The poly-A containing mRNA molecules were purified using Poly-T oligo-attached magnetic beads. Following purification, the mRNA is fragmented into small pieces using divalent cations under 94°C for 8 min. The cleaved RNA fragments are copied into first-strand cDNA using reverse transcriptase and random primers. This is followed by second-strand cDNA synthesis using DNA polymerase I and RNase H. These cDNA fragments then go through an end repair process, the addition of a single “A” base, and then ligation of the adapters. The products are then purified and enriched with PCR to create the final cDNA library. Purified libraries were quantified by Qubit® 2.0 Fluorometer (Thermo Scientific, USA) and validated by Agilent 2100 bioanalyzer (Agilent Technologies, USA) to confirm the insert size and calculate the mole concentration. Cluster was generated by cBot with the library diluted to 10 pM and then were sequenced on the Illumina HiSeq (Illumina, USA).
Proteomic analysis
The proteomic analysis was performed by the Shanghai Applied Protein Technology Company (Shanghai, China).
Proteins isolation and identification
The isolated EVs (400 μl, obtained from 240 ml of conditioned media) were digested in SDT lysis buffer (4% SDS, 100 mM Tris-HCl, 1 mM DTT, pH7.6). The lysate was sonicated for 15 min and then heated to 95°C for 15 min. After centrifuged at 14000 × g for 40 min, the supernatant was quantified with the BCA Protein Assay Kit (Bio-Rad, USA). 20 μg of proteins for each sample were mixed with 5× loading buffer, respectively, and boiled for 5 min. The proteins were separated on 12.5% SDS-PAGE gel (constant current 14 mA, 90 min). Protein bands were visualized by Coomassie Blue R-250 staining.
Filter-Aided Sample Preparation (FASP)
100 μg of proteins for each sample were incorporated into 30 μl SDT buffer (4% SDS, 100 mM DTT, 150 mM Tris-HCl pH 8.0). The detergent, DTT and other low-molecular-weight components were removed using UA buffer (8 M Urea, 150 mM Tris-HCl pH 8.0) by repeated ultrafiltration (Microcon units, 10 kD). Then, 100 μl iodoacetamide (100 mM IAA in UA buffer) was added to block reduced cysteine residues and the samples were incubated for 30 min in darkness. The filters were washed with 100 μl UA buffer three times and then 100 μl 25 mM NH4HCO3 buffer twice. Finally, the protein suspensions were digested with 4 μg trypsin (Promega) in 40 μl 25 mM NH4HCO3 buffer overnight at 37°C, and the resulting peptides were collected as a filtrate. The peptides of each sample were desalted on C18 Cartridges (EmporeTM SPE Cartridges C18 (standard density), bed I.D. 7 mm, volume 3 ml, Sigma), concentrated by vacuum centrifugation and reconstituted in 40 μl of 0.1% (v/v) formic acid. The peptide content was estimated by UV light spectral density at 280 nm using an extinction coefficient of 1.1 of 0.1% (g/l) solution.
Peptide fractionation with high pH reversed-phased
To increase the depth of protein identification, peptide fractionation was performed using Pierce High pH Reversed-Phased Peptide Fractionation kit (Thermo Scientific, 84868). Digest samples were fractionated into five fractions (F1-F5) by an increasing acetonitrile step-gradient elution according to the kit instructions.
MS data acquisition
The MS analyses were performed using an Easy-nLC 1000 nano-UPLC chromatography (Thermo Scientific) interfaced with a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific) with a nano-electrospray ion source. The sample was loaded onto an Acclaim PepMap100 C18 reverse-phase trap column (100 μm×2 cm) with nanoViper fittings (Thermo Scientific) connected to the C18-reversed-phase analytical column (Thermo Scientific Easy Column, 10 cm long, 75 μm inner diameter, 3 μm resin) in buffer A (0.1% Formic acid) and separated with a linear gradient of buffer B (84% acetonitrile and 0.1% formic acid) at a flow rate of 300 nL/min controlled by IntelliFlow technology. The mass spectrometer was operated in the positive ion mode. MS data were acquired using a data-dependent top10 method that dynamically selected the most abundant precursor ions from the survey scan (300–1800 m/z) for HCD fragmentation. The automatic gain control (AGC) target was set to 1e6, and the maximum inject time was set to 50 ms. The dynamic exclusion duration was 60.0 s. Survey scans were acquired at a resolution of 70,000 at 200 m/z, the resolution for HCD spectra was set to 17,500 at 200 m/z, and the isolation window was 2 m/z. The normalized collision energy was 30 eV, and the underfill ratio was defined as 0.1%. The instrument was run with the peptide recognition mode enabled.
Data processing
All data were analysed using MaxQuant software version 1.5.3.17. Proteins were identified by comparing all the spectra with the human proteome reference database (Swissprot_human_20422_20190522.fasta). Enzymatic digestion was performed using trypsin, and the maximum number of missed cleavages allowed was two. In addition, the oxidation of methionine was selected as a variable modification, while the carbamidomethylation of cysteine was selected as a fixed modification. Razor and unique peptides were used for protein quantification. In the MS mode, an initial mass tolerance of 6 ppm was selected, and the MS/MS tolerance was set to 20 ppm for fragmentation data. The false discovery rate (FDR) was specified as 0.01 for proteins and peptides. Intensity-based absolute quantification (iBAQ) was performed using the default parameters in MaxQuant. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018655.
Bioinformatics analysis
The identified proteins were compared with the results from the EVpedia database of published exosomal proteins. The gene ontology (GO) enrichment analysis was determined using FunRich software (Version 3.1.3) [33]. Canonical pathway analysis was conducted utilizing the Ingenuity Pathway Analysis (IPA, Qiagen). The protein–protein interaction was analysed in the Ingenuity Pathway Knowledge Base (IPKB, Qiagen).
Statistical analysis
All data are presented as mean±s.e.m. Student’s unpaired t-test or one-way ANOVA was used for comparisons between two groups unless otherwise noted. A value of P < 0.05 was considered significant difference.
Results
Isolation and characterization of hESC-sEVs
First, we characterized the human embryonic stem cells (hESCs) used in this study. As shown in Figure 1(a), hESC colonies positively expressed pluripotency-related markers, including OCT4, NANOG, SSEA4, TRA-1-60 and TRA-1-81. sEVs were then isolated from hESC-derived conditioned media and characterized in terms of morphology, particle size and concentration, and surface markers. The purified hESC-sEVs displayed a size around 100 nm and a cup-shaped morphology, as shown by TEM (Figure 1(b)). qNano analysis revealed that the mean diameter was 133.2 ± 11.2 nm, and the mean particle concentration is 2.1 × 1011 ± 1.5 × 1010 particles/mL (Figure 1(c)). Western blotting analysis showed that hESC-sEVs were positive for CD9, CD63 and TSG101, but negative for the Golgi-associated protein GM130 (Figure 1(d)). Additionally, the yield of hESC-sEVs was evaluated in terms of particle concentration and protein concentration. As shown in Figure 1(e-h), the mean particle concentration was 5.45 × 108 ± 7.24 × 107 particles/mL CM (Figure 1(e)), or 861.40 ± 74.61 particles per cell (Figure 1(f)). The mean protein concentration was 1114.04 ± 42.66 ng/mL CM (Figure 1(g)), or 2.07 × 10−6 ± 2.76 × 10−5 ng per particle (Figure 1(h)).
Figure 1.
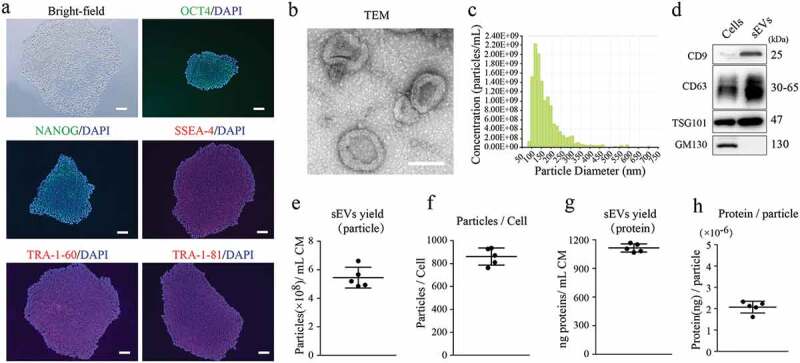
Characterization of hESCs and hESC-sEVs.
(a) Representative bright-field image and OCT4, NANOG, SSEA-4, TRA-1-60, TRA-1-81 immunostaining micrographs of hESCs. Nucleus was stained with DAPI. Scale bar, 50 μm. (b) Representative TEM images of hESC-sEVs. Scale bar, 100 nm. (c) Average particle size distribution and concentration of hESC-sEVs measured by qNano. (d) Representative western blot analysis showing the presence of exosomal markers including CD9, CD63, and TSG101, but negative for GM130 in hESC-sEVs. (e,f) Evaluation of hESC-sEVs yield in term of particle concentration. (g,h) Evaluation of hESC-sEVs yield in term of protein concentration. Each experiment was repeated three to five times independently. Data are represented as mean±s.e.m.
Aged-related bone loss and senescent properties of BM-MSCs in SAMP8 mice
In this study, the senescence-accelerated SAMP8 mouse model was used to evaluate the effect of hESC-sEVs in preventing age-related bone loss. Micro-CT analysis revealed a gradual loss of trabecular bone mass in the femur of SAMP8 mice aged 2 to 12 months (Supplementary Figure 1a). Quantitative analysis showed that the most dramatic trabecular bone loss occurred at 6 to 12 months of age, as indicated by gradually decreased bone mineral density (BMD), bone volume/tissue volume ratio (BV/TV), trabecular number (Tb. N), and trabecular thickness (Tb. Th), and increased trabecular separation (Tb. Sp) (Supplementary Figure 1b). Moreover, quantitative analysis of cortical bone showed no significant differences in SAMP8 mice of different ages (Supplementary Figure 1c, d).
As BM-MSCs represent a promising cellular source to maintain bone homoeostasis, while the senescence of BM-MSCs may lead to bone loss [8]. We then investigated the causative role of BM-MSC senescence in mediating age-related trabecular bone loss in SAMP8 mice. BM-MSCs were negative for CD34, CD11b and CD45 and positive for CD29, CD90, and Sca-1 (Supplementary Figure 2a). We found that the expression of ageing-related markers, including p16, p21, IL-6, IL-1b, and Ccl4, gradually increased in BM-MSCs that were isolated from SAMP8 mice of different ages (Supplementary Figure 2b), while the proliferation capacity and number of colony units gradually decreased (Supplementary Figure 2c, d). Moreover, primary BM-MSCs exhibited significantly reduced osteogenic potential, as evidenced by ALP staining (Supplementary Figure 2e) and Alizarin red staining (Supplementary Figure 2f). RT-qPCR analysis also showed that the expression of the osteogenic genes (Fzd9, Rbpj, Hgf, Fgr, and Dmp1) gradually decreased in BM-MSCs isolated from SAMP8 mice of different ages (Supplementary Figure 2g). These results indicated that BM-MSCs undergo a gradual decline in proliferation and osteogenic differentiation beginning at 6 months of age, and BM-MSC senescence is closely correlated with the progression of bone loss in SAMP8 mice.
hESC-sEVs prevent age-related bone loss in SAMP8 mice
We then explored the role of hESC-sEVs in preventing age-related bone loss. To track the biodistribution of hESC-sEVs in vivo, DiR-labelled hESC-sEVs (1 × 1010 particles/mL, 100 μl) were administered to SAMP8 mice, followed by euthanasia and ex vivo imaging of dissected organs at 6, 12, 24, and 48 hours after administration. The fluorescent signal could be first observed in the femurs and tibias after 6 hours of administration. With extended observation time, the fluorescent signal in the bone gradually increased, and reached a high point at 48 hours after administration. Meanwhile, the high signal was observed in the GI tract, liver, spleen, kidney, heart and lung. Fluorescent signal can not be detected in naïve SAMP8 mice (Supplementary Figure 3).
Next, hESC-sEVs were administrated to 6-month-old male SAMP8 mice for 6 months. The mice were randomized to either the vehicle or hESC-sEVs treatment group. After 6 months of treatment, the mice were sacrificed, and the femurs were harvested for micro-CT and histological analyses (Figure 2(a)). As shown in Figure 2(b), compared with the vehicle-treated mice, the sEVs-treated mice showed a healthier appearance and status. Micro-CT (Figure 2(c)) and quantitative analyses of distal femoral trabecular bone revealed that hESC-sEVs treatment increased BMD (Figure 2(d)), BV/TV (Figure 2(e)), and Tb. N (Figure 2(f)) and decreased Tb. Sp (Figure 2(h)), while Tb. Th (Figure 2(g)) was not significantly changed. Micro-CT analysis of cortical bone (Figure 2(i)) revealed that cortical thickness (Figure 2(j)), cortical area (Figure 2(k)), and total bone area (Figure 2(l)) were not significantly altered after treated with hESC-sEVs. Notably, H&E staining of femurs revealed that the amount of trabecular bones increased in sEVs-treated mice compared with that in vehicle-treated mice (Figure 2(m,n)). These results demonstrated that hESC-sEVs treatment prevents age-related bone loss in vivo.
Figure 2.
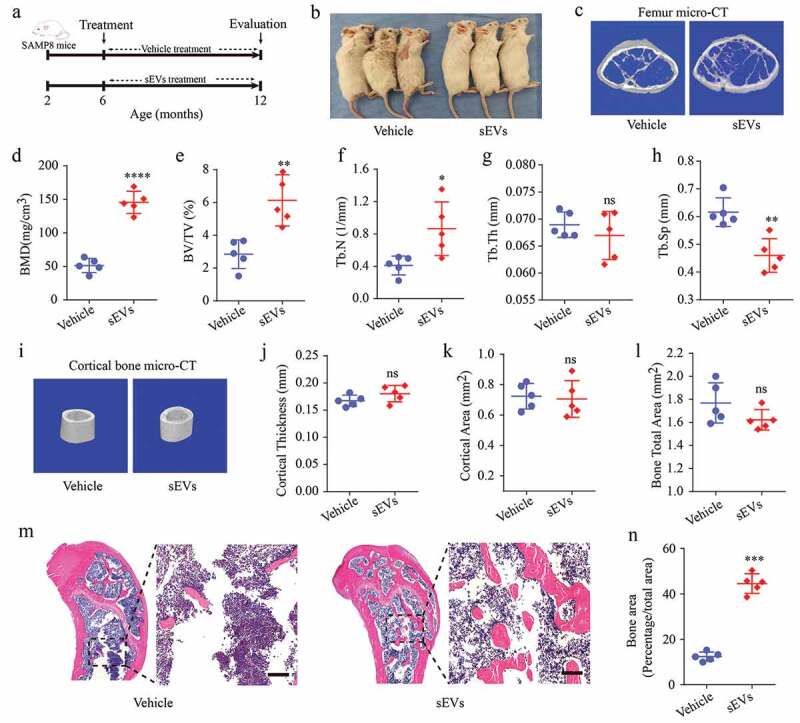
hESC-sEVs treatment prevents age-related bone loss in SAMP8 mice.
(a) Experimental design for testing the effect of hESC-sEVs on preventing age-related bone loss: 6-month-old male SAMP8 mice were randomized to either vehicle or hESC-sEVs treatment for 6 months. (b) Representative images of aged male mice applied with vehicle or hESC-sEVs. The mouse images were taken after 6 months of treatment. (c) Representative micro-CT images of bone microarchitecture at the femur. (d–h) Quantification of micro-CT derived bone mineral density (BMD; mg/cm3) (d), bone volume fraction (BV/TV; %) (e), trabecular number (Tb. N; 1/mm) (f), trabecular thickness (Tb. Th; mm) (g), trabecular separation (Tb. Sp; mm) (h) at the femur. Data from n = 5 mice per group are represented. (i) Representative micro-CT images of cortical bone. (j–l) Quantification of cortical thickness (mm) (j), cortical area (mm2) (k), bone total area (mm2) (l). Data from n = 5 mice per group are represented. (m) H&E staining of distal femoral metaphyseal regions from mice treated with vehicle or hESC-sEVs. Scale bars, 200 μm. (n) Quantification of bone area. Data from n = 5 mice per group are represented. All statistical data are represented as means±s.e.m. P-value is indicated as sEVs group versus vehicle group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant (P > 0.05). All imaging was performed and analysed in a blinded fashion.
hESC-sEVs ameliorate the senescent phenotypes, and promote the proliferation and osteogenic differentiation of BM-MSCs in vivo
It has been recognized that BM-MSC senescence is putatively responsible for osteoporotic disorders [7,8]. We then tested whether the preventative effect of hESC-sEVs on bone loss was due to decreased BM-MSC senescence. Primary BM-MSCs were isolated from vehic
le- and sEVs-treated mice and analysed ex vivo. We found that the number of SA-β-gal-positive BM-MSCs in sEVs-treated mice was significantly reduced (Figure 3(a)). Next, we performed IF staining for ageing markers and found that the protein expression of P21 and γ-H2AX in BM-MSCs was much lower in the sEVs treatment group than in the control group (Figure 3(b,c)). RT-qPCR analysis also showed that hESC-sEVs treatment significantly decreased the mRNA expression of the SASP-related genes (IL-6, IL-1b, Ccl4, Ccl2, and Timp-1) in BM-MSCs (Figure 3(d)).
Figure 3.
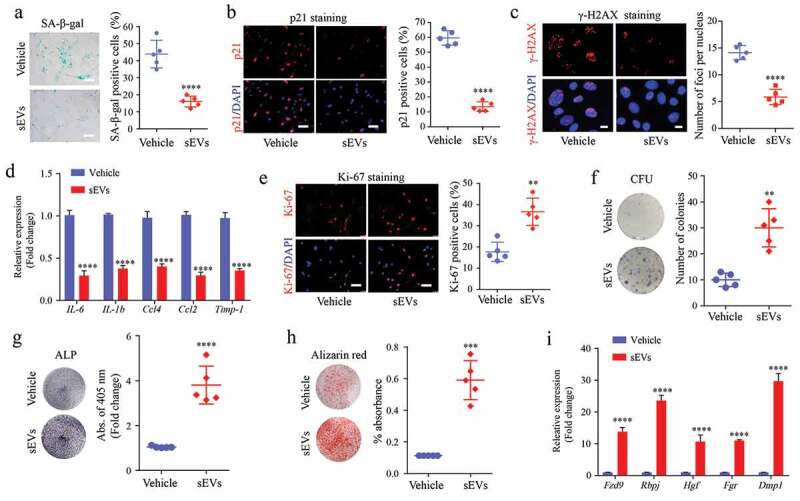
hESC-sEVs ameliorate the senescent phenotypes, and promote the proliferation and osteogenic differentiation of BM-MSCs in vivo.
(a) SA-β-gal staining of BM-MSCs from vehicle- or sEV-treated mice. Left panel, representative pictures of SA-β-gal staining. Right panel, quantification of SA-β-gal positive BM-MSCs. Scale bar, 50 μm. (b) P21 immunostaining of BM-MSCs. Left panel, representative pictures of P21 immunostaining. Right panel, quantification of P21 positive BM-MSCs. Scale bar, 50 μm. (c) γ-H2AX immunostaining of BM-MSCs. Left panel, representative pictures of γ-H2AX immunostaining. Right panel, quantification of foci per nucleus. Scale bar, 10 μm. (d) RT-qPCR analysis of SASP-related gene expression in BM-MSCs. (e) Ki67 immunostaining of BM-MSCs from vehicle- or sEVs-treated mice. Left, representative pictures of Ki67 immunostaining. Right, quantification of Ki67 positive BM-MSCs. Scale bar, 50 μm. (f) Crystal violet staining for the CFU colonies of BM-MSCs from vehicle- or sEVs-treated mice. Left, representative pictures of crystal violet staining. Right, quantification of the number of CFU colonies. (g) ALP staining of BM-MSCs from vehicle- or sEVs-treated mice after osteogenic induction. Left, representative pictures of ALP staining. Right, quantification of ALP activity. (h) Alizarin red staining of BM-MSCs from vehicle- or sEVs-treated mice after osteogenic induction. Left, representative pictures Alizarin red staining. Right, quantification of mineralized absorbance. (i) RT-qPCR analysis of osteogenic gene expression in BM-MSCs. Each experiment was repeated three times independently. Data from n = 5 independent samples per group are represented. P-value is indicated as sEVs group versus vehicle group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Next, we assessed the effect of hESC-sEVs treatment on the proliferation and osteogenic capacity of BM-MSCs. Ki67 staining revealed that the percentage of proliferating BM-MSCs significantly increased in the sEVs-treated group (Figure 3(e)). Colony-forming unit assay showed enhanced self-renewal capacity of BM-MSCs from the hESC-sEVs treatment group (Figure 3(f)). Moreover, BM-MSCs from sEVs-treated mice showed improved osteogenic potential, as evidenced by ALP (Figure 3(g)) and Alizarin red staining (Figure 3(h)). The expression of osteogenic genes (Fzd9, Rbpj, Hgf, Fgr, and Dmp1) in BM-MSCs from sEVs-treated mice was also significantly increased compared to that of vehicle-treated mice (Figure 3(i)). These results suggested that hESC-sEVs are effective in alleviating BM-MSC senescence, and promoting their proliferation and osteogenic differentiation capacity.
hESC-sEVs attenuate BM-MSC senescence in a dose-dependent manner
To further validate the effect of hESC-sEVs in reversing BM-MSC senescence, we isolated BM-MSCs from 6-month-old SAMP8 mice and incubated them with hESC-sEVs for 48 hours at a range of concentrations (1 × 108, 1 × 109, and 1 × 1010 particles/mL) in vitro. The results showed that the number of SA-β-gal-positive cells substantially decreased in the presence of sEVs in a dose-dependent manner (Figure 4(a)). Consistent with these data, IF staining of P21 and γ-H2AX (Figure 4(b,c)), and RT-qPCR analysis (Figure 4(d)) showed that the expression level of senescence-related genes decreased gradually with increasing sEVs concentrations. These results demonstrated that hESC-sEVs treatment significantly alleviated the senescent phenotypes of BM-MSCs in a dose-dependent manner.
Figure 4.
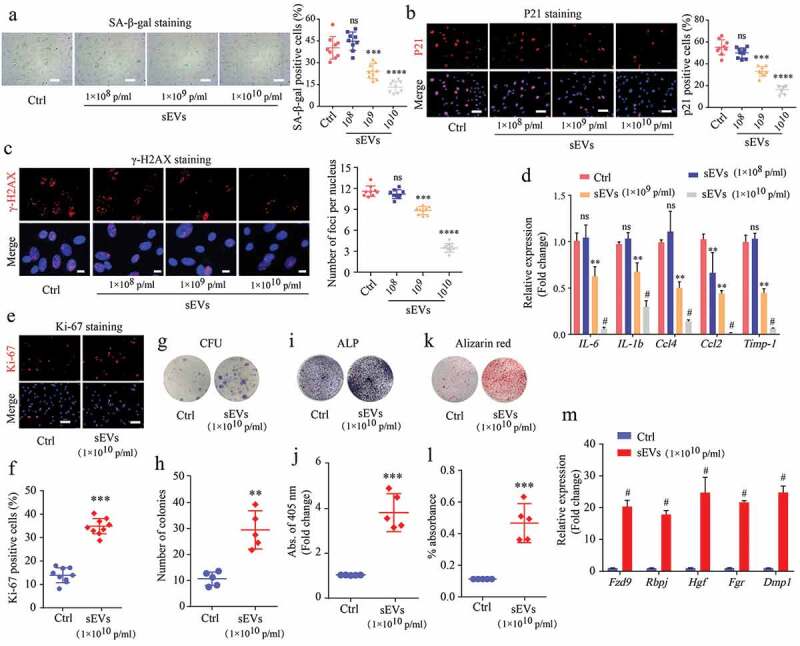
hESC-sEVs protect against senescent phenotypes of BM-MSCs in a dose-dependent manner.
(a) SA-β-gal staining of BM-MSCs treated as indicated. Left panel, representative pictures of SA-β-gal staining. Right panel, quantification of SA-β-gal positive BM-MSCs. Scale bar, 50 μm. (b) P21 immunostaining of BM-MSCs treated as indicated. Left panel, representative pictures of P21 immunostaining. Right panel, quantification of P21 positive BM-MSCs. Scale bar, 50 μm. (c) γ-H2AX immunostaining of BM-MSCs treated as indicated. Left panel, representative pictures of γ-H2AX immunostaining. Right panel, quantification of γ-H2AX positive BM-MSCs. Scale bar, 100 μm. (d) RT-qPCR analysis of age-related gene expression in BM- MSCs. (e,f) Ki67 immunostaining of BM-MSCs treated as indicated. Representative pictures (e) and quantification of Ki67 positive BM-MSCs (f). Scale bar, 50 μm. (g,h) Crystal violet staining for the CFU colonies of BM-MSCs treated as indicated. Representative images (g) and quantification of the number of CFU colonies (h). (i,j) ALP staining of BM-MSCs treated as indicated after osteogenic induction. Representative images (i) and quantification of ALP activity (j). (k,l) Alizarin red staining of BM-MSCs treated as indicated after osteogenic induction. Representative pictures (k) and quantification of mineralized absorbance (l). (m) RT-qPCR analysis of osteogenic gene expression in BM-MSCs. Each experiment was repeated three to five times independently. All statistical data are represented as means±s.e.m. P-value is indicated as sEVs group versus ctrl group. *P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.0001, ns, not significant (P > 0.05).
We then evaluated the effects of hESC-sEVs treatment on the proliferation and osteogenic differentiation of BM-MSCs at a concentration of 1 × 1010 particles/mL. Ki67 staining (Figure 4(e,f)) and colony-forming unit assays (Figure 4(g,h)) showed that the hESC-sEVs treatment promoted BM-MSC proliferation and self-renewal potential. Moreover, hESC-sEVs treatment enhanced the osteogenic potential of BM-MSCs, as assessed by ALP staining and Alizarin red staining (Figure 4(i-l)). The expression of osteogenic genes (Fzd9, Rbpj, Hgf, Fgr, and Dmp1) in BM-MSCs from the sEVs-treated group was also significantly increased compared to that of the control group (Figure 4(m)). Furthermore, the chondrogenic and adipogenic differentiation potential of BM-MSCs was also enhanced after hESC-sEVs treatment (Supplementary Figure 4a-d). These in vitro results demonstrated that hESC-sEVs treatment significantly promotes the proliferation and differentiation of BM-MSCs.
hESC-sEVs modulate the gene expression pattern of BM-MSCs
To elucidate the underlying molecular mechanism by which hESC-sEVs rescue BM-MSCs function, we performed RNA-seq analysis to clarify the changes in gene expression in BM-MSCs. We isolated BM-MSCs from 6-month-old and 12-month-old SAMP8 mice, respectively, and treated them with hESC-sEVs (1 × 1010 particles/mL) or PBS for 48 hours. Meanwhile, BM-MSCs from 2-month-old SAMP8 mice were used as the young control group. We identified 931 up-regulated genes (> 2-fold, p < 0.05) and 582 down-regulated genes (< 0.5-fold, p < 0.05) after sEVs treatment (Figure 5(a)). Next, we performed functional and pathway analyses by IPA. Functional analysis showed that many differentially expressed genes were involved in bone homoeostasis (Figure 5(b)). In addition, pathway analysis revealed that hESC-sEVs exposure significantly activated pathways involved in the regulation of ageing and osteogenesis, including the “STAT3 pathway”, “Wnt/β-catenin signaling”, “MAPK signaling”, and “AMPK signaling”. etc. (Figure 5(c)). Subsequently, we utilized gene set enrichment analysis (GSEA) to mine the RNA-seq data, and the results showed that the hESC-sEVs treatment increased the enrichment score for the module of “negative regulation of cellular aging” (Figure 5(d)), “stem cell proliferation” (Figure 5(e)), and “osteogenic differentiation” (Figure 5(f)) in BM-MSCs. Genes that showed a significant difference in expression (fold change >2) in the GSEA analysis were visualized by heatmap (Figure 5(g,h)). Furthermore, RT-qPCR analysis confirmed that the expression of antiaging genes in BM-MSCs was increased after hESC-sEVs treatment (Figure 5(i)). Taken together, these data clearly revealed that alleviation of senescent phenotypes in BM-MSCs after hESC-sEVs treatment is associated with genetic programmes and pathways that control critical aspects of the antiaging process, stem cell proliferation and osteogenic differentiation.
Figure 5.
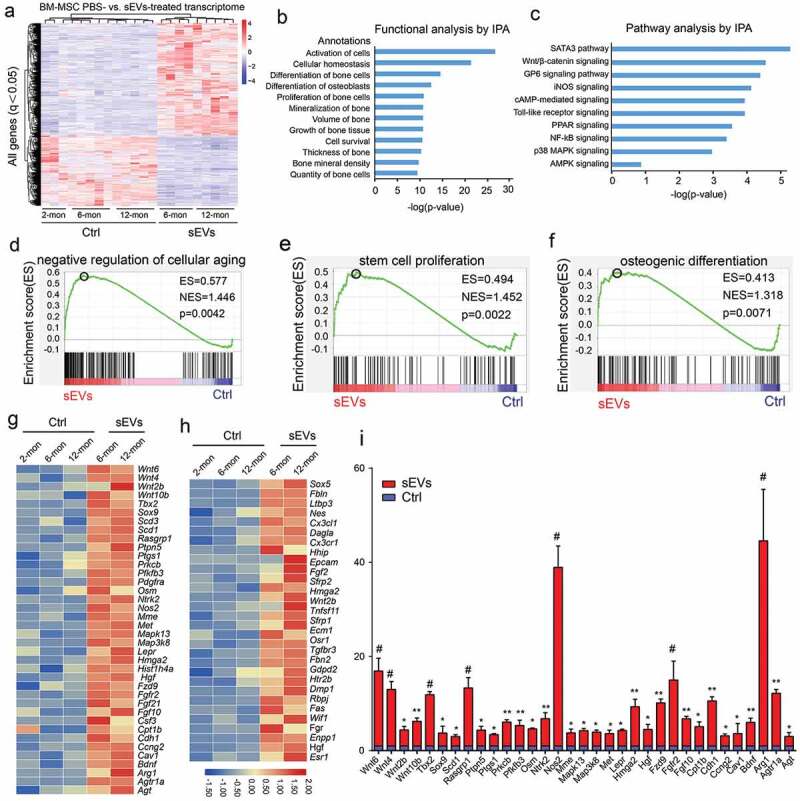
Identification of BM-MSCs transcriptome changes after hESC-sEVs treatment by RNA-seq.
(a). Heatmap representing the hierarchical clustering of significant differential expression genes in different aged BM-MSCs treated with hESC-sEVs. (b). IPA exhibiting the functional classification of differentially expressed genes in BM-MSCs. (c). IPA showing canonical pathways of the differentially expressed genes in BM-MSCs. (d-f) GSEA analysis identifies enrichment score for modules of negative regulation of cellular ageing (d), stem cell proliferation (e), and osteogenic differentiation (f) in BM-MSCs after hESC-sEVs treatment. (g,h) Heatmap representation of differentially expressed genes (fold change >2) in the GSEA analysis associated with negative regulation of cellular ageing (g), stem cell proliferation and osteogenic differentiation (h). (i) RT-qPCR analysis of anti-ageing gene expression in BM-MSCs treated with hESC-sEVs. The experiment was repeated three times independently. P-value is indicated as sEV group versus ctrl group. *P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.0001.
Proteomics analysis of hESC-sEVs
Previous studies reported that EVs can affect the biological behaviours of recipient cells by transferring the encapsulated proteins [34,35]. Therefore, we prepared proteins from hESC-sEVs (Supplementary Figure 5), and then performed LC-MS/MS analysis to identify the protein contents of hESC-sEVs. In total, 4122 proteins were identified from hESC-sEVs. 98.4% of the identified proteins could be aligned to the EV proteome database (Figure 6(a)). Enrichment analysis of GO terms by FunRich software showed that 30.8% of the proteins identified in hESC-sEVs were derived from exosomes (Figure 6(b)). The GO analysis of biological processes revealed an enrichment of proteins involved in “protein metabolism” (12.8%), “metabolism” (12.4%), “energy pathways” (12.1%), “transport” (8.6%), “cell growth and/or maintenance” (8%), and “protein modification” (0.3%) (Figure 6(c)). The GO analysis of molecular functions showed that “RNA binding” (4.3%) was a major category for hESC-sEVs proteins set. In line, “translation on regulator activity” (1.6%), “structural constituent of ribosome” (2.1%), “transporter activity” (5.5%), “GTPase activity” (2.7%) and “ubiquitin-specific protease activity” (3.6%) were the top enriched for the hESC-sEVs proteins (Figure 6(d)).
Figure 6.
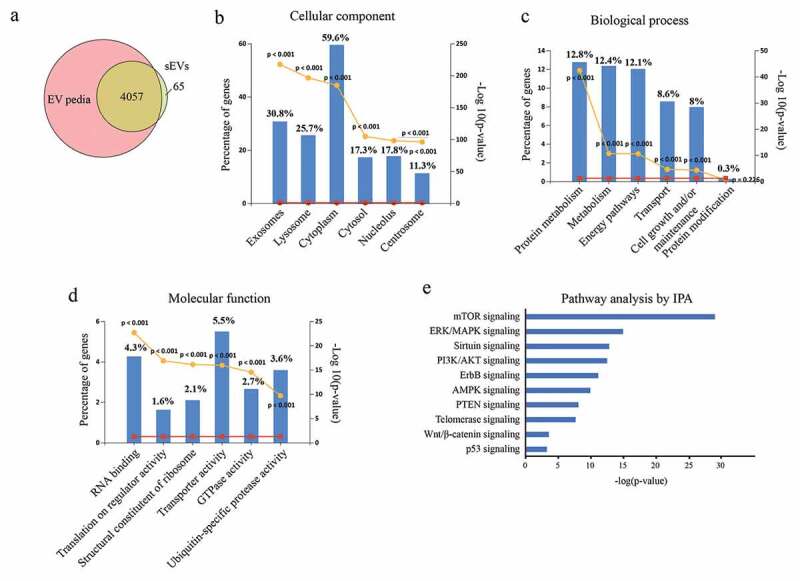
Proteomic analysis of hESC-sEVs.
(a). Venn diagram displaying the proportion of shared and unique proteins in hESC-sEVs relative to EVpedia database. (b). Gene Ontology Cellular Component enrichment analysis of proteins identified in hESC-sEVs. (c). Gene Ontology Biological Process enrichment analysis of proteins identified in hESC-sEVs. (d). Gene Ontology Molecular Function enrichment analysis of proteins identified in hESC-sEVs. (e). IPA showing canonical pathway enrichment of hESC-sEV proteins.
Next, ingenuity canonical pathway analysis of identified proteins in hESC-sEVs was performed. IPA pathway analysis indicated that the proteins in hESC-sEVs are linked to a variety of signalling pathways involved in the regulation of cellular ageing and osteogenic differentiation, including “mTOR signaling”, “ERK/MAPK signaling”, “Sirtuin signaling”, “PI3K/AKT signaling”, “ErbB signaling” “AMPK signaling”, “PTEN signaling”, “Telomerase signaling”, “Wnt/β-catenin signaling” and “p53 signaling” (Figure 6(e)). Western blot analysis further confirmed the proteome results (Figure 7, Supplementary Figure 6). These results suggested that the proteins in hESC-sEVs are linked to a variety of function and signalling pathways involved in the regulation of cellular ageing and osteogenic differentiation.
Figure 7.
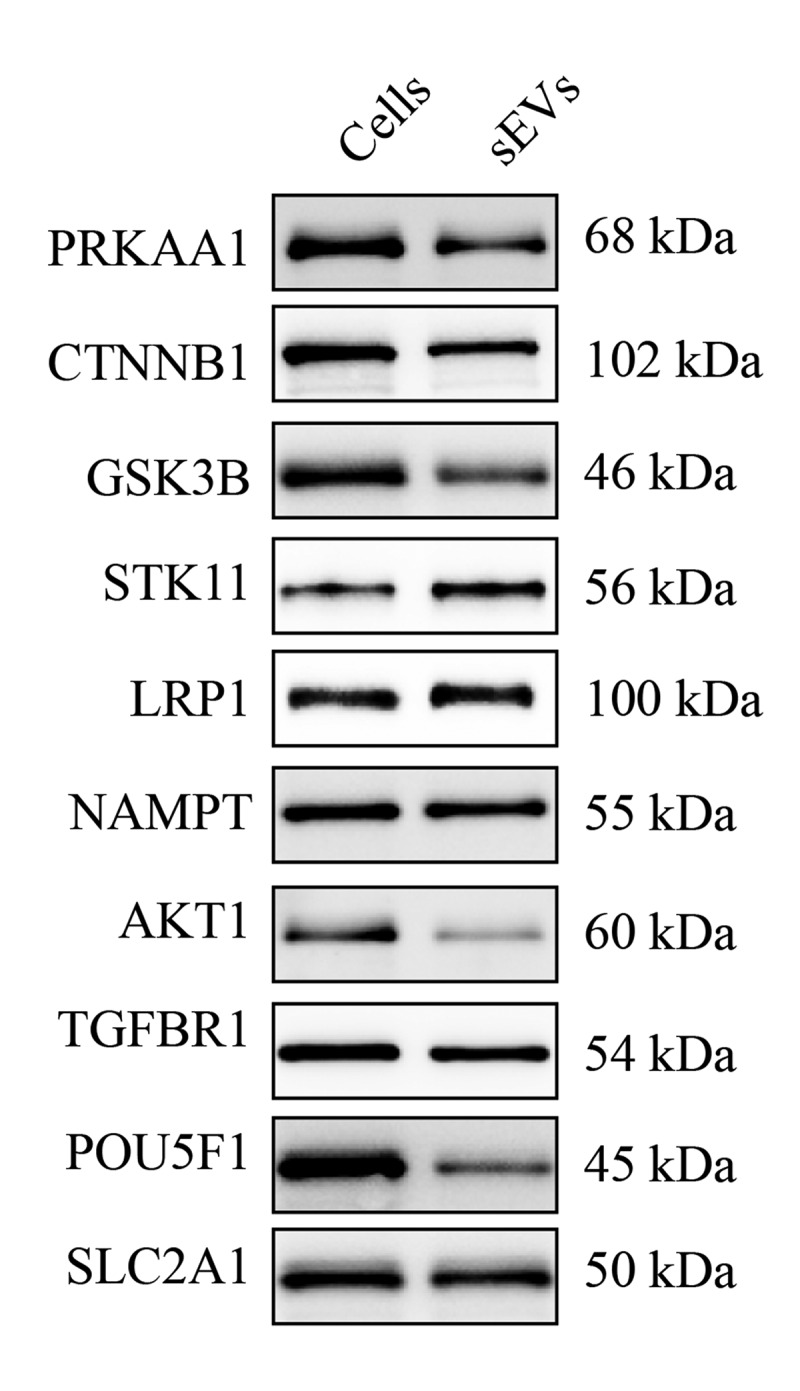
Western blot analysis of proteins in hESC-sEVs.
Representative images of western blot analysis. Cells (hESCs) were used as control. The experiment was repeated three times independently.
hESC-sEVs proteins synergistically modulate the expression of antiaging genes in BM-MSCs
To clarify how the proteins in hESC-sEVs regulate the expression of antiaging genes in BM-MSCs, we performed a regulatory network analysis using the IPA database. The results showed complex regulatory relationships between proteins identified in hESC-sEVs and the up-regulated antiaging genes in BM-MSCs (Figure 8). As shown in Figure 8, many kinds of proteins in the hESC-sEVs, such as kinase (FGFR3, MTOR, PAK1, PAK2, SRC, and STK11), enzyme (CDC42, ATG7, ACSS2, ACLY, ACACB, and SOD1), phosphatase (PPP2CA, PPP1CC), transcription regulator (RPSA, EEF2, and EIF4E) and cytokine (NAMPT) were predicted to regulate the expression of antiaging genes through direct or indirect interactions (Supplementary table 2).
Figure 8.
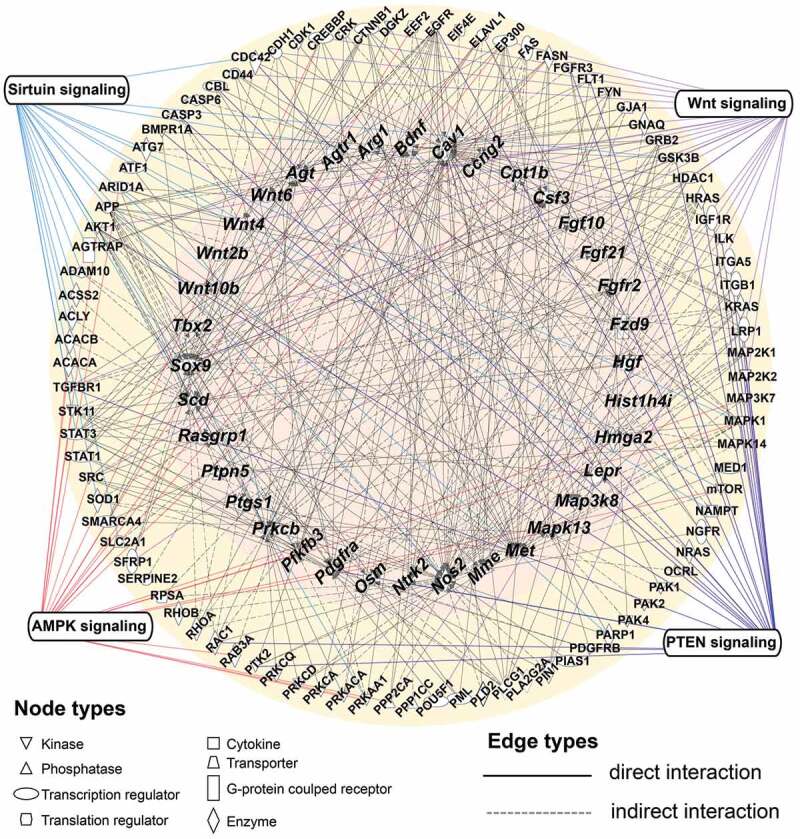
Regulatory gene and protein interaction network.
Computational molecular interaction network prediction based on anti-ageing genes and hESC-sEVs proteins associated with the significant pathways in the Ingenuity Pathways Knowledge Base (IPKB). Node properties are indicated by shape. Interactions between the different nodes are given as solid (direct interaction) and dashed (indirect interaction) lines (edges) for the different interaction types. This network received a high score by IPA and identified a synergistic and complex regulatory relationship between hESC-sEVs proteins (outer ring) and the up-regulated anti-ageing genes (inner ring). Several types of proteins (kinase, cytokine, phosphatase, transporter, transcription regulator, G-protein coupled receptor, translation regulator, enzyme) can regulate the expression of anti-ageing genes via direct or indirect interaction, or through the activation of canonical signalling pathways (Wnt, AMPK, PTEN, Sirtuin signalling).
In addition, IPA results revealed that some hESC-sEVs proteins and antiaging genes in the regulatory network are mapped to several canonical signalling pathways, including the Wnt, AMPK, PTEN and sirtuin pathways (Figure 8). These pathways have been demonstrated to be involved in counteracting ageing process, and promoting cell proliferation and osteogenesis [36–41]. The proteins and antiaging genes mapped to the pathways are listed in Table 1. Overall, these findings demonstrated that proteins in hESC-sEVs may modulate the expression of antiaging genes in BM-MSCs in a synergistic and comprehensive manner, partially through the activation of Wnt, Sirtuin, AMPK and PTEN signalling pathways.
Table 1.
List of hESC-sEVs proteins and antiaging genes associated with signalling pathways.
| Signalling pathway | Proteins components in hESCs-sEVs | Upregulated antiaging genes |
|---|---|---|
| Wnt signalling pathway | SRC, TGFBR1, AKT1, CD44, CDH1, CREBBP, CTNNB1, EP300, CJA1, GNAQ, ILK, LRP1, MAP3K7, PIN1, POU5F1, PPP2CA, SFRP1 |
Sox9, Wnt10b, Wnt2b, Wnt4, Wnt6, Fzd9 |
| Sirtuin signalling pathway | SLC2A1, SOD1, STAT3, STK11, ACLY, ACSS2, ADAM10, AGTRAP, AKT1, APP, CDH1, GSK3B, MAPK1, MTOR, NAMPT, PARP1, PRKAA1, |
Pfkfb3, Nos2, Cpt1b |
| AMPK signalling pathway | ACACA, ACACB, STK11, SRC, SMARCA4, SLC2A1, RAB3A, PRKCA, PRKAA1, PPP2CA, MTOR, MAPK1, MAP3K7, FASN, EP300, ELAVL1, EEF2, CREBBP, ARID1A, AKT1 |
Cpt1b, Mapk13 |
| PTEN signalling pathway | RAC1, PTK2, PDGFRB, OCRL, NRAS, NGFR, MAP2K1, MAP2K2, LRP1, ITGB1, ITGA5, ILK, IGF1R, HRAS, GSK3B, FLT1, FGFR3, EGFR, CDC42, CBL, CASP3, BMPR1A, AKT1, TGFBR1, |
Pdgfra, Fgfr2 |
Discussion
With advancing age, stem cells in many tissues have been found to exhibit senescent phenotypes, including blunted responsiveness to tissue injury, dysregulation of proliferation and declines in functional capacities [42,43]. These changes translate into reduced effectiveness of cell replacement and tissue regeneration in aged organisms. Accordingly, therapeutic strategies, such as cellular reprogramming [44], stem cell transplantation [45] and senolytics (i.e., ABT263 [46]), have been explored to rejuvenate or eliminate senescent tissue-specific stem cells to alleviate age-related disorders. EVs were small particles with lipid bilayer, which can be secreted by almost all type of cells [11]. There are many kinds of EVs including exosomes, microvesicles, apoptotic bodies, etc. with different functions due to their different content and function style [47]. According to MISEV 2018 guideline, EVs can also be classified into small EVs (<200 nm, pelleted at 100,000 g) and medium/large EVs (>200 nm, pelleted at 10,000 g) by the size of the particles since the subcellular origin may not be determined [28]. Recently, small EVs have been reported to be therapeutically efficacious in various preclinical studies, and are now being explored for clinical translation (Clinical Trials. gov identifiers: NCT02432287 and NCT02874924) [48]. In this study, we aimed to clarify the function of small EVs derived from human ESCs on ageing related bone loss, the large EVs were removed by successive centrifugations at increasing speeds to minimize their effects. We found that chronic application of hESC-sEVs was effective in alleviating age-related bone loss. Further studies revealed that hESC-sEVs treatment significantly ameliorated BM-MSC senescence both in vivo and in vitro, as well as increased BM-MSC proliferation and osteogenic differentiation capacity. These results mean that the therapeutic effects of hESC-sEVs on age-related bone loss may be attributed to rejuvenate BM-MSCs senescence. To the best of our knowledge, we provide the first evidence that hESC-derived sEVs can rejuvenate senescent tissue-resident stem cells and thereby prevent age-related disorders.
RNA-seq and bioinformatics analyses of sequencing data after hESC-sEVs treatment identified upregulated genes involved in bone cell proliferation and differentiation. This finding is consistent with the therapeutic effect of hESC-sEVs in promoting the proliferation and osteogenic differentiation of BM-MSCs. Importantly, GSEA analysis showed that hESC-sEVs treatment significantly activated the expression of genes involved in antiaging, stem cell proliferation and osteogenic differentiation. Among these genes, Sox9, Wnt4, Wnt10b, Wnt2b, Wnt6, and Fzd9 are associated with the Wnt signalling pathway and have been reported to enhance self-renewal and differentiation of MSCs as well as prevent skeletal ageing [37–39,49,50]. Pfkfb3, Nos and Cpt1b are Sirtuin signalling-related genes that have been demonstrated to promote cell cycle progression, suppress apoptosis, inhibit cellular ageing and stimulate bone regeneration [51–53]. Hmga2, Fgf21, Met and Tbx2 have been shown to protect against cellular senescence and promote stem cell proliferation [54–56]. In addition, Pdgfra and Fgfr2 are known to be essential for the osteogenic differentiation of BM-MSCs and bone formation [57,58]. Taken together, this evidence suggests that the therapeutic effect of hESC-sEVs in attenuating BM-MSC senescence is attributed to the activation of these antiaging and osteogenesis associated genes.
EVs are reported to contain lipids, proteins, and RNAs of the source cell and mediate intercellular communication by transferring these bioactive components [13,14]. Therefore, we hypothesized that hESC-sEVs ameliorate BM-MSC senescence by delivering their components. To the best of our knowledge, the protein contents of hESC-derived sEVs have never been elucidated before. In this study, LC-MS/MS was performed to identify the proteins enriched in hESC-sEVs. Functional enrichment analysis of the identified proteins showed a high proportion of proteins involved in cell survival, cell viability and DNA repair, suggesting the important role of hESC-sEVs proteins in promoting cell proliferation and reversing cellular ageing. Consistent with previous study reporting that components that are enriched in EVs synergistically contribute to the functional modulation of recipient cells [34], the interaction analysis by IPA in the present study revealed complex and synergistic relationships between hESC-sEVs proteins and the antiaging genes. Among them, several proteins (CREBBP, CTNNB1, LRP1, CDH1 and SFRP1) are predicted to be the regulators of Wnt4, Wnt6, Wnt10 and Sox9 through the activation of the Wnt signalling pathway, which is a central pathway involved in regulating cell proliferation and survival, suppressing the ageing process, and promoting BM-MSC differentiation [6,36,37,59]. Similarly, some proteins (CDH1, PRKAA1, APP, STK11, STAT3, SOD1, PARP1, MTOR, MAPK1, GSK3B and AKT1) are involved in the Sirtuin signalling pathway, which is reported to delay cellular senescence and extend the organismal lifespan through the regulation of diverse cellular processes [40,41,60]. These proteins are thought to activate the expression of the Sirtuin signalling-related antiaging genes, including Pffkb3, Nos and Cpt1b. Collectively, our study provides a close link between proteins enriched in hESC-sEVs and the anti-ageing genes in BM-MSCs, and highlights the critical role of hESC-sEVs proteins in reversing BM-MSC senescence.
In conclusion, we demonstrated that hESC-sEVs have positive effect in reversing BM-MSCs senescence as well as promoting their proliferation and osteogenic differentiation potential through the transfer of encapsulated proteins. Protein components in hESC-sEVs function to modulate antiaging gene expression via the direct or indirect interactions or through the activation of several canonical signalling pathways involved in decelerating cellular senescence and promoting osteogenesis. These findings not only highlight the effectiveness of EVs derived from stem cells in preventing age-related bone loss but also open an avenue for rejuvenating senescent resident stem cells and hopefully treating age-related disorders.
Supplementary Material
Acknowledgments
We thank Prof. Kunshan Zhang from Tongji University School of Medicine for his assistance with bioinformatics analysis.
Author contributions
L.Z.G and B.C did most of the experiments, collected and analyzed the data and prepared the manuscript. B.C and J.T.Z performed animal experiment, collected and analyzed the data. Y.J.S, J.Y, X.N, G.W.H and Y.C provided experimental guidance. Z.P.X and Z.F.D provided funding to support the study. Q.L and Y.W conceptualized the project, provided funding, supported study material, and approved the manuscript.
Declaration of interest statement
All the authors listed above declare that they have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153(6):1194–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Goodell MA, Rando TA.. Stem cells and healthy aging. Science (New York, NY). 2015;350:1199–1204. [DOI] [PubMed] [Google Scholar]
- [3].Oh J, Lee YD, Wagers AJ. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med. 2014;20:870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science (New York, NY). 1999;284:143–147. [DOI] [PubMed] [Google Scholar]
- [5].Zhou S, Greenberger JS, Epperly MW, et al. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell. 2008;7:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yu B, Wang CY. Osteoporosis: the result of an ‘aged’ bone microenvironment. Trends Mol Med. 2016;22:641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kim M, Kim C, Choi YS, et al. Age-related alterations in mesenchymal stem cells related to shift in differentiation from osteogenic to adipogenic potential: implication to age-associated bone diseases and defects. Mech Ageing Dev. 2012;133:215–225. [DOI] [PubMed] [Google Scholar]
- [8].Ganguly P, El-Jawhari JJ, Giannoudis PV, et al. Age-related changes in bone marrow mesenchymal stromal cells: a potential impact on osteoporosis and osteoarthritis development. Cell Transplant. 2017;26:1520–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu S, Liu D, Chen C, et al. MSC Transplantation improves osteopenia via epigenetic regulation of notch signaling in lupus. Cell Metab. 2015;22:606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kiernan J, Hu S, Grynpas MD, et al. Systemic mesenchymal stromal cell transplantation prevents functional bone loss in a mouse model of age-related osteoporosis. Stem Cells Transl Med. 2016;5:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. [DOI] [PubMed] [Google Scholar]
- [12].Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–1232. [DOI] [PubMed] [Google Scholar]
- [13].Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- [14].Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. [DOI] [PubMed] [Google Scholar]
- [15].Lai RC, Arslan F, Lee MM, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214–222. [DOI] [PubMed] [Google Scholar]
- [16].Bruno S, Grange C, Deregibus MC, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20:1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gatti S, Bruno S, Deregibus MC, et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dialysis Trans. 2011;26:1474–1483. [DOI] [PubMed] [Google Scholar]
- [18].Haga H, Yan IK, Takahashi K, et al. Extracellular vesicles from bone marrow-derived mesenchymal stem cells improve survival from lethal hepatic failure in mice. Stem Cells Transl Med. 2017;6:1262–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hu GW, Li Q, Niu X, et al. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res Ther. 2015;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cosenza S, Toupet K, Maumus M, et al. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics. 2018;8:1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cosenza S, Ruiz M, Toupet K, et al. Mesenchymal stem cells derived exosomes and microparticles protect cartilage and bone from degradation in osteoarthritis. Sci Rep. 2017;7:16214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang S, Chu WC, Lai RC, et al. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthritis Cartilage. 2016;24:2135–2140. [DOI] [PubMed] [Google Scholar]
- [23].Qi X, Zhang J, Yuan H, et al. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells repair critical-sized bone defects through enhanced angiogenesis and osteogenesis in osteoporotic rats. Int J Biol Sci. 2016;12:836–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yabut O, Bernstein HS. The promise of human embryonic stem cells in aging-associated diseases. Aging (Albany NY). 2011;3:494–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Basu J, Ludlow JW. Exosomes for repair, regeneration and rejuvenation. Expert Opin Biol Ther. 2016;16:489–506. [DOI] [PubMed] [Google Scholar]
- [26].Ratajczak J, Miekus K, Kucia M, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006;20:847–856. [DOI] [PubMed] [Google Scholar]
- [27].Chen B, Sun Y, Zhang J, et al. Human embryonic stem cell-derived exosomes promote pressure ulcer healing in aged mice by rejuvenating senescent endothelial cells. Stem Cell Res Ther. 2019;10:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Théry C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. Chapter 3:Unit 3.22. [DOI] [PubMed] [Google Scholar]
- [30].Van Deun J, Mestdagh P, Agostinis P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14:228–232. [DOI] [PubMed] [Google Scholar]
- [31].Liu X, Yang Y, Li Y, et al. Integration of stem cell-derived exosomes with in situ hydrogel glue as a promising tissue patch for articular cartilage regeneration. Nanoscale. 2017;9:4430–4438. [DOI] [PubMed] [Google Scholar]
- [32].Wiklander OP, Nordin JZ, O’Loughlin A, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pathan M, Keerthikumar S, Chisanga D, et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracell Vesicles. 2017;6:1321455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Choi JS, Cho WL, Choi YJ, et al. Functional recovery in photo-damaged human dermal fibroblasts by human adipose-derived stem cell extracellular vesicles. J Extracell Vesicles. 2019;8:1565885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].You Y, Borgmann K, Edara VV, et al. Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. J Extracell Vesicles. 2020;9:1706801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nalapareddy K, Nattamai KJ, Kumar RS, et al. Canonical Wnt signaling ameliorates aging of intestinal stem cells. Cell Rep. 2017;18:2608–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bennett CN, Longo KA, Wright WS, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102:3324–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yu B, Chang J, Liu Y, et al. Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-κB. Nat Med. 2014;20:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Simic P, Zainabadi K, Bell E, et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating β-catenin. EMBO Mol Med. 2013;5:430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen H, Liu X, Chen H, et al. Role of SIRT1 and AMPK in mesenchymal stem cells differentiation. Ageing Res Rev. 2014;13:55–64. [DOI] [PubMed] [Google Scholar]
- [42].Ermolaeva M, Neri F, Ori A, et al. Cellular and epigenetic drivers of stem cell ageing. Nat Rev Mol Cell Biol. 2018;19:594–610. [DOI] [PubMed] [Google Scholar]
- [43].Maryanovich M, Zahalka AH, Pierce H, et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat Med. 2018;24:782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ocampo A, Reddy P, Belmonte JCI. Anti-aging strategies based on cellular reprogramming. Trends Mol Med. 2016;22:725–738. [DOI] [PubMed] [Google Scholar]
- [45].Neves J, Sousa-Victor P, Jasper H. Rejuvenating strategies for stem cell-based therapies in aging. Cell Stem Cell. 2017;20:161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367 (6478). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wiklander OPB, Brennan MÁ, Lötvall J, et al. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med. 2019;11(492). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Iezaki T, Horie T, Fukasawa K, et al. Translational control of Sox9 RNA by mTORC1 contributes to skeletogenesis. Stem Cell Reports. 2018;11:228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chang J, Sonoyama W, Wang Z, et al. Noncanonical Wnt-4 signaling enhances bone regeneration of mesenchymal stem cells in craniofacial defects through activation of p38 MAPK. J Biol Chem. 2007;282:30938–30948. [DOI] [PubMed] [Google Scholar]
- [51].Montesanto A, Crocco P, Tallaro F, et al. Common polymorphisms in nitric oxide synthase (NOS) genes influence quality of aging and longevity in humans. Biogerontology. 2013;14:177–186. [DOI] [PubMed] [Google Scholar]
- [52].Bétry C, Meugnier E, Pflieger M, et al. High expression of CPT1b in skeletal muscle in metabolically healthy older subjects. Diabetes Metab. 2019;45:152–159. [DOI] [PubMed] [Google Scholar]
- [53].Hamanaka RB, Mutlu GM. PFKFB3, a direct target of p63, is required for proliferation and inhibits differentiation in epidermal keratinocytes. J Invest Dermatol. 2017;137:1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nishino J, Kim I, Chada K, et al. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wan Y. Bone marrow mesenchymal stem cells: fat on and blast off by FGF21. Int J Biochem Cell Biol. 2013;45:546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Youm YH, Horvath TL, Mangelsdorf DJ, et al. Prolongevity hormone FGF21 protects against immune senescence by delaying age-related thymic involution. Proc Natl Acad Sci U S A. 2016;113:1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Popielarczyk TL, Huckle WR, Barrett JG. Human bone marrow-derived mesenchymal stem cells home via the PI3K-Akt, MAPK, and Jak/Stat signaling pathways in response to platelet-derived growth factor. Stem Cells Dev. 2019;28:1191–1202. [DOI] [PubMed] [Google Scholar]
- [58].Eswarakumar VP, Monsonego-Ornan E, Pines M, et al. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129:3783–3793. [DOI] [PubMed] [Google Scholar]
- [59].Ye X, Zerlanko B, Kennedy A, et al. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell. 2007;27:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD(+)/Sirtuin Pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.