

ABSTRACT
In the past decade, the study of the major DNA double strand break (DSB) repair pathways, homologous recombination (HR) and classical non-homologous end joining (C-NHEJ), has revealed a vast and intricate network of regulation. The choice between HR and C-NHEJ is largely controlled at the step of DNA end-resection. A pro-C-NHEJ cascade commencing with 53BP1 and culminating in the newly discovered REV7-Shieldin complex impedes end resection and therefore HR. Importantly, loss of any component of this pathway confers PARP inhibitor resistance in BRCA1-deficient cells; hence, their study is of great clinical importance. The newest entrant on the scene of end resection regulation is the ATPase TRIP13 that disables the pro-C-NHEJ cascade by promoting a novel conformational change of the HORMA protein REV7. Here, we tie these new findings and factors with previous research on the regulation of DSB repair and HORMA proteins, and suggest testable hypotheses for how TRIP13 could specifically inactivate REV7-Shieldin to promote HR. We also discuss these biological questions in the context of clinical therapeutics.
KEYWORDS: Keywords REV7, Shieldin, TRIP13DNA repair, homologous recombination, PARP inhibitor, HORMA protein, fanconi anemia
Many human cancers, such as ovarian, breast, prostate, and pancreatic cancers, have mutations in homologous recombination (HR) pathway genes. The most commonly altered HR genes are BRCA1 and BRCA2, followed by other genes in the Fanconi Anemia (FA) pathway. Loss of HR causes genomic instability, hyper-dependence on alternative DNA repair mechanisms, and enhanced sensitivity to PARP inhibitors (PARPi) [1,2]. This synthetic lethal interaction with PARPi has been exploited therapeutically, most notably in ovarian cancer, where several PARPis have recently received FDA approval. PARPis are oral drugs and relatively well-tolerated, and they can be combined with other anti-cancer therapies. Unfortunately, PARPi resistance has emerged as a vexing clinical problem for the treatment of BRCA1/2-deficient carcinomas.
The most prevalent mechanism of PARPi resistance is secondary events, such as somatic reversion of a germline BRCA1/2 mutation, leading to restoration of HR repair [3,4]. Another important mechanism of HR restoration in BRCA1-deficient cells involves the loss of factors which normally inhibit 5ʹ-3ʹ DNA end resection at a double strand break (DSB), an important first step in HR (Figure 1a) [5]. The first gene identified in this pathway was 53BP1, whose loss was found to restore viability in BRCA1-/- mice and render BRCA1-deficient cancers resistant to DNA damaging treatment, including PARPi [6,7]. Subsequent work showed that upon DNA damage, 53BP1 is phosphorylated by the DNA damage responsive kinase ATM at numerous sites which recruit the downstream effector proteins, RIF1 and PTIP, to promote classical non-homologous end joining (C-NHEJ), a DSB repair pathway that competes with and is more error-prone than HR [8–11]. Loss of RIF1 and PTIP also induces PARPi resistance in BRCA1/2-deficient backgrounds, similarly to loss of 53BP1. In a surprising discovery, the small DNA polymerase ζ subunit, REV7, was uncovered as an essential downstream effector of RIF1, independently of any known Pol ζ-related binding partners [12,13].
Figure 1.
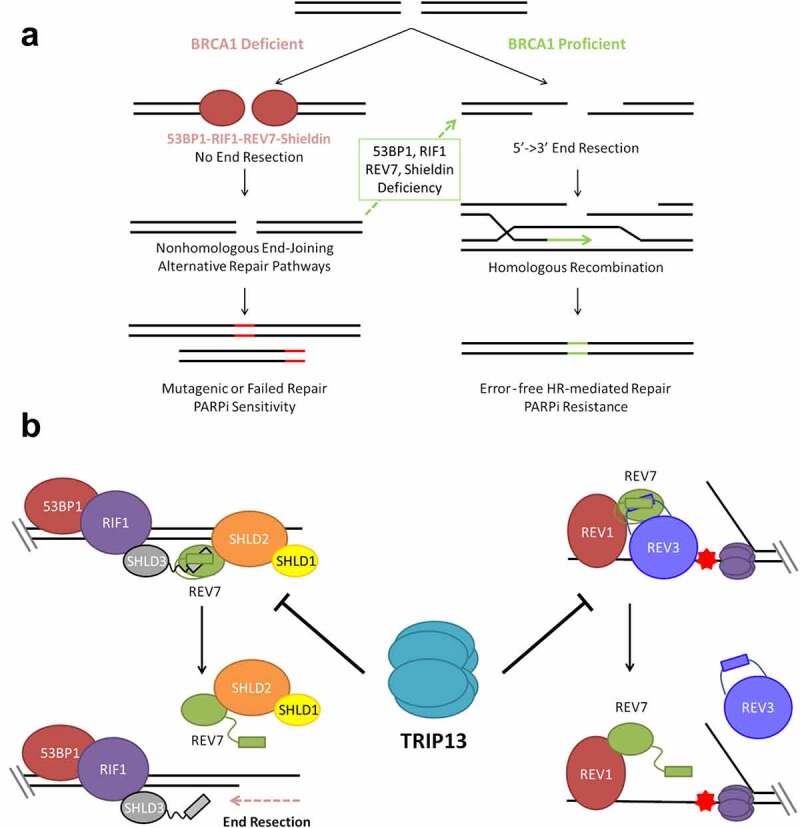
TRIP13 promotes PARP inhibitor sensitivity by disassembling REV7-Shieldin. (a) BRCA1 promotes 5ʹ-3ʹ DNA end resection, the critical first step in double strand break (DSB) repair through homologous recombination. In the absence of BRCA1; 53BP1, RIF1, REV7 and Shieldin inhibit DNA end resection, preventing repair by HR. End resection and HR can be restored in BRCA1-deficient cells by concurrent deficiency of any component of this anti-resection axis. Without HR repair, cells are unable to properly repair all DSBs, particularly those that arise during S phase leading to hypersensitivity to PARP inhibition and other DNA damaging therapies. (b) The Shieldin complex assembles at the break site through sequential recruitment of 53BP1, RIF1, SHLD3, REV7 and SHLD1-SHLD2 (left). REV7 is also recruited to sites of replication blockages, where it interacts with REV1 and REV3 as part of the DNA Polymerase ζ complex (right). TRIP13 catalyzes an inactivating conformational change of REV7, thereby inhibiting both the Shieldin complex and the Polymerase ζ complex.
The Polymerase ζ complex, consisting of REV7 and REV3, has long been known to catalyze mutagenic translesion DNA synthesis (TLS), a pathway that allows cells to replicate DNA across damaged bases [14]. All of the catalytic activity of the complex is contained within the large REV3 subunit, and REV7 has primarily been considered an accessory subunit that may function only to bridge the interaction between Pol ζ and the TLS factor, REV1 [15].
Given the known function of REV7 as an accessory subunit, these studies strongly implied the existence of novel REV7 interaction partners mediating its newly found role in HR regulation. Indeed, a flurry of studies using complementary techniques of CRISPR screening and physical interactome analysis identified three novel interaction partners – SHLD1, SHLD2 and SHLD3 [16–23]. Together with REV7, these four proteins form a complex known as Shieldin. The entire cascade, starting with 53BP1 and culminating in the Shieldin complex, inhibits end resection-dependent HR and promotes C-NHEJ, such that loss of any of them in the absence of BRCA1 relieves the inhibition and allows HR to occur. This is clinically relevant because these doubly-deficient cells are resistant to PARPis.
Our laboratory has recently demonstrated that tumor cells can also achieve a state of PARPi resistance by upregulating the expression of the ATPase, TRIP13, and that this mode is linked to REV7 [24]. Importantly, BRCA1-deficient tumor cells often have high TRIP13 expression, leading to PARPi resistance. We showed that REV7 exists in active “closed” and inactive “open” conformations (Figure 1b). TRIP13, through ATP hydrolysis, catalyzes an inactivating conformational change in REV7. In doing so, it dissociates the REV7-Shieldin complex, promoting end resection and thereby HR. TRIP13 similarly disassembles the REV7-REV3 TLS complex. Thus, TRIP13 overexpression simultaneously inactivates mutagenic TLS and C-NHEJ and promotes high fidelity HR. Importantly, since TRIP13 is an enzyme, an inhibitor of TRIP13 may be useful in re-sensitizing BRCA1-deficient tumor cells to PARPis. Despite some of the more obvious biological and clinical implications of TRIP13 overexpression, such as increased HR and the acquisition of PARPi resistance, there are several unanswered questions regarding its regulation and mechanism of action. This review addresses emerging questions in the field of regulation of end resection, with a brief background on the better known apical players and a detailed look at the newest and most downstream components, REV7 and TRIP13.
Regulation of the 53BP1-RIF1-REV7-Shieldin anti-resection pathway
As end resection is a critical determinant of DSB repair pathway and outcome, it is not surprising that nearly every participating factor is regulated. 53BP1, the apical component of the anti-resection pathway, has ubiquitin binding [25] and methyl-lysine binding domains [26,27], both of which aid in its recruitment. The DNA damage specificity of 53BP1 recruitment is thought to be mediated by its binding to H2AK15 ubiquitylation, catalyzed by the DNA damage responsive ubiquitin ligase, RNF168 [28]. RNF168 recruitment is in turn regulated by a DNA damage cascade comprising ATM, MDC1 and RNF8 [29–33]. The ATM kinase further phosphorylates 53BP1 itself, which is required for the recruitment of RIF1, REV7 and Shieldin.
The methyl-lysine binding tudor domains bind specifically to methylated histone H4K20, a ubiquitous histone mark [34]. This interaction is normally inhibited by the protein TIRR, which binds directly to the tudor domain, but is released upon DNA damage [35,36]. It has been suggested that H4K20 methylation accumulates slowly following histone deposition on newly replicated DNA, thus favoring 53BP1 binding to unreplicated DNA [37,38]. This is a sensible model, as HR requires the presence of a sister chromatid, and should therefore be repressed in unreplicated DNA. 53BP1 is also inhibited by phosphorylation during M-phase [39–41], when it can cause aberrant DNA repair. It is unclear whether this aberrancy is related to unscheduled resection, which can occur to some degree during M-phase [42].
Only recently have we learned of the downstream REV7-Shieldin components, named so due to their functional and sequence level homology to the telomeric Shelterin complex. Like Shelterin, the Shieldin complex localizes to DNA ends and binds to single stranded DNA (ssDNA), preventing resection, accumulation of the ssDNA binding protein RPA and subsequent ATR signaling. However, unlike Shelterin, which exhibits sequence-specific DNA binding, Shieldin is recruited to DNA damage through 53BP1-RIF1 interaction, with SHLD3 acting as a bridge between RIF1 and REV7, which in turn recruits SHLD1 and SHLD2. Nothing is known about the regulation of these downstream proteins other than the role of TRIP13 and REV7 conformation, the function and regulation of which still present many mysteries.
The HORMA family of proteins
REV7 is a founding member of the HORMA protein family, so-named for HOp1, REV7 and MAD2 [43]. In addition to the founding members, recent studies have identified the autophagy proteins Atg13 and Atg101 as HORMA family members [44–46]. Intriguingly, HORMA proteins and a TRIP13 orthologue have recently been identified in bacteria, suggesting that this family has ancient origins [47]. Proteins of the HORMA family are unified by modest primary sequence homology, but striking homology in tertiary structure (Figure 2a).
Figure 2.
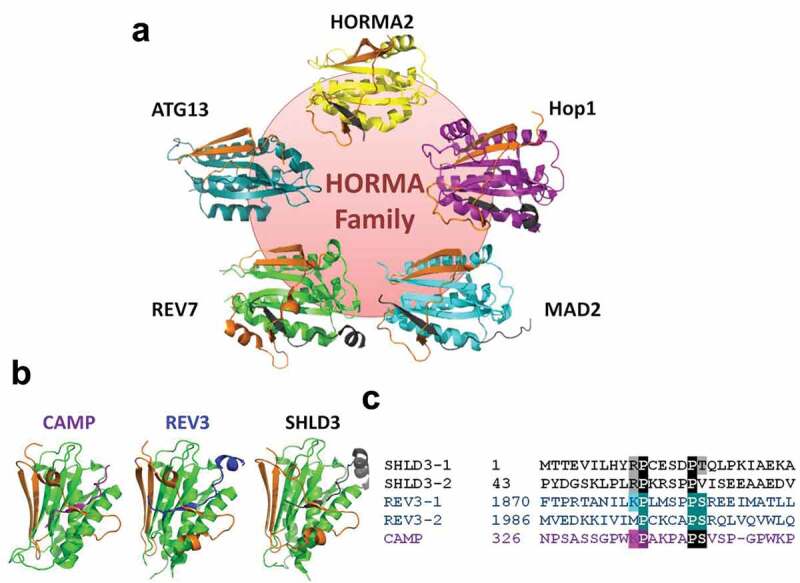
REV7 is a member of the functionally diverse HORMA family and interacts with multiple seatbelt binding partners. (a) The HORMA family is highly structurally conserved across domains of life. Shown are examples of HORMA protein structures in the closed conformation with the seatbelt subdomain highlighted in orange; clockwise from top: HORMA2 – P. aeruginosa, PDB: 6P8 U [47]; HIM-3 (Hop1 orthologue) – C. elegans, PDB: 4TZJ [60]; MAD2 – S. pombe, PDB: 4AEZ [86]; REV7 – H. sapiens, PDB: 3VU7 [87], ATG13 – H. sapiens, PDB: 5C50 [55]. (b) Structures of closed REV7 bound to seatbelt binding partners: CAMP, REV3 and SHLD3. PDB: 5XPT [62], 3VU7 [87], 6K07 [88] respectively. (c) Primary sequence alignment of REV7 seatbelt binding regions of SHLD3, REV3 and CAMP highlighting conserved amino acids constituting the REV7 seatbelt binding motif (SBM).
Like REV7, Hop1, MAD2 and the bacterial HORMA proteins activity are also regulated by stable conformational changes between open and closed conformations [47–49]. The major structural transition between the open and closed conformation occurs in the C-terminal “seatbelt” subdomain. In the closed conformation, the seatbelt latches tightly to binding partners at so-called seatbelt binding motifs (SBMs), whereas this interaction is not stable in the open conformation. It is this critical binding to SBM-containing partners that is controlled by HORMA conformational transitions. Intriguingly, Atg13 and Atg101 seem to have non-canonical HORMA function as they appear to be locked in the closed and open states respectively and form a heterodimer [46].
While the HORMA proteins share structural and regulatory features, they function in very distinct capacities. MAD2 plays a clearly defined role in the spindle assembly checkpoint (SAC). During mitosis, pools of inactive, open MAD2 are activated at unattached kinetochores through seatbelt-SBM interactions with MAD1 [50–52]. MAD1 promotes the closing of MAD2 which is propagated in a chain reaction by dimerization of open and closed MAD2 conformers [53,54]. It is noteworthy that the Atg13-Atg101 conformationally-locked heterodimer, resembles the assymetric closed-open MAD2 dimer [55], suggesting that asymmetric HORMA protein dimers may be a common feature of regulation. The target of MAD2 is the anaphase promoting complex (APC) activator, Cdc20, another SBM-containing protein [56,57]. Closed MAD2 rapidly binds and sequesters Cdc20, preventing APC activation until each chromatid is attached to the spindle.
Hop1 is a conserved member of the meiosis-specific chromosome axis complex, which organizes homologous chromosomes and promotes crossover formation [58,59]. Unlike MAD2, which forms discrete soluble MAD2:Cdc20 complexes, Hop1 and the related mammalian proteins, HORMAD1 and HORMAD2, appear to form large chromosome-bound chains. Hop1-type proteins have both a HORMA domain and SBMs, allowing for the formation of complex higher-order structures with one Hop1 monomer’s seatbelt bound over the adjacent Hop1 monomer’s SBM and so on [60].
REV7 is unique among the HORMA proteins in having multiple distinct SBM-containing partners (Figure 2b,c). SHLD3 has two tandem SBMs in its N-terminus, similar to REV3 which has two functionally important SBMs [61]. It is not known if REV7 can bind to both SBMs simultaneously or if this is functionally relevant. In addition to the well-characterized interaction with REV3 and to a lesser extent SHLD3, the zinc-finger protein CAMP is also a confirmed seatbelt interactor [62]. High-throughput interaction profiling of REV7 has revealed numerous additional binding partners, but whether any of these are also seatbelt-mediated remains to be elucidated [17,24]. Why REV7 is involved in these distinct processes is not understood, but is suggestive of some sort of coordinated regulation of REV7-dependent pathways. MAD2 also has two seatbelt-mediated interaction partners, namely MAD1 and Cdc20; however there is a clear distinction, as MAD1 stimulates MAD2 closing, while Cdc20 can only be bound following MAD1 activity, and both these partners are related to the same (and only) function of MAD2. Whether the known REV7 binding partners can play activator functions, analogous to MAD1, or are mainly targets of REV7 activity, like Cdc20, is not yet clear. It is also not known whether REV7 activation is similarly propagated through asymmetric dimerization, like with MAD2. REV7 homodimerization through a similar interface has recently been shown to be functionally important [63], lending support to this hypothesis; however, more extensive research is still required.
One unifying feature of the HORMA family of proteins is their regulation by the AAA+ ATPase TRIP13. Like REV7, Hop1 and MAD2 are both known to be inactivated by TRIP13 which is thought to remodel closed conformers into the inactive open conformation [64–66]. The involvement of TRIP13 in at least three very distinct cellular processes raises several interesting questions. Is TRIP13 activity somehow directed to specific substrates in the appropriate context, such as through substrate-specific adaptor proteins? Or is the regulation of these seemingly distinct proteins actually coordinated, perhaps at specific cell cycle phases? For instance, TRIP13 may regulate the choice between HR and NHEJ during S phase, and the activity of the SAC in mitosis.
Regulation of TRIP13 activity on HORMA proteins
TRIP13 is a highly conserved AAA+ ATPase that forms single homohexameric rings. It was first linked to the HORMA protein family when studies in yeast revealed a role in meiotic checkpoint and recombination [67–69]. This role is evolutionarily conserved as both yeast Pch2 and mouse Trip13 regulate their respective HORMA substrates, Hop1 and Hormad1 [64,68]. In both organisms, Hop1/Hormad1 monomers bind to DNA in a cooperative manner, and addition of Pch2/Trip13 alters their DNA binding or chromatin association [64,70]. Thus, Pch2/Trip13 has been proposed to remodel Hop1/Hormad1 oligomers already on chromatin, promoting their displacement from DNA and turnover. How Pch2 is targeted to Hop1 is not entirely clear, but it has been shown to interact with the BRCT domains in Xrs2, part of the MRX (Mre11-Rad50-Xrs2) damage sensor in yeast (MRN or MRE11-RAD50-NBS1 in humans), and this interaction is functionally important [68]. Mouse Hormad1 mutants lacking the N terminal region are refractory to TRIP13 action, indicating that this region is required for the disassembly of HORMAD1 from chromatin.
How TRIP13 is targeted to and inactivates MAD2 is better understood. Like in the case of Hormad1, the highly conserved N-terminal region of MAD2 is necessary for TRIP13-mediated inactivation [71]. Elegant structural studies have provided insight into why this is so. TRIP13 grasps the N terminus of MAD2, which is fed through a central pore. ATP hydrolysis by TRIP13 causes a limited unfolding of this region of MAD2, which destabilizes the overall closed MAD2 structure by disrupting a conserved hydrogen bond network between residues at the N-terminus and C-terminal seatbelt domain [71]. Interestingly, while TRIP13 can elicit this effect alone, its activity on MAD2 is greatly enhanced by another HORMA-like protein, p31, which directly interacts with both TRIP13 and MAD2, forming a bridge between them [65,72] (Figure 3). p31 binds to MAD2 at the latter’s homodimerization interface, and this structural mimicry further attenuates MAD2 signaling by preventing activation through the asymmetric closed-open dimerization pathway [73]. Whether p31 similarly coordinates the interaction of TRIP13 and REV7 is unknown.
Figure 3.
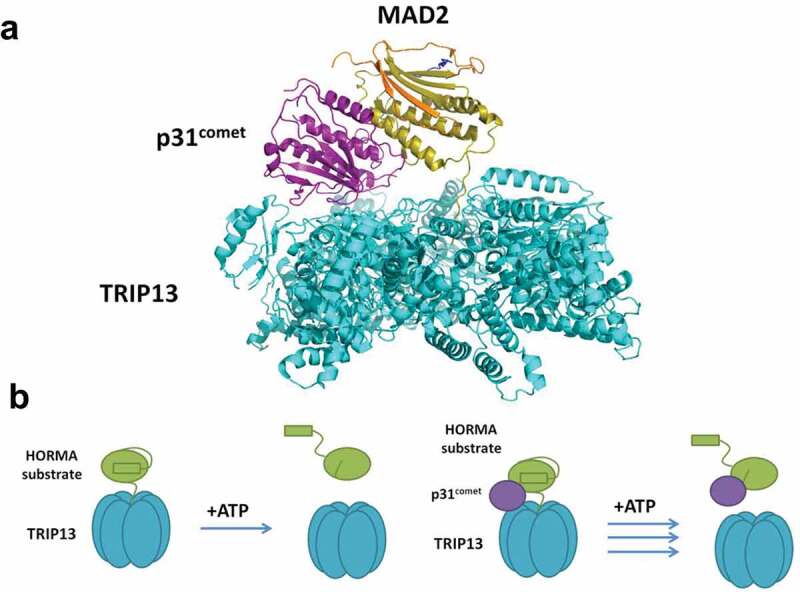
TRIP13 remodels HORMA substrates through N-terminal engagement and is stimulated by an adaptor protein. (a) Cryo-EM structure of TRIP13 acting on C-MAD2, showing bridging by the adaptor protein, p31comet, PDB: 6F0X [89]. (b) Schematic of N-terminal engagement of HORMA substrate by TRIP13 and stimulation by an adaptor, p31comet, which bridges the interaction. Following engagement of the HORMA protein, TRIP13 catalyzes HORMA remodeling coupled to ATP hydrolysis.
Models for TRIP13 activity toward REV7
Given the very recent discovery of TRIP13 action on REV7, we know little about the mechanism and regulation of this interaction; however, using evidence from other HORMA proteins, we can suggest several testable hypotheses. One critical question is whether there is any mechanism for TRIP13 substrate specificity or if all substrates present in the cell are regulated concurrently. A possible mechanism of substrate specificity is through adaptor proteins like p31, which has only been definitively shown to interact with MAD2. If there are alternative p31-like adapters for REV7 and HORMAD1, this could dictate substrate specificity (Figure 4, Model 1). There is some evidence suggesting that REV7 does in fact interact with p31, contradicting this model; however, more research is required on this topic [63]. The potential universality of the p31 adaptor is also supported by reports that the rice orthologue, CRC1, localizes to the chromosome axis and is implicated in meiotic HR [74], suggestive of a HORMAD1 regulatory role. On the other hand, there are some organisms, such as budding yeast, that possess TRIP13 but not p31, and in these cases, TRIP13 appears to have only a meiotic function through Hop1 [75]. If p31 is in fact a universal adaptor, substrate specificity may instead be dictated through posttranslational modifications of TRIP13, p31 or the HORMA substrate (Figure 4, Model 2). Indeed, Plk1-dependent phosphorylation of p31 decreases its affinity for MAD2, thereby dialing down TRIP13-mediated inactivation of MAD2 [76]. Perhaps DNA damage-dependent phosphorylation of p31 or REV7-Shieldin by ATM similarly modulates TRIP13 activity to promote certain types of repair.
Figure 4.
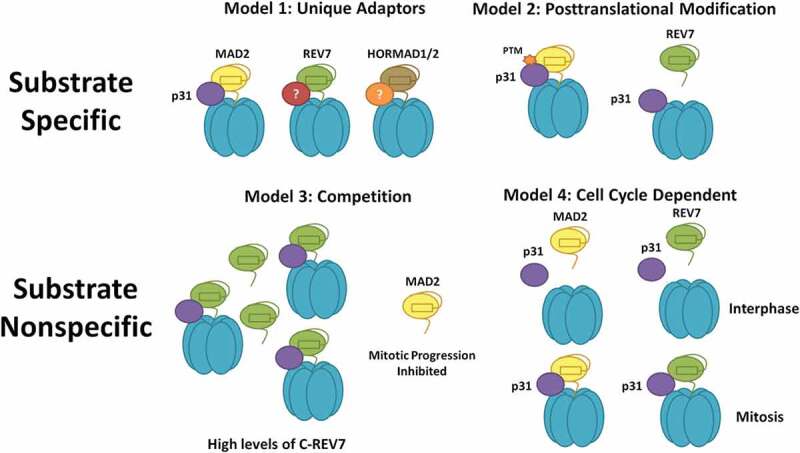
Models for TRIP13 regulation of diverse HORMA proteins. Substrate Specific Models (1): Substrate specificity is enforced by unique adaptors for each HORMA protein; (2): Substrate specificity is enforced by post-translational modification of the appropriate substrate. Substrate Nonspecific Models (3): Each HORMA protein competes for a limited supply of TRIP13-p31. High levels of DNA damage would thereby inhibit anaphase progression through TRIP13-p31 sequestration. C-REV7 indicates closed REV7; (4): TRIP13 acts on all substrates during a specific cell cycle phase or phases. TRIP13 is known to be active in mitosis and may represent another mechanism to suppress DNA repair in mitosis.
It is also possible that TRIP13 does not show substrate specificity; however, this does not mean that TRIP13 activity is not regulated. TRIP13 activity could be controlled by the relative abundance of the closed conformations of each substrate, such that they compete for limited TRIP13-p31 (Figure 4, Model 3). This would have interesting consequences as high levels of DNA damage, and therefore closed REV7, would effectively sequester TRIP13, preventing inactivation of the SAC. This would represent a novel mechanism of cell cycle arrest in response to DNA damage.
Yet another possibility is that TRIP13 and p31 are sufficiently abundant to prevent any competition. In this case, they would regulate all of their HORMA substrates concurrently. It is noteworthy that p31 protein expression is up-regulated specifically in S and G2/M phases, when REV7 and MAD2 activities need to be regulated. Thus, cells could specifically promote the efficient inactivation of REV7 and MAD2 by limiting p31 expression to these cell cycle phases (Figure 4, Model 4). Apart from these possibilities but perhaps related to them, TRIP13 cellular localization could dictate its activity. Support for this idea comes from studies showing that TRIP13 is normally cytoplasmic, but forms foci at kinetochores during mitosis [77] and on chromosome axes during meiosis [78]. Whether TRIP13 similarly forms foci after DSB induction that co-localize with DNA damage factors would be useful to pinpoint when and where TRIP13 inactivates REV7.
Clinical Implications of the TRIP13-REV7 Interaction
TRIP13 activity is not only biologically interesting, it is also clinically relevant. Previous studies have noted that TRIP13 is overexpressed in various cancers and is a negative prognostic marker [79–81]. One report examining TRIP13 in the context of head and neck cancer similarly found that TRIP13 promotes DSB repair and chemoresistance, but concluded that TRIP13 promotes C-NHEJ, not HR [82]. That study identified a physical interaction between TRIP13 and DNA-PK subunits; however, these are common contaminants that appear in nearly 50% of AP-MS experiments [83]. Additionally, there are no HORMA domains in DNA-PK and no mechanism was proposed for TRIP13 regulation of DNA-PK. It remains possible that TRIP13 affects C-NHEJ in the cancer cell lines used in that study; however, our results demonstrate that the major function of TRIP13 in DSB repair is the promotion of HR through REV7 inactivation. Interestingly, biallelic loss-of-function TRIP13 mutations have been seen in Wilms tumor cells, which not surprisingly have an SAC defect [84]. It would be clinically useful to establish whether they are additionally HR-deficient, and therefore sensitive to PARPis.
Alternative Roles of TRIP13 in Regulating Genomic Stability
TRIP13 overexpression has previously been proposed to engender genomic instability by weakening the SAC, allowing cells to replicate more quickly and become aneuploid, promoting oncogenesis [79]. This idea has been challenged by the finding that TRIP13 expression is highly correlated with that of MAD2, suggesting a homeostatic relationship between the two in maintaining a normal SAC [85]. Given the genomic proximity of TRIP13 to hTERT, a strong oncogene, TRIP13 amplification may often occur as a passenger mutation. Regardless of the mechanism of TRIP13 overexpression in cancer, our studies have shown that the consequences are clear: high TRIP13 levels enhance HR and engender chemoresistance.
While elucidating PARP inhibitor resistance mechanisms through the discovery of 53BP1, RIF1, and the Shieldin complex has been a significant advance, none of those factors are druggable. TRIP13, as the newest entrant to this field and an enzymatic, negative regulator of the Shieldin complex, emerges as a critical player whose inhibition could be used in the clinic to extend the utility of PARP inhibitors.
Funding Statement
This work was supported by the Foundation for the National Institutes of Health [R37HL052725, P01HL048546]; Leukemia and Lymphoma Society [5440-16]; DOD (Department of Defense) [BC151331P1].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. [DOI] [PubMed] [Google Scholar]
- [2].Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. [DOI] [PubMed] [Google Scholar]
- [3].Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. [DOI] [PubMed] [Google Scholar]
- [4].Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heyer WD, Ehmsen KT, Liu J.. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44(1):113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bunting SF, Callén E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zimmermann M, Lottersberger F, Buonomo SB, et al. 53BP1 regulates DSB repair using Rif1 to control 5ʹ end resection. Science. 2013;339(6120):700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49(5):872–883. [DOI] [PubMed] [Google Scholar]
- [10].Chapman JR, Barral P, Vannier J-B, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49(5):858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Callen E, Di Virgilio M, Kruhlak M, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153(6):1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521(7553):541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Boersma V, Moatti N, Segura-Bayona S, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5ʹ end resection. Nature. 2015;521(7553):537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5(10):a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hara K, Hashimoto H, Murakumo Y, et al. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J Biol Chem. 2010;285(16):12299–12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gupta R, Somyajit K, Narita T, et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018;173(4):972–988 e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Noordermeer SM, Adam S, Setiaputra D, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560(7716):117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mirman Z, Lottersberger F, Takai H, et al. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature. 2018;560(7716):112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ghezraoui H, Oliveira C, Becker JR, et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018;560(7716):122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dev H, Chiang T-WW, Lescale C, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20(8):954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tomida J, Takata K-I, Bhetawal S, et al. FAM 35A associates with REV 7 and modulates DNA damage responses of normal and BRCA 1-defective cells. Embo J. 2018;37(12). DOI: 10.15252/embj.201899543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Findlay S, Heath J, Luo VM, et al. SHLD 2/ FAM 35A co-operates with REV 7 to coordinate DNA double-strand break repair pathway choice. Embo J. 2018;37(18). DOI: 10.15252/embj.2018100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gao S, Feng S, Ning S, et al. An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nat Commun. 2018;9(1):3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Clairmont CS, Sarangi P, Ponnienselvan K, et al. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat Cell Biol. 2020;22(1):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fradet-Turcotte A, Canny MD, Escribano-Díaz C, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499(7456):50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sanders SL, Portoso M, Mata J, et al. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119(5):603–614. [DOI] [PubMed] [Google Scholar]
- [27].Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mattiroli F, Vissers JA, van Dijk W, et al. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150(6):1182–1195. [DOI] [PubMed] [Google Scholar]
- [29].Kolas NK, Chapman JR, Nakada S, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318(5856):1637–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huen MS, Grant R, Manke I, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131(5):901–914. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mailand N, Bekker-Jensen S, Faustrup H, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131(5):887–900. [DOI] [PubMed] [Google Scholar]
- [32].Doil C, Mailand N, Bekker-Jensen S, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136(3):435–446. [DOI] [PubMed] [Google Scholar]
- [33].Stewart GS, Panier S, Townsend K, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136(3):420–434. [DOI] [PubMed] [Google Scholar]
- [34].Pesavento JJ, Yang H, Kelleher NL, et al. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol. 2008;28(1):468–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Drane P, Brault M-E, Cui G, et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature. 2017;543(7644):211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang A, Peng B, Huang P, et al. The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response. J Biol Chem. 2017;292(16):6461–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pellegrino S, Michelena J, Teloni F, et al. Replication-coupled dilution of H4K20me2 guides 53BP1 to pre-replicative chromatin. Cell Rep. 2017;19(9):1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Simonetta M, de Krijger I, Serrat J, et al. H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell Cycle. 2018;17(1):124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nelson G, Buhmann M, von Zglinicki T. DNA damage foci in mitosis are devoid of 53BP1. Cell Cycle. 2009;8(20):3379–3383. [DOI] [PubMed] [Google Scholar]
- [40].van Vugt MA, Gardino AK, Linding R, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 2010;8(1):e1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Orthwein A, Fradet-Turcotte A, Noordermeer SM, et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344(6180):189–193. [DOI] [PubMed] [Google Scholar]
- [42].Peterson SE, Li Y, Chait BT, et al. Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J Cell Biol. 2011;194(5):705–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Aravind L, Koonin EV. The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci. 1998;23(8):284–286. [DOI] [PubMed] [Google Scholar]
- [44].Suzuki SW, Yamamoto H, Oikawa Y, et al. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci U S A. 2015;112(11):3350–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jao CC, Ragusa MJ, Stanley RE, et al. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A. 2013;110(14):5486–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Michel M, Schwarten M, Decker C, et al. The mammalian autophagy initiator complex contains 2 HORMA domain proteins. Autophagy. 2015;11(12):2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ye Q, Lau RK, Mathews IT, et al. HORMA domain proteins and a Trip13-like ATPase regulate bacterial cGAS-like enzymes to mediate bacteriophage immunity. Mol Cell. 2020;77(4):709–722 e707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Luo X, Tang Z, Xia G, et al. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol. 2004;11(4):338–345. [DOI] [PubMed] [Google Scholar]
- [49].West AMV, Komives EA, Corbett KD. Conformational dynamics of the Hop1 HORMA domain reveal a common mechanism with the spindle checkpoint protein Mad2. Nucleic Acids Res. 2018;46(1):279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen RH, Shevchenko A, Mann M, et al. Spindle checkpoint protein XMAD1 recruits Xmad2 to unattached kinetochores. J Cell Biol. 1998;143(2):283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Luo X, Tang Z, Rizo J, et al. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either MAD1 or Cdc20. Mol Cell. 2002;9(1):59–71. [DOI] [PubMed] [Google Scholar]
- [52].Sironi L, Mapelli M, Knapp S, et al. Crystal structure of the tetrameric MAD1-Mad2 core complex: implications of a ‘safety belt’ binding mechanism for the spindle checkpoint. Embo J. 2002;21(10):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].De Antoni A, Pearson CG, Cimini D, et al. The MAD1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol. 2005;15(3):214–225. [DOI] [PubMed] [Google Scholar]
- [54].Mapelli M, Massimiliano L, Santaguida S, et al. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131(4):730–743. [DOI] [PubMed] [Google Scholar]
- [55].Qi S, Kim DJ, Stjepanovic G, et al. Structure of the Human Atg13-Atg101 HORMA Heterodimer: an Interaction Hub within the ULK1 Complex. Structure. 2015;23(10):1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hwang LH, Lau LF, Smith DL, et al. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 1998;279(5353):1041–1044. [DOI] [PubMed] [Google Scholar]
- [57].Luo X, Wagner G, Luo X, et al. Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol. 2000;7(3):224–229. [DOI] [PubMed] [Google Scholar]
- [58].Hollingsworth NM, Byers B. HOP1: a yeast meiotic pairing gene. Genetics. 1989;121(3):445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hollingsworth NM, Goetsch L, Byers B. The HOP1 gene encodes a meiosis-specific component of yeast chromosomes. Cell. 1990;61(1):73–84. [DOI] [PubMed] [Google Scholar]
- [60].Kim Y, Rosenberg S, Kugel C, et al. The chromosome axis controls meiotic events through a hierarchical assembly of HORMA domain proteins. Dev Cell. 2014;31(4):487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tomida J, Takata K-I, Lange SS, et al. REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase ζ. Nucleic Acids Res. 2015;43(2):1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hara K, Taharazako S, Ikeda M, et al. Dynamic feature of mitotic arrest deficient 2-like protein 2 (MAD2L2) and structural basis for its interaction with chromosome alignment-maintaining phosphoprotein (CAMP). J Biol Chem. 2017;292(43):17658–17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rizzo AA, Vassel F-M, Chatterjee N, et al. Rev7 dimerization is important for assembly and function of the Rev1/Polζ translesion synthesis complex. Proc Natl Acad Sci U S A. 2018;115(35):E8191–E8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wojtasz L, Daniel K, Roig I, et al. Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 2009;5(10):e1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ye Q, Rosenberg SC, Moeller A, et al. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife. 2015;4. DOI: 10.7554/eLife.07367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ma HT, Poon RYC. TRIP13 regulates both the activation and inactivation of the spindle-assembly checkpoint. Cell Rep. 2016;14(5):1086–1099. [DOI] [PubMed] [Google Scholar]
- [67].San-Segundo PA, Roeder GS. Pch2 links chromatin silencing to meiotic checkpoint control. Cell. 1999;97(3):313–324. [DOI] [PubMed] [Google Scholar]
- [68].Ho HC, Burgess SM, McKim KS. Pch2 acts through Xrs2 and Tel1/ATM to modulate interhomolog bias and checkpoint function during meiosis. PLoS Genet. 2011;7(11):e1002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li XC, Schimenti JC. Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet. 2007;3(8):e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen C, Jomaa A, Ortega J, et al. Pch2 is a hexameric ring ATPase that remodels the chromosome axis protein Hop1. Proc Natl Acad Sci U S A. 2014;111(1):E44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ye Q, Kim DH, Dereli I, et al. The AAA + ATP ase TRIP 13 remodels HORMA domains through N-terminal engagement and unfolding. Embo J. 2017;36(16):2419–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Eytan E, Wang K, Miniowitz-Shemtov S, et al. Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31(comet). Proc Natl Acad Sci U S A. 2014;111(33):12019–12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yang M, Li B, Tomchick DR, et al. p31comet blocks Mad2 activation through structural mimicry. Cell. 2007;131(4):744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ji J, Tang D, Shen Y, et al. P31 comet, a member of the synaptonemal complex, participates in meiotic DSB formation in rice. Proc Natl Acad Sci U S A. 2016;113(38):10577–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].van Hooff JJ, Tromer E, van Wijk LM, et al. Evolutionary dynamics of the kinetochore network in eukaryotes as revealed by comparative genomics. EMBO Rep. 2017;18(9):1559–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kaisari S, Shomer P, Ziv T, et al. Role of Polo-like kinase 1 in the regulation of the action of p31(comet) in the disassembly of mitotic checkpoint complexes. Proc Natl Acad Sci U S A. 2019;116(24):11725–11730. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nelson CR, Hwang T, Chen PH, et al. TRIP13PCH-2 promotes Mad2 localization to unattached kinetochores in the spindle checkpoint response. J Cell Biol. 2015;211(3):503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Joshi N, Barot A, Jamison C, et al. Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet. 2009;5(7):e1000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang K, Sturt-Gillespie B, Hittle JC, et al. Thyroid hormone receptor interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic checkpoint-silencing protein. J Biol Chem. 2014;289(34):23928–23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kurita K, Maeda M, Mansour MA, et al. TRIP13 is expressed in colorectal cancer and promotes cancer cell invasion. Oncol Lett. 2016;12(6):5240–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Li W, Zhang G, Li X, et al. Thyroid hormone receptor interactor 13 (TRIP13) overexpression associated with tumor progression and poor prognosis in lung adenocarcinoma. Biochem Biophys Res Commun. 2018;499(3):416–424. [DOI] [PubMed] [Google Scholar]
- [82].Banerjee R, Russo N, Liu M, et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nat Commun. 2014;5(1):4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mellacheruvu D, Wright Z, Couzens AL, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods. 2013;10(8):730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yost S, de Wolf B, Hanks S, et al. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat Genet. 2017;49(1):1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Marks DH, Thomas R, Chin Y, et al. Mad2 overexpression uncovers a critical role for TRIP13 in mitotic exit. Cell Rep. 2017;19(9):1832–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Chao WC, Kulkarni K, Zhang Z, et al. Structure of the mitotic checkpoint complex. Nature. 2012;484(7393):208–213. [DOI] [PubMed] [Google Scholar]
- [87].Kikuchi S, Hara K, Shimizu T, et al. Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J Biol Chem. 2012;287(40):33847–33852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Dai Y, Zhang F, Wang L, et al. Structural basis for shieldin complex subunit 3-mediated recruitment of the checkpoint protein REV7 during DNA double-strand break repair. J Biol Chem. 2020;295(1):250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Alfieri C, Chang L, Barford D. Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature. 2018;559(7713):274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]