

Visual Abstract

Keywords: Alternative complement pathway, C3 glomerulopathy, mycophenolate mofetil
Abstract
Background and objectives
C3 glomerulopathy is a complement-mediated disease arising from abnormalities in complement genes and/or antibodies against complement components. Previous studies showed that treatment with corticosteroids plus mycophenolate mofetil (MMF) was associated with improved outcomes, although the genetic profile of these patients was not systematically analyzed. This study aims to analyze the main determinants of disease progression and response to this therapeutic regimen.
Design, setting, participants, & measurements
We conducted a retrospective, multicenter, observational cohort study in 35 nephrology departments belonging to the Spanish Group for the Study of Glomerular Diseases. Patients diagnosed with C3 glomerulopathy (n=81) or dense deposit disease (n=16) between January 1995 and March 2018 were enrolled. Multivariable and propensity score matching analyses were used to evaluate the association of clinical and genetic factors with response to treatment with corticosteroids and MMF as measured by proportion of patients with disease remission and kidney survival (status free of kidney failure).
Results
The study group comprised 97 patients (84% C3 glomerulopathy, 16% dense deposit disease). Forty-two patients were treated with corticosteroids plus MMF, and this treatment was associated with a higher rate of remission and lower probability of kidney failure (79% and 14%, respectively) compared with patients treated with other immunosuppressives (24% and 59%, respectively), or ecluzimab (33% and 67%, respectively), or conservative management (18% and 65%, respectively). The therapeutic superiority of corticosteroids plus MMF was observed both in patients with complement abnormalities and with autoantibodies. However, patients with pathogenic variants in complement genes only achieved partial remission, whereas complete remissions were common among patients with autoantibody-mediated forms. The main determinant of no remission was baseline proteinuria. Relapses occurred after treatment discontinuation in 33% of the patients who had achieved remission with corticosteroids plus MMF, and a longer treatment length of MMF was associated with a lower risk of relapse.
Conclusions
The beneficial response to corticosteroids plus MMF treatment in C3 glomerulopathy appears independent of the pathogenic drivers analyzed in this study.
Introduction
C3 glomerulopathies are a rare and heterogeneous group of glomerular diseases linked pathogenetically to a dysregulation of the alternative complement pathway, mainly on the fluid phase and glomerular microenvironment (1–3). The diagnosis relies on histopathological examination and immunofluorescence criteria, and two different clinicopathological entities can be distinguished: C3 glomerulopathy and dense deposit diseases (2).
The disease may be driven by genetic factors, involving pathogenic variants in several complement genes (4–8), or by acquired factors, namely autoantibodies against complement pathway regulators and/or C3 nephritic factors targeting the C3 convertases (1,9,10). Furthermore, there is a broad consensus to consider C3 glomerulopathy associated with monoclonal gammopathies (MGUS) as a distinct subtype of the disease because of its peculiar pathogenesis (11).
The overall prognosis of C3 glomerulopathy is poor, with progression to kidney failure in up to 40%–50% of the cases (6,12,13). There is a scarcity of data about treatment strategies in C3 glomerulopathy. Eculizumab, a humanized mAb against complement protein C5, has been tested in C3 glomerulopathy with limited results (14–16), suggesting a particular benefit among patients with rapidly progressive presentations.
In a previous study, we reported that the combination of corticosteroids plus mycophenolate mofetil (MMF) was associated with better kidney outcomes as compared with other different therapeutic regimens (17). These results were corroborated by some groups (18), although discrepant results were also reported by others (19,20). However, the low number of cases with complement analysis in our original study hindered the possibility to precisely assess the relevance of genetic and acquired abnormalities in the clinical response to this regimen.
Therefore, this study aims to analyze the main determinants of disease progression and response to this therapeutic regimen in a large series of patients with C3 glomerulopathy in whom a comprehensive analysis of the complement genes, determination of nephritic factors, and autoantibodies against complement regulatory factors was performed.
Materials and Methods
Study Population
Patients diagnosed with C3 glomerulopathy between January 1995 and March 2018 in 35 nephrology departments belonging to the Spanish Group for the Study of Glomerular Diseases were enrolled.
The diagnosis was on the basis of the 2013 Consensus Guidelines criteria (21), which required C3 staining on immunofluorescence at least two orders of magnitude greater than any Ig staining. Patients with concomitant MGUS, other autoimmune diseases, or infected with hepatitis B, C, or HIV were excluded.
The study was approved by the institutional review board of Hospital 12 de Octubre, and was conducted in accordance with the Declaration of Helsinki.
Clinical, Laboratory, and Histopathologic Data
Baseline and follow-up data were compiled from the medical records of all participating centers, following a uniform protocol that included demographics, type of clinical presentation, serum creatinine, eGFR (estimated using the CKD Epidemiology Collaboration equation for adults and the bedside Schwartz equation modified in 2009 for children), C3 and C4, serum albumin, and 24-hour proteinuria. All biochemical parameters were analyzed using routine laboratory methods.
Kidney biopsy specimens were examined in the pathology departments of participating hospitals, and the following histopathological features were recorded: percentage of sclerosed glomeruli, light microscopy pattern, degree of interstitial fibrosis/tubular atrophy, and arteriosclerosis. All samples were further evaluated with immunofluorescence staining for C3, C1q, IgG, IgA, IgM, Igκ, and Igλ (graded as 0–3+).
Patients were considered to have dense deposit disease when highly electron-dense intramembranous deposits were observed on electron microscopy, and C3 glomerulopathy when deposits did not fulfill this criterion.
Complement Genetics and Molecular Studies
A detailed description of the methods used for the search and categorization of the genetic variants and for the detection of autoantibodies is provided in Supplemental Appendix 1.
Treatment Regimens
Information about the medications prescribed was obtained from medical records at baseline and throughout the follow-up period. These treatment regimens were categorized into four subgroups: corticosteroids plus MMF, other immunosuppressive regimens (including corticosteroids alone, cyclophosphamide, azathioprine, tacrolimus, cyclosporine, and rituximab), treatment with eculizumab, and conservative management (including exclusive treatment with renin-angiotensin system blockade).
Definitions and Outcomes
Baseline was defined as the time at which the kidney biopsy was performed, and follow-up period as the interval between kidney biopsy and last outpatient visit or kidney failure (eGFR<15 ml/min per 1.73 m2 by the CKD Epidemiology Collaboration equation or the modified bedside Schwartz equation, maintenance dialysis, or preemptive kidney transplantation).
Nephrotic syndrome was defined as a proteinuria of >3.5 g/d along with serum albumin <3 g/dl. Nephritic syndrome was defined as the combination of hematuria, non-nephrotic proteinuria, hypertension, and kidney function impairment. Asymptomatic urinary abnormalities were defined by the presence of non-nephrotic proteinuria and/or persistent microscopic hematuria defined as the presence of five or more erythrocytes per high-power field.
Complete remission was defined as eGFR>60 ml/min per 1.73 m2 and proteinuria <0.5 g/24 h. Partial remission was defined as a reduction of proteinuria >50% (and a proteinuria value of <3.5 g/d in patients with nephrotic range proteinuria at baseline), plus stabilization (±25%) or improvement in eGFR. Proteinuria was adjusted to a standard body surface of 1.73 m2 in children. Relapse was defined as the return of pretreatment proteinuria and/or declining kidney function after any remission (18).
The main outcomes analyzed were disease remission (either partial or complete) and kidney survival (defined as a status free of kidney failure). The study patients were further analyzed according to C3 glomerulopathy clustering analysis, as described by Iatropoulos et al. (22) on the basis of their proposed algorithm.
Statistical Analyses
We conducted a retrospective, multicenter, observational cohort study. Descriptive statistics are presented as mean±SD, or median and interquartile range (IQR) for continuous variables, and absolute values and percentages for categorical variables. Parametric and nonparametric tests were chosen as appropriate for descriptive comparisons of continuous variables, and chi-squared test for categorical variables. For the comparisons in smaller groups, we performed a Fisher exact test. Cox proportional hazards regression and logistic regression models were used to analyze the main determinants of outcomes. Additionally, a 1:1 nearest-neighbor propensity score matching analysis was applied to compare kidney outcomes of patients treated with or without MMF. More detailed information about statistical methods is provided in Supplemental Appendix 1.
Results
Baseline Characteristics
During the study period, data from 167 patients were retrieved, of whom 70 were excluded because of nonfulfillment of the diagnostic criteria, presence of MGUS, or absence of blood sample for genetic analysis (Figure 1). Thus, the study group consisted of 97 patients (81 with C3 glomerulopathy [84%] and 16 with dense deposit disease [16%]), whose main characteristics are detailed in Table 1.
Figure 1.

Flowchart of patients included in the study.
Table 1.
Baseline clinical and histologic characteristics
| Variable | Total, n=97 | C3 GN, n=81 | Dense Deposit Disease, n=16 |
|---|---|---|---|
| Baseline | |||
| Age at diagnosis, yr | 32±21 | 34±21 | 22±15 |
| Adult/pediatric, % | 74/26 | 74/26 | 69/31 |
| Men, n (%) | 54 (56) | 49 (61) | 5 (31) |
| Antecedent infection, n (%) | 26 (27) | 21 (26) | 5 (31) |
| Clinical presentation, n (%) | |||
| Nephrotic syndrome | 39 (40) | 32 (39) | 7 (44) |
| Nephritic syndrome | 29 (30) | 26 (32) | 3 (19) |
| Asymptomatic urinary abnormalities | 29 (30) | 23 (28) | 6 (38) |
| Serum creatinine, mg/dl | 1.5 [0.8–3] | 1.5 [0.8–3] | 1.1 [0.6–3.1] |
| eGFR at baseline, ml/min per 1.73 m2 | 55 [20–120] | 53 [20–116] | 85 [26–134] |
| eGFR ranges, ml/min per 1.73 m2, n (%) | |||
| ≥60 | 41 (42) | 32 (40) | 9 (56) |
| 30–59 | 21 (22) | 18 (22) | 13 (19) |
| <30 | 35 (36) | 31 (38) | 4 (25) |
| Albumin, g/dl | 3±0.8 | 3.1±0.8 | 3±0.8 |
| Proteinuria, g/24 h | 3 [1.6–6.8] | 3 [1.5–6.8] | 3.6 [1.8–7.9] |
| Serum C3, mg/dla | 61±40 | 63±41 | 48±35 |
| Low serum C3, <75 mg/dl, n (%) | 66 (68) | 52 (64) | 14 (88) |
| Serum C4, mg/dlb | 24±9 | 25±9 | 23±9 |
| Serum C5b–9, mg/Lc | 360 (170–828) | 294 (160–781) | 497 (329–1276) |
| Elevated serum C5b-9, >100 mg/L, n (%) | 82 (84) | 67 (82) | 15 (94) |
| Histopathology | |||
| Light microscopy pattern, n (%) | |||
| Membranoproliferative GN | 70 (73) | 55 (68) | 15 (94) |
| Diffuse endocapillary proliferative GN | 7 (7) | 7 (9) | 0 (0) |
| Mesangial proliferative GN | 15 (15) | 14 (17) | 1 (6) |
| Diffuse sclerosing GN | 5 (5) | 5 (6) | 0 (0) |
| Globally sclerotic glomeruli, % | 7 [0–24] | 6 [0–24] | 12 [0–35] |
| Segmental sclerotic glomeruli, n (%) | 15 (15) | 11 (14) | 4 (25) |
| Cellular or fibrocellular crescents, n (%) | 24 (25) | 21 (26) | 3 (19) |
| Tubular atrophy/interstitial fibrosis, n % | |||
| Absence | 36 (37) | 30 (37) | 6 (38) |
| Mild | 32 (33) | 29 (36) | 3 (19) |
| Moderate | 19 (20) | 14 (17) | 5 (31) |
| Severe | 10 (10) | 8 (10) | 2 (13) |
| Arterio- and arteriolosclerosis, n (%) | 24 (25) | 19 (24) | 5 (31) |
Continuous variables are presented as mean±SD, or median [interquartile range].
Reference values: 75–135.
Reference values: 14–60.
Reference values: <100.
The majority of patients (92%) were White, and patients with dense deposit disease were significantly younger compared with patients with C3 glomerulopathy.
The membranoproliferative pattern (membranoproliferative GN) was the most common glomerular pattern of injury in both groups, accounting for over 94% of patients with dense deposit disease versus 68% of patients with C3 glomerulopathy. Supplemental Table 1 displays the main characteristics according to age groups.
Complement Genetic and Molecular Findings
Analysis of the complement genes by next-generation sequencing identified a total of 115 rare variants with a minor allele frequency <1%. In addition, three CFHR genomic rearrangements were identified by Multiplex Ligation-dependent Probe Amplification. From these 118 rare variants, 18 were considered pathogenic (19% of study patients), 58 were of unknown significance, and 42 were benign. Pathogenic variants were mainly found in the prototypical genes CFH, CFI, C3, CFB, and CFHRs (Supplemental Table 2 displays the type of complement pathogenic variants found in the study patients).
Acquired factors were detected in 29 patients (30%), with 23 patients (24%) positive for C3 nephritic factor and eight cases positive for anti-factor H (FH; 8%). Two patients presented concomitant positivity for both types of antibodies. The presence of C3 nephritic factor was significantly associated with patients with dense deposit disease (P=0.02). In contrast, anti-FH antibodies were equally distributed between subgroups. As shown in Supplemental Table 3, patients with autoantibodies had significantly lower serum levels of C3, together with higher serum C5b–9 levels at baseline.
Treatment
A total of 85% of patients were initially treated with renin-angiotensin system blockade, including angiotensin-converting enzyme inhibitors in 63%, angiotensin receptor blockers in 12%, or both in 9%.
Seventeen patients (18%) were never treated with immunosuppressants; 42 (43%) patients received corticosteroids plus MMF; whereas 29 (30%) patients were treated with other conventional immunosuppressants, including corticosteroids alone in 13 (13%) patients, cyclophosphamide in eight (8%) patients, calcineurin inhibitors in two (2%) patients, azathioprine in one (1%) patient, and rituximab in five (5%) patients. Only nine (9%) patients were treated with eculizumab.
Patients treated with corticosteroids alone received a median initial dose of 0.9 mg/kg per day followed by a tapering phase over a median period of 9 months (IQR, 3–18 months). Patients treated with corticosteroids plus MMF received the same median initial dose of corticosteroid therapy followed by a tapering phase during a median period of 9 months (IQR, 6–17 months). The median initial dose of MMF received was 1000 mg (IQR, 750–1500 mg), although the maintenance dose was titrated individually according to tolerance. Patients treated with cyclophosphamide received intravenous pulses with a median dose of 8 mg/kg over a period of 5 months (IQR, 1–10). Rituximab doses were adjusted by body surface area (375 mg/m2), and the median doses received were 3 (IQR, 2–4). Eculizumab was prescribed according to guidelines, during a median period of 7 months (IQR, 4–30 months).
Patients treated conservatively were significantly older, with worse baseline kidney function and higher albumin levels, than those treated with immunosuppression (Table 2). Patients treated with eculizumab showed a nonsignificant trend to have a higher percentage of complement pathogenic variants, whereas the presence of autoantibodies against complement components was significantly higher in patients treated with other immunosuppressants.
Table 2.
Clinical characteristics according to therapeutic regimen
| Variable | CS+MMF, n=42 | Other IS Therapy,a n=29 | Eculizumab, n=9 | No IS Therapy, n=17 |
|---|---|---|---|---|
| Baseline | ||||
| Age at diagnosis, yr | 30±19 | 27±22 | 28±15 | 46±19 |
| Adult/pediatric, % | 69/31 | 62/38 | 75/25 | 17/0 |
| Men, n (%) | 23 (55) | 16 (55) | 5 (56) | 10 (59) |
| Clinical presentation, n (%) | ||||
| Nephrotic syndrome | 18 (43) | 12 (41) | 4 (44) | 5 (29) |
| Nephritic syndrome | 11 (26) | 9 (31) | 2 (22) | 7 (41) |
| Asymptomatic urinary abnormalities | 13 (31) | 8 (28) | 3 (33) | 5 (29) |
| Serum creatinine, mg/dl | 1.3 [0.7–2.4] | 1.2 [0.8–3.4] | 1.5 [0.9–3.7] | 3 [1.1–3.8] |
| eGFR at diagnosis, ml/min per 1,73 m2 | 57 [30–124] | 63 [15–132] | 47 [19–99] | 20 [12–76] |
| Albumin, g/dl | 3.1±0.8 | 2.8±0.7 | 3.1±0.9 | 3.5±0.9 |
| Proteinuria, g/24 h | 3 [1.8–4.6] | 3 [1.6–9] | 3 [2.7–12] | 1.8 [0.7–5.3] |
| Serum C3, mg/dlb | 61±45 | 56±38 | 56±33 | 72±32 |
| Serum C5b–9, mg/Lc | 394 (206–849) | 396 (153–840) | 770 (278–1130) | 191 (124–409) |
| Complement pathogenic variants, n (%) | 5 (12) | 8 (28) | 3 (33) | 2 (12) |
| Autoantibodies, n (%)d | 10 (24) | 13 (45) | 4 (44) | 2 (12) |
| RAS blockade, n (%) | 36 (86) | 24 (83) | 8 (89) | 17 (100) |
| Treatment-related adverse events, n (%) | ||||
| Infectious complicationse | 5 (12) | 6 (21) | 5 (56) | 1 (6) |
| Diabetes mellitus | 1 (2) | 2 (7) | 0 (0) | 0 (0) |
| Cytopenia | 4 (9) | 5 (17) | 0 (0) | 0 (0) |
| Cardiovascular events | 4 (9) | 4 (14) | 1 (6) | 1 (11) |
| Other adverse eventsf | 4 (9) | 2 (7) | 0 (0) | 0 (0) |
| Outcomes | ||||
| Follow-up, mo | 49 [23–97] | 43 [5–81] | 46 [22–85] | 44 [22–67] |
| Partial remission, n (%) | 18 (43) | 4 (14) | 3 (33) | 3 (18) |
| Complete remission, n (%) | 15 (36) | 3 (10) | 0 (0) | 0 (0) |
| Time to remission, mo | 14 [6–25] | 22 [9–35] | 40 [15–60] | 19 [14–30] |
| Kidney failure, n (%) | 6 (14) | 17 (59) | 6 (67) | 11 (65) |
Continuous variables are presented as mean±SD, or median [interquartile range]. CS, corticosteroid therapy; MMF, mycophenolate mofetil; IS, immunosuppressive; RAS, renin-angiotensin system.
Including corticosteroids alone, cyclophosphamide, azathioprine, tacrolimus, cyclosporine, and rituximab.
Reference values: 75–135.
Reference values: <100.
Including C3 nephritic factor and anti-factor H.
Including pneumonia (6), flu (3), urinary tract infection pyelonephritis (3), cytomegalovirus infection (3), abdominal sepsis (1), and herpes zoster virus infection (1).
Including gastrointestinal intolerance, cataracts, and avascular necrosis of the hip.
The overall tolerance to therapeutic regimens was fairly good (Table 2). Infectious complications were the most common, accounting for 18% of cases, followed by cardiovascular events (10%) and cytopenias (9%). Infectious complications were significantly lower in patients treated with corticosteroids plus MMF than those of patients treated with other therapies.
Outcomes: Disease Remission
During a median follow-up time of 46 months (IQR, 22–112 months), 46 (47%) patients achieved remission; 18 (19%) achieved complete remission, and 28 (29%) achieved partial remission. Median time to remission was 14 months (IQR, 8–28 months). Remission rates were significantly higher among patients treated with corticosteroids plus MMF (Table 2).
Table 3 displays the main characteristics of patients according to remission status. Patients who achieved remission were significantly younger, with better baseline kidney function and lower chronicity on kidney biopsy (Supplemental Table 4 displays the characteristics of these patients according to baseline eGFR). Furthermore, patients who achieved complete remission had a significantly lower percentage of complement pathogenic variants, along with a significant higher percentage of C3 nephritic factor, compared with patients with partial remission.
Table 3.
Clinical, histopathologic, and genetic characteristics of patients according to remission
| Variable | Remission (Partial+ Complete), n=46 | No Remission, n=51 | P Value | Partial Remission, n=28 | Complete Remission, n=18 | P Value |
|---|---|---|---|---|---|---|
| Baseline | ||||||
| Age at diagnosis, yr | 26±19 | 37±21 | 0.01 | 27±19 | 25±19 | 0.75 |
| Adult/pediatric, % | 61/39 | 84/16 | 0.009 | 68/32 | 50/50 | 0.22 |
| Men, n (%) | 21 (46) | 33 (65) | 0.06 | 14 (50) | 7 (39) | 0.46 |
| C3 GN/dense deposit disease, n | 42/ 4 | 39/ 12 | 0.04 | 26/ 2 | 16/ 2 | 0.51 |
| Antecedent infection, n (%) | 10 (22) | 16 (31) | 0.28 | 5 (18) | 5 (28) | 0.43 |
| Clinical presentation, n (%) | 0.40 | 0.05 | ||||
| Nephrotic syndrome | 17 (37) | 22 (43) | 9 (32) | 8 (44) | ||
| Nephritic syndrome | 14 (30) | 15 (29) | 6 (21) | 8 (44) | ||
| Asymptomatic urinary abnormalities | 15 (33) | 14 (27) | 13 (18) | 2 (11) | ||
| Serum creatinine, mg/dl | 1 [0.6–2] | 2 [1–3.5] | 0.002 | 0.9 [0.7–1.8] | 1 [0.6–2.6] | 0.82 |
| eGFR ranges, ml/min per 1.73 m2, n (%) | 0.02 | 0.56 | ||||
| ≥60 | 25 (54) | 16 (31) | 15 (54) | 10 (55) | ||
| 30–59 | 11 (24) | 10 (20) | 8 (28) | 3 (17) | ||
| <30 | 10 (22) | 25 (49) | 5 (18) | 5 (28) | ||
| Albumin, g/dl | 3.2±0.8 | 3±0.8 | 0.14 | 3.3±0.7 | 3±0.9 | 0.31 |
| Serum C3, mg/dla | 54±40 | 67±39 | 0.10 | 60±41 | 45±38 | 0.23 |
| Serum C4, mg/dlb | 23±10 | 25±9 | 0.32 | 24±10 | 21±9 | 0.32 |
| Proteinuria, g/24 h | 2.6 [1.3–4] | 4.1 [1.7–9] | 0.04 | 2.2 [1.3–3.8] | 2.9 [1.7–4] | 0.63 |
| Histopathology | ||||||
| Globally sclerotic glomeruli, % | 4 [0–8] | 20 [0–40] | 0.001 | 2 [0–8] | 4 [0–7] | 0.88 |
| Tubular atrophy/interstitial fibrosis, n % | 0.002 | 0.11 | ||||
| Absence | 22 (48) | 14 (28) | 10 (36) | 12 (67) | ||
| Mild | 19 (41) | 13 (26) | 14 (50) | 5 (28) | ||
| Moderate | 3 (7) | 16 (31) | 3 (11) | 0 (0) | ||
| Severe | 2 (4) | 8 (16) | 1 (4) | 1 (5) | ||
| Arterio- and arteriolosclerosis, n (%) | 6 (13) | 18 (35) | 0.01 | 5 (18) | 1 (6) | 0.23 |
| Complement genetic analysisc | ||||||
| Complement abnormalities, n (%) | ||||||
| None | 19 (41) | 18 (35) | 0.54 | 11 (39) | 8 (44) | 0.73 |
| Variants unknown significance | 21 (46) | 21 (41) | 0.63 | 11 (39) | 10 (56) | 0.28 |
| Pathogenic variants | 6 (13) | 12 (23) | 0.16 | 6 (21) | 0 (0) | 0.03 |
| Autoantibodies, n (%) | ||||||
| C3 nephritic factor | 11 (24) | 12 (24) | 0.95 | 3 (11) | 8 (44) | 0.01 |
| Anti-factor H | 5 (11) | 3 (6) | 0.37 | 3 (11) | 2 (11) | 0.97 |
Continuous variables are presented as mean±SD, or median [interquartile range].
Reference values: 75–135.
Reference values: 14–60.
Information regarding complement abnormalities is provided in Supplemental Table 2.
By multivariable logistic regression analysis, the main determinants of remission (complete or partial) were female sex, younger age at diagnosis, lower baseline proteinuria, and treatment with corticosteroids plus MMF (Supplemental Table 5).
Outcomes: Kidney Survival
During the follow-up period, 40 (41%) patients reached kidney failure. The clinical profile of patients who reached kidney failure was older men presenting with lower eGFR and higher proteinuria at baseline, together with greater percentage of glomerulosclerosis and tubulointerstitial fibrosis (Supplemental Table 6).
A total of 42% of patients who eventually developed kidney failure had received an immunosuppressive regimen different than corticosteroids plus MMF, followed by 28% of patients prescribed conservative management. By contrast, the majority of patients who did not reach kidney failure had been treated with corticosteroids plus MMF (Supplemental Table 6, Table 2). Over 3 years, treatment with corticosteroids plus MMF was associated with a lower chance of reaching kidney failure by 77% compared with other regimens (14% versus 62%).
By Cox regression analysis, the main determinants of kidney failure were baseline eGFR, degree of interstitial fibrosis and tubular atrophy, and treatments other than corticosteroids plus MMF (Supplemental Table 7). Figure 2 shows Kaplan–Meier curves for kidney survival according to therapeutic regimens.
Figure 2.

Kaplan–Meier curves for kidney survival according to therapeutic regimens. The time point 0 represents the onset of treatment. MMF, mycophenolate mofetil.
Propensity Score Matching Analysis
A propensity score matching analysis was further applied to analyze the potential therapeutic superiority of corticosteroids plus MMF in C3 glomerulopathy.
Before propensity score matching analysis, patients treated with corticosteroids plus MMF were characterized by a better baseline kidney function, lower percentage of glomerulosclerosis, lower degree of chronicity on kidney biopsy, and a significantly lower proportion of dense deposit disease (Supplemental Table 8). After this analysis, 34 patients treated with corticosteroids plus MMF were compared with 34 patients treated with other immunosuppressants: 13 (38%) with prednisone, eight (23%) with cyclophosphamide, five (15%) with rituximab, six (18%) with eculizumab, and two (6%) with calcineurin inhibitors.
The main prognostic covariables were properly balanced between subgroups after propensity score matching analysis, and no significant differences were observed (Supplemental Figure 1, Supplemental Table 8). Consistent with the results for the entire cohort, kidney survival was significantly better in patients treated with corticosteroids plus MMF even after adjusting for potential confounders. Figure 3 shows Kaplan–Meier curves for kidney survival according to treatment with or without corticosteroids plus MMF in this matched cohort.
Figure 3.

Kaplan–Meier curves for kidney survival according to treatment with mycophenolate mofetil plus corticosteroids versus other immunosuppression in the propensity matched cohort.
Treatment with Corticosteroids plus MMF
The therapeutic benefit of corticosteroids plus MMF was found both in patients with pathogenic variants and in those with autoantibodies (Figure 4). The main pathogenic variants were located in C3 (40%), CFH (20%), CFI (20%), and CFB (20%).
Figure 4.
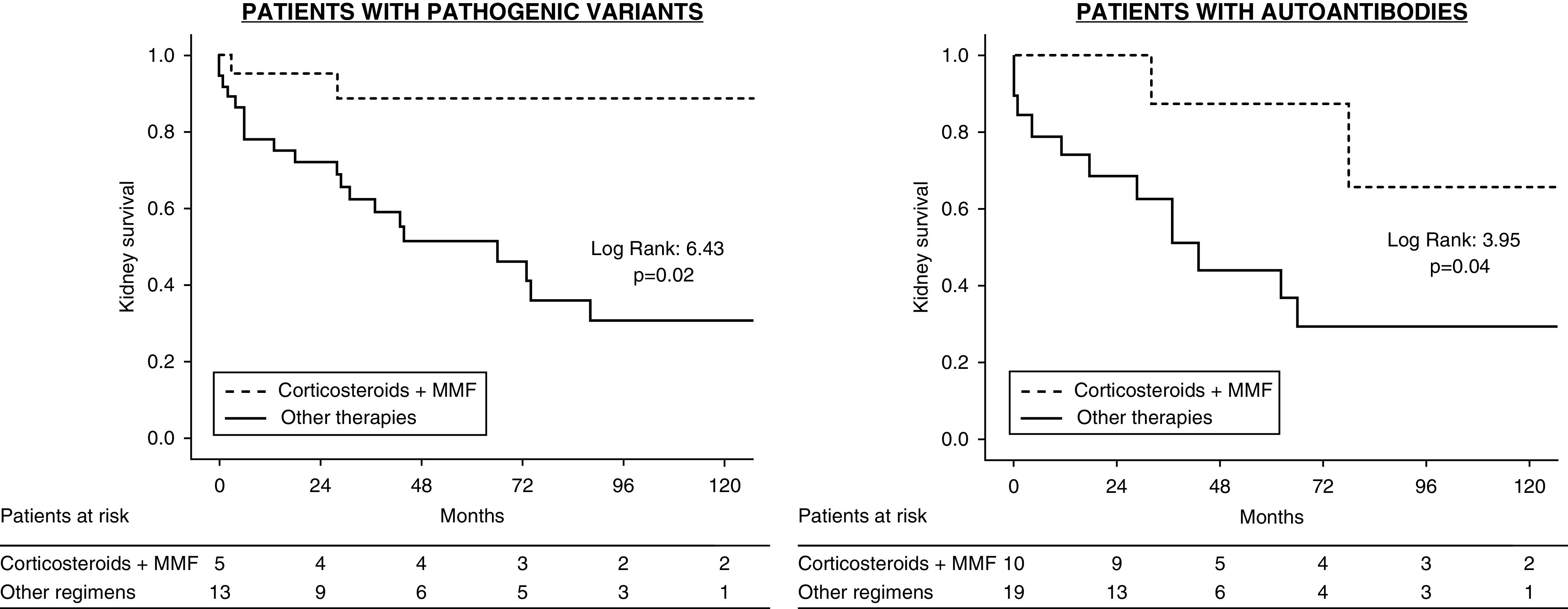
Kidney survival in patients treated with corticosteroids plus mycophenolate mofetil versus other regimens according to the presence of pathogenic variants or autoantibodies.
Table 4 shows the main characteristics of patients treated with MMF plus corticosteroids according to remission status. No significant differences were observed between groups, except for higher proteinuria in patients who did not respond to this regimen. The median initial dose of MMF received was 1000 mg (IQR, 750–1500 mg), although this dose was significantly higher in patients who did not achieve remission.
Table 4.
Clinical, histopathologic, and genetic characteristics of patients treated with corticosteroids plus mycophenolate mofetil, according to remission (partial or complete)
| Variable | Responders | Nonresponders | P Value |
|---|---|---|---|
| Baseline | |||
| Patients, n (%) | 33 (79) | 9 (21) | |
| Age at diagnosis, yr | 30±20 | 31±15 | 0.92 |
| Men, n (%) | 16 (49) | 7 (78) | 0.15 |
| C3 GN/dense deposit disease, n | 32/1 | 8/1 | 0.39 |
| Antecedent infection, n (%) | 7 (21) | 1 (11) | 0.44 |
| Clinical presentation, n (%) | 0.22 | ||
| Nephrotic syndrome | 11 (33) | 7 (78) | |
| Nephritic syndrome | 11 (33) | 0 (0) | |
| Asymptomatic urinary abnormalities | 11 (33) | 2 (22) | |
| Serum creatinine, mg/dl | 1.3 [0.6–2.4] | 1.2 [0.9–2.5] | 0.78 |
| Albumin, g/dl | 3.2±0.8 | 2.8±0.6 | 0.12 |
| Proteinuria, g/24 h | 2.7 [1.5–4] | 5 [3–10] | 0.03 |
| Serum C3, mg/dla | 60±43 | 63±55 | 0.83 |
| Serum C5b–9, mg/Lb | 344 (200–792) | 681 (268–1298) | 0.21 |
| Histopathology | |||
| Globally sclerotic glomeruli, % | 4 [0–8] | 6 [0–11] | 0.45 |
| Tubular atrophy/interstitial fibrosis, n % | 0.47 | ||
| Absence | 14 (42) | 4 (44) | |
| Mild | 15 (46) | 2 (22) | |
| Moderate | 2 (6) | 2 (22) | |
| Severe | 2 (6) | 1 (11) | |
| Arterio- and arteriolosclerosis, n (%) | 5 (15) | 2 (22) | 0.63 |
| Immunosuppressive regimen | |||
| Initial dose of prednisone, mg/kg per d | 0.9±0.4 | 0.9±0.2 | 0.97 |
| Length of treatment with prednisone, mo | 15±6 | 10±7 | 0.14 |
| Mean initial dose of MMF, mg/d | 1000±500 | 1500±500 | 0.01 |
| Length of treatment with MMF, mo | 14 [7–37] | 18 [9–26] | 0.61 |
| Complement genetics and molecular analysisc | |||
| Complement abnormalities, n (%) | |||
| None | 15 (46) | 4 (44) | 0.96 |
| Variants of unknown significance | 15 (46) | 3 (33) | 0.51 |
| Pathogenic variants | 3 (9) | 2 (22) | 0.28 |
| Autoantibodies, n (%) | |||
| C3 nephritic factor | 6 (18) | 2 (22) | 0.85 |
| Anti-factor H | 5 (15) | 0 (0) | 0.22 |
Continuous variables are presented as mean±SD, or median [interquartile range]. MMF, mycophenolate mofetil.
Reference values: 75–135.
Reference values: <100.
Information regarding complement pathogenic abnormalities is provided in Supplemental Table 2.
By Cox regression analysis, the main determinant of no remission with this therapeutic regimen was baseline proteinuria (Supplemental Table 9).
Finally, the therapeutic benefit of corticosteroids plus MMF was further analyzed in patients according to clusters, showing a significant trend toward a better kidney survival in clusters 1, 2, and 4 and a nonsignificant similar trend in cluster 3 with this therapy (Supplemental Figure 2).
Relapses after Discontinuation of Corticosteroids plus MMF
Eleven patients out of 33 (33%) who had achieved remission with corticosteroids plus MMF experienced a relapse in a median of 26 months (IQR, 15–79 months). As shown in Supplemental Table 10, patients who presented a relapse were significantly younger, with better kidney function at baseline.
Median doses of immunosuppressants received in each group were similar. However, a nonsignificant trend toward a shorter length of treatment was observed in patients who eventually relapsed (10±7 in nonresponders versus 15±6 months in responders).
By Cox regression analysis, a longer length of treatment with MMF was associated with lower risk of relapse (Supplemental Table 11). None of the patients with a relapse reached kidney failure at the end of follow-up.
Discussion
In this study of a large cohort of patients with C3 glomerulopathy with a comprehensive genetic and molecular analysis of the complement pathway, we confirmed the therapeutic efficacy of corticosteroid plus MMF that we had previously reported in a smaller study (17). Treatment with corticosteroids plus MMF induced a significantly higher number of remissions as compared with other therapies. Similarly, the occurrence of kidney failure was significantly lower in patients treated with this regimen. By multivariable analysis, treatment with corticosteroids plus MMF was significantly associated with both remission of the disease and a lower risk of kidney failure.
Patients treated with corticosteroids plus MMF had a better kidney function at baseline and lower chronicity on kidney biopsy. To eliminate these imbalances, we performed a propensity score matching analysis that compared patients treated with corticosteroids plus MMF and patients treated with other therapies, after balancing both subgroups for the main prognostic covariables. The therapeutic superiority of corticosteroids plus MMF was also evident after this analysis.
Another major finding of our study was that its beneficial effect was independent of the pathogenic drivers analyzed. Kidney survival and remission were higher both in patients with pathogenic variants in complement genes, and in whom the disease was mediated by autoantibodies. However, these results should be interpreted with caution considering the relatively small number of cases with pathogenic variants in complement genes, and that other potential factors could also drive the disease process (23). It is noteworthy that the percentage of complement pathogenic variants was higher in patients who were treated with eculizumab, but only three out of nine achieved partial remission.
The number of patients who tested positive for autoantibodies was somewhat lower in our study, as compared with previous reports (6,20). We hypothesize that the determination of autoantibodies in some patients when they were already receiving immunosuppressive treatments could account for this discrepancy (24,25).
Recently, a new pathogenic classification of immune complex–mediated membranoproliferative GN and C3 glomerulopathy has been proposed, using an unsupervised hierarchical clustering of histologic, genetic, and clinical data (22). According to this classification, patients are grouped into four distinct clusters characterized by specific clinical features and survival (22). We classified our patients into these four different clusters, following the diagnostic algorithm proposed by the authors. Notably, kidney survival was better in patients treated with corticosteroids plus MMF, as compared with other therapeutic regimens, independently of cluster classification.
Taken together, these data point to a nonspecific favorable effect of this regimen, independent of the pathogenic pathway of the disease and its histologic presentation. However, it should be noted that in patients with pathogenic variants in complement genes, only partial remissions were achieved, whereas in patients with autoantibody-mediated forms of the disease, complete remissions were more common than partial remissions. Notably, the amount of proteinuria was the only factor significantly associated with a lack of response to corticosteroids plus MMF.
MMF is a prodrug of mycophenolic acid, which inhibits the enzyme inosine monophosphate dehydrogenase, thus blocking purine synthesis in B and T lymphocytes (26). Although the immunosuppressive effects of MMF lie in its ability to inhibit the proliferation of lymphocytes, several other properties have been found both in vitro and in vivo (26). For instance, MMF impairs the expression of glycoproteins and adhesion molecules on endothelial cells, responsible for the recruitment of leukocytes to the site of inflammation (27–29). In addition, MMF can inhibit the maturation of dendritic cells (30), the proliferation of mesangial cells (31), and the hypertrophy and apoptosis of podocytes (32). MMF has also been shown to have antifibrotic effects through the inhibition of fibroblasts proliferation (33) and the upregulation of neutral endopeptidase, which degrades angiotensin II (34). The alternative complement pathway activation that occurs in C3 glomerulopathy leads to inflammation in endothelial cells and consequent neutrophil/macrophage recruitment in the glomerular microenvironment (35–38), which ultimately results in podocyte injury and dysfunction (39). In this setting, MMF could reduce inflammation in the glomerular microenvironment in a nonspecific manner.
New complement-targeted therapies are being developed and their possible efficacy in C3 glomerulopathy studied in clinical trials (40). Pending these more specific, and perhaps more effective, therapeutic alternatives, our data show that treatment with corticosteroids plus MMF induces a favorable effect on the disease.
The results of this study are in line with those of a prior study conducted by Columbia University (18) in which proteinuria was higher among nonresponders to corticosteroids plus MMF. The latter study, however, included a multiethnic cohort with younger patients, and the authors found a trend toward higher serum C5b–9 levels among responders, findings that we could not reproduce in our study.
Conversely, in other previously published observational studies, the therapeutic benefit of corticosteroids plus MMF could not be proven (19,20). Unlike these cohorts, in our study, patients with underlying MGUS, those who tested positive for other autoantibodies, or those suspected to have other autoimmune diseases were excluded, and all cases had genetic analyses performed, which helped us to analyze a probably more homogenous sample. Some other differences, such as the mean older age, the lower number of dense deposit disease, the smaller number of treated patients, or the shorter follow-up, in the aforementioned studies could also have an influence in these discrepant results (19,20). Nevertheless, prospective and controlled clinical trials are needed to confirm the beneficial effects of corticosteroids plus MMF in C3 glomerulopathy.
Treatment with corticosteroids plus MMF was well tolerated in the majority of patients, and the incidence of infections was lower than that of other immunosuppressive medications. The mean initial dose of MMF in those patients who presented a disease remission was 1000 mg/d, although this dose was adjusted during follow-up at the discretion of the treating physician, according to tolerance. This dose is somewhat lower than that used as the maintenance therapy in other autoimmune diseases such as lupus nephritis, although the appropriate dose and duration of both corticosteroids and MMF in C3 glomerulopathy should be defined by prospective studies. Our data suggest, however, that a longer duration of the treatment could induce better results. Relapses were significantly associated with a shorter duration of this treatment; however, experience in the use of MMF in organ transplantation and other autoimmune diseases such as lupus nephritis has shown that this drug can be administered for long periods of time with a good safety profile (41).
Several limitations should be noted in our study. Because of the observational and retrospective nature of the study, no causal relationships could be established. Patients were not randomly assigned to each therapeutic regimen, although we performed a propensity score matching analysis to control for confounding by indication. The detection of autoantibodies was limited to C3 nephritic factor and anti-FH, and in some cases, the screening of autoantibodies and molecular analysis were not performed at baseline. The predominance of White patients and patients with C3 glomerulopathy could limit the generalizability of some results. The definition used for partial remission has not been contrasted in prospective studies, and the long-term prognostic significance of an isolated partial remission was not analyzed in detail in this study. Despite these limitations, the study has evident strengths: a relatively large and well characterized cohort of patients with C3 glomerulopathy was gathered, with a regular follow-up that helped to analyze the clinical characteristics, response to therapies, and outcomes. Furthermore, genetic studies were performed in all participants.
In conclusion, our results show that treatment with corticosteroids plus MMF was associated with a higher probability of remission and lower probability of kidney failure in patients with C3 glomerulopathy. This favorable effect was observed both in patients with abnormalities in complement genes and in patients who tested positive for autoantibodies, as well as in the different clusters recently proposed. Prospective controlled studies are warranted to confirm these results.
Disclosures
T. Cavero reports receiving grants and nonfinancial support from Alexion, outside the submitted work. G. Fraga reports receiving speaker honoraria from Alexion Pharmaceutical, outside the submitted work. A. Huerta reports receiving personal fees from Alexion and Sanofi, outside the submitted work. M. Praga reports receiving personal fees from Otsuka, grants and personal fees from Alexion, personal fees from Fresenius, and personal fees from Retrophin, outside the submitted work. All remaining authors have nothing to disclose.
Funding
Work in this study was supported by the Instituto de Salud Carlos III/ Fondo Europeo de Desarrollo Regional (ISCIII/FEDER) grant PI16/01685 and Red de Investigación Renal (RedInRen) (RD12/0021/0029) (to M. Praga), and the Autonomous Region of Madrid (S2017/BMD-3673) (to S. Rodríguez de Córdoba and M. Praga). E. Goicoechea de Jorge is supported by the Spanish “Ministerio de Ciencia, Innovación y Universidades” (RYC-2013-13395 and RTI2018-095955-B-100). S. Rodríguez de Córdoba is supported by Ministerio de Economía y Competitividad/FEDER grant SAF2015-66287R and Autonomous Region of Madrid grant S2017/BMD3673.
Supplementary Material
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
See related editorial, “Mycophenolate Mofetil Treatment of C3 Glomerulopathy,” on pages 1234–1236.
Supplemental Material
This article contains the following supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.15241219/-/DCSupplemental.
Supplemental Appendix 1. Supplemental methods, complement genetics and molecular studies, statistical analysis, and supplemental references.
Supplemental Table 1. Baseline clinical and histologic characteristics according to age groups.
Supplemental Table 2. Complement pathogenic gene variants in the study patients.
Supplemental Table 3. Clinical characteristics according to the presence of complement genetic variants and/or autoantibodies against complement components.
Supplemental Table 4. Clinical characteristics of patients who achieved or not remission, according to baseline eGFR.
Supplemental Table 5. Multivariable logistic regression analysis for predictors of remission of the disease.
Supplemental Table 6. Clinical characteristics according to the development of kidney failure.
Supplemental Table 7. Cox proportional hazard regression analysis for association between covariables and kidney failure.
Supplemental Table 8. Clinical and histopathologic characteristics of patients before and after propensity score matching analysis.
Supplemental Table 9. Cox proportional hazard regression analysis for predictors of no response to corticosteroids plus mycophenolate mofetil.
Supplemental Table 10. Clinical, histopathologic, and genetic characteristics of patients treated with mycophenolate mofetil plus corticosteroids according to relapse of the disease.
Supplemental Table 11. Cox proportional hazard regression analysis for predictors of relapse in patients treated with corticosteroids plus mycophenolate mofetil.
Supplemental Figure 1. Dot plot for the propensity scores of patients in the mycophenolate mofetil and nonmycophenolate mofetil groups showing individual units in the data set and whether they were matched or not.
Supplemental Figure 2. Kaplan–Meier curves for kidney survival according to clustering analysis in patients treated with corticosteroids plus mycophenolate mofetil versus other regimens.
References
- 1.Smith RJH, Appel GB, Blom AM, Cook HT, D’Agati VD, Fakhouri F, Fremeaux-Bacchi V, Józsi M, Kavanagh D, Lambris JD, Noris M, Pickering MC, Remuzzi G, de Córdoba SR, Sethi S, Van der Vlag J, Zipfel PF, Nester CM: C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol 15: 129–143, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bomback AS, Appel GB: Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 8: 634–642, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Cook HT: Evolving complexity of complement-related diseases: C3 glomerulopathy and atypical haemolytic uremic syndrome. Curr Opin Nephrol Hypertens 27: 165–170, 2018. [DOI] [PubMed] [Google Scholar]
- 4.Zhao W, Ding Y, Lu J, Zhang T, Chen D, Zhang H, Zeng C, Liu Z, Chen H: Genetic analysis of the complement pathway in C3 glomerulopathy. Nephrol Dial Transplant 33: 1919–1927, 2018. [DOI] [PubMed] [Google Scholar]
- 5.Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, Curreri M, Mondo E, Zito A, Gamba S, Bettoni S, Murer L, Fremeaux-Bacchi V, Vivarelli M, Emma F, Daina E, Remuzzi G: Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol 71: 131–142, 2016. [DOI] [PubMed] [Google Scholar]
- 6.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grünfeld JP, Niaudet P, Lesavre P, Frémeaux-Bacchi V: Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 82: 454–464, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Noris M, Remuzzi G: Genetics of immune-mediated glomerular diseases: Focus on complement. Semin Nephrol 37: 447–463, 2017. [DOI] [PubMed] [Google Scholar]
- 8.Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, Fremeaux-Bacchi V, Nester C, de Córdoba SR, Noris M, Pickering M, Smith R: The role of complement in C3 glomerulopathy. Mol Immunol 67: 21–30, 2015. [DOI] [PubMed] [Google Scholar]
- 9.Donadelli R, Pulieri P, Piras R, Iatropoulos P, Valoti E, Benigni A, Remuzzi G, Noris M: Unraveling the molecular mechanisms underlying complement dysregulation by nephritic factors in C3G and IC-MPGN. Front Immunol 9: 2329, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corvillo F, Okrój M, Nozal P, Melgosa M, Sánchez-Corral P, López-Trascasa M: Nephritic factors: An overview of classification, diagnostic tools and clinical associations. Front Immunol 10: 886, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravindran A, Fervenza FC, Smith RJH, Sethi S: C3 glomerulopathy associated with monoclonal Ig is a distinct subtype. Kidney Int 94: 178–186, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Meyer NC, Wang K, Nishimura C, Frees K, Jones M, Katz LM, Sethi S, Smith RJH: Causes of alternative pathway dysregulation in dense deposit disease. Clin J Am Soc Nephrol 7: 265–274, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bomback AS, Santoriello D, Avasare RS, Regunathan-Shenk R, Canetta PA, Ahn W, Radhakrishnan J, Marasa M, Rosenstiel PE, Herlitz LC, Markowitz GS, D’Agati VD, Appel GB: C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int 93: 977–985, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D’Agati VD, Canetta PA, Radhakrishnan J, Appel GB: Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol 7: 748–756, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Quintrec M, Lapeyraque AL, Lionet A, Sellier-Leclerc AL, Delmas Y, Baudouin V, Daugas E, Decramer S, Tricot L, Cailliez M, Dubot P, Servais A, Mourey-Epron C, Pourcine F, Loirat C, Frémeaux-Bacchi V, Fakhouri F: Patterns of clinical response to eculizumab in patients with C3 glomerulopathy. Am J Kidney Dis 72: 84–92, 2018. [DOI] [PubMed] [Google Scholar]
- 16.Ruggenenti P, Daina E, Gennarini A, Carrara C, Gamba S, Noris M, Rubis N, Peraro F, Gaspari F, Pasini A, Rigotti A, Lerchner RM, Santoro D, Pisani A, Pasi A, Remuzzi G; EAGLE Study Group : C5 convertase blockade in membranoproliferative glomerulonephritis: A single-arm clinical trial. Am J Kidney Dis 74: 224–238, 2019. [DOI] [PubMed] [Google Scholar]
- 17.Rabasco C, Cavero T, Román E, Rojas-Rivera J, Olea T, Espinosa M, Cabello V, Fernández-Juarez G, González F, Ávila A, Baltar JM, Díaz M, Alegre R, Elías S, Antón M, Frutos MA, Pobes A, Blasco M, Martín F, Bernis C, Macías M, Barroso S, de Lorenzo A, Ariceta G, López-Mendoza M, Rivas B, López-Revuelta K, Campistol JM, Mendizábal S, de Córdoba SR, Praga M; Spanish Group for the Study of Glomerular Diseases (GLOSEN) : Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int 88: 1153–1160, 2015. [DOI] [PubMed] [Google Scholar]
- 18.Avasare RS, Canetta PA, Bomback AS, Marasa M, Caliskan Y, Ozluk Y, Li Y, Gharavi AG, Appel GB: Mycophenolate mofetil in combination with steroids for treatment of C3 glomerulopathy: A case series. Clin J Am Soc Nephrol 13: 406–413, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caliskan Y, Torun ES, Tiryaki TO, Oruc A, Ozluk Y, Akgul SU, Temurhan S, Oztop N, Kilicaslan I, Sever MS: Immunosuppressive treatment in C3 glomerulopathy: Is it really effective? Am J Nephrol 46: 96–107, 2017. [DOI] [PubMed] [Google Scholar]
- 20.Ravindran A, Fervenza FC, Smith RJH, De Vriese AS, Sethi S: C3 glomerulopathy: Ten years’ experience at mayo clinic. Mayo Clin Proc 93: 991–1008, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou J, Markowitz GS, Bomback AS, Appel GB, Herlitz LC, Barry Stokes M, D’Agati VD: Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int 85: 450–456, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Iatropoulos P, Daina E, Curreri M, Piras R, Valoti E, Mele C, Bresin E, Gamba S, Alberti M, Breno M, Perna A, Bettoni S, Sabadini E, Murer L, Vivarelli M, Noris M, Remuzzi G; Registry of Membranoproliferative Glomerulonephritis/C3 Glomerulopathy; Nastasi : Cluster Analysis identifies distinct pathogenetic patterns in C3 glomerulopathies/immune complex-mediated membranoproliferative GN. J Am Soc Nephrol 29: 283–294, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine AP, Chan MMY, Sadeghi-Alavijeh O, Wong EKS, Cook HT, Ashford S, Carss K, Christian MT, Hall M, Harris CL, McAlinden P, Marchbank KJ, Marks SD, Maxwell H, Megy K, Penkett CJ, Mozere M, Stirrups KE, Tuna S, Wessels J, Whitehorn D, Johnson SA, Gale DP; MPGN/DDD/C3 Glomerulopathy Rare Disease Group; NIHR BioResource : Large-scale whole-genome sequencing reveals the genetic architecture of primary membranoproliferative GN and C3 glomerulopathy. J Am Soc Nephrol 31: 365–373, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michels MAHM, Volokhina EB, van de Kar NCAJ, van den Heuvel LPWJ: The role of properdin in complement-mediated renal diseases: A new player in complement-inhibiting therapy? Pediatr Nephrol 34: 1349–1367, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frémeaux-Bacchi V, Weiss L, Brun P, Kazatchkine MD: Selective disappearance of C3NeF IgG autoantibody in the plasma of a patient with membranoproliferative glomerulonephritis following renal transplantation. Nephrol Dial Transplant 9: 811–814, 1994. [PubMed] [Google Scholar]
- 26.Allison AC, Eugui EM: Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 47: 85–118, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Blaheta RA, Leckel K, Wittig B, Zenker D, Oppermann E, Harder S, Scholz M, Weber S, Encke A, Markus BH: Mycophenolate mofetil impairs transendothelial migration of allogeneic CD4 and CD8 T-cells. Transplant Proc 31: 1250–1252, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Laurent AF, Dumont S, Poindron P, Muller CD: Mycophenolic acid suppresses protein N-linked glycosylation in human monocytes and their adhesion to endothelial cells and to some substrates. Exp Hematol 24: 59–67, 1996. [PubMed] [Google Scholar]
- 29.Glomsda BA, Blaheta RA, Hailer NP: Inhibition of monocyte/endothelial cell interactions and monocyte adhesion molecule expression by the immunosuppressant mycophenolate mofetil. Spinal Cord 41: 610–619, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Faugaret D, Lemoine R, Baron C, Lebranchu Y, Velge-Roussel F: Mycophenolic acid differentially affects dendritic cell maturation induced by tumor necrosis factor-α and lipopolysaccharide through a different modulation of MAPK signaling. Mol Immunol 47: 1848–1859, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Hauser IA, Renders L, Radeke HH, Sterzel RB, Goppelt-Struebe M: Mycophenolate mofetil inhibits rat and human mesangial cell proliferation by guanosine depletion. Nephrol Dial Transplant 14: 58–63, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Lv W, Lou J, Zhang Y, Lian P, Qi D, Wang J: Mycophenolate mofetil inhibits hypertrophy and apoptosis of podocyte in vivo and in vitro. Int J Clin Exp Med 8: 19781–19790, 2015. [PMC free article] [PubMed] [Google Scholar]
- 33.Morath C, Reuter H, Simon V, Krautkramer E, Muranyi W, Schwenger V, Goulimari P, Grosse R, Hahn M, Lichter P, Zeier M: Effects of mycophenolic acid on human fibroblast proliferation, migration and adhesion in vitro and in vivo. Am J Transplant 8: 1786–1797, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Dell’Oglio MP, Zaza G, Rossini M, Divella C, Pontrelli P, Verrienti R, Rutigliano M, Ditonno P, Stifanelli P, Ancona N, Schena FP, Grandaliano G: The anti-fibrotic effect of mycophenolic acid-induced neutral endopeptidase. J Am Soc Nephrol 21: 2157–2168, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bossi F, Rizzi L, Bulla R, Debeus A, Tripodo C, Picotti P, Betto E, Macor P, Pucillo C, Würzner R, Tedesco F: C7 is expressed on endothelial cells as a trap for the assembling terminal complement complex and may exert anti-inflammatory function. Blood 113: 3640–3648, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Bossi F, Fischetti F, Pellis V, Bulla R, Ferrero E, Mollnes TE, Regoli D, Tedesco F: Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. J Immunol 173: 6921–6927, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Dobrina A, Pausa M, Fischetti F, Bulla R, Vecile E, Ferrero E, Mantovani A, Tedesco F: Cytolytically inactive terminal complement complex causes transendothelial migration of polymorphonuclear leukocytes in vitro and in vivo. Blood 99: 185–192, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Fogo AB: Talking back: The podocytes and endothelial cells duke it out. Kidney Int 90: 1157–1159, 2016. [DOI] [PubMed] [Google Scholar]
- 39.Noris M, Mele C, Remuzzi G: Podocyte dysfunction in atypical haemolytic uraemic syndrome. Nat Rev Nephrol 11: 245–252, 2015. [DOI] [PubMed] [Google Scholar]
- 40.Ricklin D, Mastellos DC, Reis ES, Lambris JD: The renaissance of complement therapeutics. Nat Rev Nephrol 14: 26–47, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dooley MA, Jayne D, Ginzler EM, Isenberg D, Olsen NJ, Wofsy D, Eitner F, Appel GB, Contreras G, Lisk L, Solomons N; ALMS Group : Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 365: 1886–1895, 2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.