

ABSTRACT
Pregnancy is a unique situation, in which placenta-derived small extracellular vesicles (sEVs) may communicate with maternal and foetal tissues. While relevant to homoeostatic and pathological functions, the mechanisms underlying sEV entry and cargo handling in target cells remain largely unknown. Using fluorescently or luminescently labelled sEVs, derived from primary human placental trophoblasts or from a placental cell line, we interrogated the endocytic pathways used by these sEVs to enter relevant target cells, including the neighbouring primary placental fibroblasts and human uterine microvascular endothelial cells. We found that trophoblastic sEVs can enter target cells, where they retain biological activity. Importantly, using a broad series of pharmacological inhibitors and siRNA-dependent silencing approaches, we showed that trophoblastic sEVs enter target cells using macropinocytosis and clathrin-mediated endocytosis pathways, but not caveolin-dependent endocytosis. Tracking their intracellular course, we localized the sEVs to early endosomes, late endosomes, and lysosomes. Finally, we used coimmunoprecipitation to demonstrate the association of the sEV microRNA (miRNA) with the P-body proteins AGO2 and GW182. Together, our data systematically detail endocytic pathways used by placental sEVs to enter relevant fibroblastic and endothelial target cells, and provide support for “endocytic escape” of sEV miRNA to P-bodies, a key site for cytoplasmic RNA regulation.
KEYWORDS: Small extracellular vesicles, placenta, miRNA, macropinocytosis, clathrin, P-bodies
Introduction
The discovery of extracellular vesicles (EVs) and their cargo has added a new dimension to our understanding of communication among nearby or distant cells and tissues. Small EVs (sEVs, which largely include exosomes [1,2]) are formed as intraluminal vesicles (30–150 nm in diameter) by the inward budding of the endosomal membrane during multivesicular body (MVB) maturation, and are released to the extracellular space after the MVB fuses with the plasma membrane [3–6]. Small EVs play an important role in diverse physiologically and pathologically relevant intercellular communication processes [7,8]. Deciphering the influence of sEVs on target (recipient) cells is germane for our understanding of assorted conditions, such as cancer, autoimmunity, metabolic syndrome, and ageing [9–20].
The cargo of sEVs is defined by cell type and biological context and comprises nucleic acids, proteins, and other bioactive molecules. Among nucleic acids, sEVs harbour tRNAs and small non-coding RNAs, including the 19- to 24-nt microRNAs (miRNAs) that commonly bind to the 3ʹ end of mRNA and modulate its stability and translation at the RNA-induced silencing complex (RISC) [9]. miRNAs can traffic between donor and target cells either within smaller or larger EVs, or when bound by plasma proteins. The delivery of sEV miRNA into target cells has been implicated in a multitude of developmental and pathological functions [9–17].
During pregnancy, the placenta is uniquely positioned to mediate biological communication between the mother and the foetus. The basic exchange unit of the human placenta is the villus, where trophoblasts (including the syncytiotrophoblasts and the subjacent cytotrophoblasts) that line the villous surface are directly bathed in maternal blood. Other major components of the villous core include fibroblasts, endothelial cells and Hofbauer macrophages [21,22]. In addition to pregnancy-related hormones and growth factors, recent data suggest that maternal-foetal communication in the form of sEVs plays an important role in many homeostatic functions [23–25]. Interestingly, the level of placenta-derived sEVs in the maternal blood gradually rises during human pregnancy [18,19,26,27]. These sEVs were shown to modulate maternal immunity by activating NK cell receptor NKG2D, and recruitment and differentiation of monocytes [28,29]. Placental sEVs have also been implicated in vascular smooth muscle response [30,31]. We found that released placental trophoblastic sEVs could attenuate viral infection in non-trophoblastic cells and that this effect was mediated, at least in part, by the unique miRNA repertoire, representing the placenta-specific miRNA transcribed from the Chromosome 19 miRNA cluster (C19MC) [32–35]. Unlike other tissues, where discrete proteins may support selective sorting of cellular miRNA species into sEVs [36–39], we did not observe an enrichment of C19MC miRNA in trophoblastic sEVs [23]. We have created a transgenic mouse humanized for the primate- and placenta-specific C19MC miRNA [40]. Tracking the trafficking of these miRNAs into target organs and using them as a surrogate for sEV uptake suggested a tissue-specific pattern of sEV uptake. Others have used fluorescently-labelled placental sEVs to detect their selective spread primarily to maternal lungs, liver, and kidneys [41].
It has been assumed that EVs, including sEVs, are internalized by acceptor target cells using distinct mechanisms that are mechanistically linked to their function in their target cells [11,42–44]. Therefore, elucidating sEV uptake mechanisms is crucial for defining their impact on cell function. In the context of placental sEVs, limited data suggested that phagocytosis and clathrin-mediated endocytosis mediate <10% of the uptake of placental sEVs into human microvascular endothelial cell line (HMEC-1) cells [41]. A potential mixture of sEV subtypes during their isolation has also been suggested [44]. Importantly, the intracellular fate of sEV-carried, exogenous miRNA cargo has not been elucidated. To interrogate the uptake mechanisms and intracellular trafficking of sEVs and their miRNA cargo, we used the gold-standard, density gradient ultracentrifugation to derive physiologically relevant sEVs from term primary human trophoblasts (PHT cells) and from the trophoblast cell line BeWo to assess sEV uptake into human primary placental fibroblasts (PPF) and primary human uterine microvascular endothelial cells (HUtMEC), representing placental neighbouring and distant putative target cells, respectively. We surmised that the small size of sEVs may enable them to be taken up by discrete endocytic pathways, and that exogenous miRNA may localize to the RISC complex in P-bodies.
Materials and methods
Cell culture
Human placenta collection, cell dispersal, and placenta-derived cell culture were reviewed and approved by the Institutional Review Board at the University of Pittsburgh. All placentas were obtained from uncomplicated term deliveries. PHT cells were isolated using the trypsin-DNase-dispase/Percoll method as described by Kliman et al. [45], with previously published modifications from our lab [46,47]. PHT cells were cultured in DMEM (Sigma-Aldrich, St. Louis, MO) containing 10% Bovine Growth Serum (HyClone, Logan, UT) and 1% P/S antibiotics from Sigma-Aldrich at 37°C in a 5% CO2-air atmosphere. The BeWo trophoblast cell line (ATCC, Manassas, VA) was maintained in F12K Kaighn’s modified medium (Gibco, Gaithersburg, MD) supplemented with 10% bovine growth serum (BGS; HyClone) and antibiotics. Human Uterine Microvascular Endothelial Cells (HUtMEC, PromoCell, Burlington, Ontario) were cultured in EBM medium (Lonza, Walkersville, MD) supplemented with EGM-2 MV SingleQuots (Lonza). PPF cells were isolated during standard placental cell isolation, as we routinely perform in our lab, with further purification of PPF cells [32] using a magnetic-activated cell sorting separation unit after the cells were incubated with a mouse anti-CD9 antibody (BS3022, Bioworld), as reported [48–50] and validated using anti-Vimentin antibody (Clone V9, #M0725, Dako, Carpinteria, CA). PPF cells were cultured in DMEM with 10% BGS and antibiotics. HeLa cells were obtained from ATCC and maintained in EMEM with 10% BGS, glutamine, non-essential amino acids, and antibiotics. All inhibitors used in the study, their source and concentrations are listed in Table 1. Unless stated otherwise, inhibitor experiments were performed in serum-free medium.
Table 1.
Inhibitors used in the experiments.
| Inhibitors | Company | Catalogue number | Stock concentration |
|---|---|---|---|
| Chlorpromazine | Sigma Aldrich | C8138-5 G | 100 mM |
| Filipin III | Sigma Aldrich | F4767-1 MG | 10 mM |
| Nystatin | Sigma Aldrich | N6261-5MU | 25 mM |
| MβCD (Methyl-β-cyclodextrin) | Sigma Aldrich | C4555-1 G | 100 mM |
| EIPA | Sigma Aldrich | A3085-25 MG | 100 mM |
| Nocodazole | Sigma Aldrich | M1404-2 mg | 20 mM |
| Cytochalasin D | Sigma Aldrich | C8273-1 mg | 10 mM |
| Wortmannin | Thermo Fisher | PHZ1301 | 1 mM |
| U73122 | Abcam | ab120998 | 5 mM |
| Bisindolylmaleimide I | Santa Cruz Biotech | sc-24003A | 5 mM |
| IPA-3 | Sigma Aldrich | I2285-5 MG | 20 mM |
| ML141 | Sigma Aldrich | SML0407-5 MG | 10 mM |
| Wiskostatin | Sigma Aldrich | W2270-5 MG | 50 mM |
| CK869 | Sigma Aldrich | 182516–25 MG-M | 100 mM |
| Simvastatin | Sigma Aldrich | 38956–10 MG | 50 mM |
Plasmids and cell transfections
For construction of pLV-EF1a-mCherry-TSG101-NanoLuc-P2A-EGFP, we PCR-amplified mCherry-TSG101 insert, derived from pEF6.mCherry-TSG101 (Addgene Cat. #38318), with flanking 3ʹ BamHI and 5ʹ BsiWI sites. The mCherry-TSG101 insert was ligated into a lentiviral backbone cut with BamHI/BsiWI to fuse the Nanoluciferase (NanoLuc) at the C-terminus of TSG101. Lentiviruses were prepared according to the standard procedures. BeWo cells were transduced with lentiviruses bearing this plasmid in the presence of 8 µg/ml polybrene (Sigma Aldrich) at 37°C overnight. Cells stably expressing mCherry-TSG101-NanoLuc-P2A-EGFP were expanded following sorting mCherry-positive fluorescence by FACS.
A plasmid pLJM1-FH-AGO2-WT with FLAG-HA tag N-terminal to AGO2 was obtained from Addgene (Cat. #91978). PPF cells were transduced with lentiviruses bearing pLJM1-FH-AGO2-WT, and positive clones were selected using puromycin (Thermo Fisher, Waltham, MA).
sEV Isolation from trophoblast conditioned medium
At 4 h after seeding, the PHT cells were washed three times with particle-free PBS (Sigma, particle-free buffer was corroborated by our NanoSight nanoparticle tracking device) and cultured in red phenol-free DMEM medium that contained 1% antibiotics and 10% vesicle-depleted foetal bovine serum (FBS, Thermo Fisher). The conditioned medium was collected for sEV isolation after 48–72 h. For BeWo conditioned medium, cells were cultured in 50/50% red phenol-free DMEM/F12K Kaighn’s modified medium (Gibco) with 1% antibiotics and 10% vesicle-depleted FBS, as above. The conditioned medium was collected after 48–72 h. Typically, 1,000 ml of conditioned medium was collected from BeWo cells after 2–3 days in culture, when the cells reach ~90% confluence (~10 million cells per 10 cm plate) or from a similar number of PHT cells at 100% confluence. For sEV isolation, performed at 4°C as previously described, using continuous iodixanol-based gradient ultracentrifugation [33]. Briefly, the medium was first centrifuged at 500 g to remove cell debris, and the supernatant was centrifuged at 2500 g to deplete apoptotic bodies, followed by centrifugation at 12,000 g to remove microvesicles. After 0.22 µm filtration and a Vivacell 100 filtration unit (100 kDa MW cut-off, Sartorius, New York, NY), the medium was diluted in PBS and ultracentrifuged at 100,000 g overnight to pellet the sEVs. This crude sEV pellet was suspended in PBS and mixed with 1.5 ml of 60% iodixanol (OptiPrep, D1556, Sigma-Aldrich). The mixture was laid at the bottom of a tube and overlaid with 10 ml of 6–40% OptiPrep gradient using a gradient formation chamber and peristaltic pump. After 20–22 h of gradient ultracentrifugation at 100,000 g, the purified sEVs were collected [33], filtered and concentrated in a Vivacell 20 filtration unit. Small EV purity was validated using a Nanoparticle Tracking Analysis System (NTA, NanoSight, Malvern Instruments, Westborough, MA, Suppl. Fig. 9A) Protein concentration was determined using the Micro BCA method, according to the manufacturer’s instructions (Thermo Fisher), and several proteins were detected using western immunoblotting to validate the quality of our preparations (Suppl. Fig. 9B). Our typical yield from 1,000 ml of medium is ~600 µg of sEVs, where we obtain 1.1 × 109 sEVs per µg of sEVs.
Labelling and electroporation of sEVs
All luciferase-based uptake experiments were performed using sEVs that were collected and purified from conditioned medium of BeWo cells stably transfected with mCherry-TSG101-NanoLuc (see above). Small EVs were also labelled, during isolation, with 10 µM SP-DiI (SP-DiIC18(3) (1,1ʹ-Dioctadecyl-6,6ʹ-Di(4-Sulfophenyl)-3,3,3ʹ,3ʹ-Tetramethylindocarbocyanine, # D7777, Thermo Fisher) or with 1 µM of Vybran SP-DiO Cell-Labelling Solution (V22886, Thermo Fisher) in PBS at 37°C for 30 min. The sEVs were purified as described above.
Electroporation was done in citric acid buffer (20 mM citric acid, 20 mM Na2HPO4, and 0.1 mM EDTA at pH 4.6). sEVs (40 µg protein) and miRNA-517A (10 µg, Thermo Fisher) were mixed in 800 µl of citric acid buffer and electroporated with 2 pulses of 200 V, 150 µF capacity, 1000 ohm resistance, using a 4-mm cuvette in a Harvard BTX (Holliston, MA) electroporator. The electroporated sEVs were left on ice for 10 min, then pelleted by ultracentrifugation at 120,000 g for 45 min. For larger preparations, sEVs from the electroporation procedure were pulled in 300 µl of PBS containing 20 mM HEPES and RNaseA 0.1 µg/ml (0.25 Unit/ml, Thermo Fisher) at room temperature for 10 min. RNAse was blocked with 100 U/ml of supRNase inhibitor (Thermo Fisher), PBS was added to a final volume of 3 ml, and the samples were ultracentrifuged at 120,000 g for 45 min to pellet the sEVs, which were resuspended in PBS.
Vesicular stomatitis virus (VSV) infection assay
For control of trophoblastic sEV function, VSV infection assay was performed as recently described [32–34]. Briefly, target cells were seeded in 24-well plates before incubation with sEVs in complete medium for 16–18 h, at 37°C. The cells were then infected with VSV, at multiplicity of infection (MOI) equivalent to 0.5–1, for 5 h, and washed with PBS. Total RNA was extracted using QIAzol (Qiagen, Germantown, MD), and VSV infection was quantified by RT-qPCR using VSV primers, as previously described [32–34].
Cell viability assays
The cells were plated in a 96-well plate at 10,000 cells/well in 100 µl complete medium, with medium without cells for control. After 48 h, the medium was changed, and inhibitors were added at the concentrations specified in the Results section. After 4 h incubation 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2 H-tetrazolium (MTS, Sigma Aldrich, 20 µl/well)) was added, and incubation continued for an additional 2 h. The absorbance was measured at 490 nm using a Versa-max microplate reader (Molecular Devices, San Jose, CA). Cell viability was calculated by comparing absorbance to the control group (DMSO only).
Quantification of primary, precursor, and mature miRNA by RT- qPCR
Quantification of mature miRNA was performed as described [32–34]. Quantification of the primary or precursor miRNA was performed by RT-qPCR using primers from Qiagen and based on the manufacturer’s detection protocol. Briefly, total cellular RNA was extracted using TRIzol (Thermo Fisher), then reverse transcribed at 37°C for 2 h for precursor miRNA with Qiagen miScript II RT Kit (#218161, Qiagen) or, for primary miRNA, with High-Capacity cDNA Reverse Transcription Kit (# 4368813, Thermo Fisher). The cDNA was diluted 10-fold for qPCR detection with the ViiA 7 system (Life Sciences Applied Biosystems, Foster City, CA). Precursor miRNA was detected by using the miScript SYBR Green PCR Kit (#218076, Qiagen), and primary miRNA was measured using TaqMan Gene Expression Master Mix (#4370048, Thermo Fisher). Gene expression was calculated using the 2-ΔΔCT method [51], normalized to either mature miR-21, precursor miR-21, or primary miR-21, where appropriate.
Protein knockdown using siRNA
All siRNA duplex constructs used in this project were obtained from Dharmacon (Lafayette, CO) using On-Target plus siRNA SMARTpool. For silencing, cultured cells (PPF or HUtMEC, 12- or 24-well plate) were transfected with 50 nM siRNA or control siRNA and lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA), 7.5 µl/ml for PPF cells and 5 µl/ml lipofectamine for HUtMEC cells. The cells were incubated overnight with the siRNA mix. Fresh medium was then added, and the cells were cultured for an additional 48–72 h before analysis. The efficiency of siRNA knockdown was determined by western immunoblotting (below.)
Western immunoblotting
The full names of all proteins, antibody information, and concentrations are provided in Table 2. Western analysis was performed as described previously [40]. Briefly, cell or sEV preparations were lysed in lysis buffer (50 mM Tris-HCI, 150 mM NaCI, and 1% Triton X-100), supplemented with protease inhibitors (Roche, Indianapolis, IN) and phosphatase inhibitor cocktail (PhosSTOP, Roche, Mannheim, Germany). The lysate was mixed with Laemmli sample buffer, heated at 95°C and electrophoretically separated (10% or 12% for small molecular weight protein detection) and transferred to PVDF membranes (#1620177, BioRad, Hercules, CA). The membranes were blocked in 10% non-fat-dried milk and immunoblotted with the respective primary antibody, all listed in Table 2. Each was followed by appropriate horseradish peroxidase (HRP) secondary antibody (details in Table 2). Signals were visualized using WesternBright Sirius (K-12043-D20, Thermo Fisher). Each immunoblotting experiment was performed at least three times.
Table 2.
Proteins and their respective antibodies, used in the experiments.
| Full protein name | Protein abbreviation primary antibody |
Research resource identifier (RRID) | Polyclonal or monoclonal | Species made in | Concentration (stock or final, if N/A, then titre) |
Company | Catalogue number |
|---|---|---|---|---|---|---|---|
| Actin | Actin | AB_2223041 | Monoclonal | Mouse | 1:1,000 dilution | SIGMA/Millipore | MAB1501 |
| Argonaute 2 | AGO2 | H-monoclonal-clone 11A9 | Rat | 0.25 mg/ml | Sigma | MABE253 | |
| Adaptor Protein, Phosphotyrosine Interacting With PH Domain And Leucine Zipper 1 | APPL1 | AB_2056989 | Monoclonal | Rabbit | 1:1,000 dilution | Cell Signalling | 3858S |
| Actin Related Protein 3 | ARP3 | AB_626700 | Monoclonal | Mouse | 200 mg/ml 1:1,000 dilution |
Santa Cruz Biotech | sc-48344 |
| Calnexin | Calnexin | AB_2228381 | Monoclonal | Rabbit | 1:1,000 | Cell signalling | 2679S |
| Caveolin-1 | Caveolin-1 | AB_628859 | Monoclonal-clone 7C8 | Mouse | 200 mg/ml 1:1,000 dilution |
Santa Cruz Biotech | sc-53564 |
| CD63 | CD63 | AB_627877 | Monoclonal | Mouse | 200 mg/ml 1:1,000 dilution |
Santa Cruz Biotech | sc-5275 |
| Cell Division Cycle 42 | CDC42 | AB_2078082 | Monoclonal-clone 11A11 | Rabbit | 1:1,000 dilution | Cell signalling | 2466S |
| Clathrin heavy chain | Clathrin heavy chain | not found | Monoclonal- clone 6F10.1 | Mouse | 1:500 dilution | Sigma-Aldrich | MABC580 |
| Early Endosome Antigen 1 | EEA1 | AB_397830 | Monoclonal | Mouse | 250 µg/ml | BD Transduction Labs | 610457 |
| Glyceraldehyde 3- Phosphate Dehydrogenase | GAPDH | AB_796208 | Polyclonal | Rabbit | 1 mg/ml 1:1,000 dilution |
Sigma-Aldrich | G9545 |
| GM130 | GM130 | AB_2797933 | Monoclonal | Rabbit | 1:1,000 dilution | Cell Signalling | 12480 |
| GW182 | GW182 | AB_1850241 | Polyclonal | Rabbit | 1 mg/mL | Thermo Fisher | A302-330A |
| Haemagglutinin | HA | AB_1549585 | Monoclonal C29F4 |
Rabbit | 1:1,000 dilution | Cell Signalling | 3724 |
| Lysosomal Associated Membrane Protein1 | LAMP1 | AB_2296838 | Monoclonal | Mouse | 1:1,000 dilution | DSHB | H4A3 |
| P21 Activated Kinase 1 | αPAK | AB_10609226 | Monoclonal-clone A-6 | Mouse | 200 mg/ml | Santa Cruz Biotech | sc-166887 |
| P21-RAC1 | RAC1 | AB_309712 | Monoclonal-clone 23A8 | Mouse | 1 mg/ml 1:1,000 dilution |
Sigma | 05–389 |
| RAB34 | RAB34 | AB_1952423 | Polyclonal | Rabbit | 1 mg/ml 1:1,000 dilution |
ABCAM | ab73383 |
| RAB5 | RAB5 | AB_2300649 | Monoclonal-clone C8B1 | Rabbit | 1:1,000 dilution | Cell signalling | 3547S |
| Tumour Susceptible Gene 101 | TSG101 | AB_10974262 | Monoclonal | Rabbit | 1:1,000 dilution | Abcam | ab125011 |
| Secondary antibodies | Research resource identifier (RRID) | Polyclonal or monoclonal | Species made in | Concentration (stock or final, if N/A, then titre) |
Company | Catalogue number | |
| DA1E mAb IgG XP® Isotype Control | AB_1550038 | Monoclonal | Rabbit | 2.5 mg/ml | Cell Signalling | 3900 | |
| Donkey anti-mouse-Cy3 | AB_2315777 | Polyclonal | Donkey | 1:500 dilution | Jackson ImmunoResearch | 715–165-151 | |
| Donkey anti-mouse-Cy5 | AB_2340820 | Polyclonal | Donkey | 1:500 dilution | Jackson ImmunoResearch | 715–175-151 | |
| Donkey anti-Rabbit-Alexa 488 | AB_2313584 | Polyclonal | Donkey | 1:500 dilution | Jackson ImmunoResearch | 711–545-152 | |
| Goat anti-mouse IgG H&L HRP | AB_955439 | Polyclonal | Goat | 2 mg/ml | Abcam | ab6789 | |
| Goat Anti-Rabbit IgG H&L HRP | AB_955447 | Polyclonal | Rabbit | 2 mg/ml | Abcam | ab6721 | |
| Goat Anti-Rat IgG Light chain | AB_2338140 | Polyclonal | Goat | Jackson ImmunoResearch | 112–035-175 | ||
| IgGκ BP-HRP | AB_2687626 | Monoclonal | Mouse | 400 mg/ml | Santa Cruz Biotech | sc-516102 | |
| Mouse anti-rabbit IgG Light chain | AB_2339149 | Polyclonal | Mouse | Jackson ImmunoResearch | 211–032-171 | ||
| Rabbit IgG (for GW182 pulldown) | AB_2337118 | Unknown | Rabbit | 11.4 mg/ml | Jackson ImmunoResearch | 011–000-003 | |
| Rabbit IgG (for HA pulldown) | AB_1031062 | Unknown | Rabbit | 0.07 ug/ul | Cell signalling | 2729s |
Small EV uptake into target cells and luciferase assays
For kinetic sEV uptake experiments, PPF or HUtMEC cells were plated in 24-well plates at 150,000 cells per well. After 48 h, sEVs were added for variable times, as indicated. For concentration-dependence experiments, inhibitors were added for 1 h before exposure to sEVs (10 µg/ml). In siRNA protein knockdown, the siRNA-transfected acceptor cells were cultured for 48–72 h, then incubated with sEVs (10 µg/ml) for 4 h in serum-free medium. To remove sEVs bound to the cell surface or culture dish, the cells were washed using cold PBS for 1 min, followed by citric acid buffer (40 mM citric acid, 135 mM NaCl and 10 mM KCl, pH 3.0) for 3 min, and cold PBS for an additional 1 min. The cells were then lysed in 150 µl NanoLuc lysis buffer (Promega, Madison, WI) at room temperature (RT) for 15 min, then at 4°C overnight. Luciferase assays were performed by mixing 50 µl of the cell lysate with 50 µl nanoluciferase solution and incubated at RT for 5 min. Luciferase was measured using a Veritas microplate luminometer (Turner BioSystems, Promega, Madison, WI), and the data, expressed as relative luminescence units (RLU) were normalized to total lysate protein, determined by the microBCA protein quantification (noted earlier). Experiments were also performed in the presence of diverse types of inhibitors, all dissolved in DMSO at the concentration shown within the Results section and with DMSO alone as vehicle control. Inhibitors were added for 1 h before exposure to sEVs (10 µg/ml) for 3 h. The cells were then washed as detailed above and processed for luciferase activity or confocal imaging, as described below. For uptake analysis using SP-DiO-fluorescent sEVs, cells were cultured in 24-well plates for 48 h, and preincubated with inhibitors or DMSO vehicle for 1 h prior to addition of sEVs. PHT-derived SP-DiO-labelled sEVs (25 µg/ml) were used, and incubation was stopped after 4 h. Cells were washed with PBS, trypsinized, and resuspended in 500 µl PBS for flow-cytometry (LSRII, BD Biosciences, San Jose, CA). Cells that were not exposed to SP-DiO-labelled sEVs were used as background control.
Confocal imaging of SP-DiI sEV uptake and miR-517a-Alexa488 intracellular localization
For SP-Dil sEV uptake, cells were plated in a 96-well optical dish (Ibidi GmbH, Martinsried, Germany, Cat. #89626) at 50–70% confluence and used in experiments when they reached 90–95% confluence (24–36 h after plating). To monitor uptake, cells were incubated with purified sEVs stained with SP-DiI dye (exo-SP-DiI, 0.1 μg protein/μl) for 90 min in complete DMEM medium at 37°C. The cells were then washed with ice-cold PBS and fixed in freshly prepared 4% paraformaldehyde for 15 min at RT. In the assay with endocytosis inhibitors, confluent cells were pre-exposed to the inhibitors, as detailed in the Results section, for 1 h at 37°C prior to incubation with exo-SP-DiI for 3 h at 37°C. After incubation, the cells were washed with PBS and fixed as described above.
For intracellular localization of miR-517a-Alexa488 (Integrated DNA Technologies, Coralville, IA), cells were plated in 12-mm glass coverslips at 50–70% confluence. After 24–36 h, the cells were transfected with 15 nM miR-517a-Alexa488, using Lipofectamine iRNAiMAX, following the manufacturer’s recommendations. At the indicated time intervals after transfection, cells were washed in PBS, fixed in freshly prepared 4% paraformaldehyde, and used for immunofluorescence labelling.
Fixed cells were permeabilized in 0.1%Triton X-100/3%BSA/PBS for 20 min at RT and incubated with the relevant primary antibodies (Table 2) for 1 h at RT. The cells were then incubated with the secondary antibodies (Table 2) for 1 h at RT. The nuclei were stained with Hoechst 33342 (Thermo Scientific) for 10 min at 37°C. Immunolabeled cells from SP-Dil sEV uptake were stored in the optical 96-well dish with PBS containing 0.02% sodium azide and imaged within 24 h. Cells transfected with miR-517a-Alexa488 were mounted in ProLong Gold antifade reagent (Invitrogen, #P36930) mounting media. The images were acquired using a spinning disk confocal imaging system based on a Zeiss Axio Observer Z1 inverted fluorescence microscope system (with 63x Plan Apo PH NA 1.4), controlled by SlideBook6 software (Intelligent Imaging Innovation, Denver, CO) as previously described [52]. Z-stacks of x–y images through 445-nm (Hoechst), 488-nm (Alexa 488), 561-nm (sp-DiI and Cy3), and 640-nm (Cy5) channels were acquired. In PPF cells pre-treated with inhibitors, a Z-stack of confocal images was acquired through 405- and 561-nm channels at 300-nm intervals. In PPF cells transfected with miRNA-517a-Alexa488, a Z-stack of confocal images was acquired through 488-, 561-, and 640-nm channels at 300-nm intervals. All image acquisition settings were identical for the experimental variables in each experiment.
Image analysis
The number of SP-DiI fluorescent puncta (detected through the 561 nm channel), the sum fluorescence intensity of each individual puncta, and the sum of fluorescence intensity per single cell were calculated based on background-subtracted 3D images, using the segmentation-based method of SlideBook6 [53]. The segment mask was generated to select SP-DiI puncta using identical fluorescence intensity threshold parameters for all experimental variants.
In PPF cells transfected with miRNA-517a-Alexa488, two segment masks were generated from 3-D images after background subtraction to select Cy3-positive puncta (FLAG-HA-AGO2) and Cy5-positive puncta (GW182) with identical threshold parameters in all experimental variables. The co-localization mask was then generated to select voxels overlapping in Cy3 and Cy5 masks. Sum fluorescence intensity through the 488-nm channel (miRNA517a-A488) was determined in the co-localization mask in individual cells.
Protein pulldown and protein-miRNA coimmunoprecipitation
PPF cells were plated in 10-cm dishes and cultured to 80–90% confluency. Then cells were transfected with 10 nM miR-517a-Alexa488 for 24 hours by using Lipofectamine iRNAiMAX following the manufacturer’s recommendations. For protein-RNA crosslinking, PPF cells were incubated with sEVs that were electroporated with miRNA-517a (50 µg/ml), washed with cold PBS, and exposed to 254 nm UV light at an intensity of 600mJ/CM2 (UV Crosslinker Select, Spectroline, Westbury, NY). The PBS solution was then replaced with 1.4 ml of ice-cold cell lysis buffer for 30 min, scraped, and centrifuged at 10,000 g for 15 min at 4°C. Cell lysates were pre-cleaned with 20 µl Pierce Protein G Magnetic Beads (Thermo Fisher) by incubating for 1 h at RT with rotation. Primary (HA and GW182) and secondary antibodies used for pulldown experiments are detailed in Table 2. Overnight incubation at 4°C was used for all primary antibodies. Pre-washed Protein G Dynabeads (10 µl) were added to the lysate with rotation for 1 h at 4°C. After application of the magnetic bar, the solution was washed in a buffer containing 50 mM Tris, pH 7.5, 300 mM NaCl, 0.5%NP-40, 5 mM EDTA, 10 U/ml SUPERaseln RNase inhibitor (Thermo Fisher) for 1 min and then, with the same solution, but with 150 mM NaCl for an additional 1 min. The proteins were eluted in 75 µl of 1x Bolt PAGE buffer (with100 mM DTT) per sample and shaken at RT for 30 min. After digestion with proteinase K at 55°C for 45 min (Sigma-Aldrich, 5 mg/ml), 700 µl Qiazol (Qiagen) was added for 5 min, and the solution was spiked with 2 µl glycogen (20 mg/ml stock), 5 µl miR-39 mimic (Qiagen, 5 nM stock), and 4 µl of yeast tRNA (Thermo Fisher, 50 ng/µl stock), used for RNA isolation and RT-qPCR detection of miRNA-517a. Ct-values were normalized to spiked miR-39. Immunoprecipitation was verified using the appropriate antibodies, all detailed in Table 2.
Statistics
All statistical analyses were performed using GraphPad Prism software (ver. 8.00; GraphPad, San Diego, CA). For comparisons of each two groups, unpaired Student’s t-test was used after verification of equal variance using F-test. Welch’s correction was performed when the variance across groups was assumed to be unequal. For multiple comparison analyses, a one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test, was used. We used two-way ANOVA when two or more variables were tested, followed by Sidak’s multiple comparison test. All experiments were performed at least three times. Differences were considered significant when the p-value was <0.05, with the specific p-values detailed within each figure legend.
Results
Trophoblast-derived sEVs deliver their miRNA cargo to non-trophoblast target cells
To examine the entry of trophoblast-derived sEVs into target cells we isolated sEVs, as we previously described [33], from PHT cells that were dispersed from term human placental villi or from the human trophoblast cell line BeWo. These sEVs uniquely harbour miRNAs from the C19MC, which thus serve to demarcate the trophoblastic origin of sEVs. For target cells, we focused on biologically relevant cells that are located near trophoblasts, namely PPF cells or HUtMEC, which are thus more likely to internalize the released trophoblastic sEVs.
To confirm that PPF cells and HUtMEC do not endogenously express C19MC miRNA, we used RT-qPCR to assess the level of primary, precursor, and mature miRNA-517a, a highly expressed member of the C19MC family [23,32]. As expected, the expression of immature or mature forms of miR-517a in HUtMEC and PPF cells was negligible (Figure 1a–c), confirming that miR-517a is not transcribed in these cells. Notably, mature miR-517a was present in freshly isolated PPF cells, but the level of this miRNA declined to background within five passages (Suppl. Fig. 1), suggesting that the initial level represented uptake of trophoblastic miR-517a. Exposure of PPF or HUtMEC cells to PHT- or BeWo-derived sEVs led to a marked increase in intracellular miR-517a (Figure 1d). Importantly, exposure to PHT- or BeWo-derived sEVs attenuated the level of transduced vesicular stomatitis virus (VSV) in the recipient cells (Figure 1e), as we have previously shown [32–35,54], thus validating the functionality of the trophoblastic sEVs.
Figure 1.
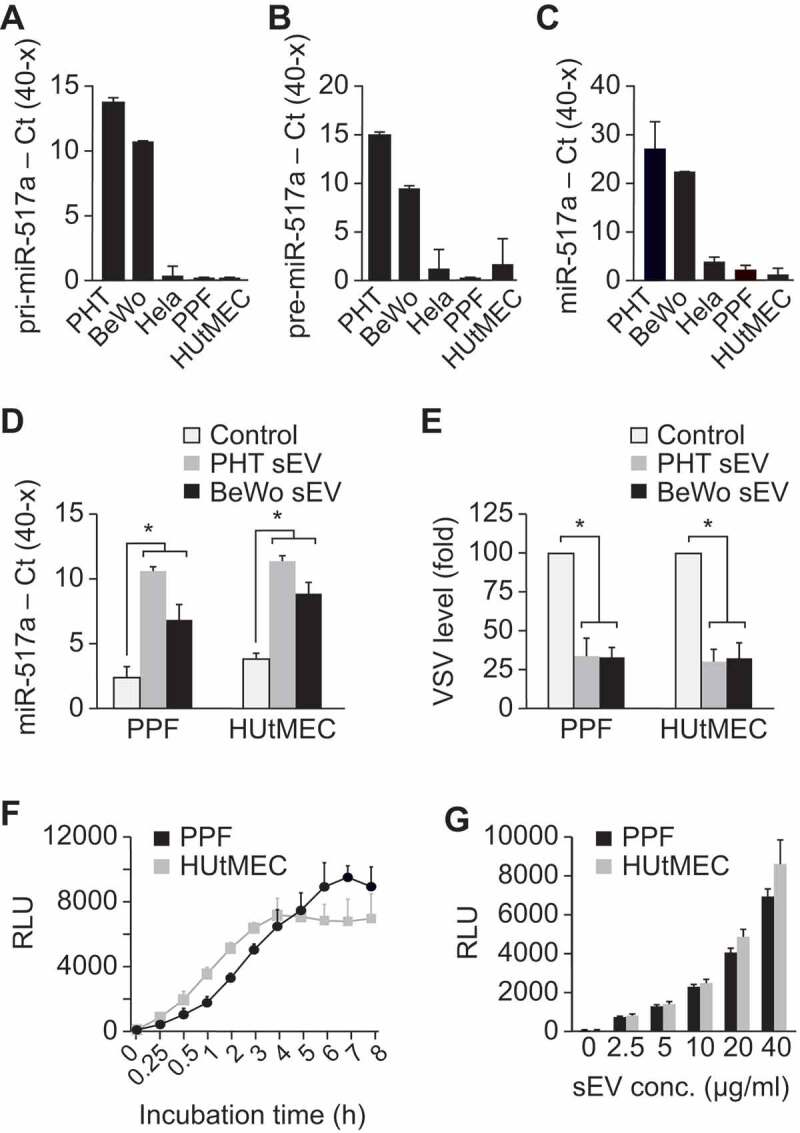
The uptake of PHT and BeWo sEVs by target cells.
The target cells PPF and HUtMEC do not express (a) primary (pri)-miRNA-517a, (b) precursor (pre)-miRNA-517a, or (c) mature miRNA-517a. PHT and BeWo cells were used as positive control, while the unrelated HeLa cells were used as negative control. Transcript levels were determined using RT-qPCR, as described in Methods, and presented as Ct(40-x) to show expression trend in the positive direction. The differences between PHT or BeWo cells and the other cell types were significant (p < 0.01) in each of the panels. (d) PHT- or BeWo-derived sEVs deliver trophoblast-specific miRNA-517a into PPF or HUtMEC target cells. (e) PHT- or BeWo-derived sEVs attenuate VSV levels in PPF or HUtMEC target cells. (f) The time-dependent pattern and (g) the concentration-dependent pattern (assayed after 3 h of sEV exposure) of sEV entry into PPF or HUtMEC cells, determined using NanoLuc activity. See text for details. The zero represents RLU reading in the absence of added sEVs. Data were analysed using one-way ANOVA with post hoc Tukey for multiple comparisons testing. * denotes p < 0.001.
To assess the time and concentration dependence of sEV uptake in our two recipient cell types, we stably transfected BeWo cells with a plasmid expressing the sEV protein TSG101, tagged with the fluorescent protein mCherry at the N-terminus and nanoluciferase (NanoLuc) at the C-terminus. Cells expressing mCherry-TSG101-NanoLuc exhibited a high level of luciferase activity, and sEVs obtained from these cells were enriched for luciferase activity (Suppl. Figure 2). Uptake of BeWo-derived mCherry-TSG101-NanoLuc sEVs by PPF or HUtMEC cells, measured by luciferase activity, was time dependent (Figure 1f) and proportional to sEV concentration (Figure 1g). Together, these data validate the use of our Nano-Luc-tagged sEVs to assess their uptake into target cells.
Macropinocytosis mediates the uptake of sEVs into target cells
We next sought to define specific endocytic pathways that mediate the uptake of trophoblastic sEVs into PPF and HUtMEC cells. To optimize and validate the sEV uptake assay, we first ensured the removal of non-internalized sEVs by cell washing with citric acid after the incubation with sEVs (Suppl. Fig. 3a), thus minimizing the contribution of non-internalized sEVs to the luciferase signal, as demonstrated when cells were incubated with sEVs at 4°C, conditions eliminating endocytic trafficking (Suppl. Fig. 3b). We also found that the sEV uptake is enhanced in the absence of serum (Suppl. Fig. 3 c); and that the use of DMSO (up to 0.5%) as a solvent for all pharmacological inhibitors had no effect on sEV uptake (Suppl. Fig. 3d). Internalization of SP-Dil-labelled sEVs was directly demonstrated, using confocal microscopy, by detection of fluorescence puncta after target cell incubation with SP-Dil-labelled sEVs at 37°C (Figure 4e). Notably, target cells that were not incubated with labelled sEVs (Figure 4e, “background” image) displayed fluorescent puncta with brightness comparable to that of some individual Dil puncta, detected in cells that were incubated with labelled sEVs (Figure 4e). However, the background autofluorescence of these vesicles displayed a wide spectral range and could be excited through multiple channels, such as 488 nm and 640 nm. Therefore, non-specific vesicles could be readily distinguished from the specific SP-Dil-containing vesicles, which only exhibited specific red fluorescence through the 561-nm channel.
Figure 4.
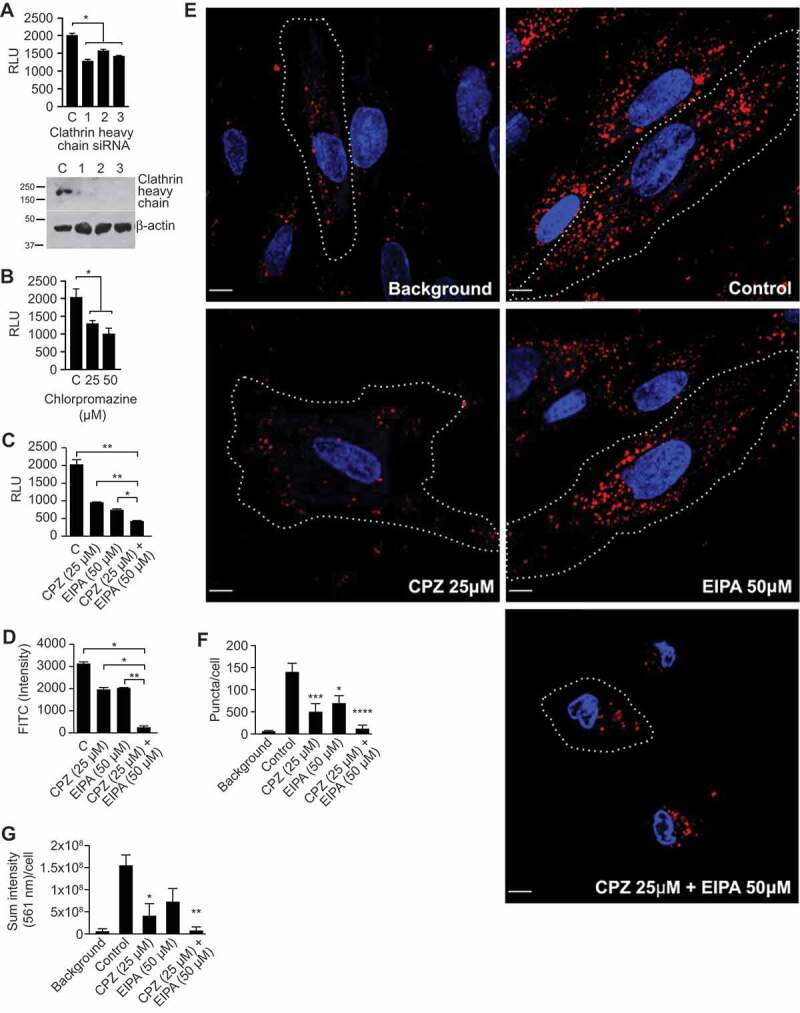
Clathrin-mediated endocytosis contributes to sEV uptake.FIGURE_SHOULD_BE_IN_COLOR_(AS_SUBMITTED)
The uptake of sEVs was determined by NanoLuc activity, as described in Methods, and expressed as RLU. (a) The effect of silencing of clathrin heavy chain by three individual siRNA duplexes in PPF cells. (b) The concentration-dependent effect of chlorpromazine in PPF cells. (c) The combined effect of chlorpromazine (CPZ) and EIPA in sEV uptake in PPF cells. (d) The combined effect of CPZ and EIPA on PPF uptake of PHT-derived sEVs that were labelled with SP-DiO, with green fluorescence detection by flow cytometry. All assays in A-D were performed at least 3 times, and data were analysed using one-way ANOVA with post hoc Tukey test. * denotes p < 0.05 and ** denotes p < 0.01. (e) PPF cells were incubated with SP-Dil-labelled BeWo-derived sEVs for 90 min and imaged through the 405-nm channel (nuclei; blue) and the 561-nm channel (SP-Dil; red) as described in Methods. Representative images of the effect of CPZ, EIPA, or both on sEV uptake are shown. Cell edges are indicated by dotted lines. Scale bar: 10 μm. Quantification of the number of SP-DiI puncta (f) and total SP-DiI fluorescence intensities in arbitrary units (g) per an individual cell from images exemplified in (e). Bar graphs show mean values (±SEM), calculated from 6 individual cells in 2 independent experiments, and data were analysed by one-way ANOVA with post hoc Tukey test. * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.005, **** denotes p < 0.0001.
Different cells use distinctive mechanisms for sEV uptake, with macropinocytosis commonly implicated in the sEV uptake mechanism for non-trophoblastic sEVs ([11,55,56] and reviewed in [44]). We therefore first assessed the effect of the Na+/H+ exchanger inhibitor EIPA, a known inhibitor of macropinocytosis [57], on the sEV uptake. Using PPF cells as targets for trophoblastic sEVs, we found that exposure to EIPA reduced sEV uptake by 50% (Figure 2a).
Figure 2.
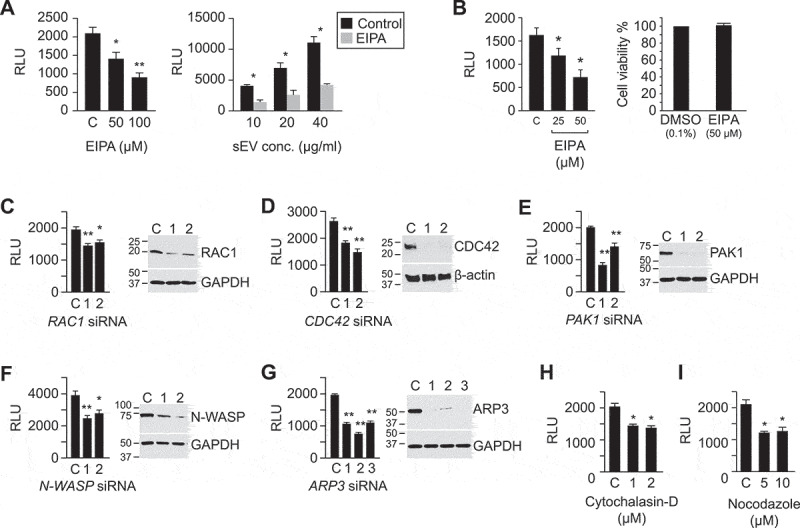
The role of macropinocytosis in the uptake of BeWo sEVs.
The figure depicts key proteins that define macropinocytosis-based uptake. The uptake of sEVs was determined by the activity of nano-luciferase (NanoLuc), as described in Methods, and expressed as relative luminescence units (RLU). (a) The left panel shows the concentration-dependent effect of EIPA on sEV uptake in PPF cells. The right panel shows the effect of EIPA (100 µM) on a range of sEV concentrations. (b) The left panel shows the concentration-dependent effect of EIPA on sEV (10 µg/ml, 1.1 x 1010/ml) uptake in HUtMEC cells, and the right panel shows the effect of EIPA on cell viability, as control. (c-g) siRNA-mediated inhibition of pivotal macropinocytotic signalling proteins attenuates sEV uptake in PPF cells. Each panel depicts the effect of silencing on RLU activity and the quality of silencing by western immunoblot by 2 or 3 distinct siRNA duplexes. (h) The effect of cytochalasin-D on luciferase activity (RLU) in PPF cells. (i) The effect of nocodazole on luciferase activity (RLU) in PPF cells. All assays were performed at least 3 times, and data were analysed using one-way ANOVA with post hoc Tukey or Sidak test, where appropriate. * denotes p < 0.05, and ** denotes p < 0.01. Note that all protein names and antibodies used for the studies are summarized in Table 2.
We confirmed the inhibitory effect of EIPA on sEV internalization by PPF cells, using flow cytometry assessment for SP-DiO-labelled sEVs derived from PHT cells (Figure 4d) and using quantitative confocal imaging for SP-Dil-labelled BeWo-derived sEVs (Figure 4e–g). Similar partial inhibition (by ~50%) of sEV uptake by EIPA was observed in HUtMEC cells (Figure 2b).
Subsequent silencing experiments for determining the role of pivotal signalling proteins in sEV uptake were performed in PPF cells, which were more amenable to siRNA-based manipulation. In light of the known requirement for RAC1/CDC42, PAK1, WAVE, and the ARP2/3 complex for actin rearrangement and formation of macropinosomes [44,58–60], we examined the effects of depletion of these proteins by RNA interference (RNAi). Depletion of RAC1 and CDC42 attenuated sEV entry into PPF cells by 25% and 40%, respectively (Figure 2c,d). The role of CDC42 was also confirmed using its pharmacological inhibitor, ML141 (Suppl. Fig. 4a). Furthermore, the inhibition (using IPA-3, Suppl. Figure 4b) or knocking down of PAK1, a kinase downstream from CDC42 and RAC1 (Figure 2e), also attenuated sEV uptake by approximately 40%. Similarly, knocking down N-WASP or ARP3, which function downstream from CDC42 to promote actin polymerization and branching, reduced sEV uptake by 30% and 50%, respectively (Figure 2f-g). This result was confirmed using wiskostatin or CK869 to inhibit N-WASP or the ARP2/3 complex, respectively (Suppl. Figure 4c,d). Finally, inhibition of actin polymerization by cytochalasin D or disruption of microtubule assembly/disassembly dynamics by nocodazole [61] also reduced sEV uptake by approximately 40% (Figure 2h,i).
The PI3K-PLC-PKC-RAB34 signalling pathway is also involved in macropinocytosis, specifically in the formation of membrane ruffles, as well as closure, trafficking, and fusion of macropinosomes with lysosomes [58,59]. To test whether the entry of trophoblastic sEVs into PPF cells involves this signalling pathway, we used wortmannin, U73122, or bisindolylmaleimide to inhibit PI3K, PLC, or PKC, respectively. As shown in Figure 3a–c, each of these inhibitors reduced the entry of trophoblastic sEVs into PPFs. Moreover, knocking down RAB34, a PKC effector protein that is associated with membrane ruffles, promotes the formation of macropinosomes, is implicated in lysosome positioning [58,62,63], and also diminishes sEV internalization (by 40%, Figure 3d). Knockdown of RAB5a-c, which has been implicated in macropinocytic cup formation and macropinosome trafficking [58,64,65], also reduced sEV uptake into PPF cells (by 50%, Figure 3e). Together, our data demonstrate that macropinocytosis plays a major role in the uptake of trophoblast-derived sEVs into PPF and (validated, in part) HUtVEC cells.
Figure 3.

The role of PI3K-PLC-PKC-RAB34 signalling pathway in mediating macropinocytosis of sEVs.
The uptake of sEVs was determined by NanoLuc, as described in Methods, and expressed as RLU. The effect of (a) the PI3K inhibitor wortmannin; (b) the PLC inhibitor U73122; and (c) the PKC inhibitor bisindolylmaleimide on sEV uptake is shown. (d) siRNA-mediated silencing of RAB34 attenuates sEV uptake in PPF cells. (e) siRNA-mediated inhibition of RAB5 proteins (Rab5a/b/c) attenuates sEV uptake in PPF cells. Each panel depicts the effect of silencing on RLU activity, and the quality of protein silencing by western immunoblot. All assays were performed at least 3 times, and data were analysed using one-way ANOVA with post hoc Tukey test. * denotes p < 0.05.
Clathrin-mediated endocytosis (CME) contributes to sEV entry, whereas caveolae/lipid raft endocytic mechanisms are dispensable
Because EIPA and other inhibitors of macropinocytosis only partially reduced sEV uptake, we next sought to examine whether the uptake of trophoblastic sEVs into PPF cells involves other endocytic mechanisms. To assess the role of clathrin-dependent endocytosis [44,66], we used RNAi depletion of clathrin heavy chain [67] and also chlorpromazine, which blocks this process by interfering with the association of clathrin with the plasma membrane [66,68]. We found that both clathrin knockdown and chlorpromazine reduced sEV uptake by 30–50% (Figure 4a,b), with a similar effect in HUtMEC (Suppl. Fig. 5a).
To test for a potential interaction between inhibitors of macropinocytosis and CME, we exposed PPFs to EIPA, chlorpromazine, or both, and assessed sEV uptake. As shown in Figure 4c, combining the two drugs reduced sEV uptake by 80–90%. We observed a similar additive effect of inhibitors of macropinocytosis and clathrin-mediated endocytosis, using SP-DiO-labelled, PHT-derived sEVs and flow cytometry (Figure 4d), and of SP-Dil-labelled BeWo sEVs under confocal fluorescence microscopic imaging (Figure 4e–g). In the latter approaches, both the number of fluorescent puncta and the total intensity of internalized sEV labels were markedly reduced by the drug combination. These experiments indicate that macropinocytosis and CME independently and additively contribute to sEV uptake into target PPF cells.
Endocytosis via caveolae, mediated by internalization of small cave-like plasma membrane invaginations, depends on cholesterol, sphingolipids, and caveolin-1 proteins and can therefore be inhibited by cholesterol sequestration or depletion [69–73]. Drugs that promote cholesterol membrane sequestration, including the polyene antibiotics filipin and nystatin [71,72,74,75], had no effect on sEV uptake into PPF cells (Figure 5a–d). Similarly, the use of methyl-β-cyclodextrin [73] to extract cholesterol from the plasma membrane or silencing of caveolin-1, a main component of the caveolae scaffold that plays a crucial role in caveolae formation and stability [71,76] did not affect sEV uptake (Figure 5e,f). Likewise, we observed no effect of filipin and nystatin on sEV uptake in HUtMEC cells (Suppl. Figure 5b–d). Importantly, none of the inhibitors used in the experiments presented in Figures 3–5, or Suppl. Figs. 3–4 had an effect on cell viability as measured by MTS assay (Suppl. Fig. 6). Together, these data indicate that macropinocytosis and clathrin-mediated endocytosis are the major pathways by which trophoblastic sEVs enter target cells, whereas caveolae/lipid raft-dependent endocytic pathways are not essential for sEV entry into PPF or HUtMEC cells.
Figure 5.
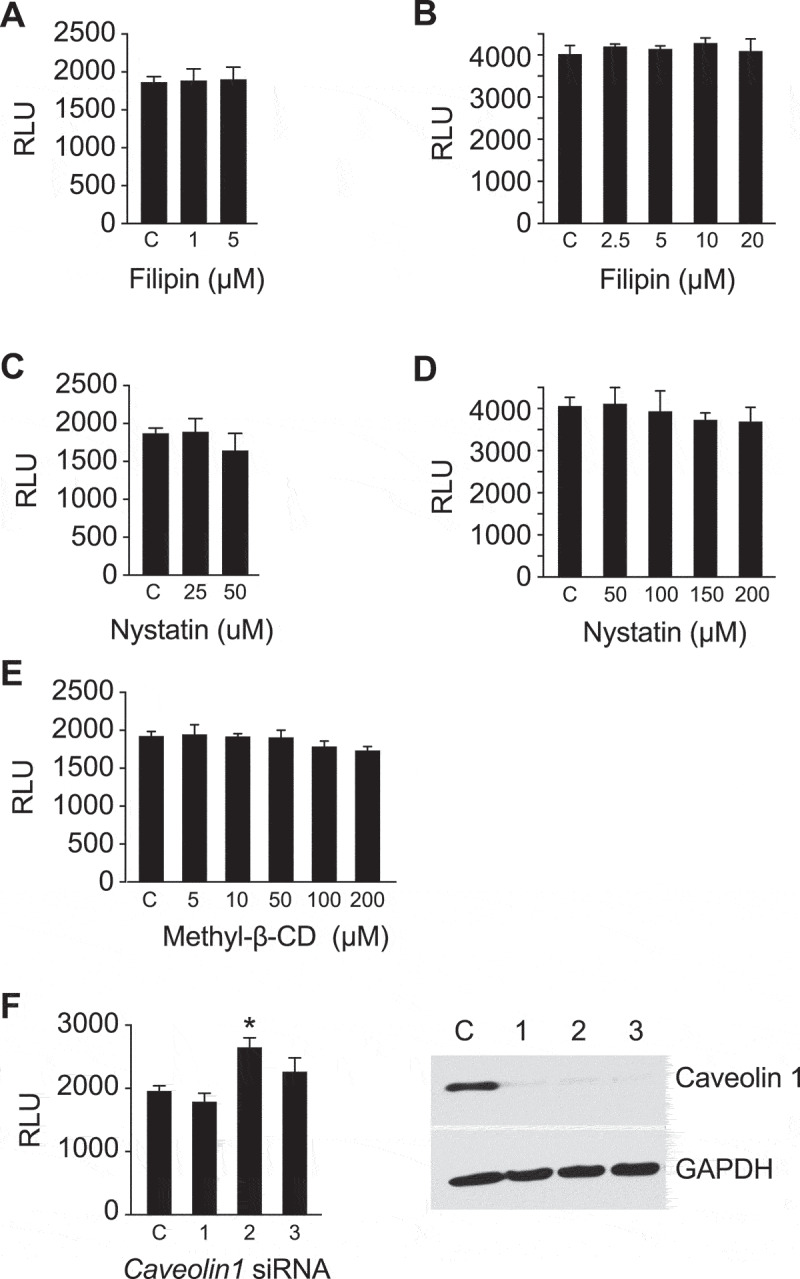
Caveolin/lipid rafts are not necessary for sEV uptake in PPF cells.
The uptake of sEVs was determined by NanoLuc activity, as described in Methods, and expressed as RLU. The effect of (a) filipin-III in serum-containing medium and (b) serum-free medium. (c) The effect of nystatin in serum-containing medium and (d) serum-free medium. (e) Cell entry of sEVs when the cells were pre-exposed to methyl-beta-cyclodextrin (methyl-β-CD) in serum-containing medium. (f) The effect of siRNA-mediated inhibition of sEV uptake into PPF cells, depicting RLU activity and the quality of silencing by western immunoblot. All assays were performed at least 3 times, and data were analysed using one-way ANOVA with post hoc Tukey test. * denotes p < 0.05.
Internalized sEV traffic to early endosomes and late endosomes/lysosomes
Having defined the endocytic pathways underlying the cellular entry of trophoblastic sEVs, we sought to define the intracellular localization of these vesicles. To this end, PPF cells were incubated with BeWo-derived SP-DiI-labelled sEVs for 90 min at 37°C, a timepoint roughly corresponding to half the maximum accumulation of the exosomal cargo in these cells (Figure 1f). A small number of SP-Dil puncta (<5%) were co-localized with APPL1-labelled vesicles (Figure 6), specialized early endosomal compartments to which some cargo is delivered immediately after clathrin-mediated endocytosis. A substantial number of SP-Dil puncta (~25%) were found to co-localize with EEA.1, a marker of early and intermediate endosomes (Figure 6). About 30% of SP-Dil-positive compartments were co-localized with LAMP1, a marker of late endosomes and lysosomes (Figure 6). Analysis of the distribution of the fluorescence intensity of individual internalized puncta revealed a single intensity peak of the diffraction-limited puncta (≥300 nm in diameter), which represents more than half of all SP-Dil labelled structures, with a right “shoulder” of a brighter puncta (Suppl. Fig. 7). Such distribution suggests that this peak corresponds to a single sEVs per puncta whereas brighter puncta (in the right shoulder) corresponds to multiple sEV aggregates.
Figure 6.
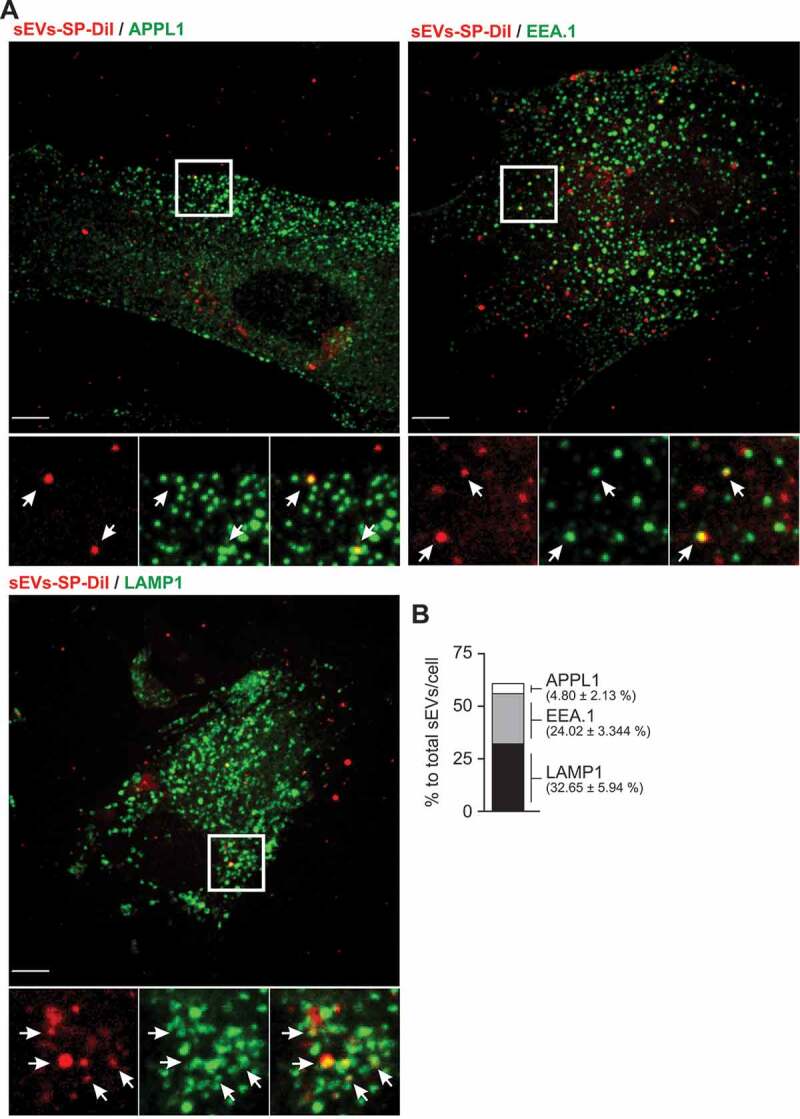
Co-localization of sEV-SP-DiI with endolysosomal markers in PPF cells.
PPF cells were incubated with SP-DiI-labelled sEVs (red) for 90 min at 37°C, fixed, permeabilized, and stained with antibodies to APPL1, EEA.1 or LAMP1 (green) as described in Methods. (a) Individual confocal sections of the merged images. Insets represent high-magnification images of each individual imaging channel and a merged image of the region marked by the white rectangle. Arrows show examples of co-localization between SP-Dil and endosomal or lysosomal markers. Scale bar: 10 μm. (b) Quantification of SP-Dil sEV co-localization with endocytic markers expressed as per cent of SP-Dil puncta co-localizing with APPL1, EEA.1, or LAMP1 (n = 5–6 cells).
There were a considerable number of SP-Dil-labelled puncta (~40%) that were not co-localized at the time of fixation with any of the endosomal/lysosomal markers used. They were found, largely, near the cell periphery. A similar localization pattern of internalized SP-Dil-labelled sEVs was observed in HUtMEC (Suppl. Fig. 8). Together, our experiments suggest that sEVs traffic through the conventional endosomal system in target cells, from early endosomes through the late endosomal compartment and to lysosomes.
Human sEV trophoblast-specific miRNA-517a is transported to P-bodies
We surmised that endosomal escape of cargo might take place prior to sEV degradation in lysosomes and that miRNA would reach the intracellular sites of their biological activity, likely RISC complexes at P-bodies, where miRNAs exhibit their RNA degradation or translational inhibition activities. As miRNA-517a is one of the most abundant C19MC miRNAs in trophoblastic sEVs, we tested whether this miRNA co-localizes with P-body proteins AGO2 and GW182. Immunofluorescence labelling of GW182 in PPF cells stably co-expressing FLAG-HA-AGO2 and fluorescently tagged ss-miR-517, allowed the direct comparison of subcellular localization of miR-517 and P-bodies (Figure 7a,b). Image analysis showed that FLAG-HA-AGO2 and endogenous GW182 were highly co-localized in relatively uniform puncta that were distributed throughout the cytoplasmic P-bodies, although few puncta containing a single marker were detected as well. A fraction of transfected ss-miR-517-A488 was co-localized with P-bodies while a substantial amount of this miRNA was seen in the cytoplasm and nucleus (Figure 7a,b). Furthermore, we showed that after UV-crosslinking of PPF cells expressing FLAG-HA-AGO2 and transfected with ss-miRNA-517a, HA antibody immunoprecipitates contained an increased level of miRNA-517a when compared to immunoprecipitates using non-specific IgG (Figure 8a).
Figure 7.
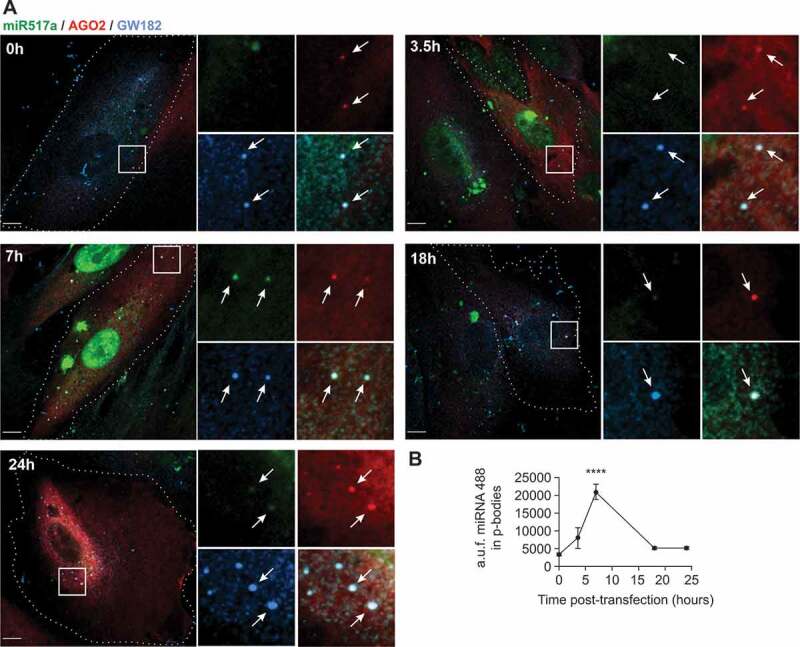
Localization of transfected miR517a-A488 in P-bodies.
PPF/FLAG-HA-AGO2 cells were transiently transfected with miR517a-A488 and fixed at 0–24 h after transfection, followed by staining with GW182 and HA antibodies, as detailed in Methods. (a) 3D imaging through 488-nm (miR517a-A488, green), 561-nm (HA, red) and 640-nm (GW182, blue) channels. Merged images are presented. Insets represent high-magnification images of each individual channel and the merged image of the region indicated by white rectangle. Arrows show examples of miR517a-A488 co-localization with the GW182/AGO2 puncta. Cell edges are indicated by dotted lines. Scale bars, 10 μm. (b) Quantification of miR517a-A488 fluorescence associated with the GW182/AGO2 puncta in images in (a). Mean values of miR517a-A488 intensity per puncta (± SEM; n = 13–36 cells) are presented. The data were analysed using one-way ANOVA. **** denotes p < 0.0001
Figure 8.

Coimmunoprecipitation of miR-517a with P-body proteins AGO2 and GW182.
RT-qPCR-based detection of miRNA-517a coimmunoprecipitated with Flag-HA-AGO2 from (a) PPF cells expressing Flag-HA-AGO2 and transiently transfected with miRNA-517a 24 h before the assay or (b) exposed to sEVs that were electroporated with miRNA-517a. Immunoprecipitation efficiency detected by western blot. (c) RT-qPCR-based detection of miRNA-517a after coimmunoprecipitation with endogenous GW182 from PPF cells following exposure to sEVs that were electroporated with miRNA-517a. The data were analysed using unpaired Student’s t-test. * denotes p < 0.05 (n = 3).
To determine whether internalization of sEVs containing miR-517a results in the transport of this miRNA to P-bodies, we first verified that P-body proteins AGO2 and GW182 are not packaged within trophoblastic sEVs (Suppl. Fig. 9B), which is consistent with the previous report [77]. To overcome the poor efficiency of immunoprecipitation of endogenous AGO2 in target cells and to ensure that AGO2-miR-517a complexes were not generated within sEVs, we overexpressed FLAG-HA-AGO2 in PPF cells prior to exposure to sEVs (Suppl. Fig. 9 C). The transfected cells retained their EIPA- and chlorpromazine-mediated inhibition of macropinocytosis and clathrin-mediated endocytosis, respectively (Suppl. Fig. 9 C-D). We enriched BeWo-derived sEVs with miR-517a by electroporation and incubated the transfected sEVs with PPF cells expressing FLAG-HA-AGO2. We used crosslinking immunoprecipitation to assess the association of miR-517a with FLAG-HA-AGO2. As shown in Figure 8b, HA antibody immunoprecipitate of HA-AGO2 contained a significantly higher level of miR-517a than did immunoprecipitate of the matched non-specific IgG. Moreover, sEV miR-517a was specifically coimmunoprecipitated with endogenous GW182 in PPF cells and incubated with miRNA-517a-electroporated BeWo-derived sEVs (Figure 8c). These data suggest that, after internalization within sEVs in target cells, miR-517a can escape endosomal shuttling to lysosomal degradation and is capable of reaching RISC proteins in P-bodies.
Discussion
The release of sEVs from donor cells to the extracellular fluid or blood may affect physiological and pathological responses of local or distant target cells. In the case of trophoblastic sEVs, these responses include resistance to viral replication, influence on cell migration, and potentially, other functions [32,34,35,78]. While the precise cellular targets of trophoblastic sEVs remain to be identified, these targets likely include neighbouring placental fibroblasts and/or the downstream uterine vein endothelial cells. Thus, a unique aspect of our study is the use of physiologically and spatially relevant primary target cells, PPFs or HUtMEC, not cell lines. We showed that the kinetics of trophoblastic sEV entry into PPF or HUtMEC cells was similar to that shown for fluorescently-labelled glioblastoma cell-derived sEVs [79] or other cell systems [80–83]. Importantly, we found that macropinocytosis and CME are the major entry pathways of trophoblastic sEVs into placental fibroblasts or uterine endothelial cells. Moreover, using a combination of biochemical and cell biological approaches, we demonstrated the trafficking of sEVs through the endosome-lysosome system and the delivery of sEV miRNA cargo to P-bodies.
The dissection of sEV uptake mechanisms is challenged by the paucity of specific small-molecule inhibitors or pathway-selective siRNA. We therefore supported our findings about the role of macropinocytosis and CME in the uptake of trophoblastic sEVs by using siRNAs for silencing pivotal proteins in these endocytic pathways, bolstered by a diverse range of pharmacological inhibitors. For macropinocytosis, we used EIPA as an established chemical inhibitor, shown to be such by others [55]. Our imaging data suggest that more than half of the visualized puncta may represent individual sEVs. It is also possible that aggregates of tethered sEVs, which are larger than 200 nm, are preferably internalized through macropinocytosis. We used a similar approach to show that caveolin-mediated pathways are unlikely to play an important role in the uptake of trophoblastic sEVs into PPF or HUtMEC cells. We recognize that the parameters that determine the uptake and internalization of sEVs largely depend on the donor cell’s sEVs properties and characteristics of the recipient cells. It is therefore not surprising that while our data are consistent with some of the publications in the field [11,84], others reported different results. For example, Costa et al. [80] used a more limited set of inhibitors to show that the entry of sEVs derived from A431 human epidermoid carcinoma cell line into HeLa cells depended on macropinocytosis and caveolar endocytosis, but not CME pathways. Similarly, phagocytosis seems to be a more prevalent sEV uptake route in phagocytes [85]. In some cases, caveolin was reported to have a negative effect on the uptake of sEVs [79]. These differences likely represent the properties of sEVs derived from diverse cell types and the nature of the target cells [6]. Interestingly, we also noted the retention of SP-Dil-labelled puncta near the cell periphery. These puncta, which were not associated with endosomes, may represent cell surface-associated sEVs that have not been internalized. They might also exhibit different cell entry kinetics, or represent a discrete population of sEVs that could not be processed by the endocytic pathways tested in our studies.
Our studies stand out in (a) our rigorous approach to isolation of sEVs using gradient-based ultracentrifugation, resulting in high-quality vesicles for the experimental assay; (b) the deployment of multiple genomic and pharmacologic approaches to relevant primary target cells; and (c) our data readouts that include Nano-Luc as a sensitive quantitative reporter system alongside fluorescently labelled sEVs, examined by confocal imaging and flow cytometry.
While not pursued in our studies, Heusermann et al. [86] showed the role of filopodia in the floating, grabbing, and pulling of sEVs to endocytic hot spots at the filopodial base, with final internalization of sEVs and sorting into lysosomes as their final intracellular lot [86]. We also did not pursue mechanisms underlying specific sEV targeting to selected tissue types. It has been suggested that such targeting might be guided by a unique “barcode,” located on the sEV surface or their target cells. A distinctive combination of exosomal surface proteins may comprise the targeting barcode, as previously found [7,8], recognized by a target cell “barcode reader.” While this remains a central question in sEV biology, in the field of placental biology it was found that the trophoblast-specific fusogenic proteins syncytin-1 and −2 are necessary for the fusion of trophoblastic sEVs into target cells [87,88]. The role of these and other placental proteins in mediating sEV entry into discrete cells remains to be elucidated.
Previous studies have shown the accumulation of labelled sEV membranes in vesicular-shaped perinuclear compartments [83,89]. Using high-resolution fluorescence imaging and analysis allowed us to demonstrate the localization of labelled sEVs in early and late endosomal/lysosomal compartments in PPF and HUtVEC cells. These data suggest the presence of endocytic escape into the cytoplasm, allowing traceable sEV cargo, such as trophoblast-specific miRNA-517a, to reach RISC complexes. Although the mechanism of such endosomal escape and the intracellular targeting of sEV miRNA remains elusive [90], our data show that transfected labelled miR-517a can be co-localized to P-bodies in target cells and that exposure of target cells to trophoblastic sEVs that have been transfected with tagged miR-517a leads to coimmunoprecipitation of this miRNA with the P-body proteins AGO2 or GW182. Our need to increase exosomal miRNA concentration in order to co-precipitate the cargo with P-body proteins likely reflects assay limitation and the overall low abundance of miRNA cargo within sEVs [38,90,91]. Together, these data are the first to suggest the delivery of sEV miRNA to the RISC complex proteins, a known site for miRNA-dependent silencing. Additional experiments utilizing sensitive assays will be required to decipher the mechanisms underlying endosomal escape and miRNA cargo localization to P-bodies.
Supplementary Material
Acknowledgments
We thank Tiffany Coon for technical assistance; Lori Rideout for assistance with manuscript preparation, and Bruce Campbell for editing.
Disclosure of interest
Y.S. is a consultant to Illumina, Inc.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Witwer KW, Thery C.. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J Extracell Vesicles. 2019;8:1648167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–19. [DOI] [PubMed] [Google Scholar]
- [4].Shao H, Im H, Castro CM, et al. New technologies for analysis of extracellular vesicles. Chem Rev. 2018;118:1917–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–228. [DOI] [PubMed] [Google Scholar]
- [6].Mathieu M, Martin-Jaular L, Lavieu G, et al. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21:9–17. [DOI] [PubMed] [Google Scholar]
- [7].Willms E, Johansson HJ, Mager I, et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci Rep. 2016;6:22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. [DOI] [PubMed] [Google Scholar]
- [10].Chen X, Liang H, Zhang J, et al. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012;22:125–132. [DOI] [PubMed] [Google Scholar]
- [11].Tian T, Zhu YL, Zhou YY, et al. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J Biol Chem. 2014;289:22258–22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fong MY, Zhou W, Liu L, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thomou T, Mori MA, Dreyfuss JM, et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 2017;542:450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ying W, Riopel M, Bandyopadhyay G, et al. Adipose tissue macrophage-derived exosomal mirRNAs can modulate in vivo and in vitro insulin sensitivity. Cell. 2017;171:372–384 e312. [DOI] [PubMed] [Google Scholar]
- [15].Guay C, Kruit JK, Rome S, et al. Lymphocyte-derived exosomal microRNAs promote pancreatic beta cell death and may contribute to type 1 diabetes development. Cell Metab. 2019;29:348–361 e346. [DOI] [PubMed] [Google Scholar]
- [16].Valadi H, Ekstrom K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- [17].Teng Y, Ren Y, Hu X, et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat Commun. 2017;8:14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Salomon C, Torres MJ, Kobayashi M, et al. A gestational profile of placental exosomes in maternal plasma and their effects on endothelial cell migration. PLoS One. 2014;9:e98667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Homer H, Rice GE, Salomon C. Embryo- and endometrium-derived exosomes and their potential role in assisted reproductive treatments-liquid biopsies for endometrial receptivity. Placenta. 2017;54:89–94. [DOI] [PubMed] [Google Scholar]
- [20].Zhang Y, Kim MS, Jia B, et al. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature. 2017;548:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Demir R, Kosanke G, Kohnen G, et al. Classification of human placental stem villi: review of structural and functional aspects. Microsc Res Tech. 1997;38:29–41. [DOI] [PubMed] [Google Scholar]
- [22].Sadovsky Y, Jansson T. Placenta and placental transport function In: Plant TM, Zeleznik AJ, editors. Knobil and Neill’s physiology of reproduction. 4th Edition ed. San Diego: Academic Press; 2015. p. 1741–1782. [Google Scholar]
- [23].Donker RB, Mouillet JF, Chu T, et al. The expression profile of C19MC microRNAs in primary human trophoblast cells and exosomes. Mol Hum Reprod. 2012;18:417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frangsmyr L, Baranov V, Nagaeva O, et al. Cytoplasmic microvesicular form of Fas ligand in human early placenta: switching the tissue immune privilege hypothesis from cellular to vesicular level. Mol Hum Reprod. 2005;11:35–41. [DOI] [PubMed] [Google Scholar]
- [25].Hedlund M, Stenqvist AC, Nagaeva O, et al. Human placenta expresses and secretes NKG2D ligands via exosomes that down-modulate the cognate receptor expression: evidence for immunosuppressive function. J Immunol. 2009;183:340–351. [DOI] [PubMed] [Google Scholar]
- [26].Sabapatha A, Gercel-Taylor C, Taylor DD. Specific isolation of placenta-derived exosomes from the circulation of pregnant women and their immunoregulatory consequences. Am J Reprod Immunol. 2006;56:345–355. [DOI] [PubMed] [Google Scholar]
- [27].Sarker S, Scholz-Romero K, Perez A, et al. Placenta-derived exosomes continuously increase in maternal circulation over the first trimester of pregnancy. J Transl Med. 2014;12:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mincheva-Nilsson L, Baranov V. The role of placental exosomes in reproduction. Am J Reprod Immunol. 2010;63:520–533. [DOI] [PubMed] [Google Scholar]
- [29].Atay S, Gercel-Taylor C, Suttles J, et al. Trophoblast-derived exosomes mediate monocyte recruitment and differentiation. Am J Reprod Immunol. 2011;65:65–77. [DOI] [PubMed] [Google Scholar]
- [30].Motta-Mejia C, Kandzija N, Zhang W, et al. Placental vesicles carry active endothelial nitric oxide synthase and their activity is reduced in preeclampsia. Hypertension. 2017;70:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Salomon C, Yee S, Scholz-Romero K, et al. Extravillous trophoblast cells-derived exosomes promote vascular smooth muscle cell migration. Front Pharmacol. 2014;5:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Delorme-Axford E, Donker RB, Mouillet JF, et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci USA. 2013;110:12048–12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ouyang Y, Bayer A, Chu T, et al. Isolation of human trophoblastic extracellular vesicles and characterization of their cargo and antiviral activity. Placenta. 2016;47:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bayer A, Lennemann NJ, Ouyang Y, et al. Chromosome 19 microRNAs exert antiviral activity independent from type III interferon signaling. Placenta. 2018;61:33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bayer A, Delorme-Axford E, Sleigher C, et al. Human trophoblasts confer resistance to viruses implicated in perinatal infection. Am J Obstet Gynecol. 2015;212:71 e71–71 e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cha DJ, Franklin JL, Dou Y, et al. KRAS-dependent sorting of miRNA to exosomes. Elife. 2015;4:e07197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mukherjee K, Ghoshal B, Ghosh S, et al. Reversible HuR-microRNA binding controls extracellular export of miR-122 and augments stress response. EMBO Rep. 2016;17:1184–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shurtleff MJ, Yao J, Qin Y, et al. Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc Natl Acad Sci USA. 2017;114:E8987–e8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Temoche-Diaz MM, Shurtleff MJ, Nottingham RM, et al. Distinct mechanisms of microRNA sorting into cancer cell-derived extracellular vesicle subtypes. Elife. 2019;8 DOI: 10.7554/eLife.47544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chang G, Mouillet JF, Mishima T, et al. Expression and trafficking of placental microRNAs at the feto-maternal interface. Faseb J. 2017;31:2760–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tong M, Stanley JL, Chen Q, et al. Placental nano-vesicles target to specific organs and modulate vascular tone In Vivo. Hum Reprod. 2017;32:2188–2198. [DOI] [PubMed] [Google Scholar]
- [42].Eguchi S, Takefuji M, Sakaguchi T, et al. Cardiomyocytes capture stem cell-derived, anti-apoptotic microRNA-214 via clathrin-mediated endocytosis in acute myocardial infarction. J Biol Chem. 2019;294:11665–11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nakase I, Kobayashi NB, Takatani-Nakase T, et al. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci Rep. 2015;5:10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3 DOI: 10.3402/jev.v3.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kliman HJ, Nestler JE, Sermasi E, et al. Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118:1567–1582. [DOI] [PubMed] [Google Scholar]
- [46].Nelson DM, Johnson RD, Smith SD, et al. Hypoxia limits differentiation and up-regulates expression and activity of prostaglandin H synthase 2 in cultured trophoblast from term human placenta. Am J Obstet Gynecol. 1999;180:896–902. [DOI] [PubMed] [Google Scholar]
- [47].Mouillet JF, Chu T, Nelson DM, et al. MiR-205 silences MED1 in hypoxic primary human trophoblasts. Faseb J. 2010;24:2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Manoussaka MS, Jackson DJ, Lock RJ, et al. Flow cytometric characterisation of cells of differing densities isolated from human term placentae and enrichment of villous trophoblast cells. Placenta. 2005;26:308–318. [DOI] [PubMed] [Google Scholar]
- [49].Nagamatsu T, Fujii T, Ishikawa T, et al. A primary cell culture system for human cytotrophoblasts of proximal cytotrophoblast cell columns enabling in vitro acquisition of the extra-villous phenotype. Placenta. 2004;25:153–165. [DOI] [PubMed] [Google Scholar]
- [50].Yui J, Garcia-Lloret M, Brown AJ, et al. Functional, long-term cultures of human term trophoblasts purified by column-elimination of CD9 expressing cells. Placenta. 1994;15:231–246. [DOI] [PubMed] [Google Scholar]
- [51].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- [52].Pinilla-Macua I, Watkins SC, Sorkin A. Endocytosis separates EGF receptors from endogenous fluorescently labeled HRas and diminishes receptor signaling to MAP kinases in endosomes. Proc Natl Acad Sci USA. 2016;113:2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Huang F, Kirkpatrick D, Jiang X, et al. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006;21:737–748. [DOI] [PubMed] [Google Scholar]
- [54].Dumont TMF, Mouillet JF, Bayer A, et al. The expression level of C19MC miRNAs in early pregnancy and in response to viral infection. Placenta. 2017;53:23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fitzner D, Schnaars M, van Rossum D, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J Cell Sci. 2011;124:447–458. [DOI] [PubMed] [Google Scholar]
- [56].Nakase I, Noguchi K, Fujii I, et al. Vectorization of biomacromolecules into cells using extracellular vesicles with enhanced internalization induced by macropinocytosis. Sci Rep. 2016;6:34937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Koivusalo M, Welch C, Hayashi H, et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188:547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mercer J, Helenius A. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol. 2012;15:490–499. [DOI] [PubMed] [Google Scholar]
- [59].Mercer J, Helenius A. Virus entry by macropinocytosis. Nat Cell Biol. 2009;11:510–520. [DOI] [PubMed] [Google Scholar]
- [60].Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10:364–371. [DOI] [PubMed] [Google Scholar]
- [61].Racoosin EL, Swanson JA. M-CSF-induced macropinocytosis increases solute endocytosis but not receptor-mediated endocytosis in mouse macrophages. J Cell Sci. 1992;102(Pt 4):867–880. [DOI] [PubMed] [Google Scholar]
- [62].Sun P, Yamamoto H, Suetsugu S, et al. Small GTPase Rah/Rab34 is associated with membrane ruffles and macropinosomes and promotes macropinosome formation. J Biol Chem. 2003;278:4063–4071. [DOI] [PubMed] [Google Scholar]
- [63].Coyne CB, Shen L, Turner JR, et al. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe. 2007;2:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Egami Y, Taguchi T, Maekawa M, et al. Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation. Front Physiol. 2014;5:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Feliciano WD, Yoshida S, Straight SW, et al. Coordination of the Rab5 cycle on macropinosomes. Traffic. 2011;12:1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. [DOI] [PubMed] [Google Scholar]
- [67].Huang F, Khvorova A, Marshall W, et al. Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J Biol Chem. 2004;279:16657–16661. [DOI] [PubMed] [Google Scholar]
- [68].Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. [DOI] [PubMed] [Google Scholar]
- [70].Kurzchalia TV, Parton RG. Membrane microdomains and caveolae. Curr Opin Cell Biol. 1999;11:424–431. [DOI] [PubMed] [Google Scholar]
- [71].Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161:673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Parton RG, Richards AA. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 2003;4:724–738. [DOI] [PubMed] [Google Scholar]
- [73].Smart EJ, Anderson RG. Alterations in membrane cholesterol that affect structure and function of caveolae. Methods Enzymol. 2002;353:131–139. [DOI] [PubMed] [Google Scholar]
- [74].Schnitzer JE, Oh P, Pinney E, et al. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Torgersen ML, Skretting G, van Deurs B, et al. Internalization of cholera toxin by different endocytic mechanisms. J Cell Sci. 2001;114:3737–3747. [DOI] [PubMed] [Google Scholar]
- [76].Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol. 2005;6:112–126. [DOI] [PubMed] [Google Scholar]
- [77].Jeppesen DK, Fenix AM, Franklin JL, et al. Reassessment of exosome composition. Cell. 2019;177:428–445 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Xie L, Mouillet JF, Chu T, et al. C19MC microRNAs regulate the migration of human trophoblasts. Endocrinology. 2014;155:4975–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Svensson KJ, Christianson HC, Wittrup A, et al. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J Biol Chem. 2013;288:17713–17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Costa Verdera H, Gitz-Francois JJ, Schiffelers RM, et al. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J Control Release. 2017;266:100–108. [DOI] [PubMed] [Google Scholar]
- [81].Franzen CA, Simms PE, Van Huis AF, et al. Characterization of uptake and internalization of exosomes by bladder cancer cells. Biomed Res Int. 2014;2014:619829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Escrevente C, Keller S, Altevogt P, et al. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer. 2011;11:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tian T, Wang Y, Wang H, et al. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J Cell Biochem. 2010;111:488–496. [DOI] [PubMed] [Google Scholar]
- [84].Yao Z, Qiao Y, Li X, et al. Exosomes exploit the virus entry machinery and pathway to transmit alpha interferon-induced antiviral activity. J Virol. 2018;92 DOI: 10.1128/JVI.01578-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Feng D, Zhao WL, Ye YY, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11:675–687. [DOI] [PubMed] [Google Scholar]
- [86].Heusermann W, Hean J, Trojer D, et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J Cell Biol. 2016;213:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Vargas A, Zhou S, Ethier-Chiasson M, et al. Syncytin proteins incorporated in placenta exosomes are important for cell uptake and show variation in abundance in serum exosomes from patients with preeclampsia. Faseb J. 2014;28:3703–3719. [DOI] [PubMed] [Google Scholar]
- [88].Record M. Intercellular communication by exosomes in placenta: a possible role in cell fusion? Placenta. 2014;35:297–302. [DOI] [PubMed] [Google Scholar]
- [89].Tian T, Zhu YL, Hu FH, et al. Dynamics of exosome internalization and trafficking. J Cell Physiol. 2013;228:1487–1495. [DOI] [PubMed] [Google Scholar]
- [90].Somiya M. Where does the cargo go?: solutions to provide experimental support for the “extracellular vesicle cargo transfer hypothesis”. J Cell Commun Signal. 2020. DOI: 10.1007/s12079-020-00552-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].He D, Wang H, Ho SL, et al. Total internal reflection-based single-vesicle in situ quantitative and stoichiometric analysis of tumor-derived exosomal microRNAs for diagnosis and treatment monitoring. Theranostics. 2019;9:4494–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.