

Abstract
Objective
We identified a novel de novo SCN2A variant (M1879T) associated with infantile‐onset epilepsy that responded dramatically to sodium channel blocker antiepileptic drugs. We analyzed the functional and pharmacological consequences of this variant to establish pathogenicity, and to correlate genotype with phenotype and clinical drug response.
Methods
The clinical and genetic features of an infant boy with epilepsy are presented. We investigated the effect of the variant using heterologously expressed recombinant human NaV1.2 channels. We performed whole‐cell patch clamp recording to determine the functional consequences and response to carbamazepine.
Results
The M1879T variant caused disturbances in channel inactivation including substantially depolarized voltage dependence of inactivation, slower time course of inactivation, and enhanced resurgent current that collectively represent a gain‐of‐function. Carbamazepine partially normalized the voltage dependence of inactivation and produced use‐dependent block of the variant channel at high pulsing frequencies. Carbamazepine also suppresses resurgent current conducted by M1879T channels, but this effect was explained primarily by reducing the peak transient current. Molecular modeling suggests that the M1879T variant disrupts contacts with nearby residues in the C‐terminal domain of the channel.
Interpretation
Our study demonstrates the value of conducting functional analyses of SCN2A variants of unknown significance to establish pathogenicity and genotype–phenotype correlations. We also show concordance of in vitro pharmacology using heterologous cells with the drug response observed clinically in a case of SCN2A‐associated epilepsy.
Introduction
Variants in the SCN2A gene are associated with a range of childhood epilepsies differing in severity with or without accompanying neurodevelopmental delay. 1 , 2 Clinical phenotypes include benign infantile seizures, developmental epileptic encephalopathy, West syndrome, and Ohtahara syndrome. SCN2A encodes the pore‐forming subunit of the voltage‐gated sodium channel NaV1.2. Gain‐of‐function variants, which result in greater NaV1.2 activity than normal, are associated with neonatal and infantile‐onset epilepsy (onset before 3 months of age). 3 In contrast, loss‐of‐function variants are associated with later‐onset epilepsy with more prominent neurodevelopmental delay as well as autism spectrum disorder. 4 More widespread genetic testing of children with epilepsy has resulted in an explosion of new SCN2A variants, 5 yet the functional consequences and clear genotype–phenotype relationships have not been determined for most variants.
The majority of NaV1.2 channels are concentrated in the axon initial segment (AIS) of cortical excitatory neurons. During early development, NaV1.2 predominates throughout the AIS, acting as an action potential initiator and summing somatodendritic inputs. As neurons mature, NaV1.6 (encoded by SCN8A) replaces NaV1.2 in the more distal segments of the AIS. Later in development, NaV1.2 contributes to the back propagation of action potentials into the somatodendritic compartment of excitatory neurons. 6 , 7 , 8 Gain‐of‐function variants in SCN2A are thought to cause early‐onset epilepsy by promoting excitability of cortical neurons during the developmental stage when NaV1.2 predominates in the AIS. 9 Clinical studies suggest that some infantile‐onset epilepsies due to gain‐of‐function variants are responsive to treatment with antiseizure drugs (ASDs) having a principal mechanism of action to block sodium channels, albeit in a nonselective manner. 3
Here we report a case of infantile‐onset epilepsy associated with a novel SCN2A variant (M1879T) of uncertain significance combined with extensive functional and pharmacological studies of the variant sodium channel. Our findings reveal a novel constellation of functional abnormalities associated with infantile‐onset epilepsy, and provide a demonstration that in vitro pharmacological properties of a variant sodium channel correlate with the clinical response to a specific drug.
Materials and Methods
Study participant
The patient was recruited to study #2015‐738, which was approved by the Institutional Review Board of Ann & Robert H. Lurie Children’s Hospital of Chicago. Following informed consent, data were collected by parental report through a REDCap database questionnaire and review of medical records.
Mutagenesis and heterologous expression of NaV1.2
The open‐reading frame of human NaV1.2 was subcloned into a custom pCMV‐IRES‐mScarlet vector. The M1879T mutant channel was constructed using site‐directed mutagenesis PCR (Forward primer: AATACAGACGGAAGAGCGATTCATGGCATCAAACCC; Reverse primer: GCTCTTCCGTCTGTATTCGAAGGGCATCCATCTCTCC) with Q5 polymerase (New England Biolabs, Ipswitch, MA). The neonatal construct differed from the adult by inclusion of the neonatal exon 6N instead of the adult exon. All constructs were verified by Sanger sequencing of the promoter through to the polyadenylation signal, inclusive.
HEK293T cells (ATCC, Manassas, VA) were stably transfected with the β1 and β2 sodium channel subunits under puromycin selection to generate a stable cell line using previously established methods. 10 Cells were maintained at passages 5–15 in DMEM media supplemented with 1% Pen/Strep, 10% FBS, and puromycin (3 μg/mL). The HEK293T β1/β2 cell line was transiently transfected using SuperFect reagent (Qiagen, Germantown, MD) with 2 µg of WT or mutant NaV1.2 plasmid along with 0.25 µg of a second plasmid (pEGFP‐IRES‐SCN2B) for use as an additional transfection marker. After 24–48 h, transfected cells were harvested with trypsin and plated on poly‐D‐lysine (Sigma‐Aldrich, St. Louis, MO) coated coverslips prior to recording. Only fluorescent cells were selected for whole‐cell patch clamp recording.
Electrophysiology
Whole‐cell patch‐clamp recordings were performed at room temperature (22–23°C). Borosilicate glass pipettes were pulled with a P‐1000 (Sutter Instruments, Novato, CA), with resistances of 1–2.5 MOhm. External solution contained (in mmol/L) 140 NaCl, 10 HEPES, 4 KCl, 1 MgCl2, 1.8 CaCl2, 10 dextrose, pH 7.35 adjusted with NaOH, osmolarity 310 mOSm. Internal solution contained (in mmol/L) 10 NaF, 105 CsF, 20 CsCl, 2 EGTA, 10 HEPES, 10 dextrose, pH 7.35 adjusted with CsOH, osmolarity 300 mOsm. For measurement of resurgent current, 200 µmol/L β4 peptide was included in the internal solution. Recordings were acquired with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) at 50 kHz and filtered at 5 kHz. Leak subtraction was performed with a P/4 protocol. Fast and slow capacitive currents were compensated. Recordings began 10 min after achieving the whole‐cell configuration to allow for equilibrium. Series resistance was compensated 80–90%, and cells with unstable series resistance or series resistance >8 MOhms were discarded from further analysis.
Protocols for activation, inactivation, recovery from inactivation, and resurgent current are depicted in the figures. To measure residual current after high‐frequency pulses, cells were held at −120 mV and depolarized to 0 mV for 5 msec for 300 pulses at the indicated frequency. The residual current was calculated as the ratio of the 300th pulse to the 1st pulse. Resurgent current was assessed by first depolarizing the cell to +30 mV, followed by repolarization steps at the indicated voltages. To control for variable resurgent current related to the time‐dependent diffusion of the β4 peptide, cells expressing wild‐type and variant channel were alternated during the recording day, and each recording used similar pipette size and the same β4 internal solution. Persistent current was determined as the average current over two intervals (45–50 msec and 190–200 msec) recorded during a 200 msec depolarizing pulse to 0 mV and normalized to the peak current from a TTX‐subtracted record.
Data analysis
Analysis of electrophysiological data was performed with ClampFit (Molecular Devices), Microsoft Excel, MatLab (Mathworks, Natick, MA), and GraphPad Prism (San Diego, CA). All data points are represented as mean ± SEM, and n is the sample size. Current densities were calculated by dividing peak currents at each potential by the membrane capacitance obtained during recording. Conductance was calculated using the equation G = I*(V − E rev), where E rev is the calculated sodium reversal potential. Activation and inactivation V1/2 values were obtained by fitting the normalized conductance (activation) or the normalized current (inactivation) to a Boltzmann equation: 1/(1 + exp[(V − V 1/2)/k]), where V 1/2 is the half‐activation voltage and k is the slope factor. Inactivation time‐course was estimated by fitting a single‐component exponential curve to the decay portion of the sodium current using ClampFit. Recovery from inactivation was fit with a biexponential curve using Prism. The data distribution was assessed for normality with a Shapiro–Wilk test. If the data were normally distributed, a parametric test (t‐test) was used to assess significance; if not, a nonparametric test was used (Mann–Whitney U‐test). Statistical significance was set at P < 0.05. When assessing three or more groups (as in the carbamazepine experiment), the data were analyzed with a repeated measures ANOVA with Dunnett’s multiple comparisons post hoc test, comparing each drug response to the control condition.
Reagents
Tetrodotoxin (TTX; Tocris Bioscience, Minneapolis, MN) was prepared as a 1 mmol/L stock solution in water, and then diluted to 500 nmol/L in the external solution. Carbamazepine (Sigma) was prepared as a 250 mmol/L stock solution in DMSO and diluted to the indicated concentration in the external solution. A β4 peptide (KKLITFILKKTREK‐OH), generously supplied by Chris Ahern at the University of Iowa, was diluted in the internal solution to a final concentration of 200 µmol/L and used immediately or frozen in aliquots.
Molecular modeling
A structure of the C‐Terminal domain of human NaV1.2 determined by X‐ray crystallography at 3.02 Angstrom resolution in complex with calmodulin and FGF13 was obtained from chain B of PDB code 4JPZ. 11 The Met‐1879 residue was changed to Thr and separately the Arg‐1882 residue was changed to Gln in silico using UCSF Chimera (RBVI, San Francisco, CA), with the highest probability rotamer selected. The M1879T and R1882Q structures and the original structure (WT) were energy minimized using 1000 steepest descent and 10 conjugate gradient steps. Contacts of nonbackbone atoms with either Met‐1879/Arg‐1882 or Thr‐1879/Gln‐1882 were identified using Chimera with default parameters.
Results
Case report
An infant boy was evaluated for a seizure disorder accompanied by mild global developmental delay with onset at age 2 months. He had a normal birth history, and no family history of seizures or epilepsy. The child initially came to medical attention for an apneic event, which was diagnosed as reflux. EEG and brain MRI were performed which were normal. Events recurred and he was referred to Lurie Children’s Hospital and diagnosed with tonic seizures accompanied by apneic spells and cyanosis. Neurological exam was notable for low axial and appendicular muscle tone. He was treated with levetiracetam, pyridoxine, and topiramate, yet continued to have daily seizures. Following a loading dose of intravenous fosphenytoin he became seizure free for 2 days, but then seizures recurred. Transition to oral phenytoin maintained seizure freedom. Genetic testing revealed a de novo SCN2A variant (c.5636T > C) predicting a novel missense mutation (p.Met1879Thr, M1879T) in the NaV1.2 voltage‐gated sodium channel. On follow‐up, there had been difficulty maintaining adequate phenytoin levels, and he was transitioned to carbamazepine monotherapy with continued seizure control. At last follow‐up at age 2 years, he exhibited developmental delay and hyperactivity with autistic features. He had minimal receptive and expressive language, can feed himself, and can run with an ataxic gait.
Functional consequences of SCN2A‐M1879T
We investigated the functional effect of the M1879T mutation using recombinant human NaV1.2 expressed in a heterologous cell line (HEK293T) stably expressing the human β1 and β2 subunits. Representative voltage‐dependent currents recorded from WT and M1879T expressing cells are depicted in Figure 1A and summary data for all functional properties are presented in Table 1. The peak current density was not significantly different between WT and M1879T channels (Fig. 1B). However, the time‐course of inactivation was significantly slower for M1879T compared to WT channels (Fig. 1C, upper). A voltage ramp protocol evoked a pronounced aberrant current in cells expressing M1879T but not in WT expressing cells (Fig. 1C, lower). Inactivation time constants reveal a significant impairment of M1879T inactivation compared to WT across the range of tested voltages (Fig. 1D). M1879T mutant channels did not exhibit a significant difference in persistent sodium current measured as a percentage of peak current at either 50 or 200 msec (Table 1). These findings are consistent with impaired fast inactivation for the mutant channel that predicts greater sodium conductance over time following channel activation.
Figure 1.
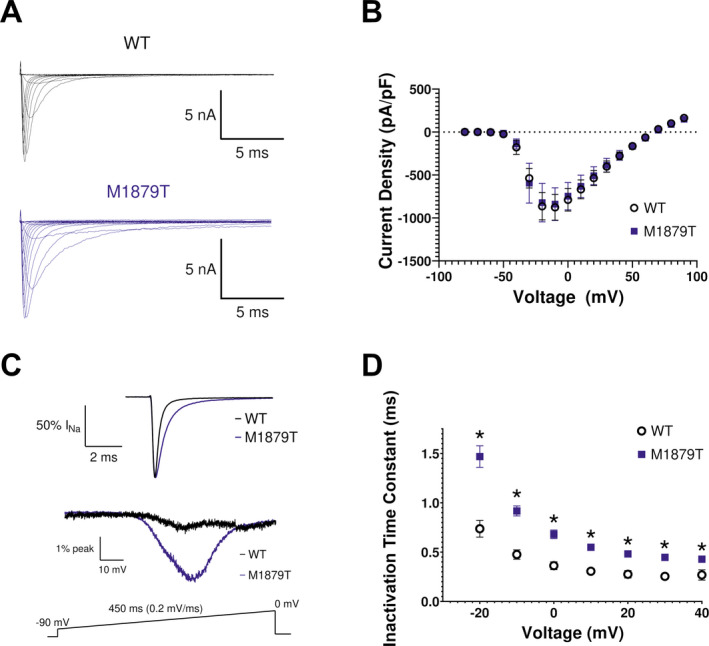
M1879T mutation alters NaV1.2 inactivation kinetics. (A) Representative current recordings from WT and M1879T NaV1.2 obtained using the voltage protocol shown in Figure 2A (inset). (B) Current density–voltage plot for WT (n = 18) and M1879T (n = 20) channels. (C) Top, average of six TTX‐subtracted recorded at 0 mV and normalized to peak current to highlight inactivation time‐course of WT (black) and M1879T (blue). Bottom, average currents for WT (black, average of 5) and M1879T (blue, average of 6) elicited using the voltage ramp protocol shown below. Traces were normalized to peak current measured at 0 mV. (D) Voltage dependence of inactivation time constants determined from single‐component exponential curve fits to the current decay. n = 14 for WT and n = 14 for M1879T, *P < 0.05 by Mann–Whitney U test.
Table 1.
Summary of functional properties for WT and M1879T channels.
| WT (n) | M1879T (n) | M1879T‐neonatal (n) | |
|---|---|---|---|
| Peak current density (pA/pF) | −972 ± 151 (18) | −908 ± 163 (20) | −654 ± 125 (12) |
| Inactivation time constant (τ, msec) 0 mV | 0.36 ± 0.04 (14) | 0.68 ± 0.04* (14) | 0.69 ± 0.05 † (12) |
| Activation voltage dependence | |||
| V 1/2 (mV) | −29.2 ± 1.7 (14) | −28.2 ± 1.8 (14) | −27.7 ± 1.2 † (14) |
| Slope factor (k) | 4.8 ± 0.5 (14) | 4.5 ± 0.4 (14) | 4.8 ± 0.4 † (14) |
| Inactivation voltage dependence | |||
| V 1/2 (mV) | −66.3 ± 1.1 (18) | −53.8 ± 1.1* (25) | −50.3 ± 1.4 † (15) |
| Slope factor (k) | −6.1 ± 0.2 (18) | −7.4 ± 0.3* (25) | −7.8 ± 0.3 † (15) |
| Recovery from inactivation | |||
| τ slow (msec) | 154 ± 88 (10) | 139 ± 35 (10) | 132 ± 28 † (11) |
| τ fast (msec) | 2.18 ± 0.30 (10) | 2.48 ± 0.32 (10) | 2.96 ± 0.44 † (11) |
| % fast component | 77.7 ± 1.8 (10) | 76.7 ± 4.4 (10) | 78.3 ± 3.1 † (11) |
| Persistent current (% of peak) | |||
| 50 msec | 0.47 ± 0.06 (9) | 0.40 ± 0.11 (9) | 0.44 ± 0.14 † (8) |
| 200 msec | 0.33 ± 0.05 (9) | 0.21 ± 0.04 (9) | 0.47 ± 0.11 † (8) |
P < 0.05 by Mann–Whitney U test.
P > 0.05 between adult and neonatal isoforms.
We compared the voltage dependence of channel activation and steady‐state inactivation between M1879T and WT channels. The voltage dependence of activation was not different between M1879T and WT channels (Fig. 2A). In contrast, cells expressing M1879T exhibited a significantly depolarized steady‐state inactivation curve compared to WT expressing cells (Fig. 2B). These results are also consistent with altered fast inactivation and predict greater sodium conductance by mutant channels evoked by membrane depolarizations within the physiological range.
Figure 2.
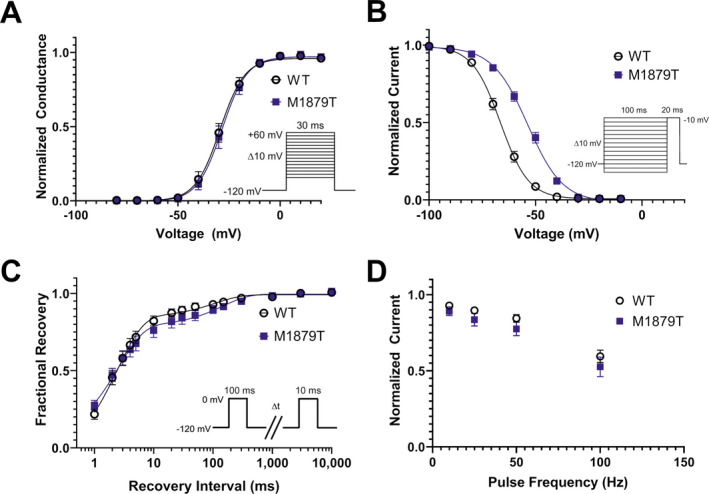
M1879T affects voltage dependence of inactivation. (A) Voltage dependence of activation of WT (n = 14) and M1879T (n = 14) channels determined using the voltage protocol shown as an inset. There was no difference in activation V 1/2 between WT and M1879T (P = 0.51 by Mann–Whitney U test). (B) Voltage dependence of inactivation of WT (n = 18) and M1879T (n = 25) channels (voltage protocol shown as an inset). There was a significant depolarizing shift in inactivation V 1/2 (P < 0.0001 by Mann–Whitney U test). (C) Time course of recovery from inactivation after 100 msec depolarization (protocol shown as inset) comparing WT (n = 10) and M1879T (n = 10). There were no significant differences in time constants for recovery from inactivation between WT and M1879T (P > 0.05 by Mann–Whitney U test; Table 1). (D) Plot of residual current (comparing 300th pulse to 1st pulse) after repetitive pulsing to 0 mV at the indicated frequency (n = 7 for WT and n = 7 for M1879T). There were no significant differences between WT and M1879T at any frequency (P > 0.05 by Mann–Whitney U test).
Recovery from inactivation, assessed using a two‐pulse protocol, followed a biexponential time course and was not significantly different between M1879T and WT channels (Fig. 2C). Similarly, sodium channel availability during depolarizing pulse trains at different frequencies (10, 25, 50, and 100 Hz) were not significantly different between mutant and WT channels (Fig. 2D).
SCN2A undergoes developmentally regulated alternative mRNA splicing that leads to incorporation of an alternate exon encoding a portion of the domain I voltage‐sensor domain (S3 and S4 helices). Because SCN2A variants associated with early onset epilepsy may exhibit more severe functional consequences in the splice variant expressed preferentially during early development, 12 we tested whether the substitution of the neonatal exon would affect the properties of the M1879T mutant channel. In this case, none of the key functional properties (Table 1, Fig. S1) were significantly different between the adult and neonatal isoforms of the M1879T channel.
M1879T channels exhibit enhanced resurgent current
We next compared the level of resurgent current between WT and M1879T channels. These experiments were motivated by a prior report indicating that the epilepsy‐associated mutation SCN2A‐R1882Q affecting a nearby residue had greater resurgent current than WT channels. 13 We recorded resurgent current in WT and M1879T expressing cells elicited by the voltage protocol shown in Figure 3A (top) with the β4 peptide in the internal solution. M1879T channels (Fig. 3A, bottom) exhibited larger resurgent currents than WT channels (Fig. 3A, middle). This difference was evident across several tested voltages with comparisons made using either current density (Fig. 3B) or the level of resurgent current measured as a percentage of the peak current elicited at −10 mV (Fig. 3C). We further analyzed resurgent current for WT and M1879T channels at three different recording time‐points after establishing the whole cell configuration (11, 18, and 26 min). At each time‐point, the maximum resurgent current of M1879T channels, measured as a percentage of peak current, was significantly greater than WT channels (11 min: WT 3.5 ± 0.4 % vs. M1879T 8.4 ± 1.0 %; 18 min: WT 4.7 ± 0.7 % vs. M1879T 10.1 ± 1.2 %; 26 min: WT 5.4 ± 0.8 % vs. M1879T 13.1 ± 2.0 %; P < 0.05 for each time point; see Fig. S2). Because the resurgent current amplitude exhibited by WT channels is small, we could not accurately determine differences in its voltage dependence compared to M1879T channels. Enhanced resurgent current exhibited by M1879T channels may contribute to the gain of sodium conductance conferred by this mutation.
Figure 3.
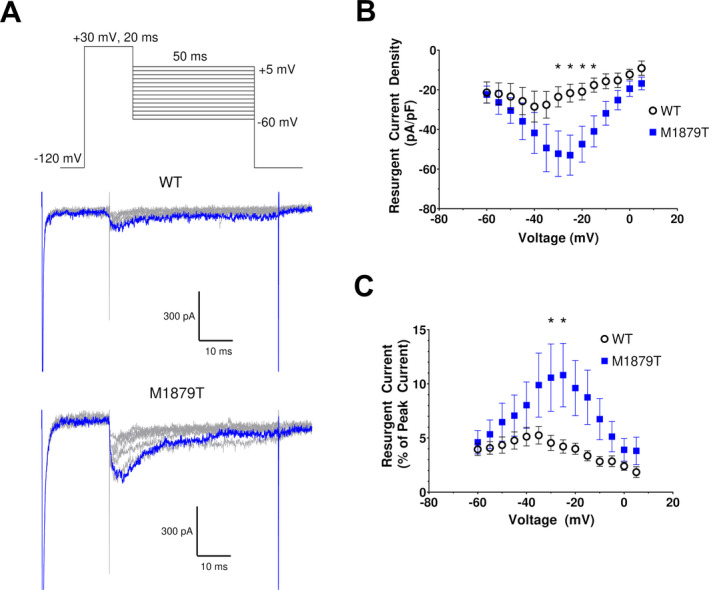
M1879T exhibits enhanced resurgent current. (A) Top, voltage protocol for eliciting resurgent current. Middle (WT) and bottom (M1879T) panels show representative current traces using protocol above (10 mV voltage steps are illustrated). The blue trace represents the peak resurgent current elicited at −20 mV. (B) Voltage dependence of resurgent current density for WT (n = 7) and M1879T (n = 9) (*P < 0.05 by Mann–Whitney U test). (C) Plot of resurgent current expressed as % of peak transient current for WT and M1879T (*P < 0.05 by Mann–Whitney U test).
Effect of carbamazepine on M1879T channels
Because carbamazepine (CBZ) was effective as monotherapy in the reported case, we examined the effects of this drug on M1879T channels (adult splice isoform). As shown in Figure 4A, application of 100 and 300 µmol/L CBZ caused a dose‐dependent hyperpolarizing shift in steady‐state inactivation (control [no drug]: V 1/2 = −48.5; 100 μmol/L CBZ: V 1/2 = −52.0; 300 μmol/L CBZ: V 1/2 = −55.8; n = 6). By comparing each treated cell to its own control value, we determined that 100 and 300 μmol/L CBZ caused 3.5 ± 0.7 mV and 8.5 ± 0.7 mV hyperpolarizing shifts in steady‐state inactivation V 1/2, respectively (Fig. 4B). Vehicle (DMSO) treatment did not cause a significant shift in steady‐state inactivation (data not shown). Additionally, CBZ treatment was associated with a significantly lower degree of channel availability after high‐frequency pulses at 10 Hz (control: 0.78 ± 0.05, 100 μmol/L CBZ: 0.61 ± 0.06, 300 μmol/L CBZ: 0.43 ± 0.06), 25 Hz (control: 0.56 ± 0.06, 100 μmol/L CBZ: 0.36 ± 0.07, 300 μmol/L CBZ: 0.18 ± 0.05), and 50 Hz (control: 0.34 ± 0.05, 100 μmol/L CBZ 0.16 ± 0.04, 300 μmol/L CBZ 0.04 ± 0.03 fractional availability; n = 6 for each frequency) (Fig. 4C). The average time course of residual current at each frequency is depicted in Figure 4D. Similar results were seen with CBZ treatment of WT channels (Fig. S3), such that CBZ treatment resulted in a concentration‐dependent hyperpolarizing shift in steady‐state inactivation (100 μmol/L CBZ: −4.2 ± 0.4 mV; 300 μmol/L CBZ: −9.3 ± 0.4 mV; n = 5) and reduction in availability after high‐frequency pulses (significant difference seen at 25 Hz [control: 0.55 ± 0.08; 100 μmol/L CBZ: 0.44 ± 0.07; 300 μmol/L CBZ: 0.28 ± 0.07] and 50 Hz [control: 0.39 ± 0.09; 100 μmol/L CBZ: 0.21 ± 0.06; 300 μmol/L CBZ: 0.09 ± 0.04; n = 5 for each frequency]). The partial normalization of voltage dependence of inactivation coupled with use‐dependent block of the M1879T channel correlate with the clinical efficacy of CBZ in the reported case.
Figure 4.
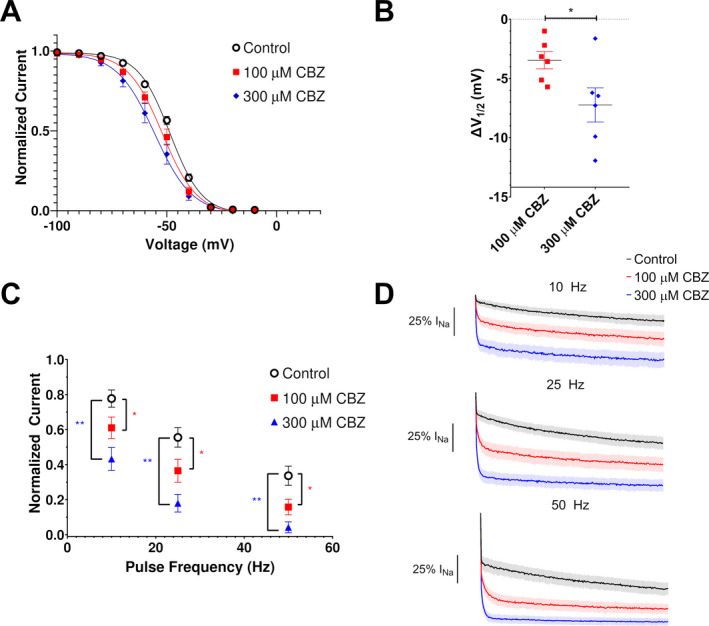
Effects of carbamazepine on M1879T channels. (A) Carbamazepine (CBZ) induces a hyperpolarizing shift of the voltage dependence of inactivation (n = 6). (B) Plot of change in inactivation V 1/2 in response to 100 and 300 µmol/L carbamazepine (n = 6; *P < 0.05 by paired t‐test). (C) Plot of residual current (comparing 300th pulse to 1st pulse) after 0 mV pulses at the indicated frequencies from a holding potential of −90 mV. There was a significant difference in residual current at 100 μmol/L (*) and 300 μmol/L (**) at 10, 25, and 50 Hz compared to the control value (P < 0.05, one‐way ANOVA with repeated measures and Dunnett’s post hoc test, n = 6). (D) Average time‐course of CBZ treated cells at 10 Hz (top), 25 Hz (middle), and 50 Hz (bottom).
We also assessed whether CBZ treatment affects resurgent current. Cells expressing M1879T were treated with either 300 µmol/L CBZ or DMSO vehicle. Cells treated with DMSO only (Fig. 5A, left) exhibited a time‐dependent rise in resurgent current, likely related to the time‐dependent diffusion of the β4 peptide into the intracellular compartment seen in control solution. In contrast, cells treated with CBZ showed lower resurgent current levels compared to the control condition (Fig. 5A and B and Fig. S4). Specifically, DMSO‐treated cells exhibited a 22.1 ± 4.6% (n = 7) rise in maximal resurgent current density compared to control, while CBZ‐treated cells had a 21.2 ± 3.1% (n = 7) fall compared to the control condition (Fig. 5C). Because CBZ affects peak current, we considered the possibility that reduced resurgent current merely reflects a proportional reduction in the total current. Indeed, 300 µmol/L CBZ treatment caused a significant change in peak current compared to DMSO treatment (Fig. 6A). When this difference was taken into account by examining the maximal resurgent current as a percentage of the peak current, there remained a significant difference between the DMSO‐treated and CBZ‐treated cells (Fig. 6B). Taken together, these data indicate that 300 μmol/L CBZ likely attenuates resurgent current independent of its effect on peak current, but this effect is modest.
Figure 5.
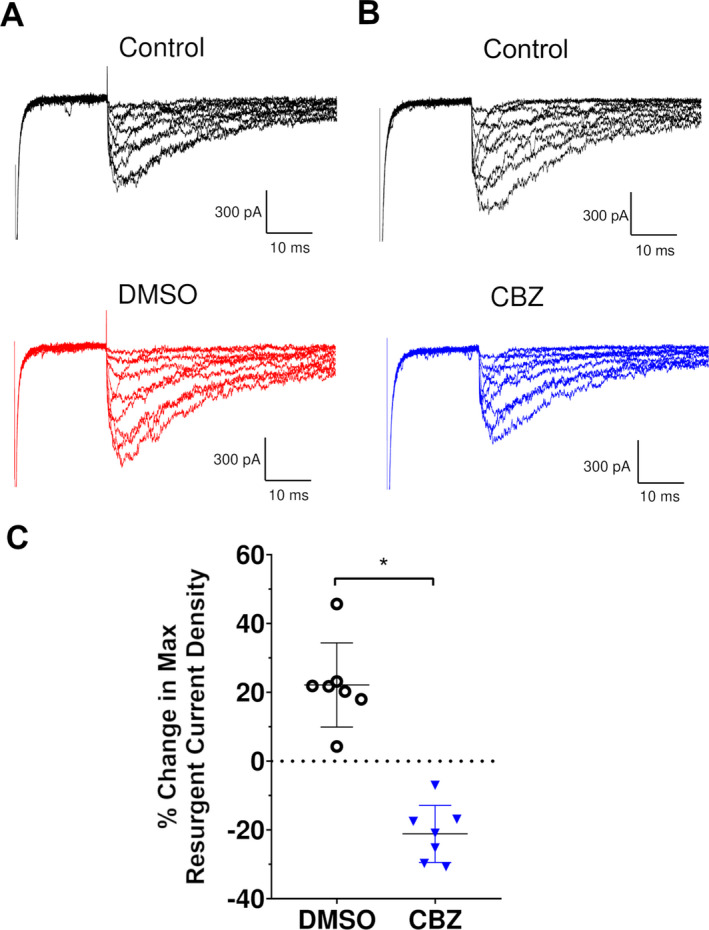
Effect of carbamazepine on resurgent current. (A) Representative M1879T resurgent current traces before (top) and after DMSO (bottom) treatment. (B) Representative M1879T resurgent current before (top) and after CBZ (bottom) treatment. (C) Percent change in maximum resurgent current density, comparing DMSO and CBZ (n = 7; *P < 0.0001 by unpaired t test).
Figure 6.
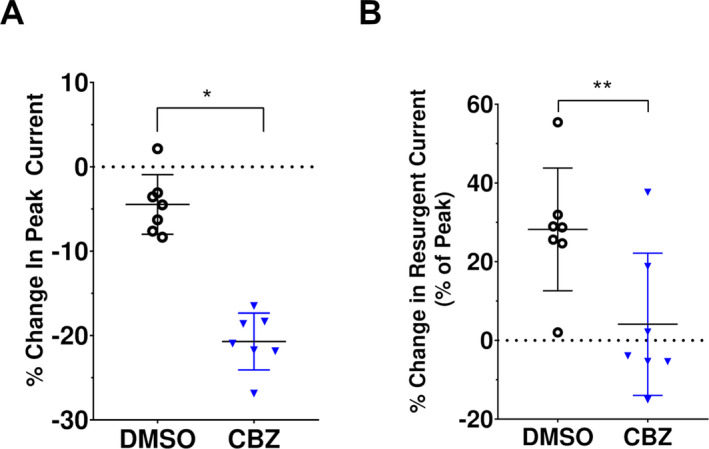
Comparison of carbamazepine effects on M1879T peak transient and resurgent current. (A) Percent change in peak transient current (DMSO: −4.5 ± 1.3% vs. CBZ: −20.7 ± 1.3%; n = 7 both groups; *P < 0.0001 by unpaired t test). (B) Percent change in resurgent current (as percent of peak transient current; DMSO: 28.2 ± 5.9% vs. CBZ: 4.1 ± 6.8% of peak, n = 7 for both groups; **P = 0.02 by unpaired t test).
Predicted structural effects of M1879T mutation
To explore possible mechanisms to explain how substitution of threonine for Met‐1879 affects inactivation, we modeled the M1879T variant in silico using the isolated human NaV1.2 C‐terminal domain (CTD) structure (PDB code 4JPZ, see Methods) and assessed its effect on local residue interactions. Detailed interactions are presented in Tables S1 and S2. In this structure, the side chain of Met‐1879 projects toward a nearby helix in the CTD where it forms contacts with a network of side chains belonging to nearby residues (Fig. 7A). Molecular modeling suggests that the M1879T variant, with its smaller side chain, eliminates several of these intramolecular contacts (Fig. 7B). We also modeled the R1882Q variant, because this residue located downstream of Met‐1879 has similar effects on inactivation and resurgent current. 13 , 14 Arg‐1882 forms multiple contacts with Glu‐1792 in the proximal CTD (Fig. 7C), which are eliminated in the R1882Q variant (Fig. 7D).
Figure 7.
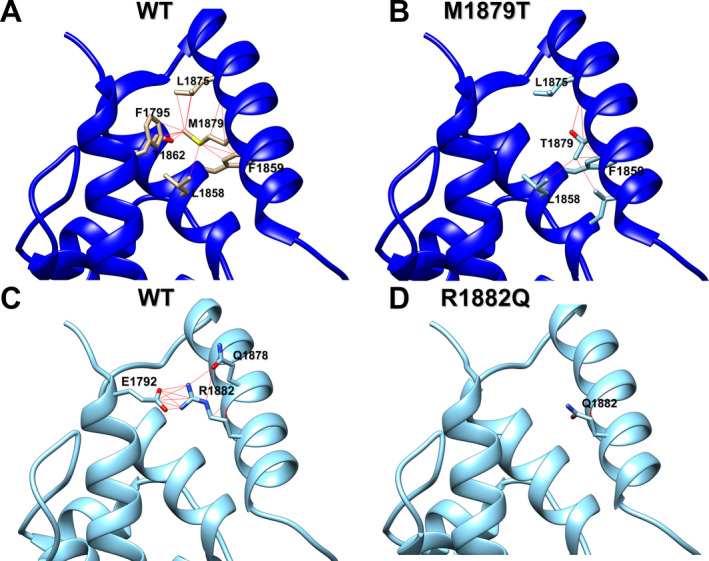
Molecular modeling of M1879T and R1882Q channel contacts in NaV1.2 C‐terminal domain compared to WT. (A) Ribbon diagram of WT channel showing specific contacts between Met‐1879 and interacting residues (red lines). (B) Ribbon diagram of M1879T channel showing specific contacts between Thr‐1879 and interacting residues (red lines). (C) Ribbon diagram of WT channel showing specific contacts between Arg‐1882 and interacting residues (red lines). (D) Ribbon diagram of R1882Q channel showing specific contacts between Gln‐1882 and interacting residues (red lines). See Tables S1 and S2 for list of atomic contacts.
Discussion
Our study investigated a de novo SCN2A coding sequence variant (p.M1879T) associated with infantile‐onset epilepsy. The child we report exhibited frequent tonic seizures complicated by apnea unresponsive to levetiracetam and topiramate who subsequently had a dramatic response to the sodium channel blockers phenytoin and later carbamazepine. Initially the variant was categorized as a variant of uncertain significance (VUS), but electrophysiological evaluation of the variant sodium channel revealed severely impaired inactivation gating consistent with a gain‐of‐function. Specifically, we observed a substantially depolarized voltage dependence of inactivation, slower time course of inactivation, and enhanced resurgent current. Unlike other SCN2A variants associated with early‐onset epilepsy, expression of the variant in the neonatal splice variant of the channel was not associated with greater dysfunction. 12 Carbamazepine partially normalized the voltage‐dependent inactivation of the mutant channel, dampened resurgent current, and exhibited potent frequency‐dependent block of the mutant channel. Therefore, our study demonstrates concordance of in vitro pharmacology using heterologous cells with the drug response observed clinically for the affected child.
The novel missense mutation we report in this paper affects residue Met‐1879 located in the distal portion of the intracellular carboxyl‐terminal domain (CTD), upstream of a conserved IQ domain necessary for calmodulin binding. 15 , 16 , 17 This methionine residue is absolutely conserved in the mammalian voltage‐gated sodium channel family. Variants affecting orthologous sodium channels predicting the same methionine to threonine amino acid substitution occur at corresponding positions in the cardiac sodium channel (NaV1.5, M1875T) associated with familial atrial fibrillation, 18 and in a peripheral nerve sodium channel (NaV1.7, M1870T) associated with painful diabetic peripheral neuropathy. 19 Both orthologous mutations exhibited similar biophysical effects as SCN2A‐M1879T in heterologous cells including a depolarizing shift in the voltage‐dependent inactivation and slowed inactivation kinetics without affecting voltage‐dependent activation or persistent current. These results along with our findings implicate Met‐1879 as important for inactivation properties, and mutation to threonine appears to destabilize the inactivated state of the channel.
Other epilepsy‐associated mutations have been identified in the NaV1.2 CTD, most notably R1882Q, which causes early‐onset epilepsy of varying severity and drug responsiveness. The R1882Q channel exhibits a depolarized voltage dependence of inactivation, as well as an increase in noninactivating persistent current compared to wild‐type channels, an attribute we did not observe for the M1879T channel. This combination of functional anomalies is predicted to promote greater neuronal excitability. 14
The molecular basis for channel dysfunction associated with the M1879T mutation can be considered in the context of recent advances in the structural biology of sodium channels. In the past few years, there has been an explosion of available structures for eukaryotic sodium channels, including human NaV1.2. 20 Unfortunately, the intracellular regions including the CTD are poorly resolved or missing in most of these structures except in the full‐length sodium channel from the American cockroach (NaVPas). 21 Fortunately, there are structural data available for the isolated NaV1.2 CTD in complex with calmodulin and FGF13. 11 In this structure, Met‐1879 resides within an alpha‐helix that forms a helix‐loop‐helix motif neighboring the IQ domain needed for calmodulin binding. The proximity of this residue to the base of the fourth voltage‐sensing domain provides a plausible explanation for how the M1879T mutation might affect inactivation driven by this domain. 22 In this study, molecular modeling of the M1879T variant demonstrates the loss of several side‐chain to side‐chain contacts (Thr‐1862, Phe‐1795, Leu‐1875) with nearby helices, an intramolecular interaction network that may work to modulate inactivation in the WT channel. The R1882Q variant, which has similar effects on inactivation and resurgent current as M1879T, loses all contacts with Glu‐1792 in the proximal CTD, a residue that extends intracellularly from the S6 helix of domain IV. Glu‐1792 maps to Glu‐1783 in the orthologous NaV1.6 channel encoded by SCN8A, for which the epilepsy‐associated variant R1872Q (analogous to R1882Q in NaV1.2) has been functionally studied. 23 Although the interaction network of Met‐1879 and Arg‐1882 does not directly overlap, Glu‐1792 is located one helical turn amino‐terminal to Phe‐1795, a residue forming contacts with Met‐1879. The similar effect on inactivation found in these two variants implies a role for contact stabilization between the two alpha‐helices containing these residues (i.e., Phe‐1795/Glu‐1792 and Met‐1879/Arg‐1882) in regulating inactivation.
An understudied functional effect of SCN2A mutations is resurgent current, and only two variants (R853Q, R1882Q) have been investigated previously for this phenomenon in heterologous cells. 13 R853Q channels exhibit less resurgent current compared to WT channels, whereas R1882Q has a much larger resurgent current compared to WT, similar to what we observed with M1879T. However, the impact of resurgent current on promoting epileptic activity remains unclear. Resurgent currents have been demonstrated for other sodium channelopathies, including those implicated in pain, myotonia, long‐QT syndrome, and SCN8A‐related epilepsy. 24 , 25 , 26
We examined the effect of carbamazepine on resurgent current to determine if suppression of this anomalous channel activity might explain the antiepileptic effects observed in this patient. Although high CBZ concentration (300 μmol/L) did significantly suppress resurgent current, the reduction we observed correlated with an overall reduction in peak current. When resurgent current was normalized to peak current, the effect of carbamazepine appeared modest. These experimental findings suggested that the principal effect of carbamazepine on reducing seizures associated with the M1879T variant more likely relates to changes in voltage‐dependent inactivation and channel inhibition at high‐frequency stimulation rather than attenuation of resurgent current.
Suppression of resurgent current by other agents has also been demonstrated. The endogenous cannabinoid anandamide inhibits resurgent current without affecting transient current, 27 whereas PRX‐330, a novel anti‐epileptic drug candidate, inhibits both persistent and resurgent currents in a mouse model of SCN8A encephalopathy. 28 Cannabidiol (CBD), a recently approved anti‐epileptic drug for the treatment of Dravet and Lennox‐Gastaut syndromes, also inhibits resurgent current more than transient current associated with two epilepsy‐associated SCN8A variants (L1331V, N1768D). 25 Further investigations of resurgent current pharmacology are warranted.
In summary, our study illustrates that functional and pharmacological investigations of SCN2A variants in heterologous cells has value in defining genotype–phenotype correlations, demonstrating molecular mechanisms of disease and drug responses, and contributing to our knowledge of structure–functional relationships for the broader voltage‐gated sodium channel family.
Conflict of Interest
Adney, DeKeyser, Abramova, and Thompson declare no conflicts of interest with the work described herein. Millichap reports personal fees from American Academy of Neurology, personal fees from Up‐To‐Date, grants from UCB Pharma, grants and personal fees from Mallinkrodt, personal fees from Esai, grants and personal fees from Xenon, personal fees from Biomarin, personal fees from Ionis, personal fees from Greenwich, personal fees from Sunovion, personal fees from Upsher‐Smith, grants from NIH, grants from Citizens United for Research in Epilepsy, personal fees from Praxis, outside the submitted work. George reports personal fees from Amgen, Inc., grants from Praxis Precision Medicines, Inc., outside the submitted work.
Authors’ Contributions
The study was conceived by SKA, JMM, and ALG. Experiments were designed by SKA and ALG with guidance from CHT. Experiments were performed and data analyzed by SKA. JMD, CHT, and TA performed mutagenesis and development of cell lines. SKA and ALG wrote the manuscript with input from all co‐authors.
Supporting information
Figure S1. Functional properties of M1879T channels in either the NaV1.2 neonatal or adult splice variant.
Figure S2. Time dependence of resurgent current in WT and M1879T NaV1.2 channels.
Figure S3. Effects of carbamazepine on WT NaV1.2 channels.
Figure S4. Effect of carbamazepine on resurgent current.
Table S1. Atomic contacts of Met‐1879 from WT and Thr‐1879 from M1879T model.
Table S2. Atomic contacts of Arg‐1882 from WT and Gln‐1882 from R1882Q model.
Acknowledgments
The authors sincerely thank the patient and his family for their inclusion in this study. This work was supported by National Institute of Neurological Disorders and Stroke grants R25‐NS070695 (S.K.A.) and U54‐NS108874 (A.L.G.), and a generous gift from the Davee Foundation (A.L.G.).
Funding Information
This work was supported by National Institute of Neurological Disorders and Stroke grants R25‐NS070695 (S.K.A.) and U54‐NS108874 (A.L.G.), and a generous gift from the Davee Foundation (A.L.G.).
Funding Statement
This work was funded by National Institute of Neurological Disorders and Stroke grants R25‐NS070695 and U54‐NS108874; Davee Foundation grant .
References
- 1. Brunklaus A, Du J, Steckler F, et al. Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 2020;61:387–399. 10.1111/epi.16438 [DOI] [PubMed] [Google Scholar]
- 2. Reynolds C, King MD, Gorman KM. The phenotypic spectrum of SCN2A‐related epilepsy. Eur J Paediatr Neurol 2020;24:117–122. 10.1016/j.ejpn.2019.12.016 [DOI] [PubMed] [Google Scholar]
- 3. Musto E, Gardella E, Møller RS. Recent advances in treatment of epilepsy‐related sodium channelopathies. Eur J Paediatr Neurol 2020;24:123–128. 10.1016/j.ejpn.2019.12.009 [DOI] [PubMed] [Google Scholar]
- 4. Ben‐Shalom R, Keeshen CM, Berrios KN, et al. Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol Psychiatry 2017;82:224–232. 10.1016/j.biopsych.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Symonds JD, McTague A. Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur J Paediatr Neurol 2020;24:15–23. 10.1016/j.ejpn.2019.12.008 [DOI] [PubMed] [Google Scholar]
- 6. Hu W, Tian C, Li T, et al. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci 2009;12:996–1002. 10.1038/nn.2359 [DOI] [PubMed] [Google Scholar]
- 7. Tian C, Wang K, Ke W, et al. Molecular identity of axonal sodium channels in human cortical pyramidal cells. Front Cell Neurosci 2014;8 10.3389/fncel.2014.00297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liao Y, Deprez L, Maljevic S, et al. Molecular correlates of age‐dependent seizures in an inherited neonatal‐infantile epilepsy. Brain 2010;133(Pt 5):1403–1414. 10.1093/brain/awq057 [DOI] [PubMed] [Google Scholar]
- 9. Sanders SJ, Campbell AJ, Cottrell JR, et al. Progress in understanding and treating SCN2A‐mediated disorders. Trends Neurosci 2018;41:442–456. 10.1016/j.tins.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kahlig KM, Saridey SK, Kaja A, et al. Multiplexed transposon‐mediated stable gene transfer in human cells. Proc Natl Acad Sci USA 2010;107:1343–1348. 10.1073/pnas.0910383107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang C, Chung BC, Yan H, et al. Structural analyses of Ca2+/CaM interaction with NaV channel C‐termini reveal mechanisms of calcium‐dependent regulation. Nat Commun 2014;5:4896 10.1038/ncomms5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thompson CH, Ben‐Shalom R, Bender KJ, George AL. Alternative splicing potentiates dysfunction of early‐onset epileptic encephalopathy SCN2A variants. J Gen Physiol 2020;152(3):e201912442 10.1085/jgp.201912442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mason ER, Wu F, Patel RR, et al. Resurgent and gating pore currents induced by de novo SCN2A epilepsy mutations. eNeuro 2019;6(5). 10.1523/ENEURO.0141-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berecki G, Howell KB, Deerasooriya YH, et al. Dynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy. Proc Natl Acad Sci USA 2018;115:E5516–E5525. 10.1073/pnas.1800077115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mori M, Konno T, Ozawa T, et al. Novel interaction of the voltage‐dependent sodium channel (VDSC) with calmodulin: Does VDSC acquire calmodulin‐mediated Ca2+‐sensitivity? Biochemistry 2000;39:1316–1323. 10.1021/bi9912600 [DOI] [PubMed] [Google Scholar]
- 16. Kim J, Ghosh S, Liu H, et al. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem 2004;279:45004–45012. 10.1074/jbc.M407286200 [DOI] [PubMed] [Google Scholar]
- 17. Theoharis NT, Sorensen BR, Theisen‐Toupal J, Shea MA. The neuronal voltage‐dependent sodium channel type II IQ motif lowers the calcium affinity of the C‐domain of calmodulin. Biochemistry 2008;47:112–123. 10.1021/bi7013129 [DOI] [PubMed] [Google Scholar]
- 18. Makiyama T, Akao M, Shizuta S, et al. A novel SCN5A Gain‐of‐function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol 2008;52:1326–1334. 10.1016/j.jacc.2008.07.013 [DOI] [PubMed] [Google Scholar]
- 19. Blesneac I, Themistocleous AC, Fratter C, et al. Rare NaV1.7 variants associated with painful diabetic peripheral neuropathy. Pain 2018;159:469–480. 10.1097/j.pain.0000000000001116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan X, Li Z, Huang X, et al. Molecular basis for pore blockade of human Na+ channel Nav1.2 by the μ‐conotoxin KIIIA. Science 2019;363:1309–1313. 10.1126/science.aaw2999 [DOI] [PubMed] [Google Scholar]
- 21. Shen H, Zhou Q, Pan X, et al. Structure of a eukaryotic voltage‐gated sodium channel at near‐atomic resolution. Science 2017;355:eaal4326 10.1126/science.aal4326 [DOI] [PubMed] [Google Scholar]
- 22. Capes DL, Goldschen‐Ohm MP, Arcisio‐Miranda M, et al. Domain IV voltage‐sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J Gen Physiol 2013;142:101–112. 10.1085/jgp.201310998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pan Y, Cummins TR. Distinct functional alterations in SCN8A epilepsy mutant channels. J Physiol 2020;598:381–401. 10.1113/JP278952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jarecki BW, Piekarz AD, Jackson JO, Cummins TR. Human voltage‐gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest 2010;120:369–378. 10.1172/JCI40801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patel RR, Barbosa C, Brustovetsky T, et al. Aberrant epilepsy‐associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 2016;139:2164–2181. 10.1093/brain/aww129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiao Y, Barbosa C, Pei Z, et al. Increased resurgent sodium currents in Nav1.8 contribute to nociceptive sensory neuron hyperexcitability associated with peripheral neuropathies. J Neurosci 2019;39:1539–1550. 10.1523/JNEUROSCI.0468-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Theile JW, Cummins TR. Inhibition of Navβ4 peptide‐mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol Pharmacol 2011;80:724–734 10.1124/mol.111.072751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wengert ER, Saga AU, Panchal PS, et al. Prax330 reduces persistent and resurgent sodium channel currents and neuronal hyperexcitability of subiculum neurons in a mouse model of SCN8A epileptic encephalopathy. Neuropharmacology 2019;158:107699 10.1016/j.neuropharm.2019.107699 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Functional properties of M1879T channels in either the NaV1.2 neonatal or adult splice variant.
Figure S2. Time dependence of resurgent current in WT and M1879T NaV1.2 channels.
Figure S3. Effects of carbamazepine on WT NaV1.2 channels.
Figure S4. Effect of carbamazepine on resurgent current.
Table S1. Atomic contacts of Met‐1879 from WT and Thr‐1879 from M1879T model.
Table S2. Atomic contacts of Arg‐1882 from WT and Gln‐1882 from R1882Q model.