

Introduction
Developing technology has allowed enormous advances in expanding our knowledge of tumor physiology and genomics, such that our understanding of hereditary cancer syndromes is evolving rapidly. These advances have the potential to individualize cancer treatment with targeted therapies, to improve prognostication, and to allow appropriate counseling for risk reduction.
Understanding tumor pathophysiology
Mutations in key cell-cycle regulators result in uncontrolled cellular growth or impaired cellular death. This is typically due to loss of tumor suppressor gene function or an upregulation of oncogenes, often requiring “two-hits” to result in malignant growth. The four key mutation types that may result in malignancy include:
Point mutations: single nucleotide variants causing missense or nonsense DNA
Complex mutations: frameshift insertions or deletions
Exon or gene copy number: large duplications or deletions changing functional domain of proteins
Structural variants: translocations or inversions from point-breaks resulting in fused proteins or non-functional proteins
These tumor mutations can occur sporadically or in an inherited fashion. Sporadic, or acquired, mutations may occur in either somatic (peripheral) cells or germ (gonadal) cells. If a mutation occurs in a somatic tissue, it may remain silent or be expressed as tumor growth. This phenotypic expression as tumor growth would occur if concurrent mutational changes or tumor micro-environmental pressures result in additional loss of cell cycle regulation. These mutations can also be thought of as random, or non-inherited.
If a mutation occurs in germ cells, it may lead to tumor growth by a similar mechanism to that described above, or remain silent and be passed down to the next generation. Inherited or germline mutations occur in germ cells, and thus allow a deleterious mutation to be transmitted to progeny in an autosomal dominant fashion. In this progeny, all cells in the body will be affected by the mutation, including both somatic and germ cells.
The Metamorphosis of Genetic Testing
The discovery of BRCA 1 and BRCA2 gene mutations were landmark discoveries in the 1990’s and have heralded a new era of understanding for cancer genetics. With the sequencing of the human genome and genetic profiling of cancers over the past decade, The Cancer Genome Atlas and other comprehensive analyses have provided many additional insights into the genomic aberrations that elevate cancer risk1-4.
There are many approaches for interrogating a tumor or patient for relevant genetic mutations and phenotypic expression patterns which may be clinically relevant, reviewed in figure 2. Molecular testing can assess any of the four major gene alterations in cancer listed above. Current molecular profiling paradigms typically use PCR, RNA or DNA sequencing technology, mass spectrometry, or fluorescence in situ hybridization to assess variants. Testing can range from a simple single-gene point mutation evaluation to a full sequencing of 20,000 genes. Emerging technology, called next-generation sequencing, allows assessment of all protein coding exosomes, the whole genome, or a customized panel of areas of interest.
Figure 2.

Testing strategies to assess tumor behavior
PCR – polymerase chain reaction, PD-L1 – programmed death ligand-1; FISH – fluorescent in-situ hybridization
In the absence of a germline mutation, several mutations isolated within the tumor itself can help to guide management decisions, particularly BRCA or loss of homologous recombination. As our understanding of genomics expands, there are hundreds of identifiable somatic mutations within a tumor, however those considered to be “hotspot” mutations in gynecologic oncology include BRCA1, BRCA2, ATM, BRIP1, RAD51C, RAD51D, EPCAM, ARID1A, BRAF, CDKN2A, CTNNB1, FBXW7, FGFR2, FGFR3, FOXL2, HRAS, KRAS, NRAS, PIK3CA, PPP2R1A and PTEN among many others5,6. Several of these genes have a matched targeted therapeutic strategies.
Germline and Somatic Testing in Ovarian Cancer
The use of somatic or germline testing is specific to each malignancy. In 2014, the Society for Gynecologic Oncology released a position paper recommending genetic testing in all women with epithelial, fallopian tube or peritoneal cancers 12. Ovarian cancer merits use of a molecular sequencing technique utilizing a commercially-available platform. Only in women with negative germline testing, or in recurrent malignancy, is somatic testing utilized. Specific hereditary syndromes that are defined by these germline or somatic mutations are reviewed in more detail below.
Once a patient has been diagnosed with a gynecologic malignancy, both somatic and germline genetic sequencing information has additional importance in both prognostication as well as targeting of new molecular therapies and immunotherapy. Specifically, a recent meta-analysis has established that women with a BRCA 1 mutation had improved overall survival (HR = 0.75) but not PFS when compared with non-BRCA controls, and BRCA2 was not associated with better PFS or OS 13.
As the clinically relevant cellular pathways have become apparent through genetic sequencing, the potential targets for therapy have been investigated. The most commonly utilized targeted agents in ovarian cancer are VEGF (vascular endothelial growth factor) inhibitors, for which personalized molecular targeting is not yet utilized, and the class of drugs called PARP (poly(ADP-ribose) polymerase) inhibitors, which are targeted at BRCA 1 and BRCA 2 mutated tumors, but also show efficacy in non-BRCA mutated cancers. Only 1% of ovarian cancers harbor a germline mismatch repair mutation resulting microsatellite instability, and only 10% a somatic mismatch repair phenotype, although MSI does have important applications for therapeutic targeting 14. An immune check-point inhibitor, pembrolizumab, is now FDA-approved specifically for unresectable or MSI-High solid tumors, independent of tumor location 15. These agents are emerging as an essential component of therapy for recurrent ovarian cancers, and are being actively studied in both the upfront and maintenance settings.
Germline and Somatic Testing in Uterine Cancer
The 2014 Society for Gynecologic Oncology position paper also recommends genetic testing in all women with endometrial cancer 12,16. There are four primary genes responsible for mismatch repair: MSH2, MLH1, PMS2 and MSH6; any mutation in these genes or somatic loss of expression is known as microsatellite instability (MSI). Normal expression of all mismatch repair proteins is known as microsatellite-stable.
Uterine cancer, as compared to ovarian, follows an alternative paradigm. Uterine cancer is evaluated first using somatic tissue testing via immunohistochemistry of the biopsy or surgical tissue17. Using MMR protein antibodies, this protein expression assessment reveals either a presence or absence of mismatch repair proteins including MLH1, MSH2, MSH6 and PMS2. Loss of expression has excellent sensitivity and specificity >90% 18.
The inactivation of mismatch repair genes can be due to germline or somatic mutation, or epigenetic silencing. The first step after immunohistochemistry has detected a loss of mismatch repair protein expression is evaluation for epigenetic hypermethylation, which allows functional silencing of these protein but does not herald an underlying somatic or germline mutation. For MLH1 and PMS2 particularly, hypermethylation is frequently the cause for loss of expression and represents a sporadic mutation. In the setting of loss of MLH2 or MLH6, or a lack of hypermethylation in MLH1 or PMS2, molecular testing should be performed. This is typically a point in management that a genetic counselor becomes involved. Molecular testing is performed on DNA from fresh, frozen, or paraffin-embedded tissue using PCR-based testing of 5 specific markers. Any confirmed mutation merits molecular germline testing.
While MSI-High represents a more favorable prognosis for colon cancer, this has not been confirmed in uterine cancer 19. MSI does have important applications for therapeutic targeting as well. Immune check-point inhibitor, pembrolizumab, for unresectable or MSI-High solid tumors can be utilized in this setting as well 15. In uterine cancer, molecular targeting of VEGF, PI3K/MTOR and HER2 mutations as a result of somatic tumor testing can also guide therapeutic choices for these women given emerging data for these agents.
Germline and Somatic Testing in Cervical and Vulvar Cancer
Currently the role of somatic tumor testing is not well established for cervical or vulvar cancer and outside of rare clinical situations, such as cervical adenoma malignum, germline testing is not warranted.
Immunohistochemical or molecular evidence of microsatellite instability is of increasing relevance, as 5-11% of cervical cancers may express this phenotype and would be candidates for targeted checkpoint-inhibitor therapy as described above 20. In cervical and vulvar cancers as well, the role of somatic tumor testing remains limited otherwise, however both VEGF-inhibitors and EGFR-inhibitors remain under investigation in the upfront and recurrent settings and the utilization of somatic tumor biomarkers to guide these therapeutic choices is needed21.
Molecular Testing Platforms
There are a number of commonly used commercially-available genetic sequencing platforms being utilized by genetic counselors and physicians some of which may include: Foundation One, Personal Genome Diagnostics, Ambry, Caris, and Myriad. Each of these offers a pre-set panel of genes to be evaluated, ranging from 2-50 genes and has its inherent strengths and limitations 7.
Interpretation of results from genetic sequencing platforms should be performed by a certified genetic counselor or gynecologic oncologist, who has experience in making evidence-based recommendations regarding appropriate screening and risk-reduction strategies. This becomes particularly essential to avoid the pitfalls of genetic sequencing, which can include limited mutation assessment with various panels, misinterpretation of somatic and germline mutations and their implications, as well as variants of uncertain significance.
Limitations of Commercial Genetic Testing
Among the commercially available gene sequencing panels above, each analyzes a pre-set list of mutations that are determined most likely to be clinically relevant. As our understanding of clinically relevant mutations expands rapidly, these panels cannot accommodate all possible relevant mutations and thus, it is essential to be aware of the limitations of testing. Additionally, less commonly detected mutations of questionable significance are found with increasing frequency. These are labeled “variants of uncertain significance” or VUS. These variants are not actionable, as the current evidence base does not support application of screening and treatment recommendations to this poorly characterized group.
Direct-to-Consumer Testing
Providers in today’s landscape must also face the challenges of direct-to-consumer genetic testing. Specifically, patients have kits mailed to their homes, typically requiring saliva collection, and the results are mailed back detailing any mutations found. Dozens of these companies exist, with specific examples including Color, Veritas, Helix, or 23andMe. This bypass of traditional health systems allows the benefit of bypassing insurance approvals for patients, but has many limitations as well. These include a lack of regulatory oversight, a limitation in the number of genes screened, and often out-of-context reporting of mutations detected.
In 2018, the FDA authorized the first direct-to-consumer testing for BRCA genes. Very specifically, this testing reports on 3 genes most common in people of Ashkenazi Jewish descent. This is in recognition that these three genes are not the most common in the general population and that there are >1,000 known BRCA mutations which are not evaluable by this testing 22. The approval clearly advises that these results should not be used to determine treatments or risk-reduction strategies, rather they should guide confirmatory testing and genetic counseling. This approval was offered to 23and Me, Inc.
Unfortunately, these details of the FDA approval are often overlooked and these tests are frequently being utilized by the general population. In May 2018, the American College of Obstetricians and Gynecologists (ACOG) published a practice advisory regarding direct-to-consumer testing. ACOG discourages direct-to-consumer genetic testing due to the absence of pre- and post-test counseling and the possibility of false-reassurance with a “negative” test result. Furthermore, providers are limited in the ability to counsel patients with a “positive” test result and may subject patients to potential intervention unnecessarily 23.
Genetic Counselor Referral Guidelines
NCCN guidelines and the Society of Gynecologic Oncology (SGO) support that all women diagnosed with ovarian and uterine cancer undergo genetic assessment, as described above 24. In those with the following high-risk characteristics, referral should be made to a genetic counselor who can guide patients on which, if any, genetic testing should be utilized. Additionally, the National Comprehensive Cancer Network (NCCN) guidelines on Genetic/Familial High-Risk Assessment: Breast and Ovarian is an excellent resource to guide providers in genetic referral practices25.
Hereditary Cancer Syndromes
Throughout this section, various syndromes will be reviewed including their genomic aberration(s) and resultant phenotype, the gynecologic cancer risks, important non-gynecologic cancer risks, as well as the recommended screening and preventative measures.
Hereditary Breast and Ovarian Cancer Syndromes
Hereditary Non-Polyposis Colorectal Cancer Syndrome/Lynch Syndrome
Polymerase Proofreading-Associated Polyposis Syndrome
Peutz-Jeghers Syndrome
Cowden Syndrome
Li-Fraumini Syndrome
Dicer Syndrome
Hereditary Ovarian Cancer Syndromes
The BRCA-Fanconi anemia DNA repair pathway controls DNA repair via homologous recombination. Numerous important genes regulate this pathway, with BRCA1 and BRCA2 being the most common. These two mutations, with their risk of breast cancer, define Hereditary Breast and Ovarian Cancer Syndrome (HBOC). An additional 2.5% of ovarian cancer patients have mutations in BRIP1, RAD51C and RAD51D, which are associated with an elevated risk of ovarian cancer alone and thus characterize Hereditary Ovarian Cancer Syndrome (HOC) 28,29. Furthermore, PALB2 and BARD1 genes are responsible for a protein which binds BRCA1 and BRCA2 at the site of DNA damage and their role in ovarian cancer risk is not yet fully understood, however the Cancer Genome Atlas project supports that PALB2 is the frequently mutated after BRCA1 and 2 29,2. Several other mutations in this pathway, such as NBN and CHEK2, have been linked to breast cancer and have been identified in women with ovarian cancer, however the causal relationship remains unclear 29.
Ovarian cancer is attributable to a BRCA-1 or BRCA-2 mutation in 24% of the 22,280 new cases of ovarian cancer each year in the United States, with 18% being germline mutation 30,28. Furthermore, numerous other mutations within the BRCA pathway of double stranded DNA repair confer an increased risk of ovarian cancer, including RAD51C, RAD51D and BRIP1 31,32,33. Emerging evidence also suggests increased risk in women with PALB2 and BARD1 and supports that loss of homologous recombination phenotype leads to a “BRCA-ness” in some women 34.
Women with BRCA1 have a 65-85% risk of breast cancer and a 39-46% risk of ovarian cancer 28. Retrospective data shows that the risk of uterine cancer in women with BRCA1 and 2 mutations is not increased compared to the general population. However, in those women with BRCA1 who develop uterine cancer, the risk of an aggressive, serous uterine cancer is elevated at 4.7% between ages 45-70. This has elicited controversy regarding the role of risk-reducing hysterectomy in this population and should be discussed with patients at the providers discretion 35. Women with BRCA2 have a 45-85% risk of breast cancer and a 10-27% risk of ovarian cancer 28. Additionally, these women carry an elevated risk of melanoma and pancreatic malignancies for which clear risk reduction and screening strategies do not current exist.
Hereditary Non-Polyposis Colorectal Cancer Syndrome
Hereditary nonpolyposis colorectal cancer (HNPCC) is also well known as Lynch Syndrome. It is characterized by colorectal, as well as endometrial, ovarian, gastric, renal and skin malignancies. Approximately 15% of these cancers have microsatellite instability, often in the form of epigenetic silencing of MLH-1 or MSH2 via hypermethylation, however both somatic and germline mutations can also exist in the four primary mismatch repair genes: MSH2, MSH6, MLH1, and PMS2.
Mutations within the MLH1 and MSH2 genes carry an elevated risk of all malignancies, including 25-60% uterine cancer, 52-82% colon cancer, 4-25% ovarian cancer, 6-13% gastric cancer, and approximately 5% risk of hepatobiliary, CNS, pancreatic, small bowel, and urothelial cancers. Conversely, the PMS2 and MLH6 mutations carry a risk of 10-22% of colon cancer, 15-26% for uterine cancer, and <5% for all other cancer listed above including ovarian cancer 28.
Polymerase Proofreading-Associated Polyposis Syndrome
Polymerase proofreading associated polyposis syndrome (PPAP) syndrome represents mutations in 2 DNA polymerases, POLD1 and POLE 28,36,37. This germline mutation results in a loss of proofreading capability and accumulation of mutations, manifesting as microsatellite instability with an elevated risk of both uterine and colonic malignancies 38. Assessment for mutations of these genes should be performed during routine genetic evaluation of those being tested for Lynch syndrome. The Cancer Genome Atlas additionally identified a subgroup of uterine cancers with an ultra-mutated phenotype that have a very favorable prognosis 2.
Peutz-Jeghers Syndrome (Resta, van Lier reviews)
This syndrome, characterized by STK11 mutation, results in an elevated risk of malignancy of several rare gynecologic malignancies 39,40. SKT11, or serine/threonine kinase 11, is a tumor suppressor gene and a germline mutation in this gene is inherited in an autosomal dominant fashion. Germline mutations result in pigmented skin and mucosal macules, gastrointestinal polyps and elevated cancer risk. Sex cord stromal tumor of the ovary with annular tubules (SCTAT) risk is elevated to 18-21% and uterine cancer risk is elevated to 9%, which merits an annual pelvic exam and consideration of pelvic ultrasound starting at age 18. There is also an elevated risk of cervical adenoma malignum at 10% - while national guidelines suggest initiating Pap surveillance at age 21, this population warrants imitation of screening at age 18-20. There are currently no risk-reducing strategies that are supported.
In addition to gynecologic cancers, women are also at risk of breast cancer in 45-50% meriting significantly more frequent and intensive breast screening and consideration of risk-reducing mastectomy. Risk of colon cancer may be as high as 39% and stomach cancer 29%, with increased surveillance utilizing colonoscopy and EGD every 2-3 years beginning by age 18 39,40.
Cowden Syndrome
The PTEN gene, which encodes the phosphatase and tensin homolog protein, is a tumor suppressor gene that, when mutated, causes an elevated risk of uterine cancer at 19-28%28. Beginning at age 30-35, women should undergo annual transvaginal ultrasound and endometrial biopsy. Given this elevated uterine cancer risk, consideration can be given to hysterectomy upon completion of childbearing. Women also carry an elevated risk of breast cancer as high as 50% and intensive and early screening are advised with annual exam, mammogram and MRI. Follicular thyroid cancer risk is up to 38% and warrants annual thyroid ultrasound. Renal cell carcinoma risk of 5% should prompt consideration of annual renal ultrasound starting at age 40 and colon cancer risk of 9% requires colonoscopy every 5 years beginning at age 35.
Li-Fraumini Syndrome
The lifetime risk of cancer with this devastating syndrome approaches 90-100% by age 70. This results from mutation of the CHEK2 tumor suppressor gene and TP53 oncogene, with resultant uncontrolled cellular growth. The most common malignancies include breast cancer at 50%, osteosarcoma and soft tissue sarcomas, as well as brain, adrenal and hematologic cancers. This additional confers an elevated risk of ovarian and uterine cancers, however no current screening guidelines or risk reduction strategies exist in this regard 28,41.
DICER 1 Syndrome
The DICER1 gene plays an essential role in microRNA function. Loss of function results in an elevated risk of both benign tumor and malignancy, including pleuropulmonary blastoma, nodular goiters and cystic nephromas 42,43. Women have an elevated risk of Sertoli-Leydig ovarian tumors, thyroid cancer and rhabdomyosarcoma. Currently, no surveillance of risk reduction strategies exist outside of routine physical exam and possible regular imaging studies.
The Future of Molecular Medicine
As the fields of genomics and proteomics continue to expand, their applicability to patient screening, tumor interrogation and prognostication, as well identification of targetable mutations for novel therapeutic are exciting. As a provider, however, this plethora of genetic information can be overwhelming and challenging to navigate. This brief summary of the most applicable technology for an OBGYN or women’s physician today should guide surveillance and risk-reduction strategies for the most common syndromes.
Figure 1.
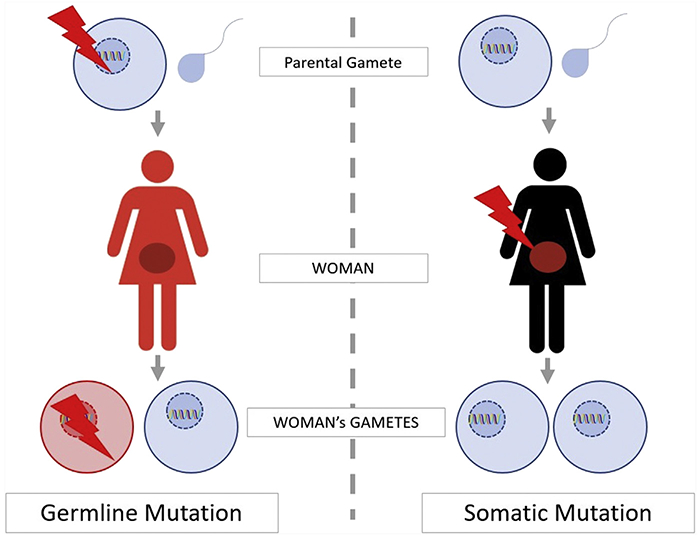
Understanding germline and somatic mutations
Figure 3.
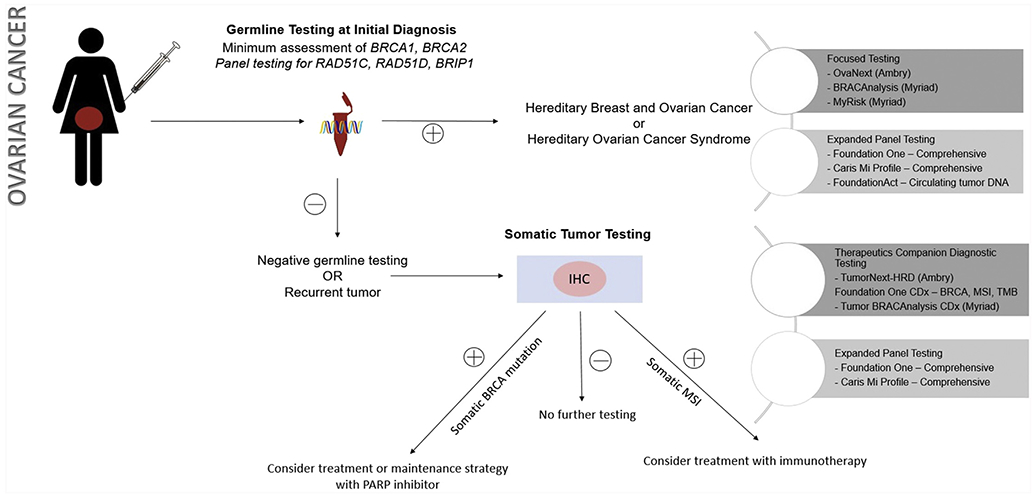
Approach to testing strategies in ovarian cancer
IHC: Immunohistochemistry; PARP: poly-ADP ribose polymerase; MSI: microsatellite instability; TMB: tumor mutational burden
Figure 4.
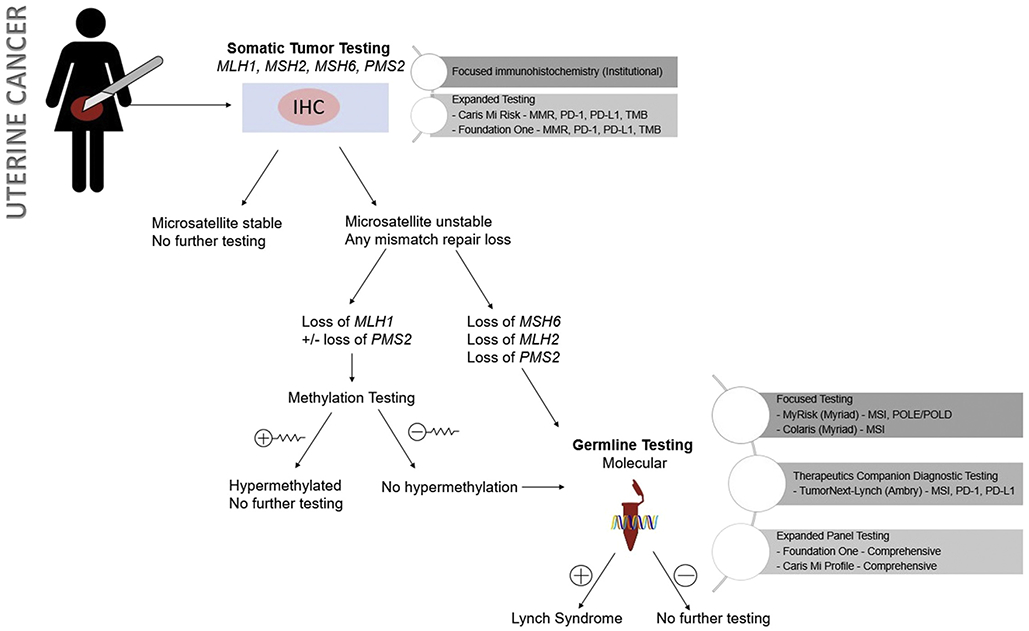
Approach to testing strategies in uterine cancer
Figure 5.
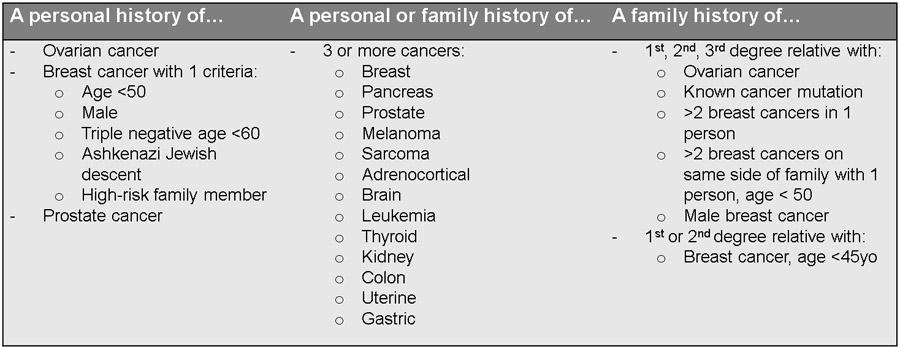
Review of criteria for genetic counseling assessment
Figure 6.
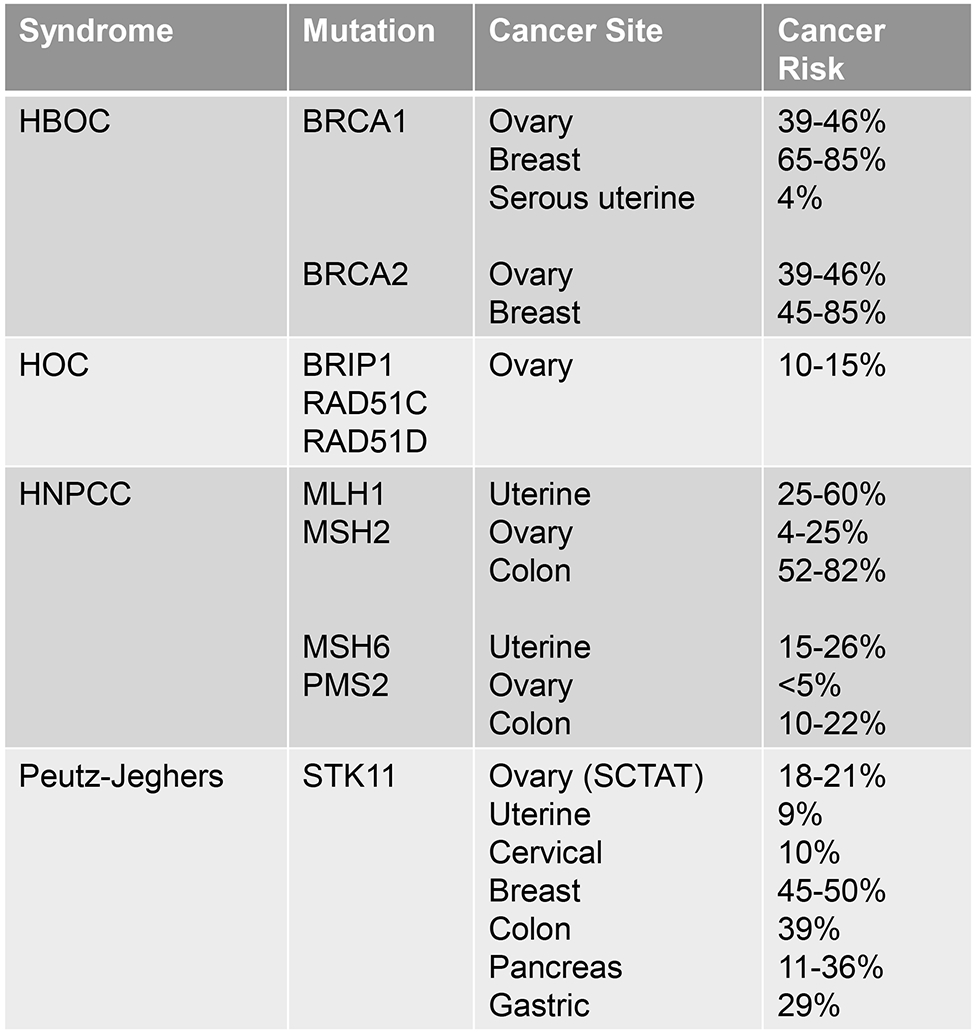
Summary of Hereditary Cancer Syndromes
HBOC-Hereditary Breast and Ovarian Cancer; HOC-Hereditary ovarian cancer; HNPCC-Hereditary non-polyposis colon cancer; SCTAT- sex cord stromal tumor with annular tubules; PPAP-polymerase proofreading-associated polyposis
Footnotes
Disclosures:
Neither author has any financial conflicts of interest to disclose.
References
- 1.Berger AC, Korkut A, Kanchi RS, et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer cell. 2018;33(4):690–705.e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research N, Albert Einstein College of M, Analytical Biological S, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543(7645):378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7(283):283ra253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr TH, McEwen R, Dougherty B, et al. Defining actionable mutations for oncology therapeutic development. Nature reviews Cancer. 2016;16(5):319–329. [DOI] [PubMed] [Google Scholar]
- 7.Kotelnikova EA, Pyatnitskiy M, Paleeva A, Kremenetskaya O, Vinogradov D. Practical aspects of NGS-based pathways analysis for personalized cancer science and medicine. Oncotarget. 2016;7(32):52493–52516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diaz LA Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadare O, D K. Molecular Profiling of Epithelial Ovarian Cancer. My Cancer Genome 2016; https://www.mycancergenome.org/content/disease/ovarian-cancer/. [Google Scholar]
- 10.Meghnani V, Mohammed N, Giauque C, Nahire R, David T. Performance Characterization and Validation of Saliva as an Alternative Specimen Source for Detecting Hereditary Breast Cancer Mutations by Next Generation Sequencing. International journal of genomics. 2016;2016:2059041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vnencak-Jones C, Berger M, Pao W. Types of Molecular Tumor Testing. My Cancer Genome 2016; https://www.mycancergenome.org/content/molecular-medicine/types-of-molecular-tumor-testing/ [Google Scholar]
- 12.Oncology SoG. SGO Clinical Practice Statement: Genetic Testing for Ovarian Cancer. October 2014; https://www.sgo.org/clinical-practice/guidelines/genetic-testing-for-ovarian-cancer/.
- 13.Huang YW. Association of BRCA1/2 mutations with ovarian cancer prognosis: An updated meta-analysis. Medicine. 2018;97(2):e9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen KC, Mariappan MR, Putcha GV, et al. Microsatellite instability and mismatch repair protein defects in ovarian epithelial neoplasms in patients 50 years of age and younger. The American journal of surgical pathology. 2008;32(7):1029–1037. [DOI] [PubMed] [Google Scholar]
- 15.Approved Drugs - FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication [press release]. http://www.fda.gov5/2017.
- 16.Oncology SoG. Clinical Practice Statement for Lynch Syndrome in endometrial cancer. March 2014; https://www.sgo.org/clinical-practice/guidelines/screening-for-lynchsyndrome-in-endometrial-cancer/. [Google Scholar]
- 17.Terdiman JP, Gum JR Jr., Conrad PG, et al. Efficient detection of hereditary nonpolyposis colorectal cancer gene carriers by screening for tumor microsatellite instability before germline genetic testing. Gastroenterology. 2001;120(1):21–30. [DOI] [PubMed] [Google Scholar]
- 18.Mills AM, Longacre TA. Lynch Syndrome Screening in the Gynecologic Tract: Current State of the Art. The American journal of surgical pathology. 2016;40(4):e35–44. [DOI] [PubMed] [Google Scholar]
- 19.Diaz-Padilla I, Romero N, Amir E, et al. Mismatch repair status and clinical outcome in endometrial cancer: a systematic review and meta-analysis. Critical reviews in oncology/hematology. 2013;88(1):154–167. [DOI] [PubMed] [Google Scholar]
- 20.Wong YF, Cheung TH, Poon KY, et al. The role of microsatellite instability in cervical intraepithelial neoplasia and squamous cell carcinoma of the cervix. Gynecol Oncol. 2003;89(3):434–439. [DOI] [PubMed] [Google Scholar]
- 21.Crafton SM, Salani R. Beyond Chemotherapy: An Overview and Review of Targeted Therapy in Cervical Cancer. Clinical therapeutics. 2016;38(3):449–458. [DOI] [PubMed] [Google Scholar]
- 22.FDA authorizes, with special controls, direct-to-consumer test that reports three mutations in the BRCA breast cancer genes [press release]. http://www.fda.gov3/2018.
- 23.Practice Advisory: Response to FDA’s Authorization of BRCA1 and BRCA2 Gene Mutation Direct-to-Consumer Testing [press release]. https://www.acog.org/Clinical-Guidance-and-Publications/Practice-Advisories/Practice-Advisory-Response-to-FDAs-Authorization-of-BRCA1-and-BRCA2-Genes-Direct-to-Consumer-TestingMay 2018.
- 24.Schorge JO, Modesitt SC, Coleman RL, et al. SGO White Paper on ovarian cancer: etiology, screening and surveillance. Gynecol Oncol. 2010;119(1):7–17. [DOI] [PubMed] [Google Scholar]
- 25.Daly MB, Axilbund JE, Bryant E, et al. Genetic/familial high-risk assessment: breast and ovarian. Journal of the National Comprehensive Cancer Network : JNCCN. 2006;4(2):156–176. [DOI] [PubMed] [Google Scholar]
- 26.Hampel H, Bennett RL, Buchanan A, Pearlman R, Wiesner GL. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(1):70–87. [DOI] [PubMed] [Google Scholar]
- 27.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. JNCI Journal of the National Cancer Institute. 2004;96(4):261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ring KL, Garcia C, Thomas MH, Modesitt SC. Current and future role of genetic screening in gynecologic malignancies. Am J Obstet Gynecol. 2017. [DOI] [PubMed] [Google Scholar]
- 29.Norquist BM, Harrell MI, Brady MF, et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA oncology. 2016;2(4):482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66(1):7–30. [DOI] [PubMed] [Google Scholar]
- 31.Loveday C, Turnbull C, Ramsay E, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nature genetics. 2011;43(9):879–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loveday C, Turnbull C, Ruark E, et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nature genetics. 2012;44(5):475–476; author reply 476. [DOI] [PubMed] [Google Scholar]
- 33.Lynch HT, Casey MJ, Snyder CL, et al. Hereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and management. Molecular oncology. 2009;3(2):97–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanday S BRCA mutations and risk of uterine cancer. The Lancet Oncology. 2016;17(8):e324. [DOI] [PubMed] [Google Scholar]
- 36.Bellido F, Pineda M, Aiza G, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genetics in medicine : official journal of the American College of Medical Genetics. 2016;18(4):325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Briggs S, Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. The Journal of pathology. 2013;230(2):148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature genetics. 2013;45(2):136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Resta N, Pierannunzio D, Lenato GM, et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2013;45(7):606–611. [DOI] [PubMed] [Google Scholar]
- 40.van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. The American journal of gastroenterology. 2010;105(6):1258–1264; author reply 1265. [DOI] [PubMed] [Google Scholar]
- 41.Amadou A, Waddington Achatz MI, Hainaut P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li-Fraumeni syndrome. Current opinion in oncology. 2018;30(1):23–29. [DOI] [PubMed] [Google Scholar]
- 42.Schultz KA, Harris A, Messinger Y, et al. Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Fam Cancer. 2016;15(1):105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schultz KA, Pacheco MC, Yang J, et al. Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol. 2011;122(2):246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]