

ABSTRACT
The vascular endothelium and smooth muscle form adjacent cellular layers that comprise part of the vascular wall. Each cell type can regulate the other’s structure and function through a variety of paracrine effectors. Extracellular vesicles (EVs) are released from and transit between cells constituting a novel means of cell–cell communication. Here, we characterized the proteome of EVs released from each vascular cell type and examined the extent to which these vesicles participate in endothelial-vascular smooth muscle cell (VSMC) communication. EVs were collected by ultracentrifugation from media of rat aortic endothelial and smooth muscle cells cultured under serum-free conditions. Vesicle morphology, size and concentration were evaluated by transmission electron microscopy and nanoparticle tracking analysis. Western blot as well as shot gun proteomic analyses revealed sets of proteins common to both endothelial- and smooth muscle-derived EVs as well as proteins unique to each vascular cell type. Functionally, endothelial-derived EVs stimulated vascular cell adhesion molecule-1 (VCAM-1) expression and enhanced leukocyte adhesion in VSMCs while smooth muscle EVs did not elicit similar effects in endothelial cells (ECs). EVs from ECs also induced protein synthesis and senescence in VSMCs. Proteomic analysis of VSMCs following exposure to EC-derived EVs revealed upregulation of several proteins including pro-inflammatory molecules, high-mobility group box (HMGB) 1 and HMGB2. Pharmacological blockade HMGB1 and HMGB2 and siRNA depletion of HMGB1 in smooth muscle cells attenuated VCAM-1 expression and leukocyte adhesion induced by EC EVs. These data suggest that EC-derived EVs can enhance signalling pathways which influence smooth muscle cell phenotype.
KEYWORDS: Extracellular vesicles, exosome, endothelial cells, vascular smooth muscle cells, high-mobility group box 1, proteomics
Introduction
Cells release variously sized vesicles to the external environment under both physiological and pathophysiological conditions. These vesicles can be generated through shedding and/or outward fission of the plasma membrane proper resulting in vesicles measuring 50 to 1,000 nm in diameter. Other vesicles form through the endosomal network during maturation of multivesicular bodies (MVB). Generally, 50–150 nm in diameter, these vesicles are released into the extracellular environment upon MVB fusion with the plasma membrane and termed exosomes. Each vesicle type carries cytosol, lipid, protein, DNA and RNA cargo that reflects the physiological state of the parent cell. Once released, these vesicles have the capacity to interact with recipient cells through protein–protein mediated surface binding, fusion with the target cell plasma membrane or endocytosis of the vesicle itself [1,2]. These events initiate signal transduction pathways and deliver vesicle contents which can influence recipient cell phenotype.
Cardiovascular disease (CVD) is a leading cause of death globally and encompasses a large group of disorders such as hypertension and arteriosclerosis [3]. The progression to CVD involves the disruption of homoeostatic interactions both within and between the cells that form the vasculature [4–6]. The arterial wall consists of three main layers, the intima, media and adventitia, which are composed mainly of endothelial, smooth muscle and fibroblast cells, respectively. The interaction between the endothelial cells (ECs) of the intima and vascular smooth muscle cells (VSMCs) of the media is especially relevant since perturbations in communication can lead to endothelial dysfunction, inflammatory cell infiltration and phenotypic switching of VSMCs, all of which contribute to the development of hypertension and/or atherosclerosis [7]. Although general processes through which ECs and VSMCs interact, such as nitric oxide signalling, are well described, the extent to which ECs and VSMCs utilize extracellular vesicle (EVs) as a means of communication is in an early stage of exploration [8].
Recent in vivo and in vitro studies demonstrate that endothelial and VSMC EVs can contribute to processes that regulate both vascular homoeostasis and propagation of pathophysiology. Circulating levels of EVs are increased in individuals at risk for cardiovascular events [9], such as those with hypertension [10] or type 2 diabetes mellitus [11,12]. Moreover, a population of these vesicles are reported to be of EC origin [13]. Cell culture studies show that both ECs and VSMCs enhance their release of EVs in response to cardiovascular risk factors, such as tumour necrosis factor- α (TNF-α) [14] and angiotensin II [15]. More importantly, the RNA contents of these vesicles are often quite different from EVs derived from unchallenged cells. For example, VSMC overexpressing Kruppel-like factor 5, a cellular model designed to mimic a pro-atherogenic VSMC phenotype, release EVs that are enriched in miR-155. These vesicles transfer miR-155 to ECs which reduce expression of tight junction proteins and enhance permeability [16]. Similarly, expression of Kruppel-like factor 2 or exposure to laminar shear stress enhances release of EC EVs which contained miR-143/145. Whether injected in an atherogenic apolipoprotein E-/- mouse or incubated with VSMC cultures, these EVs protected against plaque formation and expression of de-differentiation-associated genes, respectively [17].
While these studies demonstrate that vesicle-mediated transfer of functional miRNAs occurs between these vascular cell type, information regarding functional protein transfer is rather limited [18]. Here, we profiled the protein content of EVs isolated from rat aortic EC and VSMC cultures using an unbiased approach [19–21]. Through proteomic analysis, we report that EVs released from EC and VSMC carry unique protein cargo. Additionally, we tested whether these EVs influence respective vascular cell phenotype. EC-derived EVs induced a pro-inflammatory, hypertrophic and senescent VSMC phenotype through a mechanism that involves high-mobility group box proteins (HMGB) while VSMC EVs had little effect on the endothelium. These findings signify that EC-derived EVs are modulators of VSMC phenotype.
Materials and methods
Reagents and antibodies
The sources and concentrations of the reagents and antibodies utilized in the present studies are described in Online Table I.
Animals
All animal procedures were performed with prior approval of the Temple University Institutional Animal Care and Use Committee and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. To obtain rat aortic VSMC, 8-week old male Sprague-Dawley rats (Charles River) were anaesthetized under sodium pentobarbital (65 mg/kg) and thoracic aortas were harvested and processed for cell isolation. For aortic organ culture, 8–10 weeks old male C57BL/6 J mice (Jackson Laboratory) were utilized. The C57BL/6 mice were sacrificed by CO2 asphyxiation followed by removal of thoracic aortas.
Cell culture
Primary aortic ECs derived from 6 to 8 week Sprague-Dawley rats (Passage 4–8, purchased from Cell Biologics RA-6052) were cultured in DMEM with 1 g/L D-glucose, 1 mmol/L sodium pyruvate and 10% FBS supplementation. Primary aortic VSMCs derived from 8-week Sprague-Dawley rats were isolated and cultured in DMEM with 4.5 g/L D-glucose, 1 mmol/L sodium pyruvate and 10% FBS supplementation as previously described [22]. The VSMCs from three rat aortas were combined and utilized for experiments from passage 4 to 10. Stocked VSMCs were not used in any experiment due to an observed enhancement in proliferation and phenotype switch upon reconstitution of these cells in culture. Both cell types were cultured on 10 cm dishes until approximately 80–90% confluent and washed once with Hank’s balanced salt solution prior to replacing with EV production media which consisted of serum-free DMEM (10 mL/dish). In some experiments, ECs were incubated with media containing EV-depleted serum. EV-depleted FBS was either produced in-house by ultracentrifugation of serum at 110,000xg for 16 hours followed by sterile filtration through a 0.2 µm filter or purchased from Thermo Scientific (A2720803). EV-depleted serum was added to DMEM at a final serum concentration of 10%.
Isolation of vascular cell-derived EVs
Ultracentrifugation was employed to concentrate EVs [23] released from rat aortic EC and VSMC. Media were collected after 48 (ECs) or 72 (VSMCs) hour incubations. Based on pilot studies, we found the rate of EV production from VSMCs was lower than that of ECs. Therefore, additional time was required to gather enough vesicles from VSMCs for characterization and experimentation. Also, we observed that EC viability under serum-free conditions declined at 72 h, limiting the use of these cells to the 24–48-h time period. Overall, we found that these collection time points allowed for the greatest accumulation of vesicles in culture media for each cell type without compromising the cells integrity.
To control for non-specific effects that might be attributed to general components contained in/on any EV, EVs were collected from neonatal cardiac fibroblasts and used in a subset of functional studies. Briefly, fibroblast (passage 1) were cultured in serum-free DMEM for 48 h to allow for the production of EVs.
In each case, the collected media was centrifuged at 300 × g for 10 min, followed by 2000 × g for 20 min to remove cellular debris. EV-containing media were then subjected to ultracentrifugation using an MLS 50 swinging bucket rotor loaded into a Beckman Coulter Optima MAX-XP Ultracentrifuge at 12,000 × g for 40 min to remove larger vesicles and apoptotic bodies. Supernatant was subsequently ultracentrifuged at 1100,000 × g for 70 min to pellet EVs. Pellets were washed with phosphate buffer saline (PBS), passed through a 0.2 µm filter (VWR International), recentrifuged at 110,000 × g for 70 min and processed for transmission electron microscopic or mass spectroscopic analyses. For mass spectroscopy, EV pellets were re-suspended in 100 µL urea lysis buffer (8 mol/L urea, 50 mmo//L NH4HCO3, 1:100 dilution of protease inhibitor mix from GE Healthcare). EV pellets used in the functional studies were not filtered but underwent two additional ultracentrifugations (110,000 × g for 70 min) to wash (PBS) and concentrate EVs. The final EV pellets were re-suspended in 50–100 µL PBS for subsequent applications. In some experiments, cells were treated with vehicle alone (0.1% DMSO) or the neutral sphingomyelinase inhibitor GW4869, which blocks exosome release.
Isolation of serum EVs
Sprague-Dawley rats, 6–8 week of age, were anaesthetized and euthanized via cervical dislocation. Upon euthanasia, blood was collected via cardiac puncture of left ventricle and transferred 15 mL Falcon tube. To generate sera, tubes were left undisturbed for 60 min to allow for clot formation. Tubes were then centrifuged at 10,000 rpm for 10 min to pellet the clot. Supernatants were collected and centrifuged again at 10,000 rpm for 10 min. Rat serum was diluted with equal volumes of PBS and centrifuged at 12,000xg to remove cell debris. Supernatants were then ultracentrifuged at 110,000 × g for 120 min. EV pellets were washed, re-suspended in PBS and subjected to four rounds of washes in PBS and ultracentrifugation to remove contaminants before a final re-suspension in urea lysis buffer for mass spectroscopy analysis. Unlike EVs collected from culture media, serum EVs were not filtered due to the small volume of starting blood sample size and concern for low EV yield. In lieu of filtration, we used an EV isolation protocol that has been shown to reduce contaminating serum proteins [24].
Electron microscopy
EV preparations were fixed in 2% paraformaldehyde solution and allowed to adhere on Formvar-coated carbon grids. Grids were washed with PBS followed by fixation in 1% glutaraldehyde and subsequently washed with deionized water. Grids were negative stained with a solution of 1% uranyl-oxalate and 4%/2% uranyl acetate/methyl cellulose. Following air drying, grids were placed in a JEM-1400 transmission electron microscope (JEOL USA) operating at 80kV and EV images were digitally captured at either 80,000x or 200,000x depending on the preparation, as indicated in the Figure Legend. Vesicle diameters were directly measured from digital images using the image analysis tracing tool and line function.
Nanoparticle tracking analysis
EV preparations were diluted in deionized water prior to nanoparticle tracking analysis (NTA) using NanoSight NS300 (Malvern Instruments, Malvern, UK). Five replicates of 60 sec videos were captured to analyse concentration and size distribution of EV preparation at threshold detection of 3–5 depending on dilution. After video capture, data analysis was performed using NanoSight analysis software. Particle number determined by the NanoSight analysis was used for a normalization between the assays. According to pilot studies, final EV concentrations were set at 0.5–1 × 109 particles/mL, which provides reproducible biological responses in vitro and allowed for multiple assays in the present study.
Western blot analysis
The concentration of total cellular proteins, prepared in standard RIPA buffer, and EV proteins were determined using a bicinchoninic acid assay (Pierce™ BCA Protein Assay Kit, Thermo Scientific). Each preparation was sonicated, diluted with 5× standard SDS loading buffer containing β-mercaptoethanol and heated at 95°C for 5 min. Equal protein loading (10 μg) of whole-cell and EV proteins were separated via SDS-PAGE on 10% acrylamide/bis-acrylamide (37.5:1) resolving gel and subsequently transferred onto 0.45 µm nitrocellulose membrane overnight at 30 V. Membranes were blocked for 2 h in 5% non-fat dry milk and Tris-buffered saline pH 7.5 with 0.1% Tween20 (TBS-T). To detect proteins, membranes were incubated with primary antibody overnight at 4°C, washed with TBS-T and incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature followed by enhanced chemiluminescence for protein detection and quantification as described [22].
Shotgun proteomics and bioinformatics
For determination of EV proteins, EVs pellets were resuspended in the urea lysis buffer and incubated with 10 mmol/L dithiothreitol for 1 h at 37°C to prevent disulphide bond formation. The samples were then treated with 25 mmol/L 2-iodoacetamide for 45 min at room temperature to alkylate cysteines. For total cell protein analysis, VSMC and EC lysates were collected with 100 µL urea lysis buffer (8 mol/L urea, 50 mmol/L NH4HCO3, 1:100 dilution of protease inhibitor mix from GE Healthcare). Samples were diluted fourfold with 50 mmol/L ammonium bicarbonate and digested overnight at room temperature with 0.05 µg/mL Trypsin Gold (Promega). Digestion was enzymatically quenched with the addition of 20% trifluoroacetic acid and the samples were lyophilized. Protein digests (1 μg) were analysed using a mass spectrometry for protein identification [25]. For protein and peptide identifications, the obtained MS/MS data were subjected to database searches using the MASCOT programme (Matrix Science Ltd., London, UK) with the following parameters: two missed cleavage sites and a peptide and MS/MS mass tolerance setting of ± 5 ppm and ± 0.05 Da, respectively, for MS/MS Ions Search. The database used for this search consisted of amino acid sequences of rat proteins, which were retrieved from a subset of the UniProtKB/SWISS-PROT database (2014_07). Variable modifications such as oxidation of methionine, carbamylation and acetylation of N-termini, and carbamidomethylation of cysteine were taken into consideration for the database searches. We used a 1% overall false discovery rate as a cut-off value to export our results from the database search using the MASCOT. Peptides that yielded a peptide ion score of greater than or equal to 30, were used for relative quantitation. In addition, peptides that were either more or less abundant in the comparison were extracted using the following parameters in Progenesis QI for proteomics (v.4.1, Nonlinear Dynamics, Durham, NC, USA): maximum fold change ≥2 or ≥1.5 and analysis of variance (ANOVA) p < 0.01.
EV release, uptake and trafficking assay
EV dynamics were monitored in either co-culture systems or direct labelling of isolated EVs. Lipilight dyes by Membright were used to label cells and EVs. Lipilights contain two amphiphilic groups of zwitterions and alkyl chains which directly and stably insert into bilayer lipid membranes [26]. For direct labelling experiments, EC EV preparations were mixed with 200 nM Lipilight (560 nm) for 1 h at room temperature in the dark. To control for dye incorporating into undefined elements within the medium or formation of dye aggregates which could be taken up by cells, Lipilight was added to non-conditioned cell culture media. Labelled EVs and dye-containing media were centrifuged 2 times at 10,000xg for 15 min each at 4°C in fixed-angle centrifuge to pellet traces of excess dye. The supernatant was retained as labelled EVs or media. EC EVs (5×108) or media (10 µL, equivalent volume to dilution of EVs) were added to VSMC cultures on coverslips and allowed to incubate for 30 min. Coverslips were then fixed with 4% paraformaldehyde solution, washed 3x with PBS and mounted on glass slides with Prolong Gold/Dapi. Images were obtained using 20x objective lens on EVOS M5000 microscope (Invitrogen). Unstained VSMC were also imaged and used as controls to correct for background fluorescence.
For the co-culture system, EC and VSMC were washed with serum-free media followed incubation with 100 nM Lipilight (640 nm for EC and 488 nm for VSMC) for 1 h at 37°C. Cells were washed 3x with serum-free media to remove excess dye, trypsinized and re-seeded at a density of 5 × 104 cells in DMEM onto coverslips. Cells were allowed to attach and co-cultured for 4 h in DMEM containing EV-depleted FBS. Cells were washed 2x with PBS, fixed in 4% paraformaldehyde solution, washed 3x with PBS, and mounted onto glass slides with Prolong Gold/Dapi. Images were obtained using 20x or 60x-oil objectives.
THP-1 cell adhesion assay
To observe leukocyte attachment to VSMCs, THP-1 adhesion assay was performed as described [22]. In brief, VSMC cultures were challenged with either EC-derived EV (5x108), TNF-α (10 ng/mL) or PBS for 16 h. THP-1 monocytes (American Type Culture Collection) cultured in Roswell Park Memorial Institute/RPMI 1640 medium with 10% FBS, penicillin and streptomycin were suspended in serum-free DMEM with 0.2% BSA and 5 μg/mL Hoechst 33342 (ThermoFisher) for 30 min at 37ºC. These cells (104 cells per cm2) were then applied to VSMC cultures for 30 min at 37ºC. In some experiments, anti-VCAM-1 antibody or rabbit IgG were added at 1 µg/mL for 1 h prior to addition of THP-1 cells. VSMC cultures were then washed in PBS to remove unattached cells and fixed in 3.7% paraformaldehyde for 10 min at room temperature. Fixed cells were washed in PBS and subsequently imaged using a fluorescent inverted microscope. Stained THP-1 nuclei were counted to quantify adhesion to VSMCs as follows. Five separate fields were captured per condition using a 10x objective lens. Images were imported into ImageJ where the image was processed for background subtraction and conversion to a binary image followed by an analysis of particle count per visual field. Data were reported as the mean value of five fields per condition and compared across groups.
Surface sensing of translation assay for new protein synthesis
Analysis of new protein synthesis in EV-treated VSMCs was performed using an adapted Surface Sensing of Translation (SUnSET) assay protocol, as previously described [27]. Briefly, VSMCs were serum-starved to synchronize cell cycle and subsequently incubated with EC EVs for indicated time points. At conclusion of the experiment, cells were pulsed with 5 µmol/L puromycin for 30 min. Cells were washed twice with PBS, harvested and cell lysates prepared for Western blotting. Proteins from each sample were separated on SDS-PAGE gels followed by transfer to nitrocellulose membranes and immunoblotted with a monoclonal antibody specific to puromycin. As a negative control, cycloheximide (10 µg/mL, 30 min treatment) was included in some experiments to inhibit protein synthesis.
Cell volume assessment
VSMC volume was measured as we reported [28] with a slight modification. In brief, after 3-day incubation with EC EVs, PBS, or angiotensin II (100 nM), VSMCs were washed twice with Hank’s balanced salt solution, detached with trypsin, and re-suspended in PBS. Cell volume was then evaluated with the Sceptre Cell Coulter Counter (MilliporeSigma).
Senescence-associated β galactosidase assay
Senescence-associated β galactosidase activity was measured using X-Gal staining system (Goldbio). In brief, VSMCs were serum-starved for 48 hours then incubated with EC EV or PBS for an additional 72 h. Cells were then rinsed twice with Hanks balanced salt solution containing calcium, fixed with a 4% paraformaldehyde solution in PBS for 10 min, washed again with the Hanks solution four times and incubated with 40 mmol/L phosphate buffer containing 1 mg/mL X-Gal in 150 mmol/L NaCl, 2 mmol/L MgCl2, 5 mmol/L K3Fe(CN)6 and 5 mmol/L K4Fe(CN)6 at 37°C for 12 h. Cells were washed twice with the Hanks solution with calcium and magnesium, and counter stained with Hoescht33342 for 5 min to stain nuclei. VSMC cultures were examined at 10x magnification to identify β galactosidase positive cells. Cells were counted in three to six individual wells per condition and values were expressed as a percentage of positive cells versus total cell number. Compiled data were expressed as fold-change between control and EV conditions.
Immunofluorescent staining
VSMC was cultured on glass coverslips until 70–75% confluent, followed by serum deprivation for a period of 48 h. VSMC was stimulated with EC EVs for a period of 24 hs followed by fixation with 3.7% paraformaldehyde solution and then washed 3x with PBS. Cells were made permeable through incubation with 0.1% Triton-PBS and blocked for 2 h at room temperature with 1% BSA-PBS-T (0.1% Tween). Cells were incubated with a rabbit antibody against Ser139 phosphorylated histone H2AX (γH2Ax) at 1 µg/mL overnight at 4°C. Cells were then washed with PBS 3x and incubated with goat anti-rabbit IgG Alexa Fluor 488 (Invitrogen A11034) at 1:1000 for 2 hours in the dark. After washing (3x with PBS), cells were mounted on glass slides with Prolong Gold w/Dapi reagent (ThermoFisher P36931). Cells were imaged at 40x magnification on EVOS M5000 microscope (Invitrogen). Accumulation of distinct foci within nucleus (>5) indicated a positive cell which were then counted as previously described [29].
Pharmacological inhibition of high-mobility group box (HMGB)
In some experiments, VSMCs were pretreated with the HMGB1 inhibitor, glycyrrhizin [30] (500 µmol/L) or the HMGB1/HMGB2 inhibitor, inflachromene [31] (25 µmol/L) for 1 h prior to incubation with EC-derived EVs.
siRNA depletion of HMGB1
In order to deplete HMGB1 in VSMC, cells were cultured until 85–90% confluent and then infected with either adenovirus encoding HMGB1 siRNA (TTGCCTCTCGGCTTC) or control non-silencing siRNA (100 MOI) for a period of 72 hours [32]. Cells were then exposed to EC EVs for indicated times.
Mouse aorta organ cultures
The thoracic aorta was isolated and cleaned of the surrounding adipose and connective tissue using HEPES buffer solution (400 mmol/L NaCl, 200 mmol/L KCl, 100 mmol/L MgCl2, 100 mmol/L HEPES, 11.5 mmol/L glucose) in 5% penicillin-streptomycin. Aortas were opened en face, cut into equal size strips and then transferred to DMEM culture media containing 4.5 g/L D-glucose, 1 mmol/L sodium pyruvate and 10% FBS and incubated at 37◦C with 5% CO2 overnight. The medium was replaced with serum-free DMEM for 24 h. Aortic organ cultures were incubated with either PBS, 10 ng/mL TNF-α or 109 EVs for 16 h. Following the conclusion of the experiment, aortic strips were SNAP-frozen in liquid nitrogen and stored at −80◦C for subsequent Western blot analysis.
Statistical analysis
Data are presented as mean±SEM. Comparisons were performed via t test for two groups, or via 1-way ANOVA with the post hoc Tukey method for multiple groups using Prism software (GraphPad, San Diego, CA). Differences were considered statistically significant at p < 0.05.
Results
Extracellular vesicles (EVs) are actively released from ECs and VSMCs
Differential ultracentrifugation was employed to concentrate the population of EVs that are released from rat aortic ECs and VSMCs cultured under serum-free conditions. Ultrastructural analysis by transmission electron microscopy showed the EVs released by each cell type were in the size range of 30–300 nm in diameter and displayed a cup-shaped morphology, which is typical of vesicles isolated by this ultracentrifugation technique and having undergone chemical fixation (Figure 1(a,b)). Dynamic light scattering analysis of EVs from both ECs and VSMCs confirmed the size distribution and showed that the majority of vesicles were approximately 150 nm in diameter (Online Figure I-A and I-B). There was no significant difference in size between EC and VSMC EVs. Vesicle production rate, as measured by a particular number, was lower for VSMCs compared to EC’s with VSMCs requiring an additional 24 h of incubation to produce roughly equivalent amounts of EVs as ECs (Online Figure I-C). Cell viability was approximately 93 ~ 96% in both cell types during the exosome secretion period (Online Figure I-D). Pretreatment of each cell type with GW4869, a neutral sphingomyelinase inhibitor reported to block ceramide-mediated inward budding of MVBs and the release of mature exosomes from MVBs [33,34], reduced the number of EV acquired by ultracentrifugation by ~50% (Figure 1(c,d)). These data suggest that a substantial portion of the EVs released by cultured vascular cells are exosomes.
Figure 1.
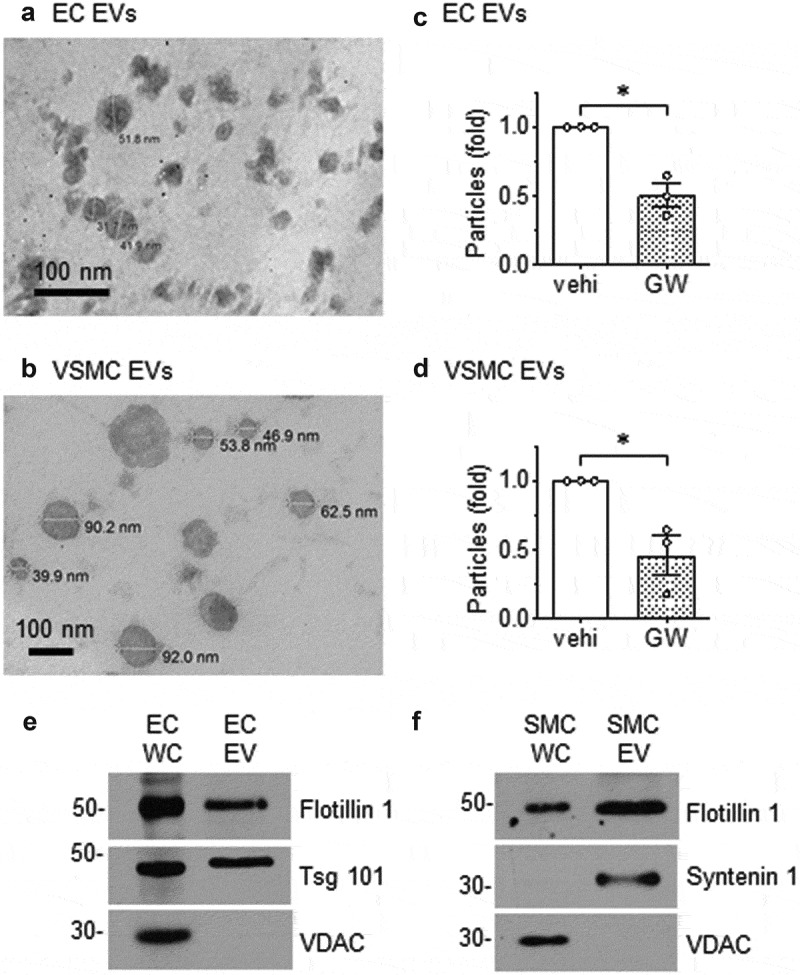
Morphology of EVs released from rat aortic EC and VSMC. Rat aortic EC (a) and VSMC (b) were cultured under serum-free conditions. EVs released into the media were isolated by ultracentrifugation, fixed to Formvar carbon-coated copper grids, stained and observed under transmission electron microscopy. Images were taken at either 200,000 (a) or 80,000 (b) magnification. (c) ECs or (d) VSMCs were cultured with serum-free DMEM containing GW4869 (GW: 20 μmol/L) or the vehicle (vehi: DMSO 0.1%) for 48h. EVs were isolated from the culture media and NanoSight (NS300 NTA) analysis was used to determine the ratio of released EV particles. The bars in the graphs show the mean±SEM from three independent experiments. (e and f) Immunoblot analysis of equal loading of EC and VSMC whole cell lysates (WC) and EV preparations. Tsg 101; tumour susceptibility gene-101, VDAC; voltage-dependent anion-selective channel-1. Representative data are shown from three independent experiments. c* indicates p < 0.05.
EVs contain characteristic sets of proteins including transmembrane proteins, proteins involved in membrane transport, metabolic enzymes, and cytoskeletal proteins [35]. Western blot analysis revealed that EC and VSMC EVs display protein markers associated with exosomes and other EVs [36] including the lipid raft protein, flotillin-1, and proteins involved in multivesicular body biogenesis and trafficking such as tumour susceptibility gene-101 and syntenin-1 (EC and VSMC, respectively) (Figure 1(e,f)). Preparations were devoid of mitochondrial outer membrane protein, voltage-dependent anion-selective channel-1, illustrating a lack of mitochondrial contaminants in the isolates.
EVs from EC and VSMC harbour common as well as unique proteomic signatures
EVs have been shown to carry unique protein and nucleic acid cargo depending on cell type and the cellular milieu in which the parent cell resides [18,35]. To further explore the protein content of our EV isolates, LC/MS analysis was performed. Shotgun proteomic analysis identified 194 total proteins in EC-derived EVs and 118 proteins in VSMC EVs in which 74 proteins are common and 120 and 44 proteins appears unique to the corresponding EVs (Figure 2(a)). Amongst the proteins in common are EV markers such as Tetraspanins, CD9 and CD82, membrane binding Annexins (1, 2, 3, 5, and 6), EV biogenesis/endosomal proteins (Syntenin, Programmed cell death 6-interacting protein/ALG-2-interacting protein X, Ras-related protein Rab-7a), and other cytosolic/signalling proteins (Online Table II). In addition, vascular EV proteins were analysed with total cell lysate proteomes from EC and VSMC. There are 30 proteins which were identified in EC EVs and 35 proteins identified in VSMC EVs that were not detected in their respective cell lysates (Figure 2(b,c)). Among these EV unique proteins, 9 proteins were present in both cell populations suggesting common as well as cell-type specific protein enrichment into the EVs. Interestingly, 54 proteins were also identified that were common in both EVs and both cell lysates (Online Figure II-A). These proteins represent a remarkable interactome with significant numbers of potential signalling functions (Online Figure II-B) suggesting common regulatory roles for vascular EVs shared by EC and VSMC. Lastly, serum EV analysis revealed proteins that were detected in EC EVs (~38%), VSMC EVs (~18%) or both EVs (~13%) (Online Figure III and Online Table III) suggesting the potential secretion of vascular EVs into circulation.
Figure 2.
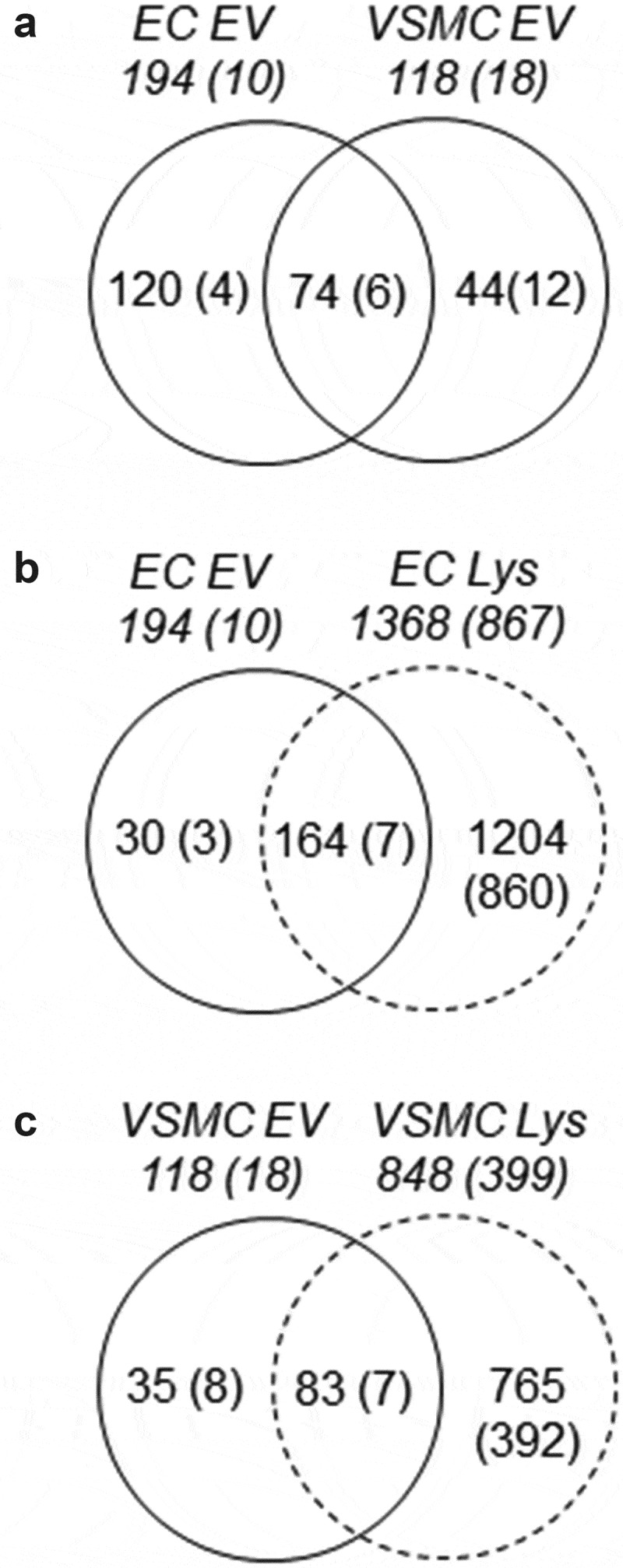
Proteomic identification of EV proteins common and unique to EC and VSMC. Shotgun proteomic analyses were performed in EV and whole-cell lysates (Lys) from serum-deprived rat aortic EC and VSMC cultures. Each population was analysed with four samples prepared from distinct culture plates. Numbers without brackets are the numbers of peptides detected in at least one sample. Numbers with brackets are the numbers of peptides detected in all four samples.
To further assess cell-type enriched EV proteins, proteins expressed greater than twofold in abundance (p < 0.05) were characterized with DAVID analysis [37]. VSMC enriched EVs possessed proteins which are implicated in complex biological processes, molecular functions, and cellular components that include cytoskeleton organization, cell adhesion, molecule binding and external side of the plasma membrane, respectively, whereas EC enriched EVs showed much simpler characteristics in Gene Ontology terms (Online Figure IV and Online Table IV). Among the identified peptide populations, interactome analysis was further performed on the cell-type specific populations with p values less than 0.01. EC EVs contained 12 associating proteins while VSMC expressed 30 proteins within their EVs. There were five common proteins identified with distinct peptide fragments (Online Table V). STRING gene interaction analysis illustrates a complex web of interactions between the proteins enriched in VSMC EVs, while EC analysis produced a smaller interactome even with the inclusion of the five common proteins (Online Figure V). Western blot analysis was performed to verify protein expression of the peptide fragments which were significantly enriched in the proteome analyses. The analysis confirmed that Clathrin heavy chain (Cltc) and Hsc70 (Hspa8) were indeed present in EC EVs and Enolase 1 (Eno1) and Calmodulin (Calm1) were present in VSMC EVs, respectively (Online Figure VI).
Endothelial EVs initiate vascular inflammation, protein synthesis and senescence in smooth muscle cells
The communication between the endothelium and vascular smooth muscle of the artery is critical in the maintenance of vascular homoeostasis as well as progression to pathology [7]. First, we verified that EC EVs can be internalized by VSMCs when the EVs were applied exogenously or in a paracrine manner when both cells were co-cultured. VSMCs were incubated with DMEM containing EC EVs labelled with Lipilight. VSMCs rapidly accumulated Liplight signal whereas no signal was detected when VSMCs were incubated with DMEM plus Lipilight alone (Online Figure VII-A). To observe EV uptake in a more physiological environment, VSMCs and ECs were prelabeled with different Liplight fluorescent colours prior to co-culture. Upon co-culture, cell reattached and spread as early as 2 hours and the Liplight fluorophore used to label ECs was detected, together with the fluorophore label from VSMC, within VSMCs by 4 hours (Online Figure VII-B). These observations suggest active uptakes of available vesicles including EVs that were secreted into the medium by ECs.
Next, we examined whether EC and VSMC derived EVs could initiate change in cell phenotype. The alteration in the expression of vascular cell adhesion molecule 1 (VCAM-1) was initially investigated since enhanced VCAM-1 expression in ECs and/or VSMCs has been implicated in vascular pathologies [38,39]. We found that EVs derived from ECs cultured under serum-free conditions were able to induce the expression of VCAM-1 in VSMC, whereas EVs derived from ECs cultured with media containing EV-depleted serum had little effect on altering VCAM-1 expression (Figure 3(a)). Similarly, rat cardiac fibroblast EVs did not enhance VCAM-1 expression in ECs (Figure 3(b)), indicating that upregulation of VCAM-1 protein expression in VSMCs was specific to EVs derived from serum starved ECs. Interestingly, VSMC EVs did not have a reciprocal effect of enhancing VCAM-1 expression in ECs (Online Figure VIII). Functionally, EC EVs enhanced leukocyte adhesion to VSMC that was comparable to that induced by TNF-α in VSMC (Figure 3(c)). Pretreatment with a VCAM-1 neutralizing antibody markedly attenuated EV-induced leukocyte adhesion compared with control IgG treatment (Figure 3(d)), indicating that VCAM-1 plays a major role in inflammatory cell adhesion following EV challenge. In an in situ model, VCAM-1 induction by EC EVs was preserved in mouse aortic segments (Figure 3(e)) lending further support to the pathophysiological relevance of EC-derived EVs in mediating vascular inflammation.
Figure 3.
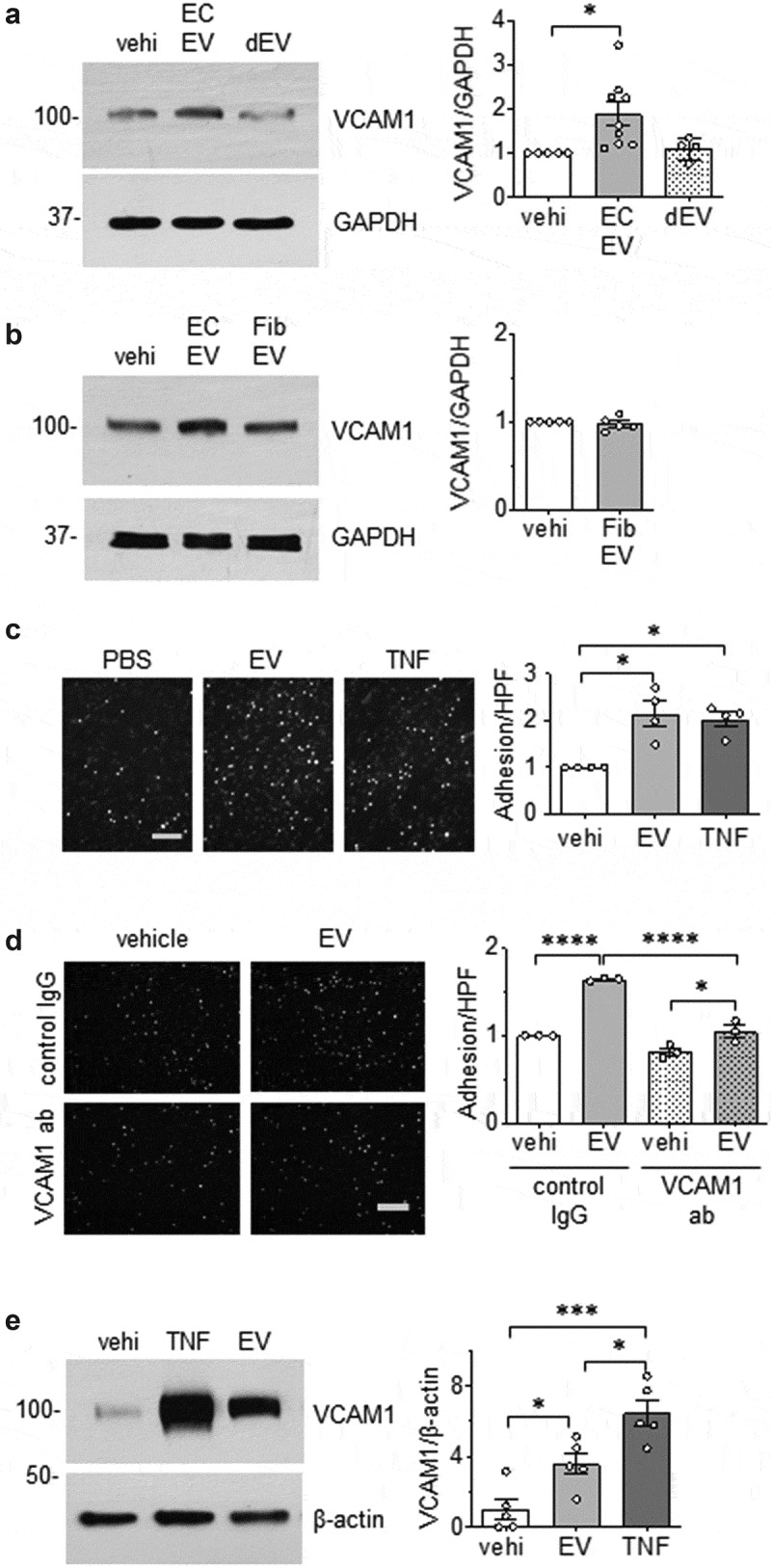
Pro-inflammatory effects of EC EVs on VSMCs. (a and b) Serum starved (48 h) rat aortic VSMCs were incubated with vehicle (vehi: PBS), EC EVs (5x108 particles) from either serum-free conditions or EV-depleted serum conditions (a) or serum-free fibroblast EVs (5 x 108 particles, b) for 6 hours and expression of VCAM-1 and GAPDH was determined by Western blotting (n = 5–8). (c) Serum starved VSMCs were incubated with vehicle, EC-derived EVs (5 x 108 particles) or TNF-α (TNF: 10 ng/mL) for 16 hours, washed and then incubated with THP-1 monocytes for 30 min. Adherent THP-1 cells were counted (n = 4). (d) VSMC were serum-starved followed by incubation with EC-derived EVs for 16 hours as in 1 C. However, prior to incubation with THP-1 cells, 1.0 µg/mL of either rabbit IgG or VCAM-1 antibody was added for 1 hour to block binding through VCAM-1 (n = 3). (e) Mouse aorta organ culture was incubated with vehicle, EC-derived EVs (109 particles) or TNF-α (TNF: 10 ng/mL) for 16 hours. Western blotting was performed to evaluate expression of VCAM-1 and β-actin (n = 5). The bars in the graphs show the mean±SEM. *, **, *** indicate p < 0.05, 0.01, and 0.001, respectively.
Since enhanced protein synthesis in VSMC has been implicated in pathological vascular hypertrophy [40], we further investigate whether EC EVs can stimulate protein synthesis. Here, we used the SUnSET assay [27], which relies on the ability of puromycin to function as a structural analog of aminoacyl tRNAs with incorporation into newly synthesized polypeptides and terminate elongation. Detection of puromycin is performed via Western blot using anti-puromycin antibody and the enhanced incorporation reflect the rate of protein synthesis [41]. We found that EC-EVs stimulate protein synthesis in VSMCs maximally at 3 hours (Figure 4). Given that enhanced protein synthesis is associated with VSMC hypertrophy, VSMC size was evaluated after a 72-h incubation with EC EVs. We observed a strong trend towards cellular enlargement in VSMC treatment with EC EVs (Online Figure IX-A), which neared the effect of the pro-hypertrophic molecule, angiotensin II [42]. In addition, expression of early growth factor-1 (EGR-1), an important transcription factor implicated in VSMC hypertrophy [43] and proliferation [44] was significantly elevated upon EC EV stimulation of VSMCs at 3 hours (Online Figure IX-B).
Figure 4.
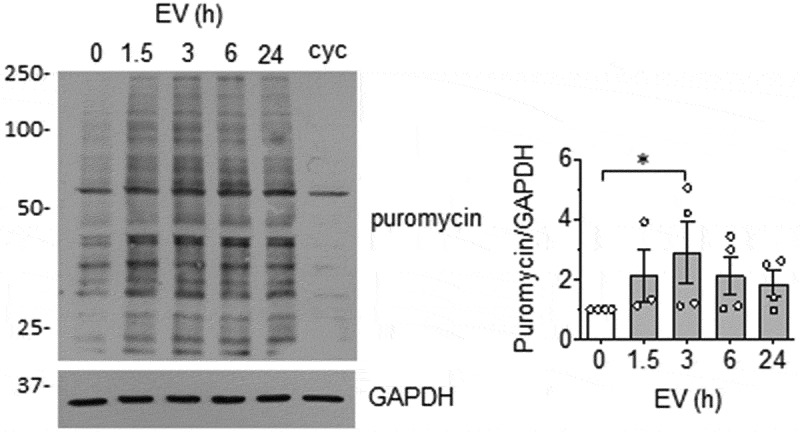
EC EVs stimulate protein synthesis in VSMCs. VSMCs were incubated with EC-derived EVs (5 x108 particles) for indicated time points. Samples were pulsed with 5 µmol/L puromycin for 30 min at the end of the experiment and processed for Western blot analysis using an anti-body against puromycin. Cycloheximide (cyc: 10 µg/mL for 30 min treatment) was used to inhibit protein synthesis as control (n = 3 for 1.5 h, n = 4 for remainder). * indicates p < 0.05.
Given that hypertrophic factors such as angiotensin II is known to cause premature vascular cell senescence [45], we further examined the effects of EVs released from serum-deprived ECs on VSMC senescence. We found that these EVs significantly increased the population of VSMC expressing β-galactosidase activity (Figure 5(a)). As a second measure of cellular senescence, we examined γH2AX which is a sensitive marker of double-stranded DNA breaks and telomere shortening. Similar to our findings with β-galactosidase activity, γH2Ax positive cells increased in VSMCs following EV challenge (Figure 5(b)).
Figure 5.
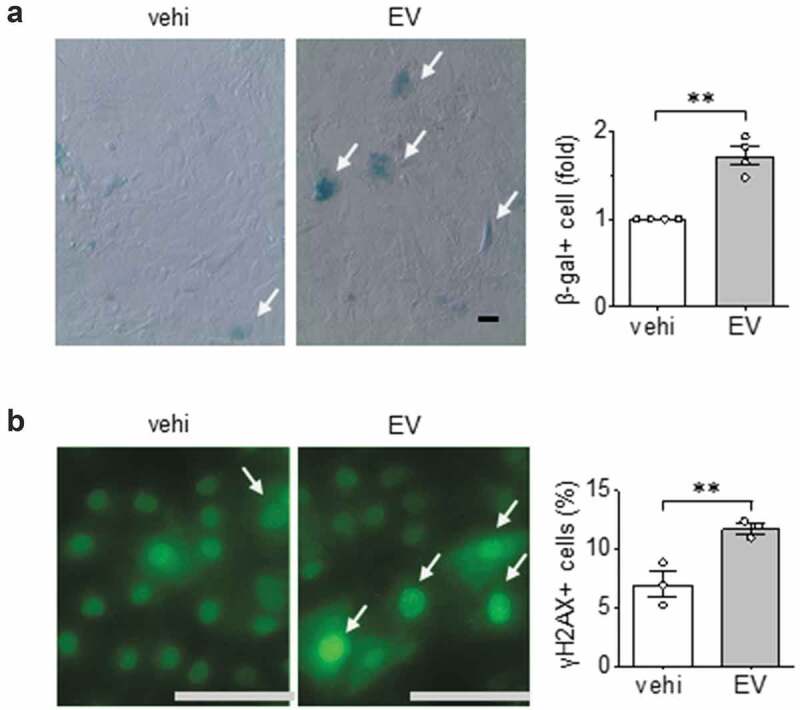
Pro-senescent effects of EC EVs on VSMCs. (a) Rat aortic VSMCs serum starved for 48 hours were incubated with EC EVs (109 particles) for 72 h and β galactosidase positive cells were counted (n = 3). (b) Rat aortic VSMC were serum-starved for 48 hours followed by incubation with EC EVs for 24 h followed by immunostaining with γH2Ax for DNA damage (n = 3). The bars in the graphs show the mean±SEM. ** indicates p < 0.01.
EC EVs induce altered proteome in VSMC
To gain a broader view of the effects of EC-derived EV on VSMC phenotype, we performed unbiased mass spectroscopy analysis on VSMCs following incubation with the EVs derived from ECs. Using a cut-off for peptide fold-change at 1.5, we identified several peptides that were up-regulated in VSMC upon EC EV incubation (Figure 6, Online Figure X and Online Table VI). Gene ontology analysis and interactome analysis suggest that EC EVs induce VSMC proteins involved in metabolic regulations including those involved in mitochondrial respiration and response to stress. Of note, among these altered proteins were the pro-hypertrophy molecule, ribosomal protein S6, and pro-inflammatory molecules, HMGB1 and HMGB2 on the peptide level. On the protein level, 4 proteins (Srprb, Maoa, Rhot2 and Kdelr2) were up-regulated (Online Table VII). Signal recognition particle receptor β (Srprb) is necessary for sorting secretory and membrane proteins to the endoplasmic reticulum membranes, suggesting up-regulation of membrane protein synthesis and excretion [46].
Figure 6.
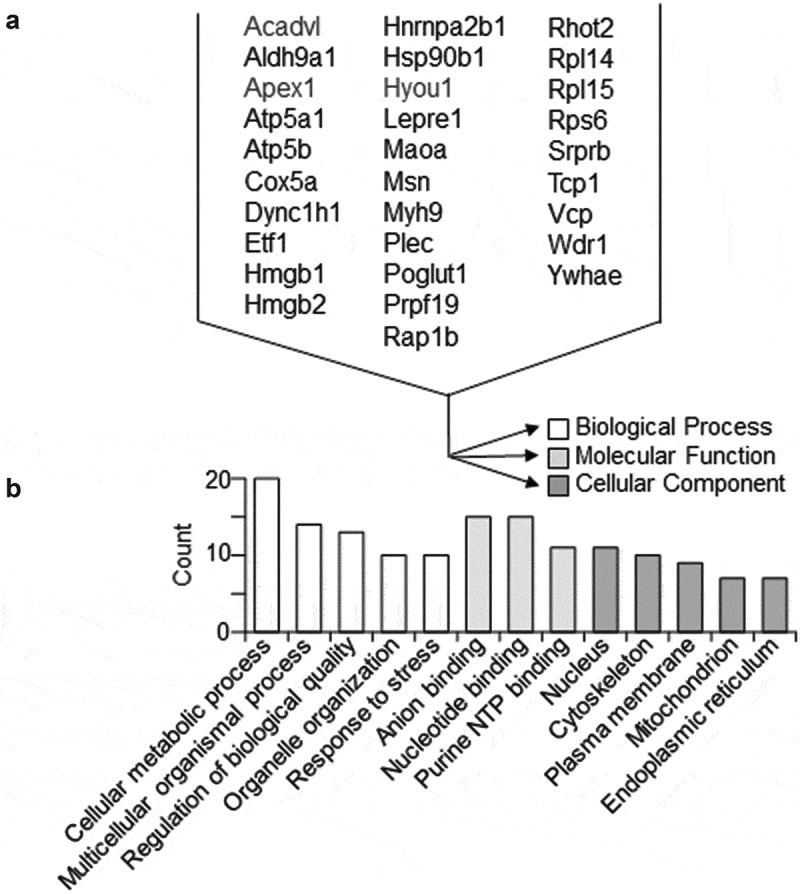
Proteomic identification of VSMC proteins significantly altered by EC EVs. Shotgun proteomic analyses were performed in whole-cell lysates from VSMCs incubated 16 h with EVs derived from serum-deprived ECs. Each population was analysed with 4 samples prepared from distinct culture plates. Rat gene names denote significantly detected peptide fragments in all 4 samples with the abundance more than 1.5 times compared with control vehicle (PBS) incubation (a). Gene ontology analysis for the 31 VSMC proteins significantly up-regulated by EC EVs (b).
Of the VSMC proteins encoded by the peptides which showed significant changes in response to EC-derived EVs, HMGB1 [47] and HMGB2 [48] play a prominent role in inflammation. To examine the extent to which EC EVs signal through these molecules to induce an inflammatory phenotype in VSMCs, VSMCs were pretreated with the HMGB1 inhibitor, glycyrrhizin [30] or the HMGB1/HMGB2 inhibitor, inflachromene [31]. Pharmacological inhibition of these HMGBs attenuated the induction of VCAM1 expression and adhesion of leukocytes in VSMCs exposed to EC-derived EVs (Figure 7(a,b)). In a separate set of experiments, VSMC were pretreated with HMGB1 siRNA prior to incubation with EC EVs. We confirmed that EC EVs up-regulated HMGB1 protein expression in VSMCs and both basal and stimulated expressions of HMGB1 was reduced by ~50% in HMGB1 siRNA treated cells. Moreover, depletion of HMGB1 attenuated VCAM1 induction and adhesion of THP1 cells in VSMCs exposed to EC EVs (Figure 8(a,b)).
Figure 7.
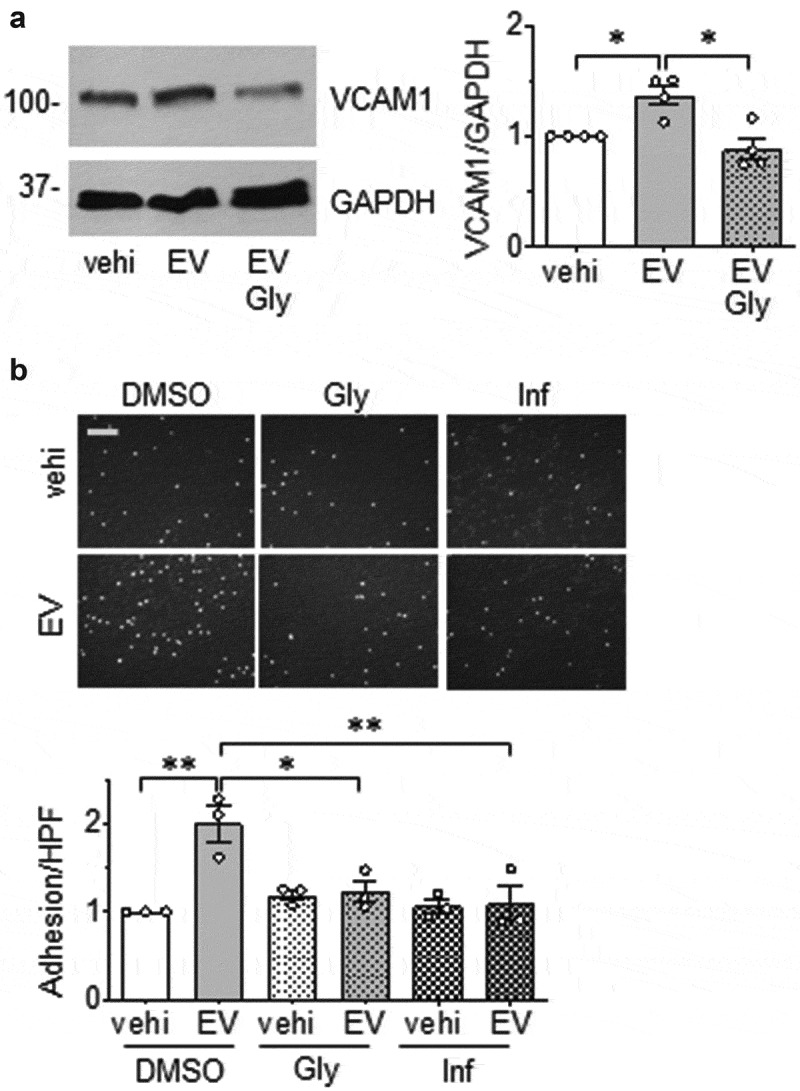
EC EVs induce an inflammatory VSMC phenotype via HMGB1 and HMGB2. (a). Serum starved (48 h) rat aortic VSMCs pretreated with or without glycyrrhizin (Gly) for 1 hour at 500 µmol/L were incubated with EC EVs (5 x 108 particles) for 6 hours and expression of VCAM-1 and GAPDH was determined by Western blotting (n = 4). (b). Serum-starved (48 h) rat aortic VSMCs were pre-treated with either DMSO, HMGB1 inhibitor glycyrrhizin (Gly), at 500 µmol/L or HMGB2/HMGB1 inhibitor inflachromene (Inf) at 25 µmol/L for 1 hour followed by stimulation with PBS or EC EVs (5x108) for 16 h. Cells were then incubated with THP-1 cells and adherent cells quantified (n = 3). The bars in the graphs show the mean±SEM. * and ** indicate p < 0.05 and 0.01, respectively.
Figure 8.
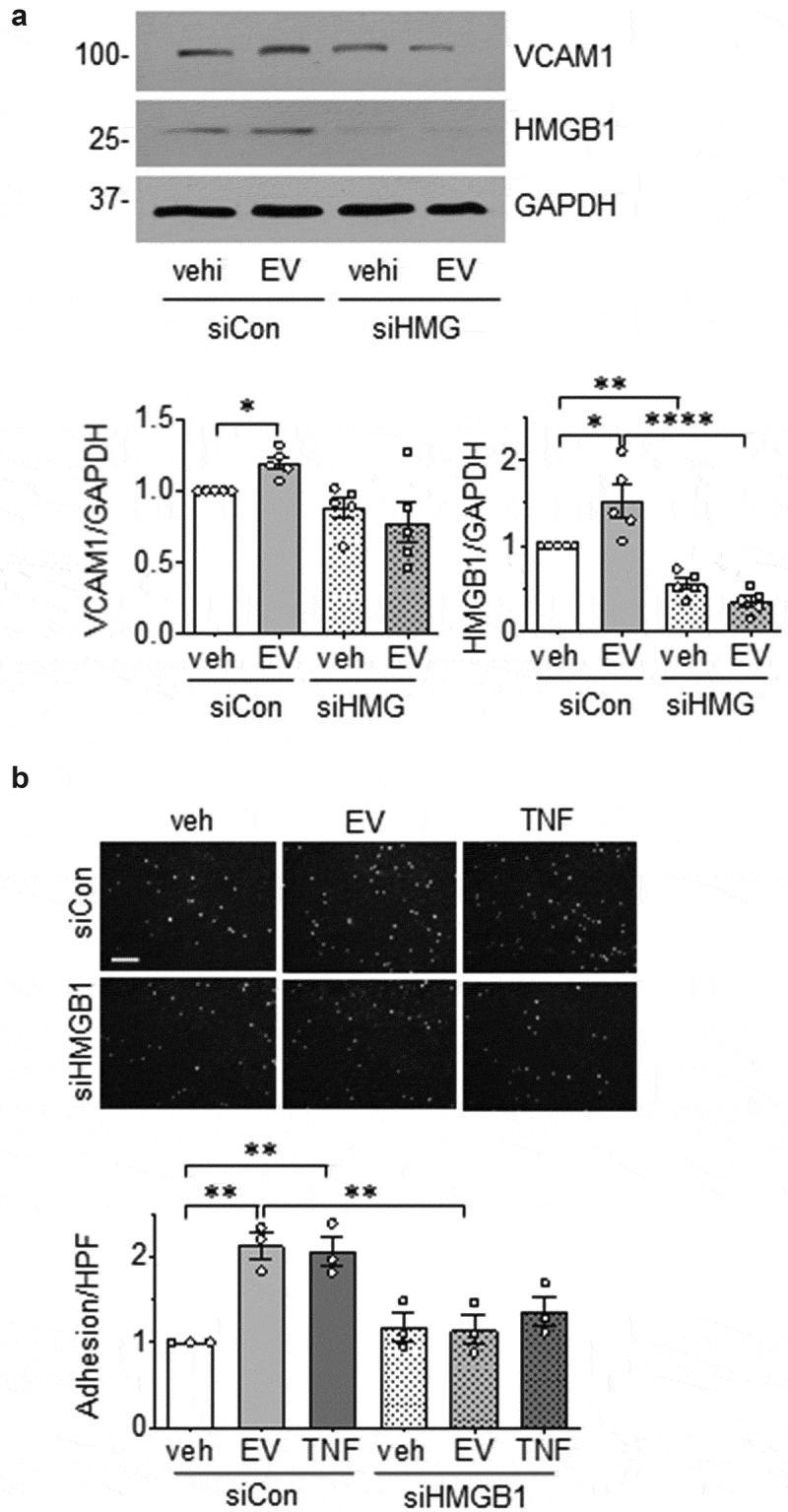
HMGB1 silencing in VSMC attenuates EC EV-induced inflammation. (a). VSMC were infected with adenovirus encoding either 100 MOI HMGB1 siRNA or 100 MOI control siRNA for a period of 72 h then incubated with EC EVs (5 x 108 particles) for 6 hours and expression of HMGB1, VCAM-1 and GAPDH was determined by Western blot analysis (n = 5). (b). VSMCs depleted of HMGB1 for at least for 48 h as above were incubated with vehicle, EC-derived EVs (5 x 108 particles) or TNF-α (10 ng/mL) for 16 h, washed and then incubated with THP-1 monocytes for 30 min. Adherent THP-1 cells were counted (n = 3). The bars in the graphs show the mean±SEM. *, **, **** indicate p < 0.05, 0.01, and 0.0001, respectively.
Discussion
In the present study, we have demonstrated that cultured endothelial and VSMCs release EVs. EVs from each cell type displayed physical characteristics (size and shape) consistent with vesicles and exosomes isolated by differential centrifugation [49]. Immunoblot analysis of EVs identified proteins considered to be “classical” markers of EVs, including exosomes. EC-derived vesicles were positive for flotillin-1 and tumour susceptibility gene-101 (TSG-101), while VSMC EVs were positive for flotillin-1 and syntenin-1. Curiously, the signal for syntenin-1 was only detected in the EV fraction and not in the whole cell lysate sample. The failure to detect this protein in the host cell likely relates to conditions that influence the sensitivity of the assay, such as the concentration of the total protein loaded into the SDS-PAGE gel which may have been below detection limits for that particular protein. Such findings have been observed in previous studies where ADAM10 and flotillin-1 were detected in EVs and not in the host cell [50] leading the authors to speculate that these proteins are greatly enrichmed in EVs relative to the whole cell. However, whether our findings indicate a similar enrichment of particular proteins in EC and VSMC EVs or a nuance of the Western blotting protocol requires further evaluation.
While we were unable to detect certain “canonical” transmembrane marker of EVs and exosomes: the tetraspanins, CD9, CD63, CD81 and CD82 [23] with immunoblotting (See Online Table I for these antibodies), mass spectroscopy revealed the presence of CD9 and CD82 in both VSMC and EC EVs confirming the expression of “canonical” tetraspanin molecules in these vesicles. A neutral sphingomyelinase inhibitor GW4869, which is known to inhibit EV release [33], reduced the number of EVs collected from EC and VSMC culture media by approximately 50% suggesting the active release of endosomal-derived EVs from these vascular cell types.
To further analyse the protein content of EVs, we performed mass spectroscopy on EVs collected from each vascular cell type. In addition to the proteins identified to be common to both cell populations, several proteins were found to be significantly abundant in either EC or VSMC EVs. This proteomic analysis marks one of the first direct comparisons of EVs from ECs and VSMCs, which provide future directions when assessing how endothelial and vascular smooth muscle EVs may function. Our analysis detected a relatively small number of proteins (<200) found in the vascular cell type EVs. It is our opinion that the number of proteins identified may be a conservative value and potentially underestimate the protein diversity in each vesicle type. The low yield is based partly on the processing of EVs (filtration and excessive washing to reduce contaminating proteins) which may have reduced the diversity of input material for mass spec analysis. In addition, we used a relatively high threshold level which may have ignored or eliminated certain peptides in the analysis. With this potential limitation in mind, characterization of both EC and VSMC EVs revealed that while 74 proteins were common between the two populations including many EV markers, there were also marked differences between them (120 and 44 unique for EC and VSMC, respectively). Of the unique proteins found in EC EVs, some include proteins involved in insulin signalling (Insulin-like growth factor-binding protein-2/IGFBP2, IGFBP4, and Insulin-like growth factor II), Lysyl oxidase homolog 2, a known elastin/collagen remodelling protein, and Lipolysis-stimulated lipoprotein receptor, a protein essential for fatty acid uptake. The presence of these proteins suggests several unique possibilities for EC EVs in biological function such as insulin signalling, fatty acid uptake, and matrix remodelling. Within the VSMC population, we observed Smooth muscle protein 22-alpha/transgelin, a smooth muscle-specific protein, and Matrix GLA protein, a protein previously identified as a marker of VSMC exosomes and involved in vascular calcification [51] as well as some adhesion molecules (VCAM-1, CD166, and Neural cell adhesion molecule 1). These proteins suggest potential roles for VSMC EVs in autocrine communication which may participate in the regulation of vascular function.
Similar to past reports demonstrating EV mediated communication between EC and VSMC [16,17], we found that EVs released from each cell type can traffic to and be internalized by the reciprocal cell type. Functionally, we surmised that the EC EVs could activate pathological signalling pathways in VSMCs since several proteins known to be involved in vascular pathology such IGFBP2, IGFBP4 [52], ICAM-1 [53], HMGB1 [54], Fibronectin [55], and Tissue inhibitor of metalloproteinase 2 [56] are present in these vesicles.
In response to EC EVs produced specifically under serum-free media conditions, VSMC significantly up-regulated the expression of VCAM-1 and concomitant increases in monocyte adhesion. This interaction with monocytes was seen to be dependent on VCAM-1 to some degree through the use of a VCAM-1 blocking antibody. The expression of VCAM-1 on VSMC has been implicated in several CVDs including atherosclerosis [57], hypertension [58] and neointimal formation [59]. Serum-free culturing of EC seems to be integral to the observed inflammatory response in VSMCs, as EVs produced from both ECs incubated in media containing EV-depleted FBS and cardiac fibroblast cultured under serum-free conditions did not replicate this response. Given the evidence that serum deprivation induces endothelial dysfunction [60] and EC dysfunction is a prominent feature of CVD [61], it appears that serum starved EC and their EVs are associated with pathological conditions. While further study will be needed to analyse what mechanistically occurs during serum deprivation that causes the release of pathological EVs, these finding reinforces the notion that the environment with which the cells reside affects the function of EVs they release. In addition to enhancing VCAM-1 expression, EVs from serum-deprived ECs increased protein synthesis in a time-dependent manner, a process that is likely associated with VSMC hypertrophy. We also found that induction of EGR-1 coincided with the increase in protein synthesis. Given that EGR-1 has been implicated in pathogenesis of atherosclerosis and VSMC dysfunction [44,62,63] through its ability to serve as a positive regulator of VCAM-1 expression [64,65], our findings suggest that EGR-1 might mediate pro-inflammatory and hypertrophic VSMC responses to EVs from serum-starved ECs.
The analysis of EC EV proteins that may be implicated in inflammation coincide with factors that are indicative of the senescence-associated secretory phenotype [66]. Stress-induced cellular senescence is a state that is characterized by increased secretion of cytokines, expression of lysosome β galactosidase activity, and increased DNA damage [67]. This state differs from terminal senescence as stress-induced senescence can occur independent of natural telomere attrition and inability to perform cell replication [68]. From this, it was hypothesized that EVs from serum-deprived ECs may serve as a mediator of a stress-induced ECs to propagate senescence. Indeed, significant increases in senescence-associated β galactosidase and the DNA damage marker γH2Ax were observed when VSMC were exposed to EVs from serum-starved ECs. These findings are in agreement with prior studies [69,70] demonstrating EVs as vehicles to transmit pro-senescent signals. Taken together, incubation of VSMC with EC EVs resulted in increases in immediate early gene expression (EGR-1), VCAM-1 expression, monocyte adherence and pro-hypertrophic/pro-senescence hallmarks, indicative of pathological vascular remodelling during disease progression [71].
To gain a broad view of how EC EVs affect overall VSMC phenotype, mass spectroscopy analysis was conducted on VSMC whole cell lysates following their exposure to EVs derived from serum-deprived ECs. We found several proteins that were significantly up-regulated given stringent cut-off analysis. We found increased abundance of ribosomal protein S6, a key regulator of cellular protein synthesis and cellular size [72] and signal recognition particle receptor β, which is essential for targeting of proteins to the endoplasmic reticulum for secretion or membrane insertion [46]. Additionally, many proteins involved in cellular metabolism and mitochondrial function were found up-regulated, including ETC Complex IV member Cox5a, ATP Synthase members ATP5a1/ATP5b, and regulator of mitochondrial movement/division Miro2. Increases in these markers may be indicative of increased mitochondrial mass and ATP demand, which, along with increases in Rps6 (indicative of cellular growth/size) are hallmarks found in cellular senescence [73]. Although rigorous analysis is warranted to confirm these targets, these findings further support a role for EC EVs in mediating VSMC protein synthesis, protein/organelle trafficking, and metabolic reprogramming.
Two additional VSMC proteins of particular interest were HMGB1 and HMGB2, which were found significantly up-regulated in VSMC by EVs from serum-starved ECs. Both act as nuclear proteins that control chromatin remodelling but have differing roles in inflammatory processes, cellular senescence and senescence-associated secretory phenotype (SASP), a state characterized with irreversible cell cycle arrest and secretion of inflammatory molecules [66]. HMGB1 is known to translocate from the nucleus to the extracellular space to act as an alarmin and is identified as a key member of the SASP [47]. HMGB2, while highly similar in amino acid identity to HMGB1, is uniquely associated with neointimal hyperplasia, pathological cell growth, cancer progression, and serves as a critical transcription factor for driving the gene expression necessary for SASP [48,74,75]. In addition, HMGB1/2 are critical non-histone chromatin binding proteins and may bind many different transcription factors to facilitate transcription or bend DNA themselves to initiate transcription [76]. Here, we found that EC EVs induce an inflammatory phenotype in VSMCs via a mechanism that includes HMGBs, as pharmacological inhibition of HMGB1 and HMGB2 and siRNA depletion of HMGB1 attenuated EC EV mediated up-regulation of VCAM1 and monocyte adhesion in VSMCs. While we observed a relatively weaker VCAM-1 response in VSMCs in these experiments compared to other experiments conducted in this study, a phenomenon that is likely due to distinct experimental condition where the inhibitor vehicle, infection of adenoviruses or GFP transduction might influence certain cellular processes to temper the robustness of the EV response, our findings are in agreement with past studies demonstrating a role of HMGB1 in activating VCAM1 through transcription factor binding directly to NF-kB family members [77].
The results of the present study enhance our understanding of the role that EVs play in the vasculature and marks the relevance of EC-EVs as an important means of intravascular communication in vascular disease. The extent to which specific proteins, or other elements such as miRNA or lipids, contained within the EC EVs contribute to the induction of VSMC dysfunctional phenotype will require further investigation.
Supplementary Material
Acknowledgments
We thank Akito Eguchi and Walter J. Koch for providing rat cardiac fibroblasts.
Author contributions
M.J.B., R.S., S.E. and V.R. designed the study. M.J.B., Y.K., T.A., A.Y.B., and K.J.P. performed the experiments. M.J.B, Y.K., S.E. and V.R. wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bang C, Thum T.. Exosomes: new players in cell-cell communication. Int J Biochem Cell Biol. 2012;44:2060–19. [DOI] [PubMed] [Google Scholar]
- [2].Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–1232. [DOI] [PubMed] [Google Scholar]
- [3].Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017 Update: a Report From the American Heart Association. Circulation. 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lacolley P, Regnault V, Segers P, et al. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol Rev. 2017;97:1555–1617. [DOI] [PubMed] [Google Scholar]
- [6].Majesky MW. Adventitia and perivascular cells. Arterioscler Thromb Vasc Biol. 2015;35:e31–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lilly B. We have contact: endothelial cell-smooth muscle cell interactions. Physiology (Bethesda). 2014;29:234–241. [DOI] [PubMed] [Google Scholar]
- [8].Hutcheson JD, Aikawa E. Extracellular vesicles in cardiovascular homeostasis and disease. Curr Opin Cardiol. 2018;33:290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jansen F, Nickenig G, Werner N. Extracellular vesicles in cardiovascular disease: potential applications in diagnosis, prognosis, and epidemiology. Circ Res. 2017;120:1649–1657. [DOI] [PubMed] [Google Scholar]
- [10].Wang JM, Su C, Wang Y, et al. Elevated circulating endothelial microparticles and brachial-ankle pulse wave velocity in well-controlled hypertensive patients. J Hum Hypertens. 2009;23:307–315. [DOI] [PubMed] [Google Scholar]
- [11].Feng B, Chen Y, Luo Y, et al. Circulating level of microparticles and their correlation with arterial elasticity and endothelium-dependent dilation in patients with type 2 diabetes mellitus. Atherosclerosis. 2010;208:264–269. [DOI] [PubMed] [Google Scholar]
- [12].Koga H, Sugiyama S, Kugiyama K, et al. Elevated levels of VE-cadherin-positive endothelial microparticles in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol. 2005;45:1622–1630. [DOI] [PubMed] [Google Scholar]
- [13].Tramontano AF, Lyubarova R, Tsiakos J, et al. Circulating endothelial microparticles in diabetes mellitus. Mediators Inflamm. 2010;2010:250476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Andrews AM, Rizzo V. Microparticle-induced activation of the vascular endothelium requires Caveolin-1/Caveolae. PLoS One. 2016;11:e0149272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Burger D, Montezano AC, Nishigaki N, et al. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol. 2011;31:1898–1907. [DOI] [PubMed] [Google Scholar]
- [16].Zheng B, Yin WN, Suzuki T, et al. Exosome-mediated miR-155 transfer from smooth muscle cells to endothelial cells induces endothelial injury and promotes atherosclerosis. Mol Ther. 2017;25:1279–1294. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [17].Hergenreider E, Heydt S, Treguer K, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14:249–256. [DOI] [PubMed] [Google Scholar]
- [18].de Jong OG, Verhaar MC, Chen Y, et al. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J Extracell Vesicles. 2012;1:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee SB, Kim JJ, Kim TW, et al. Serum deprivation-induced reactive oxygen species production is mediated by Romo1. Apoptosis. 2010;15:204–218. [DOI] [PubMed] [Google Scholar]
- [20].Russell FD, Hamilton KD. Nutrient deprivation increases vulnerability of endothelial cells to proinflammatory insults. Free Radic Biol Med. 2014;67:408–415. [DOI] [PubMed] [Google Scholar]
- [21].Rashid MU, Coombs KM. Serum-reduced media impacts on cell viability and protein expression in human lung epithelial cells. J Cell Physiol. 2019;234:7718–7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Forrester SJ, Elliott KJ, Kawai T, et al. Caveolin-1 deletion prevents hypertensive vascular remodeling induced by angiotensin II. Hypertension. 2017;69:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Thery C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;30(1):3.22.1–3.22.29 Chapter 3:Unit 3 22. [DOI] [PubMed] [Google Scholar]
- [24].An M, Lohse I, Tan Z, et al. Quantitative proteomic analysis of serum exosomes from patients with locally advanced pancreatic cancer undergoing chemoradiotherapy. J Proteome Res. 2017;16:1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nakamura H, Takahashi-Jitsuki A, Makihara H, et al. Proteome and behavioral alterations in phosphorylation-deficient mutant collapsin response mediator Protein2 knock-in mice. Neurochem Int. 2018;119:207–217. [DOI] [PubMed] [Google Scholar]
- [26].Collot M, Ashokkumar P, Anton H, et al. MemBright: A family of fluorescent membrane probes for advanced cellular imaging and neuroscience. Cell Chem Biol. 2019;26:600–614.e7. DOI: 10.1016/j.chembiol.2019.01.009 [DOI] [PubMed] [Google Scholar]
- [27].Schmidt EK, Clavarino G, Ceppi M, et al. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. [DOI] [PubMed] [Google Scholar]
- [28].Nakashima H, Frank GD, Shirai H, et al. Novel role of protein kinase C-delta Tyr 311 phosphorylation in vascular smooth muscle cell hypertrophy by angiotensin II. Hypertension. 2008;51:232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grudzenski S, Raths A, Conrad S, et al. Inducible response required for repair of low-dose radiation damage in human fibroblasts. Proc Natl Acad Sci U S A. 2010;107:14205–14210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mollica L, De Marchis F, Spitaleri A, et al. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol. 2007;14:431–441. [DOI] [PubMed] [Google Scholar]
- [31].Lee S, Nam Y, Koo JY, et al. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat Chem Biol. 2014;10:1055–1060. [DOI] [PubMed] [Google Scholar]
- [32].Elliott KJ, Bourne AM, Takayanagi T, et al. ADAM17 silencing by adenovirus encoding miRNA-embedded siRNA revealed essential signal transduction by angiotensin II in vascular smooth muscle cells. J Mol Cell Cardiol. 2013;62:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Trajkovic K, Hsu C, Chiantia S, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. [DOI] [PubMed] [Google Scholar]
- [34].Kosaka N, Iguchi H, Yoshioka Y, et al. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vlassov AV, Magdaleno S, Setterquist R, et al. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820:940–948. [DOI] [PubMed] [Google Scholar]
- [36].Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907–1920. [DOI] [PubMed] [Google Scholar]
- [37].Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. DOI: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- [38].Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. [DOI] [PubMed] [Google Scholar]
- [39].Barks JL, McQuillan JJ, Iademarco MF. TNF-alpha and IL-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J Immunol. 1997;159:4532–4538. [PubMed] [Google Scholar]
- [40].Touyz RM, Alves-Lopes R, Rios FJ, et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Iwasaki S, Ingolia NT. The growing toolbox for protein synthesis studies. Trends Biochem Sci. 2017;42:612–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Forrester SJ, Booz GW, Sigmund CD, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98:1627–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bouallegue A, Simo Cheyou ER, Anand-Srivastava MB, et al. ET-1-induced growth promoting responses involving ERK1/2 and PKB signaling and Egr-1 expression are mediated by Ca2+/CaM-dependent protein kinase-II in vascular smooth muscle cells. Cell Calcium. 2013;54:428–435. [DOI] [PubMed] [Google Scholar]
- [44].Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circ Res. 2006;98:186–191. [DOI] [PubMed] [Google Scholar]
- [45].Kunieda T, Minamino T, Nishi J, et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114:953–960. [DOI] [PubMed] [Google Scholar]
- [46].Akopian D, Shen K, Zhang X, et al. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem. 2013;82:693–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Davalos AR, Kawahara M, Malhotra GK, et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201:613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Aird KM, Iwasaki O, Kossenkov AV, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol. 2016;215:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–228. [DOI] [PubMed] [Google Scholar]
- [50].Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kapustin AN, Shanahan CM. Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J Physiol. 2016;594:2905–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hoeflich A, David R, Hjortebjerg R. Current IGFBP-Related biomarker research in cardiovascular disease-we need more structural and functional information in clinical studies. Front Endocrinol (Lausanne). 2018;9:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep. 2009;61:22–32. [DOI] [PubMed] [Google Scholar]
- [54].Li W, Sama AE, Wang H. Role of HMGB1 in cardiovascular diseases. Curr Opin Pharmacol. 2006;6:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Moore KJ, Fisher EA. The double-edged sword of fibronectin in atherosclerosis. EMBO Mol Med. 2012;4:561–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Di Gregoli K, George SJ, Jackson CL, et al. Differential effects of tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 on atherosclerosis and monocyte/macrophage invasion. Cardiovasc Res. 2016;109:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Braun M, Pietsch P, Schror K. Cellular adhesion molecules on vascular smooth muscle cells. Cardiovasc Res. 1999;41:395–401. [DOI] [PubMed] [Google Scholar]
- [58].Tummala PE, Chen XL, Sundell CL, et al. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation. 1999;100:1223–1229. [DOI] [PubMed] [Google Scholar]
- [59].Ellison S, Gabunia K, Richards JM, et al. IL-19 reduces ligation-mediated neointimal hyperplasia by reducing vascular smooth muscle cell activation. Am J Pathol. 2014;184:2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Scioli MG, Bielli A, Agostinelli S, et al. Antioxidant treatment prevents serum deprivation- and TNF-alpha-induced endothelial dysfunction through the inhibition of NADPH oxidase 4 and the restoration of beta-oxidation. J Vasc Res. 2014;51:327–337. [DOI] [PubMed] [Google Scholar]
- [61].Daiber A, Steven S, Weber A, et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. 2017;174:1591–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Harja E, Bucciarelli LG, Lu Y, et al. Early growth response-1 promotes atherogenesis: mice deficient in early growth response-1 and apolipoprotein E display decreased atherosclerosis and vascular inflammation. Circ Res. 2004;94:333–339. [DOI] [PubMed] [Google Scholar]
- [63].Wang CC, Sharma G, Draznin B. Early growth response gene-1 expression in vascular smooth muscle cells effects of insulin and oxidant stress. Am J Hypertens. 2006;19:366–372. [DOI] [PubMed] [Google Scholar]
- [64].Wada Y, Fujimori M, Suzuki J, et al. Egr-1 in vascular smooth muscle cell proliferation in response to allo-antigen. J Surg Res. 2003;115:294–302. [DOI] [PubMed] [Google Scholar]
- [65].Albrecht C, Preusch MR, Hofmann G, et al. Egr-1 deficiency in bone marrow-derived cells reduces atherosclerotic lesion formation in a hyperlipidaemic mouse model. Cardiovasc Res. 2010;86:321–329. [DOI] [PubMed] [Google Scholar]
- [66].Coppe JP, Desprez PY, Krtolica A, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Katsuumi G, Shimizu I, Yoshida Y, et al. Vascular senescence in cardiovascular and metabolic diseases. Front Cardiovasc Med. 2018;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pascal T, Debacq-Chainiaux F, Chretien A, et al. Comparison of replicative senescence and stress-induced premature senescence combining differential display and low-density DNA arrays. FEBS Lett. 2005;579:3651–3659. [DOI] [PubMed] [Google Scholar]
- [69].Borghesan M, Fafian-Labora J, Eleftheriadou O, et al. Small extracellular vesicles are key regulators of non-cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep. 2019;27:3956–3971.e6. DOI: 10.1016/j.celrep.2019.05.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Mensa E, Guescini M, Giuliani A, et al. Small extracellular vesicles deliver miR-21 and miR-217 as pro-senescence effectors to endothelial cells. J Extracell Vesicles. 2020;9:1725285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–348. [DOI] [PubMed] [Google Scholar]
- [73].Wiley CD, Campisi J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 2016;23:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Franklin S, Chen H, Mitchell-Jordan S, et al. Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth. Mol Cell Proteomics. 2012;11(M111):014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kapustin AN, Schoppet M, Schurgers LJ, et al. Prothrombin loading of vascular smooth muscle cell-derived exosomes regulates coagulation and calcification. Arterioscler Thromb Vasc Biol. 2017;37:e22–e32. [DOI] [PubMed] [Google Scholar]
- [76].Kang R, Chen R, Zhang Q, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Agresti A, Lupo R, Bianchi ME, et al. HMGB1 interacts differentially with members of the Rel family of transcription factors. Biochem Biophys Res Commun. 2003;302:421–426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.