

ABSTRACT
Most patients with acute myeloid leukaemia (AML) experience disease recurrence after chemotherapy largely due to the development of drug resistance. Small extracellular vesicles (sEVs) are known to play a significant role in leukaemia drug resistance by delivery of anti-apoptotic proteins and genes conferring resistance to recipient cells. sEV levels are elevated in AML patients’ plasma at the time of diagnosis and remain elevated in complete remission after chemotherapy. The mechanism of enhanced sEV secretion in AML is unknown. We speculated that cholesterol synthesis by AML blasts may be related to elevated sEV secretion. Intracellular levels of cholesterol and of HMGCR (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase), the rate-limiting enzyme in cholesterol synthesizing mevalonate pathway, significantly increased in cultured AML cells or primary human non-malignant cells treated with cytarabine or decitabine. Concomitantly, levels of sEVs produced by these cells also increased. Treatment with an HMGCR inhibitor, Simvastatin, or siRNAs targeting HMGCR blocked the chemotherapy-induced enhancement of sEV secretion in AML cells. sEVs carry HMGCR and chemotherapy enhances HMGCR levels in sEVs. HMGCR+ sEVs upregulate intracellular cholesterol and promote AML cell proliferation. A pharmacologic blockade of HMGCR emerges as a potential future therapeutic option for disrupting sEV signalling leading to cholesterol-driven chemo-resistance in AML.
KEYWORDS: Leukaemia, chemotherapy, cholesterol, HMGCR, small extracellular vesicles
Introduction
Although recent advances in acute myeloid leukaemia (AML) therapy have considerably improved initial response rates and patients’ quality of life, the 5-year event-free survival rates remain low, only 35–40% in adults and 60–70% in children [1]. These low response rates are largely due to chemo-resistance of leukaemic blasts resulting in disease relapse. The development of leukaemic cell resistance to chemotherapy is known to involve numerous molecular mechanisms, including alterations in apoptosis-regulating genes or in drug-transport genes, but the molecular and genetic mechanisms underpinning chemo-resistance of cancer cells remain largely unknown [2]. Recently, AML blast-derived small extracellular vesicle (sEVs) have been shown to contribute to chemo-resistance [3,4]. sEVs secreted by chemo-resistant AML blasts deliver global regulatory anti-apoptotic gene clusters that mediate mRNA splicing, protein translation and chromatin remodelling in neighbouring chemo-sensitive blasts, changing them into chemo-resistant cells.
sEVs are about 30–150 nm particles which are generated by inward membrane budding in multivesicular bodies (MVBs). MVBs traffic to and fuse with the plasma membrane, releasing sEVs into the extracellular space. sEVs carry a cargo of proteins/lipids/nucleic acids that mimic the content of the parental cells [5–7]. Tumour-derived sEVs are enriched in molecules which play critical roles in tumour proliferation, immunosuppression and metastasis [8]. We have previously shown that protein levels of AML plasma-derived sEVs and their molecular profiles dramatically change in the course of chemotherapy, suggesting that AML sEVs could serve as biomarkers of responses to therapy [9]. Plasma of newly diagnosed AML patients contains elevated levels of sEVs (in ug protein/mL plasma) compared to normal controls’ plasma. The level of sEVs remains elevated even after chemotherapy, when the patients reach complete remission. Our data suggest that chemotherapy exerts profound effects on sEVs, increasing their production not only by AML blasts but also by non-malignant cells.
Mechanisms responsible for production and release of sEVs from cells have been extensively investigated but remain poorly understood. Proteins associated with MVBs such as tetraspanins (CD9, CD63, CD82) are involved in regulating sEV packaging and release from cells [10]. In addition to proteins, lipids are important components of the sEV secretion pathway. For example, inhibition of enzymes synthesizing ceramides from sphingomyelin, such as e.g., neutral sphingomyelinase (nSMase2), reduces sEV secretion [11]. In addition, changes of cellular homoeostasis also affect sEV release [12]. sEV secretion is known to ameliorate intracellular cholesterol burden in Niemann-Pick type C1 disease [13]. sEV secretion was enhanced upon treatments inducing intracellular cholesterol accumulation [14].
AML cells have been reported to have abnormal cholesterol homoeostasis. Cultured AML cells are hyperactive in cholesterol synthesis and low-density lipoprotein (LDL) processing [15]. Treatments with sublethal doses of radiation or chemotherapy increase cholesterol levels in AML cells [16]. These studies suggest that elevated levels of sEV secretion seen in AML patients’ plasma at diagnosis as well as post-chemotherapy could be related to abnormalities in cholesterol homoeostasis. In this study, we test the hypothesis that sEV levels in AML plasma and sEV secretion from AML blasts are related to intracellular cholesterol accumulations after chemotherapy, specifically to the presence and levels of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), the key enzyme regulating cholesterol synthesis (see Supplementary Fig 1). We present evidence that elevated intracellular cholesterol levels in AML cells after chemotherapy promote sEV production and might contribute to the development of chemo-resistance by autocrine, juxtracrine or paracrine mechanisms. We suggest that blocking of cholesterol synthesis in AML cells would sensitize them to chemotherapy by reducing circulating levels of HMGCR+ sEVs.
Materials and methods
Materials
Cholesterol (C4951) and Simvastatin (S6196) were all obtained from Sigma-Aldrich (St. Louis, MO, USA). Water-soluble cholesterol as the Methyl-β-cyclodextrin (MβCD) complex was dissolved at 15 mg/mL in ethanol. Activated Simvastatin was prepared by dissolving Simvastatin at 50 mg/mL in ethanol. Then, 1 N NaOH was added to the solution at a final concentration of 450mN, and subsequently the solution was incubated at 50°C for 2 hours. The pH was brought to 7.0 by adding 1 N HCl. Cytarabine (Ara-C) and decitabine were dissolved at 100 mM in DMSO (Sigma-Aldrich, St. Louis, MO, USA).
Cells and cell cultures
AML cell lines, Kasumi-1 and Thp-1 were purchased from American Type Culture Collection (Manassas, VA, USA). Peripheral blood mononuclear cells (PBMC) were isolated from healthy donors’ blood using Ficoll Histopaque (Sigma-Aldrich, St. Louis, MO, USA). Cells were cultured in RPMI medium (Lonza, Wayne, PA, USA) supplemented with 2 mM L-glutamine, 10% (v/v) foetal bovine serum, 100 U/ml penicillin, 100ug/ml streptomycin. All reagents were purchased from Invitrogen (Grand Island, NY, USA). Foetal bovine serum was depleted of exosomes by ultracentrifugation at 100,000xg for 3 h. Cell viabilities were assessed using a Trypan Blue dye exclusion method (0.4% Trypan Blue solution, Thermo Fisher). Conditioned media (CM) were collected from 24 h cultures after centrifugation at 10,000xg for 30 min, filtered using a 0.22um syringe-filter unit (EMD Millipore, Cleveland, OH, USA) and concentrated to 1 mL using a Vivaspin 6, 100,000 MWCO (Sartorius Corp, Bohemia, NY, USA) for sEV isolation.
Plasma samples
sEVs were obtained from AML plasma samples drawn serially at diagnosis on day 5 and day 10 after starting induction chemotherapy and from age-matched healthy volunteers. All subjects participating in this study signed an informed consent approved by the Institutional Review Board of the University of Pittsburgh (IRB #960,279, IRB #0403105 and IRB #0506140). Peripheral blood was collected into heparinized vacutainer tubes and was centrifuged at 1000xg for 10 min to remove blood cells and cell fragments. Clear plasma was collected and aliquoted into 1 mL vials, which were stored in liquid N2. Frozen and thawed plasma was centrifuged at 10,000 g for 30 min and filtered using a 0.22um syringe-filter unit before its use for sEV isolation.
sEV isolation
sEVs were isolated from plasma or cell culture supernatants prepared as described above using mini size-exclusion chromatography (mini-SEC) as previously described [17]. Briefly,10-mL Sepharose 2B agarose gels (Sigma-Aldrich, St. Louis, MO) were packed into mini columns (Bio-Rad Laboratories, Hercules, CA, USA). Then, the pre-cleared samples were loaded onto the columns. The void volume fraction #4 contained the bulk of eluted sEVs. The sEVs were characterized for the protein content, size, nanoparticle numbers, morphology and molecular profiles as described by us [17].
sEV labelling and flow cytometry
Isolated sEVs were labelled with PKH26 red fluorescent cell linker according to manufacturer’s instruction (Sigma-Aldrich, St. Louis, MO). Briefly, about 30ug of sEVs were mixed with an equal volume of Diluent C containing 2uL of PKH26 dye for 5 min at room temperature and then 1% BSA/PBS solution was added to stop the reaction. To remove the unincorporated dye, sEVs were washed five volumes of PBS using Amicon Ultra filter (100,000 MWCO). For carboxyfluorescein succinimidyl ester (CFSE) labelling, sEVs were incubated with 10 µM CFSE for 30 min at 37°C, stopped with 1% BSA/PBS solution and then washed using Amicon Ultra filter (100,000 MWCO) same as in PKH26 labelling. PKH26 or CFSE dye alone in PBS was used as control. For flow cytometry analysis, sEVs were captured with ExoCap streptavidin microbeads coated with antibodies against tetraspanins according to the manufacturer’s instruction (MBL, Woburn, MA). Then, the microbeads were stained with APC conjugated anti-CD81 antibody and analysed with LSRFortessa flow cytometer (BD Biosciences).
Transmission electron microscopy (TEM)
Transmission electron microscopy was performed at the Centre for Biologic Imaging at the University of Pittsburgh as described previously [17]. sEVs were visualized by the transmission electron microscope JEOL JEM-1011.
sEV size and concentration assessment by tunable resistive pulse sensing (TRPS) using qNano
Size ranges and concentrations of sEVs isolated from the cell culture supernatants were measured using TRPS as recommended by the manufacturer (Izon qNano, Cambridge, MA, USA). The measurement conditions and details were previously described by us [17].
Western blots
sEVs or cell lysates (10ug) were separated on 7–15% SDS/PAGE gels and transferred onto PVDF membrane (Millipore, Billerica, MA, USA) for western blot analysis. Membranes were incubated overnight at 4°C with antibodies specific for: CD34 (1:2000, ab81289, Abcam), CD81 (1:200, PA5-13,582, Thermo Fisher), HMGCR (1:2000, ab174830, Abcam), SREBP-2 (1:500, sc-271,616, Santa Cruz), LDLR (1:500, sc-18,823, Santa Cruz), GAPDH (1:500, FL-335, Santa Cruz), CD123 (1:1000, AF841,R&D), CLL-1 (1:2000, AF2946, R&D), β-Actin (1:500, sc-47,778, Santa Cruz), TSG101 (1:500, PA5-31,260, Thermo Fisher), ApoB (1:2000, 20,578-1-AP, Thermo Fisher), Calnexin (1:1000, #2433, Cell Signalling), Grp94 (1:1000, #20,292, Cell Signalling). Next, the HRP-conjugated secondary antibody (1:10,000, Pierce, Thermo Fisher) was added for 1 h at room temperature (RT) and blots were developed with ECL detection reagents (GE Healthcare Biosciences, Pittsburgh, PA, USA). The intensities of the bands on exposed films were quantified using Image J software (NIH, USA).
qRT-PCR
Total RNAs were isolated using an RNA isolation kit (Exiqon miRCURY RNA isolation kit, Qiagen, MD, USA). The expression levels of HMGCR mRNA were evaluated in AML cell lines cultured with/without Ara-C using a qRT-PCR kit (QIAGEN OneStep RT-PCR Kit, Qiagen, MD, USA), with GAPDH internal mRNA control. Primers of HMGCR and GAPDH were purchased from Bio-Rad (qHsaCID0008700 and qHsaCED0038674, respectively). Relative CT values were collected and analysed using StepOnePlus software (Applied Biosystems, Thermofisher, Pittsburgh, PA).
Cholesterol quantification
Cholesterols from cells and sEVs were extracted using methanol and measured using a Cholesterol Quantitation kit (Sigma-Aldrich, St. Louis, MO, USA) according to manufacturer’s instructions.
HMGCR ELISA
In preparation for ELISA assay, the isolated sEVs were concentrated by centrifugation on a 100 K Amicon Ultra 0.5 mL centrifugal filter (EMD Millipore, Billerica, MA, USA) at 5,000x g and then sonicated for five 2 s bursts using Vibra Cell sonicator (Sonics, Newtown, CT, USA). HMGCR concentration in sEVs was measured using HMGCR sandwich ELISA (Life Span Biosciences, Seattle, WA, USA) according to manufacturer’s instruction.
Transfer of HMGCR-specific siRNA into cells
To block the expression of HMGCR in cells, we used PCI13 tumour cells, because transfection of non-adherent leukaemia cells is difficult. 5×105 PCI13 cells in a 6-well plate were transfected with 100pmole of siRNA targeting HMGCR or non-specific siRNA (Santa Cruz sc-43,838 or sc-37,007, respectively). After 24 h transfection, the cells were cultured in fresh DMEM medium for 48 h, and the culture supernatants were collected for sEV isolation and cell lysates were analysed for HMGCR protein expression by western blotting.
Filipin staining
Kasumi-1 cells were fixed with 4% (w/v) paraformaldehyde for 15 min, washed with PBS and stained with 1 µL of Filipin (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at RT. Then, cells were washed twice with PBS and analysed with LSR Fortessa flow cytometer (BD Biosciences).
Data analysis
Data were summarized by descriptive statistics (IBM SPSS, v.23) using means and standard errors (SE). A p-value of <0.05 was considered to be statistically significant.
Results
Characteristics of sEVs isolated from AML plasma
We analysed sEVs isolated from AML plasma samples drawn serially at diagnosis on day 5 and day 10 after starting induction chemotherapy. The data presented in Figure 1 indicate that the isolated sEVs range in size from 30 to 100 nm in diameter (Figure 1(a)) and carry sEV characteristic molecules such as CD81, CD9, TSG101 and Alix (Figure 1(b)). However, sEVs do not contain non-endocytic proteins (Grp94, Calnexin) or lipoprotein (ApoB). Since in the mini-SEC, sEVs are collected in fraction #4, we show the absence of ApoB in fraction #4 by western blots (Figure 1(b)). In contrast, fraction #5, which is not harvested as a source of sEVs in our experiments, contains non-sEV makers including ApoB (Supplementary Fig.2). The data indicate that the sEVs we isolated were minimally contaminated with non-sEV components. These AML sEVs also carried leukaemia associated antigens (LAAs), including CD34, CD123 and CLL-1 (Figure 1(b)). These data correspond to our previously published results [9,18]. Total sEV protein concentrations were compared among the follow-up samples (Figure 1(c)). Mean protein concentration of AML plasma-derived sEVs was significantly higher on days 5 and 10 post-induction chemotherapy than at diagnosis (p < 0.02 and p < 0.001, respectively). The measurement of total sEV protein concentration correlated with the sEV counts, as determined by qNano analyses (Supplementary Fig. 3). The data show that chemotherapy induces an increase in sEV accumulation in AML plasma.
Figure 1.

sEV characteristics. (a) Image of transmission electron microscope of sEVs isolated by min-SEC. (b) Western blot analysis of sEVs showing sEV markers as well as leukaemia associated antigens (LAA). (c) Increase of sEV production in plasma after chemotherapy. sEVs were isolated from series of plasma samples collected at diagnosis (d0) and consecutively at day5 and day10 after starting induction chemotherapy (d5 and d10, respectively). Levels of sEV proteins (n = 13, mean (green bar): d0: 49 ± 19 (mean±SEM), d5: 71 ± 20, d10: 83 ± 20, * p < 0.02, ** p < 0.001.
Ara-C treatment enhances sEV secretion in AML cells
AML cell lines were initially cultured with Ara-C, a common induction chemotherapy for AML, using increasing concentrations of Ara-C (0.0uM to 0.8uM) for 24 h and were tested for cell viability. We selected 0.2uM of Ara-C for further studies, because at this Ara-C level, the viability of cells was not compromised (Supplementary Fig. 4). sEVs were isolated from cell supernatants and their protein concentrations and the number of particles per 10 × 106 cells were determined. After 24 h culture in the presence of 0.2 µM Ara-C, Kasumi-1 cells produced significantly more sEVs (proteins: p < 0.02, particles: p < 0.05) than control cells (Figure 2). Increases in levels of secreted sEVs were dose-dependent (Supplementary Fig. 5).
Figure 2.

Changes in sEV production by Kasumi-1 cells. Cells were left untreated (Con) or were treated with 0.2 µM Ara-C, 5 µM Simvastatin (Sim) or 10 µg/mL Cholesterol (CHO) cultured for 24 h and then the cell culture supernatants were collected for sEV isolation. The cell viabilities were not affected by the treatments for 24 h (supplementary Figure 4). (A) Total sEV protein concentrations per 10 × 106 cells were plotted. * p < 0.02 over control, # p < 0.03 over Ara-C, Δ p < 0.04 over Sim. (B, C) sEV particles were counted using qNano and plotted over particle diameters. ** p < 0.05 over control.
To investigate whether cholesterol metabolism is involved in the enhanced sEV production in Ara-C-treated cells, Kasumi-1 cells were cultured for 24 h in the presence of agents modulating intracellular cholesterol concentrations, including Simvastatin and exogenous cholesterol (Figure 2(a)). The data showed that an excess of exogenous cholesterol in culture media enhanced sEV production, while Simvastatin, an inhibitor of HMGCR, reduced sEV secretion (p < 0.02). In addition, Simvastatin blocked Ara-C-induced production of sEVs (p < 0.03). These results indicated that increased intracellular cholesterol levels enhanced sEV production in AML cells, while blocking of intracellular cholesterol synthesis decreased sEV production. In aggregate, the results showed that in vitro treatment of leukaemia cells with Ara-C altered their cholesterol metabolism.
Cholesterol metabolism in AML cells treated with chemotherapy
To elucidate whether treatment with Ara-C altered cholesterol synthesis or import mechanisms, Kasumi-1 cells were cultured in the absence or presence of Ara-C and assessed for the intracellular cholesterol and protein concentrations. Flow cytometry analysis for Filipin, a fluorescent compound that binds to free cholesterol, showed that Ara-C treatment enhanced binding of Filipin to AML cells (Figure 3(a)). Concomitantly, intracellular total cholesterol levels were significantly increased (p < 0.005) upon Ara-C treatment (Figure 3(b)). Western blot analysis of cell lysates showed that Ara-C treatment significantly enhances HMGCR protein level compared to the control (p < 0.05, Figure 3(c)). The mRNA level of HMGCR assessed by real-time RT-PCR was also increased about 1.5-fold (p < 0.001) by Ara-C treatment (Figure 3(d)). Ara-C treatment also increases protein expression levels of sterol regulatory element-binding protein 2 (SREBP-2), a transcription factor for activating cholesterol synthesis and LDL receptor for cholesterol import, compared to controls. These results showed that Ara-C treatment enhanced cholesterol synthesis and uptake in these cells. To further illustrate the involvement of HMGCR in Ara-C-induced exosome secretion, we transfected HMGCR-specific siRNA into PCI-13 tumour cells and showed that blocking of HMGCR activity significantly reduced exosomes secretion by the cells (Figure 4).
Figure 3.

Leukaemia cell lines increase cholesterol synthesis after Ara-C treatment. Kasumi-1 cells were treated with or without 0.2 µM Ara-C for 24 h. (a, b) Increased level of intracellular cholesterols was measured using flow cytometry of Filipin binding or cholesterol quantification kit, respectively. * p < 0.005 over control. (c) Two representative western blots of cell lysates are shown. Relative fold increase of protein expression for HMGCR, SREBP2 and LDLR relative to control were calculated from three repeated experiments. ** p < 0.05 over control. (d) HMGCR mRNA levels were determined by real-time qPCR and normalized to GAPDH mRNA. Fold increase in HMGCR expression relative to the control is shown. *** p < 0.001 over control.
Figure 4.
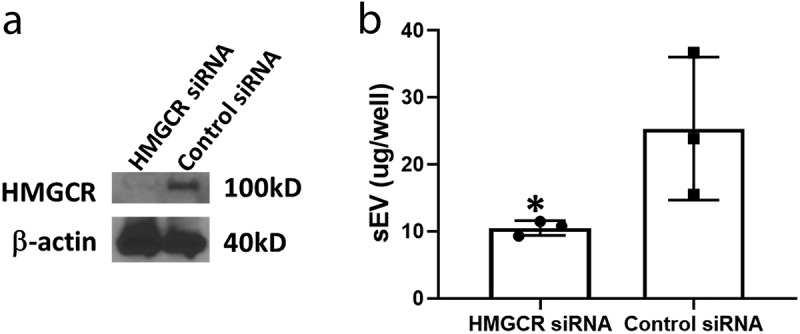
Blocking HMGCR by transfection with siRNA into PCI-13 cells. Cells were transfected with HMGCR-specific siRNA or control siRNA, supernatants were collected and sEVs were isolated. (a) Western blot of HMGCR expression in siRNA transfected PCI-13 cell lysates. (b) Levels of sEV protein isolated from the transfected PCI-13 cells in culture. * p < 0.05 over control.
Chemotherapy enhances production of sEVs carrying HMGCR
sEVs isolated from supernatants of Kasumi-1 cells also carried HMGCR as indicated by western blot analysis of sEV lysates (Figure 5(a)). Furthermore, HMGCR expression levels in sEV lysates were increased upon Ara-C treatment of cultured cells. This result corresponds to the post-chemotherapy plasma-derived sEVs (Figure 5(b)). sEVs were isolated from paired pre- and post-chemotherapy AML plasma samples. Post-chemotherapy plasma samples were drawn on day 10 after the patients received 5 days of decitabine and then 5 days of Ara-C therapy. Decitabine, similar to Ara-C, is a cytosine analogue and is commonly used as induction chemotherapy for AML. As shown in Figure 5(b), AML sEVs were HMGCR+. sEVs from post-chemotherapy plasma contained higher levels of HMGCR compared to the sEVs from paired pre-chemotherapy plasma samples (p < 0.04). The graph shows that the total sEV protein levels per 1 mL plasma are higher in post-chemotherapy than in pre-chemotherapy samples.
Figure 5.

Chemotherapy enhances expression of HMGCR in sEVs. (a) Kasumi-1 cells were cultured with or without 0.2uM Ara-C for 24 h. sEVs were isolated from the supernatants. 10 µg of sEV lysates were separated on the gels and analysed by western blotting. TSG101 is used as an sEV marker. Note that the sEVs in fraction#4 contain minimal ApoB, while the fraction #5 are rich in ApoB. (b) sEVs isolated from AML follow-up plasma drawn at diagnosis [D] and at day10 during the induction chemotherapy [C] of decitabine and Ara-C were analysed using western blotting. Expression of HMGCR appears stronger in plasma after chemotherapy than at diagnosis. The graph below shows total levels of sEV proteins per 1 mL plasma. Fold change of the HMGCR/GAPDH ratio between diagnosis and chemotherapy is shown on the right. * p < 0.04 over diagnosis. ND, normal donor plasma.
Autocrine effect of HMGCR+ exosomes
Since AML leukaemia cells are not only avid producers of sEVs but also actively up-takes EVs, we investigated whether sEVs could deliver additional HMGCR protein into recipient cells. As shown in Figure 6(a,b), PKH26 or CFSE-labelled Kasumi-1 sEVs were abundantly taken up by Kasumi-1 cells. The Kasumi cell lysates contained increased HMGCR levels upon co-incubation of cells with sEVs in a dose-dependent manner (Figure 6(c)). As expected, cells co-incubated with sEVs isolated from Ara-C treated cell culture supernatants bind more Filipin (Figure 6(d), p < 0.02) and tend to contain more intracellular cholesterol (Figure 6(e), p = 0.058) than control cultures. Interestingly, they also proliferate better than control cells (Figure 6(f), p < 0.01). The data suggest that the chemotherapy-induced increase in HMGCR+ sEVs changes cell signalling in recipient cells, leading to tumour-promoting effects.
Figure 6.

Autocrine effect by HMGCR+ sEVs in Kasumi-1 cells. (a, b, c) Kasumi-1 cells were co-incubated with PKH26 (red: sEVs+PHK26) or CFSE (green: sEVs+CFSE) labelled sEVs isolated from Kasumi-1 culture supernatant for 1 hr, then washed with pH3 acid buffer and analysed by flow cytometry (a) or by confocal microscopy (b, red: PKH26, blue: DAPI). Dyes alone without sEVs (PBS+PKH26 or PBS+CFSE) are controls. (c) Cell lysates were analysed in western blots for HMGCR protein. (d, e) Kasumi-1 cells were co-incubated with sEVs isolated from Kasumi-1 cultures with/without treatment of Ara-C for 48 hr. Then cells were analysed for Filipin binding (d), total cholesterol measurement (e) and cell counts (f). * p < 0.02, ** p = 0.058, ***p < 0.01.
Effects of chemotherapy on sEV production by non-malignant cells
Since AML plasma contains sEVs secreted from leukaemic blasts and from non-malignant cells, we investigated whether PBMC obtained from normal donors and incubated for 24 h in the presence of Ara-C or decitabine also produced more sEVs. As shown in Figure 7, both treatments enhanced expression levels of HMGCR in cell lysates as well as intracellular cholesterol concentrations. Consistently, levels of sEVs isolated from cell supernatants were increased as was the HMGCR concentration in sEVs. The results indicated that chemotherapy enhanced secretion of HMGCR+ sEVs from PBMCs of normal donors.
Figure 7.

Chemotherapy treatment enhances sEV production from PBMC. PBMC isolated from normal donors were cultured without (Control) or with 0.2uM Ara-C (Ara-C) or 0.5uM Decitabine (Dec) for 24 h. (a) PBMC lysates were analysed by western blotting. (b) Levels of intracellular cholesterols in PBMC were measured using cholesterol quantification kit. * p < 0.02 over control. (c) Levels of sEV proteins isolated from 10 million PBMC culture supernatants. ** p < 0.02 over control. (d) ELISA assay of HMGCR concentration on sEVs. sEVs isolated from PBMC cultures were sonicated and analysed using sandwich ELISA for HMGCR protein. *** p < 0.05 over control.
Discussion
EVs, including sEVs, mediate intercellular communication [5–8]. sEVs are produced by all cells and are present in all body fluids. Cell-free circulating sEVs carry and deliver messages to cells throughout the body. They participate in physiological as well as pathological processes. The biology of sEVs, especially their biogenesis and secretion by normal and malignant cells, has been intensively investigated. However, it remains unclear how sEV production is regulated in health or disease. Here, we evaluate a cellular mechanism that appears to regulate sEV production by modulating intracellular cholesterol levels in cells exposed to chemotherapy. Following chemotherapy, AML cells increased synthesis of cholesterol and released more sEVs into culture media. The excess secretion of sEVs induced by chemotherapy was ameliorated by blocking cholesterol synthesis with Statin or siRNA targeting HMGCR, the key enzyme for cholesterol synthesis. Further, these sEVs carried HMGCR, and there was significantly more HMGCR in sEVs released by chemotherapy-treated cells than in control cells. Comparisons of sEVs isolated from pre- and post-chemotherapy AML patient plasma showed that: (a) sEVprotein levels were elevated and (b) sEVs were enriched in HMGCR in plasma obtained after induction chemotherapy. Thus, chemotherapy was shown to enhance sEV release from cells as well as sEV content of HMGCR, consequently converting AML sEVs into highly effective stimulators of cholesterol synthesis.
Although the importance of cholesterol and lipid in cancer progression has been studied for a long time, their connection to sEVs, specifically cancer-derived sEVs, has not been previously reported. In cancer patients, including those with AML, blood cholesterol levels are usually lower than those in non-cancer patients [19,20]. This phenomenon has been explained by a high demand of malignant cells for cholesterol and lipids that are necessary for proliferation, migration and metastatic activity of tumour cells. Furthermore, it has been shown that cellular stress, including chemotherapy, affects cholesterol metabolism. Doxorubicin treatment in breast cancer was reported to increase levels of intracellular cholesterol [21]. Chemotherapy resistant leukaemia cells down-regulate miR-27a expression which was known as a key regulator of cholesterol homoeostasis [22,23]. Metabolism of intracellular cholesterol is tightly regulated by complex multi-step networks. For example, sterol regulatory element-binding protein 2 (SREBP2) is a key sensor of cholesterol in the endoplasmic reticulum and, at low levels of cholesterol, activates transcription of genes for cholesterol synthesis such as HMGCR [24]. On the other hand, excess levels of intracellular cholesterol facilitate its efflux via increasing expression of cholesterol export proteins such as ATP-binding cassette transporter (ABCA1) [25]. Recent data show that ABCA1 is involved in the sEV biogenesis during high-density lipoprotein (HDL) formation [26]. In addition, it has been reported that cholesterol accumulations in late endosomes via knocking down NPC1, a mediator of intracellular cholesterol trafficking, induces sEV secretion and ameliorates intracellular cholesterol levels [13]. Consistent with this mechanism, our results indicate that elevated sEV secretion in response to the increased cholesterol synthesis sustains the cholesterol homoeostasis. This may be particularly important during chemotherapy, where high cholesterol levels promote leukaemia cell survival.
We have shown that sEVs in AML plasma carry many immunosuppressive molecules such as TGFβ-1, PD-1/PD-L1, Fas/FasL and CD39/CD73 [18]. AML sEVs reprogram NK-92 cells, interfering with their anti-leukaemia functions and reducing the therapeutic potential of adoptive cell transfers. Since levels of sEVs which also carry immunosuppressive molecules remain high in AML plasma after post-induction chemotherapy [9,18], it could be surmised that these sEVs are involved in chemoresistance of residual leukaemic blasts post-chemotherapy, contributing to leukaemia relapse. In breast cancer, sEVs from chemo-resistant cancer stem cells transfer miR-155 to chemo-sensitive cells, promoting to the epithelial to mesenchymal transition in the sensitive cells and contributing to increase in chemo-resistance [27]. However, sEV-mediated chemoresistance seems not to be limited to tumour-derived sEVs. In cancer patients, non-malignant cells also secrete sEVs, interfere with chemotherapy and promote cancer cell proliferation. For example, cancer-associated fibroblasts (CAFs) secrete sEVs and promote chemoresistance in colorectal cancer cells upon treatment with 5-fluorouracil or oxaliplatin [28]. In our study, post-induction chemotherapy plasma specimens drawn at the time when the number of leukaemic blasts was very low or undetectable contained significantly elevated level of sEVs carrying immunosuppressive markers and containing high levels of HMGCR. In addition, delivering high levels of HMGCR, a key enzyme for cholesterol synthesis via sEVs to immune cells could be disruptive to their functions. A recent study shows that activity of SREBP, a cholesterol sensor, is important for growth, proliferation and effector functions of NK cells [29]. High cholesterol inhibits SREBP activity and subsequently leads to dysfunction of NK cell in interferon-γ production and anti-tumour cytotoxicity. Therefore, sEVs in post-chemotherapy plasma set up a vicious cycle of chemo-resistance and leukaemia recurrence.
In various cancer types, inhibiting cholesterol synthesis by, for example, the use of statins has been shown to induce apoptosis of tumour cells and increase their sensitivity to therapy. Treatment of AML cells with statin reduces the cholesterol level and sensitizes the cells to radiochemotherapy [15]. Recent phase 2 study of Idarubicin and Cytarabine in combination with Pravastatin in relapsed AML patients showed significantly improved complete remission rates [30,31]. Primary leukaemia and lymphoma cells were efficiently killed by co-treatment of statins and BCL2 inhibitor venetoclax compared to venetoclax alone [32]. To our knowledge, no studies have reported a connection between statin efficacy and sEVs in cancer treatment. Our results indicate that the statin treatment might potentially reduce harmful sEV burden and so improve the efficacy of chemotherapy.
Supplementary Material
Declaration of interest
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am. J. Hematol. 2013;88(4):317–327. [DOI] [PubMed] [Google Scholar]
- [2].Yeung CCS, Radich J. Predicting chemotherapy resistance in AML. Curr Hematol Malig Rep. 2017;12(6):530–9. [DOI] [PubMed] [Google Scholar]
- [3].Chen T, Zhang G, Kong L, et al. Leukemia-derived exosomes induced IL-8 production in bone marrow stromal cells to protect the leukemia cells against chemotherapy. Life Sci. 2019;221:187–195. [DOI] [PubMed] [Google Scholar]
- [4].Wojtuszkiewicz A, Schuurhuis GJ, Kessler FL, et al. Exosomes secreted by apoptosis-resistant acute myeloid leukemia (AML) blasts harbor regulatory network proteins potentially involved in antagonism of apoptosis. Mol Cell Proteomics. 2016;15(4):1281–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Whiteside TL. Exosomes and tumor-mediated immune suppression. J Clin Invest. 2016;126(4):1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Boyiadzis M, Whiteside TL. The emerging roles of tumor-derived exosomes in hematological malignancies. Leukemia. 2017;31(6):1259–1268. [DOI] [PubMed] [Google Scholar]
- [7].Mathieu M, Martin-Jaular L, Lavieu G, et al. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21(1):9–17. [DOI] [PubMed] [Google Scholar]
- [8].Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hong CS, Muller L, Whiteside TL, et al. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front Immunol. 2014;5:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lang JK, Young RF, Ashraf H, et al. Inhibiting extracellular vesicle release from human cardiosphere derived cells with lentiviral knockdown of nSMase2 differentially effects proliferation and apoptosis in cardiomyocytes, fibroblasts and endothelial cells in vitro. PLoS One. 2016;11(11):e0165926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Baixauli F, Lopez-Otin C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014;5:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Strauss K, Goebel C, Runz H, et al. Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J Biol Chem. 2010;285(34):26279–26288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pfrieger FW, Vitale N. Cholesterol and the journey of extracellular vesicles. J Lipid Res. 2018;59(12):2255–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li HY, Appelbaum FR, Willman CL, et al. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood. 2003;101(9):3628–3634. [DOI] [PubMed] [Google Scholar]
- [16].Banker DE, Mayer SJ, Li HY, et al. Cholesterol synthesis and import contribute to protective cholesterol increments in acute myeloid leukemia cells. Blood. 2004;104(6):1816–1824. [DOI] [PubMed] [Google Scholar]
- [17].Hong CS, Funk S, Muller L, et al. Isolation of biologically active and morphologically intact exosomes from plasma of patients with cancer. J Extracell Vesicles. 2016;5(1):29289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hong CS, Sharma P, Yerneni SS, et al. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci Rep. 2017;7(1):14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cruz PM, Mo H, McConathy WJ, et al. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol. 2013;4:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kuzu OF, Noory MA, Robertson GP. The Role of Cholesterol in Cancer. Cancer Res. 2016;76(8):2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sharma M, Tuaine J, McLaren B, et al. Chemotherapy agents alter plasma lipids in breast cancer patients and show differential effects on lipid metabolism genes in liver cells. PLoS One. 2016;11(1):e0148049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Feng DD, Zhang H, Zhang P, et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J Cell Mol Med. 2011;15(10):2164–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Khan AA, Gupta V, Ananthamohan K, et al. Identification of microRNA-27a as a key regulator of cholesterol homeostasis. bioRxiv. 2018;383448 DOI: 10.1101/383448 [DOI] [Google Scholar]
- [24].Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem. 2013;288(26):18707–18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289(35):24020–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hafiane A, Genest J. ATP binding cassette A1 (ABCA1) mediates microparticle formation during high-density lipoprotein (HDL) biogenesis. Atherosclerosis. 2017;257:90–99. [DOI] [PubMed] [Google Scholar]
- [27].Santos JC, Lima NDS, Sarian LO, et al. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci Rep. 2018;8(1):829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hu Y, Yan C, Mu L, et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One. 2015;10(5):e0125625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Assmann N, O’Brien KL, Donnelly RP, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. 2017;18(11):1197–1206. [DOI] [PubMed] [Google Scholar]
- [30].Advani AS, McDonough S, Copelan E, et al. SWOG0919: a Phase 2 study of idarubicin and cytarabine in combination with pravastatin for relapsed acute myeloid leukaemia. Br J Haematol. 2014;167(2):233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Advani AS, Li H, Michaelis LC, et al. Report of the relapsed/refractory cohort of SWOG S0919: A phase 2 study of idarubicin and cytarabine in combination with pravastatin for acute myelogenous leukemia (AML). Leuk Res. 2018;67:17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee JS, Roberts A, Juarez D, et al. Statins enhance efficacy of venetoclax in blood cancers. Sci. Transl. Med. 2018;10(445):eaaq1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.