

Abstract
Objective
Neuroinflammation is considered a key driver for neurodegeneration in several neurological diseases, including amyotrophic lateral sclerosis (ALS). SOD1 mutations cause about 20% of familial ALS, and related pathology might generate microglial activation triggering neurodegeneration. 11C‐PK11195 is the prototypical and most validated PET radiotracer, targeting the 18‐kDa translocator protein which is overexpressed in activated microglia. In this study, we investigated microglia activation in asymptomatic (ASYM) and symptomatic (SYM) SOD1 mutated carriers, by using 11C‐PK11195 and PET imaging.
Methods
We included 20 subjects: 4 ASYM‐carriers, neurologically normal, 6 SYM‐carriers with probable ALS, and 10 healthy controls. A receptor parametric mapping procedure estimated 11C‐PK11195 binding potentials and voxel‐wise statistical comparisons were performed at group and single‐subject levels.
Results
Both the SYM‐ and ASYM‐carriers showed significant microglia activation in cortical and subcortical structures, with variable patterns at individual level. Clusters of activation were present in occipital and temporal regions, cerebellum, thalamus, and medulla oblongata. Notably, SYM‐carriers showed microglia activation also in supplementary and primary motor cortices and in the somatosensory regions.
Interpretation
In vivo neuroinflammation occurred in all SOD1 mutated cases since the presymptomatic stages, as shown by a significant cortical and subcortical microglia activation. The involvement of sensorimotor cortex became evident at the symptomatic disease stage. Although our data indicate the role of in vivo PET imaging for assessing resident microglia in the investigation of SOD1‐ALS pathophysiology, further studies are needed to clarify the temporal and spatial dynamics of microglia activation and its relationship with neurodegeneration.
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult‐onset devastating neurodegenerative disease characterized by progressive loss of motor neurons in the cerebral cortex, brainstem, and spinal cord. ALS leads to progressive paralysis, muscle atrophy, and death from respiratory failure, typically within 3–5 years after symptoms onset. 1 , 2 Cognitive and behavioral disturbances due to frontal dysfunction, involving about 50% of ALS patients 3 , 4 can be associated with motor impairment. Currently, no treatment is available to stop or reverse ALS, and the identification of reliable biomarkers for presymptomatic diagnosis as well as for monitoring disease progression is of utmost importance, favoring the understanding of pathophysiology and the development and evaluation of potential disease‐modifying drugs. 5
Although the majority of ALS cases are sporadic (sALS), about 5–10% are familial (fALS) and are caused by specific pathogenic mutations, mostly with dominant inheritance. 6 , 7 , 8 Since the discovery of the first mutations in superoxide dismutase gene (SOD1), 9 more than 25 ALS causative genes and more than 100 gene mutations increasing susceptibility or influencing ALS development were identified. 10 , 11 , 12 In addition to SOD1, mutations in the TAR DNA‐binding protein (TARDBP) gene, fused in sarcoma (FUS), C9ORF72, Ubiquilin2 (UBQLN2), CCNF, TIA1, and TBK1 make up the majority of fALS cases. 13 , 14 Similar genetic alterations are present in the 5–10% of the sporadic cases. Thus, a genetic component may play a causative role in sporadic cases as well, making the distinction between fALS and sALS not straightforward. 15 The penetrance of many ALS genes is age dependent and a large variability in clinical manifestations and prognosis is observed in fALS 16 , 17 Recent evidence suggests that in the multistep pathogenic process which leads to the development of ALS, genetic background is dynamically interacting with specific environmental factors, reducing the number of steps necessary to start neurodegeneration and favoring disease onset. 18
An important role in ALS is played by SOD1 mutations, accounting for 20% of fALS and 1‐2% of sALS. 10 , 12 , 19 , 20 At present, more than 180 SOD1 mutations have been identified. ALS due to SOD1 mutations (SOD1‐ALS) is typically autosomal‐dominant inherited and shows an age‐dependent penetrance, reaching 90% by the age of seventy. 21 SOD1‐mutated symptomatic cases usually show a spinal phenotype, with a more likely lower than upper limb onset. Bulbar ALS is less common in SOD1‐ALS and cognitive impairment usually is not present. 22 , 23 , 24 Overall, SOD1‐ALS clinical phenotypes can be heterogeneous in regard to age of onset, severity and prognosis, which could be modulated by other determinants such as central nervous system dynamics, as well as by environmental factors. 25
How SOD1 pathogenic mutations trigger neurodegeneration is still a matter of debate. SOD1 is an antioxidant enzyme which binds copper and zinc ions catalyzing the deactivation of toxic superoxide radicals. 26 Consequently, detrimental mutations in SOD1 gene modify the protein activity, leading to accumulation of toxic hydroxyl radicals. 27 Oxidative stress is considered one of the potential pathogenic mechanisms, but misfolding protein aggregation, mitochondrial dysfunction, glutamate excitotoxicity, inflammation, and microglia activation have also been hypothesized to play a role. 13 , 20 , 28
Microglia activation, a proxy of neuroinflammation, seems to play a key role in the pathogenic mechanisms leading to neurodegeneration in several disorders, including ALS. 29 , 30 ALS post mortem studies have shown that neurodegeneration of both upper and lower motor neurons is associated with activation of neuroimmune cells, including microglia, astrocytes, and oligodendrocytes. 31 , 32 At the neuropathological level, microglia activation is increased in motor cortex, brainstem, and spinal cord, and the extent of activation seems to be associated with disease severity. 33
SOD1 mutations are considered as a potent trigger of microglia cells, and deleterious effects of microglial activation have been reported in SOD1‐ALS animal models. 34
In vivo PET imaging represents a useful tool to quantify and localize microglial activity and possibly to assess response to novel therapeutic interventions in neurodegeneration. 11C‐PK11195 is the first, prototypical PET tracer for the in vivo detection of the 18‐kDa translocator protein (TSPO), an outer mitochondrial membrane protein which is overexpressed in activated microglia and reactive astrocytes, thus becoming a sensitive marker of neuroinflammation. 35 A TSPO overexpression as detected by 11C‐PK11195 PET has been reported in several neurodegenerative disorders, such as Alzheimer’s disease and other neurodegenerative dementias, prion diseases, Parkinson’s disease, as well as in sALS. 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 In addition, 11C‐PK11195 PET studies showed microglia activation also in preclinical stages of neurodegenerative conditions, 43 , 46 , 47 shedding light on the possible early molecular mechanisms of neurodegeneration.
At present, no evidence exists on in vivo measures of microglia activation in SOD1‐ALS patients or in asymptomatic carriers of the mutation. The present work aimed to assess by 11C‐PK11195 PET imaging the brain TSPO expression in SOD1‐mutated cases, either in the symptomatic (clinically affected) or asymptomatic (unaffected cases) phase. We thus tested the hypothesis of a possible early microglia activation, and of an involvement of the brain most vulnerable regions.
Methods
Ethics statement
The study was approved by the San Raffaele Hospital Ethical Committee and all the procedures involving human participants performed in this study were in accordance with the Declaration of Helsinki. Informed consent was obtained from each subject involved in the study.
Participants
SOD1‐mutated subjects were recruited from the Bellaria Hospital, IRCCS of Neurological Sciences, Bologna, Italy and from the NeuroMuscular Omnicentre, Serena Onlus Foundation, Milan, Italy.
Participants included six SOD1‐ALS patients (SYM‐carriers) fulfilling the revised El Escorial criteria 48 and four asymptomatic SOD1 mutation carriers (ASYM‐carriers) who were neurologically and cognitively normal. Demographic, genetic, and clinical data are reported in Table 1. Mean age at time of inclusion in this study was 61.2 ± 14.5 for SYM‐carriers (mean disease duration 3.8 ± 3) and 48 ± 19.8 for ASYM‐carriers. The six SYM‐carriers underwent an in‐depth neurological evaluation, including the ALS Functional Rating Scale‐Revised (ALSFRS‐r) for the assessment of severity of ALS and neuropsychological evaluation to evaluate cognitive efficiency. Ten healthy controls were included for statistical comparison (female/male 6/4; mean age 44.2 ± 10.5 years), previously recruited for different research studies. 43 , 44 , 49
Table 1.
Demographic, clinical, and genetic data of study participants
| Subject | Sex | Age | ALS onset | Disease Duration | ALSFRS‐r | SOD1 mutation |
|---|---|---|---|---|---|---|
| SYM‐carrier01 | F | 52 | Spinal | 9 years | 44 | A95T |
| SYM‐carrier02 | M | 76 | Spinal | 2 years | 37 | N66T |
| SYM‐carrier03 | F | 50 | Spinal | 2 years | 30 | L144F |
| SYM‐carrier04 | F | 71 | Spinal | 2 years | 21 | V5M |
| SYM‐carrier05 | F | 75 | Spinal | 6 years | 36 | T137A |
| SYM‐carrier06 | F | 43 | Spinal | 2 years | 34 | L84F |
| ASYM‐carrier01 | F | 46 | ‐‐ | ‐‐ | ‐‐ | V6M |
| ASYM‐carrier02 | M | 30 | ‐‐ | ‐‐ | ‐‐ | 133del |
| ASYM‐carrier03 | F | 76 | ‐‐ | ‐‐ | ‐‐ | A95T |
| ASYM‐carrier04 | F | 40 | ‐‐ | ‐‐ | ‐‐ | T137A |
11C‐PK11195 PET acquisition
All participants underwent 11C‐PK11195 PET imaging for the assessment of the extent and spatial distribution of microglia activation. All PET scans were performed at the Nuclear Medicine Unit, San Raffaele Hospital (Milan, Italy), and participants were randomly scanned with two multi‐ring 3D PET tomographs, either PET‐CT system “Discovery LS” or “Discovery 690” General Electric Medical Systems. 11C‐(R)‐PK11195 was produced in the Cyclotron Unit of the Nuclear Medicine of the San Raffaele Hospital as previously described obtaining optimal radiochemical purity> 95%. 50 Mean 11C‐PK11195 injected dose was 341 ± 51 MBq for SYM‐carriers, 312 ± 29 MBq for ASYM‐carriers, and 375 ± 64 MBq for healthy controls. The acquisition protocol consisted of a dynamic scan of 58 minutes (15 frames), i.e. 6 x 30 s/ 2 x 1 m/ 1 x 3 m/ 3 x 5 m/ 2 x 10 m/ 1 x 15 m. Transaxial images were reconstructed using a Shepp–Logan filter (cutoff 5 mm) in the transaxial plane, and a Shepp–Logan filter (cutoff 8.5 mm) in the axial direction. Corrections were applied for attenuation artifacts, radioactive decay, and scatter. Individual frames for each scan were realigned over time with statistical parametric mapping (SPM) software (http://www.fil.ion.ucl.ac.uk/spm/software) to minimize the effect of subject movement during data acquisition.
Imaging processing and data analysis
In 11C‐PK11195 PET scans, the selection of a reference region might be difficult, due to the heterogeneous distribution of the tracer across the whole brain in neurodegenerative conditions, hindering the identification of an anatomically defined region. 51 To overcome this methodological caveat, clustering methods have been proposed for the in vivo PET quantification of microglia activation. In our study, we employed the curve distance clustering algorithm (CDCA), 49 an adaptation of the well‐validated SuperVised Clustering Algorithm. 52 The CDCA algorithm estimates the similarity of the time activity curves (TACs) of each voxel with four predefined TACs (i.e., tracer delivery in blood, white matter, gray matter with nonspecific binding and gray matter with high specific binding), resulting in a pseudo‐reference region of cluster of voxels were the tracer kinetic is devoid of specific uptake, which can be used for subsequent parametric analysis. 49
11C‐PK11195 binding potentials (BPs) images were estimated adopting a receptor parametric mapping (RPM) procedure, 53 a basis function implementation of the simplified reference tissue modelling (SRTM) method, 54 using the following parameters: 0.04 min−1 lower bound, 1.0 min−1 upper bound, 30 basis functions.
The clustering maps obtained with the CDCA procedure were spatially normalized to the standard Montreal Neurological Institute (MNI) space. Spatial normalization was performed using a specific previously described PET template after masking to remove extracranial components (see 44 for full details). Warped 11C‐PK11195 BPs images were then smoothed with 8 mm Full Width at Half Maximum‐(FWHM) Gaussian‐Kernel.
To compare 11C‐PK11195 BPs across groups (SYM‐carriers vs. controls and ASYM‐carriers vs. controls), group level voxel‐wise statistical comparisons were performed, covarying for age as nuisance factor. W‐maps were used to perform voxel‐wise single‐subject analysis, comparing each subject in the SYM‐carriers and in the ASYM‐carriers group with the group of controls by the means of a two‐sample t test. A liberal statistical threshold (set at P < 0.01, uncorrected for multiple comparisons, considering significant those clusters containing more than 100 voxels) was chosen for both group and single‐subject level analyses, due to the small number of subjects included in the study, a limit imposed by the rare occurrence of the mutation.
Results
Group level analysis
11C‐PK11195 BPs between‐group comparisons demonstrated clusters of significant microglia activation in both SYM‐carriers and ASYM‐carriers compared with healthy controls. Specifically, SYM‐carriers showed significant microglia activation involving inferior occipital, lateral and inferior temporal cortex, premotor frontal and postcentral parietal cortex, plus thalamus, cerebellum, and medulla oblongata. A comparable significant increase in microglia activation in ASYM‐carriers compared with normal controls was present in the inferior occipital and lateral and inferior temporal cortex, with additional small clusters in frontal premotor and supplementary motor cortex, postcentral parietal cortex, and cerebellum (see Figure 1).
Figure 1.
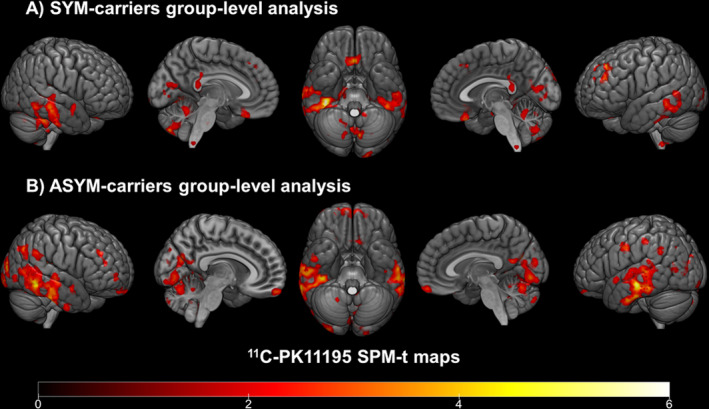
Group‐level 11C‐PK11195 BP maps in SYM‐ and ASYM‐carriers. (A) Clusters of increased 11C‐PK11195 bindings in SYM‐carriers compared to controls and (B) in ASYM‐carriers compared to controls (from red to yellow). Statistical threshold of P < 0.01 (uncorrected for multiple comparisons, minimum cluster extent k = 100 voxels).
Single‐subject analysis
At the single‐subject level, W‐maps revealed in both SYM‐ and ASYM‐carrier clusters of significantly higher 11C‐PK11195 binding than in healthy controls (see Figure 2 and Figure 3 for evidence in single individuals). In detail, the SYM‐carriers had a consistent microglia activation at single‐subject level in the temporal and occipital lobes, in the sensorimotor cortex and in the cerebellum. Medial temporal lobes and insula were involved in three patients. As for subcortical structures, thalamus was involved in four patients, three patients showed small clusters of microglia activation in the medulla oblongata, and two patients had higher 11C‐PK11195 binding in the basal ganglia, with a remarkable microglia activation in SYM‐carrier02 (Figure 2 and Figure 4). In the ASYM‐carriers microglia activation peaked with some clusters in the temporal lobe, the occipital lobe, and the cerebellum. Clusters in parietal lobe and frontal lobe were less frequent. In two subjects, microglia activation was present also in medial temporal lobe structures and insula. The same two subjects had additional subcortical activations, involving thalamus, basal ganglia, and medulla oblongata (ASYM‐carrier01 and ASYM‐carrier02) (Figure 3 and Figure 4).
Figure 2.
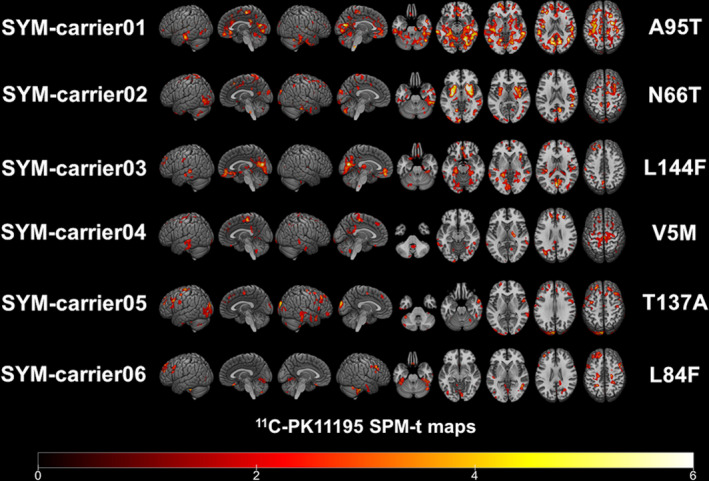
Single‐subject 11C‐PK11195 BP maps in SYM‐carriers. Single‐subject 11C‐PK11195 Binding Potentials maps are presented in each symptomatic carrier showing the corresponding SOD1 mutation. Statistical threshold of P < 0.01 (uncorrected for multiple comparisons, minimum cluster extent k = 100 voxels).
Figure 3.
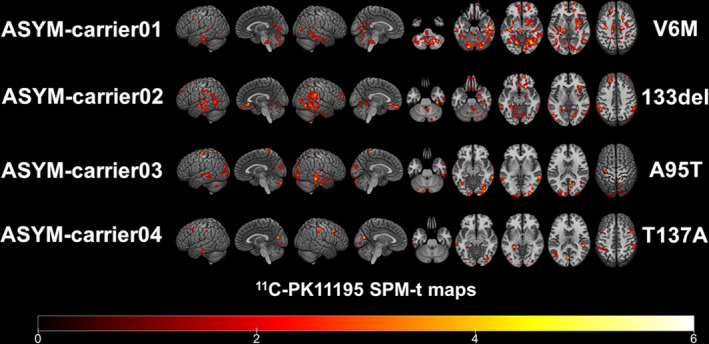
Single‐subject 11C‐PK11195 BP maps in ASYM‐carriers. Single‐subject 11C‐PK11195 Binding Potentials maps are presented in each of the four asymptomatic carriers showing the corresponding SOD1 mutation. Statistical threshold of P < 0.01 (uncorrected for multiple comparisons, minimum cluster extent k = 100 voxels).
Figure 4.
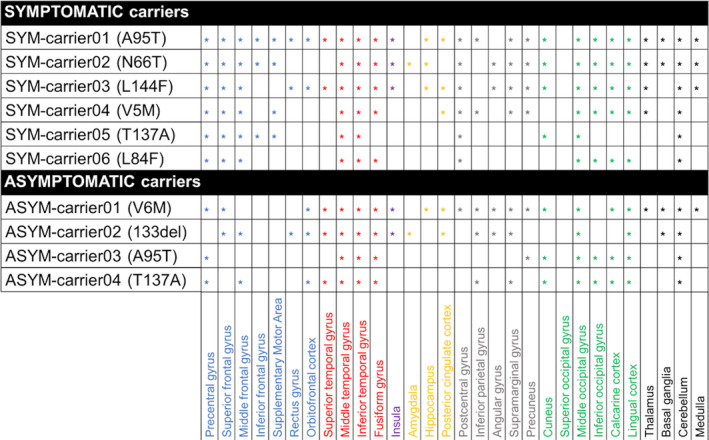
Peaks of microglia activation in different brain regions. Regions with increased 11C‐PK11195 binding potentials in each symptomatic (SYM) and asymptomatic (ASYM) carrier (the corresponding SOD1 mutation in brackets). Blue: frontal lobe; red: temporal lobe; yellow: medial temporal structures; violet: insula; grey: parietal lobe; green: occipital lobe; black: subcortical structures and cerebellum.
Discussion
The present study provides the first in vivo evidence of brain microglia activation in fALS SOD1 mutation, both in symptomatic and asymptomatic carriers, expanding the previous evidence in ALS. These results add novel knowledge in the understanding of ALS pathophysiology, with possible relevant implications for future therapeutic strategies.
Neuroinflammation is recognized as a crucial component of the pathophysiological mechanisms involved in neurodegeneration. Microglia represents, together with astrocytes, the major phagocytic cells in the brain and spinal cord that are involved not only in proinflammatory, but also in anti‐inflammatory responses, as well as in synaptic modulation, clearance of neuronal debris and tissue homeostasis. 55 Neuroinflammation may be both beneficial and detrimental, and a possible early protective response can be overcome by deleterious effects resulting in synaptic impairment and neurodegeneration in the case of persistent neuroinflammation. 35 , 56 , 57
Widespread microglia activation was present in SOD1G93A mice, as showed by animal‐PET imaging. 58 Glial cells activation in the spinal cord of SOD1G93A mice was reported near the motor neurons before symptoms onset and correlated with disease progression. 59 The modulation of microglia activity in SOD1 ‐mutated mice showed to be effective in slowing disease progression, reducing motor neuron degeneration, and improving survival. 30 , 34 , 60 A neuroprotective role of microglia cells expressing anti‐inflammatory cytokines was also suggested in the early phase of SOD1‐ALS in mice, 61 confirming the complexity of neuroinflammatory responses depending on the phase of the neurodegenerative process.
There are multiple reports the presence of neuroinflammation in ALS, both in postmortem and in in vivo PET studies. 31 , 33 , 39 , 62 , 63 , 64 , 65
Our study provides new and remarkable findings on the in vivo TSPO overexpression, both in symptomatic and asymptomatic carriers of SOD1 mutations. SYM‐carriers showed microglia activation in the occipital and temporal lobes, in the precentral, postcentral gyri, and the supplementary motor area, in the cerebellum. Most of them had increased 11C‐PK11195 binding also in the thalamus and in the medulla oblongata. Clusters of significant microglia activation were also evident in ASYM‐carriers, involving again the occipital and temporal cortex and cerebellum, suggesting that an early neuroinflammatory activity might be present even in an asymptomatic phase of disease.
Other research groups sought to investigate microglia activation in vivo in sALS, using PET imaging with both first‐ and second‐generation TSPO tracers. 39 , 62 , 63 , 64 , 65 In the first 11C‐PK11195 PET study in ALS, Turner and colleagues showed significant microglial activation in the motor cortex, pons, dorsolateral prefrontal cortex, and thalamus in 10 ALS patients compared to controls. 39 More recent studies involving second‐generation TSPO tracers, that is, 18F‐DPA‐714 and 11C‐PBR28, confirmed increased microglial activation in primary motor and supplementary motor cortices, 62 , 63 , 64 which correlated with markers of degeneration in the same regions, including atrophy and white matter changes, 65 and with the severity of symptoms. 63 Crucially, microglia activation not restricted to brain motor regions, and especially involving temporal regions, has been reported and related to the spread of the neurodegenerative process. 62 However, several remarkable differences between the present and previous studies limit the comparability of the findings. Only the study of Turner and colleagues investigated microglia activation in ALS patients using 11C‐PK11195 PET, whereas all the others used second‐generation TSPO tracers, which implies different quantification methods and some additional caveats, such as the possible influence of genotype in tracer binding affinity which may increase interindividual variability. 51 Currently, 11C‐PK11195 remains the prototypical and most validated tracer to investigate microglia activation in vivo, its cortical binding is not influenced by genotype, and it has been applied in several neurodegenerative condition, showing reproducible results. 66
The involvement of both motor and extra‐motor cortical and subcortical brain regions is in line with the suggested pattern of progression for staging ALS, which is consistent with autoptic studies hypothesizing the spread of the pathogenic processes from spinal cord and brainstem to the occipital and temporal lobes, prefrontal and postcentral cortices, the cerebellum, and the striatum. 67 A large body of literature exploring neuroimaging features in ALS patients confirmed the spread of neuronal pathological changes also to nonmotor regions. For example, structural MRI studies in sALS investigating cortical thickness, reported significant thinning primarily in the precentral gyrus, but involving also extra‐motor regions, including frontal, temporal, parietal, and occipital areas. 68 Notably, an atrophy spreading to the temporal cortex was correlated to the progression of the disease. 69 , 70 In addition, voxel‐based morphometry studies confirmed a widespread grey matter atrophy in sALS patients, peaking in the motor cortices and extending to the basal ganglia, thalamus, and cerebellum. 71 , 72 The brainstem involvement was also reported in a large population of sALS and primary lateral sclerosis patients, dominated by medulla oblongata atrophy primarily due to the flattening of the medullary pyramids. 73 Structural neuroimaging studies in SOD1‐ALS are sparse, supporting, however, the hypothesis that a specific pattern of neuronal changes may characterize fALS. SOD1‐ALS patients showed indeed higher cervical cord atrophy in comparison with sALS patients, suggesting that neurodegenerative processes in SOD1‐ALS are likely to start in the spinal cord. 74 More pronounced cortical changes in the frontal lobes in SOD1‐ALS than in sALS were reported in a voxel‐based morphometry study. 75 Furthermore, a distinctive pattern of neuronal loss, as assessed by decreased 11C‐flumazenil binding, involving the left frontotemporal junction and anterior cingulate gyrus, was revealed by a PET study comparing SOD1‐ASL and sALS patients. Crucially, a small cluster of reduced 11C‐flumazenil binding, similar to the pattern of the clinically affected patients, was seen also in two asymptomatic SOD1 mutation carriers. 76
In our sample, despite some variability, microglia activation in temporal and occipital lobes, in sensorimotor cortex, and in the cerebellum, represents the most consistent finding in the SYM‐carriers, together with the TSPO overexpression in the thalamus and medulla oblongata (see Figure 4 for details). Of note, previous 18F‐FDG PET studies consistently reported, in both sporadic and genetic ALS cases, along with motor cortical and frontal hypometabolism, a relative increase in glucose metabolism in the brainstem and cerebellum. 77 , 78 An increase in cerebral metabolism was found also in the occipital 79 , 80 , 81 and temporal regions. 82 , 83 A possible explanation for these metabolic increases relies on the presence of activated microglia or reactive astrocytes in the same regions, which has been shown in regions of neuronal dysfunction in inherited ALS. 84 Glial cells activities, related either to the neuroinflammatory response or to the clearance of cellular debris and death neurons, depending on disease phase and severity, may thus justify the regional brain hypermetabolism reported by FDG‐PET studies in ALS patients. 77
Overall, our study shows that in SOD1‐ALS patients with a moderate disease duration (mean disease duration 3.8 ± 2.9 years), microglia activation involves key pathology regions for ALS, that is, sensorimotor and associative motor areas, the temporal and occipital cortices, the basal ganglia, cerebellum, and medulla oblongata. Due to the rarity of the mutations, which limited the recruitment of patients, we could not provide an exhaustive correlation between microglia activation and geno/phenotype findings. We can argue a lower microglia activation and the lack of brainstem involvement in both the symptomatic and asymptomatic carrier of the T137A SOD1 mutation (SYMcarrier05 and ASYMcarrier04), which has been associated with a slower disease progression without bulbar involvement. 85 We observed a widespread microglia activation at cortical and subcortical levels and also in the cerebellum and medulla oblongata in SYMcarrier02, carrying the SOD1 mutation N66T. N66T SOD1 mutation has been recently described with an early severe bulbar, respiratory, and cognitive involvement. 86 The massive microglia activation shown in this patient might reflect a complex endophenotype, unusual in SOD1‐ALS. Both the L144F and V5M SOD1 mutations, whose symptomatic carriers showed a moderate microglia activation in our study (SYM‐carrie03 and SYMcarrier04), were associated with ALS spinal onset, involving the lower limbs and the upper limbs, respectively. 87 Finally, the A95T variant was associated with an unclear pathological tract, 88 which again hampers solid imaging to geno/phenotype correlation. Further studies with larger samples and extensive clinical characterization and follow‐up are needed to explore the role of microglia activation in SOD1‐ALS pathophysiology.
In conclusion, our findings support the presence of an early neuroinflammatory processes in SOD1‐ALS, with significant microglia activity already present in the asymptomatic carriers, evolving over disease progression in symptomatic patients. In vivo PET studies of brain resident glia processes represent a unique approach to the understanding the pathogenic mechanisms of neurodegeneration in ALS. In the future, these measures can help monitoring the effect of potential novel therapeutic strategies targeting neuroinflammation.
Author Contributions
GT, LI, CC, IB, CL, and DP were involved in the conception and design of the study. CC, GEV, LP, VM, AC, IB, FS, LM, and CL performed recruitment of subjects and acquisition of data. GT, LI, LP, FS, LM, and DP performed the analysis and interpretation of data. GT, LI, CC, LP, and DP drafted the manuscript, GEV, FS, LM, VM, AC, IB, and CL revised the manuscript adding important intellectual content. All the coauthors revised and approved the final version of the manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest relevant for this article.
Acknowledgment
We acknowledge all subjects and their families for participating in the study. This work was supported by the EU FP7 Imaging of Neuroinflammation in Neurodegenerative diseases (INMIND) Project (FP7‐HEALTH‐2011, Grant No. 278850).
Funding Information
This work was supported by the EU FP7 Imaging of Neuroinflammation in Neurodegenerative diseases (INMIND) Project (FP7‐HEALTH‐2011, Grant No. 278850).
Funding Statement
This work was funded by INMIND grants 278850 and FP7‐HEALTH‐2011.
References
- 1. Robberecht W., Philips T.. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013;14:248–264. [DOI] [PubMed] [Google Scholar]
- 2. Kiernan M.C., Vucic S., Cheah B.C., et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–955. [DOI] [PubMed] [Google Scholar]
- 3. Burke T., Pinto‐Grau M., Lonergan K., et al. A cross‐sectional population‐based investigation into behavioral change in amyotrophic lateral sclerosis: Subphenotypes, staging, cognitive predictors, and survival. Ann Clin Transl Neurol 2017;4:305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Marchi F., Sarnelli M.F., Solara V., et al. Depression and risk of cognitive dysfunctions in amyotrophic lateral sclerosis. Acta Neurol Scand 2019;139:438–445. [DOI] [PubMed] [Google Scholar]
- 5. Chiò A., Mazzini L., Mora G.. Disease‐modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 2020;107986. [DOI] [PubMed] [Google Scholar]
- 6. Talbott E.O., Malek A.M., Lacomis D.. The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol 2016;138:225–238. [DOI] [PubMed] [Google Scholar]
- 7. Swinnen B., Robberecht W.. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014;10:661. [DOI] [PubMed] [Google Scholar]
- 8. Andersen P.M., Al‐Chalabi A.. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7:603. [DOI] [PubMed] [Google Scholar]
- 9. Rosen D.R., Siddique T., Patterson D., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59–62. [DOI] [PubMed] [Google Scholar]
- 10. Ghasemi M., Brown R.H.. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med. 2017;a024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Renton A.E., Chiò A., Traynor B.J.. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chia R., Chiò A., Traynor B.J.. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol 2018;17:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen S., Sayana P., Zhang X., Le W.. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener 2013;8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang J.. Novel Aspects on Motor Neuron Disease: The Recent Genetic Studies on ALS. In: Novel Aspects on Motor Neuron Disease. IntechOpen; 2019.
- 15. Turner M.R., Al‐Chalabi A., Chio A., et al. Genetic screening in sporadic ALS and FTD. J Neurol Neurosurg Psychiatry 2017;88:1042–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roggenbuck J., Quick A., Kolb S.J.. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians. Genet Med 2017;19:267–274. [DOI] [PubMed] [Google Scholar]
- 17. Chiò A., Moglia C., Canosa A., et al. ALS phenotype is influenced by age, sex, and genetics: a population‐based study. Neurology 2020;94:e802–e810. [DOI] [PubMed] [Google Scholar]
- 18. Chiò A., Mazzini L., D’Alfonso S., et al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology 2018;91:e635–e642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abel O., Powell J.F., Andersen P.M., Al‐Chalabi A.. ALSoD: A user‐friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat 2012;33:1345–1351. [DOI] [PubMed] [Google Scholar]
- 20. Pansarasa O., Bordoni M., Diamanti L., et al. SOD1 in amyotrophic lateral sclerosis:“ambivalent” behavior connected to the disease. Int J Mol Sci 2018;19:1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cudkowicz M.E., McKenna‐Yasek D., Sapp P.E., et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateal sclerosis. Ann Neurol 1997;41:210–221. [DOI] [PubMed] [Google Scholar]
- 22. Kenna K.P., McLaughlin R.L., Byrne S., et al. Delineating the genetic heterogeneity of ALS using targeted high‐throughput sequencing. J Med Genet 2013;50:776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiò A., Calvo A., Mazzini L., et al. Extensive genetics of ALS: a population‐based study in Italy. Neurology 2012;79:1983–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamashita S., Ando Y.. Genotype‐phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl Neurodegener 2015;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nowicka N., Juranek J., Juranek J.K., Wojtkiewicz J.. Risk factors and emerging therapies in amyotrophic lateral sclerosis. Int J Mol Sci 2019;20:2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCord J.M., Fridovich I.. Superoxide dismutase an enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969;244:6049–6055. [PubMed] [Google Scholar]
- 27. Kaur S.J., McKeown S.R., Rashid S.. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene 2016;577:109–118. [DOI] [PubMed] [Google Scholar]
- 28. Pasinelli P., Brown R.H.. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006;7:710–723. [DOI] [PubMed] [Google Scholar]
- 29. Schain M., Kreisl W.C.. Neuroinflammation in Neurodegenerative Disorders—a Review. Curr Neurol Neurosci Rep 2017;17 10.1007/s11910-017-0733-2. [DOI] [PubMed] [Google Scholar]
- 30. Liu J., Wang F.. Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front Immunol 2017;8:1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Philips T., Rothstein J.D.. Glial cells in amyotrophic lateral sclerosis. Exp Neurol. 2014;262:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang S.H., Li Y., Fukaya M., et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 2013;16:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brettschneider J., Toledo J.B., Van Deerlin V.M., et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One 2012;7:e39216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boillée S., Yamanaka K., Lobsiger C.S., et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science (80‐ ) 2006;312:1389–1392. [DOI] [PubMed] [Google Scholar]
- 35. Werry E.L., Bright F.M., Piguet O., et al. Recent developments in TSPO PET imaging as a biomarker of neuroinflammation in neurodegenerative disorders. Int J Mol Sci 2019;20:3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Passamonti L., Tsvetanov K., Jones P.S., et al. Neuroinflammation and functional connectivity in Alzheimer’s disease: interactive influences on cognitive performance. bioRxiv 2019:532291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Surendranathan A., Su L., Mak E., et al. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 2018;141:3415–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang Y., Mozley P.D., Verma A., et al. Noninvasive PK11195‐PET image analysis techniques can detect abnormal cerebral microglial activation in Parkinson’s disease. J Neuroimaging 2018;28:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Turner M.R., Cagnin A., Turkheimer F.E., et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)‐PK11195 positron emission tomography study. Neurobiol Dis 2004;15:601–609. [DOI] [PubMed] [Google Scholar]
- 40. Cagnin A., Rossor M., Sampson E.L., et al. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol 2004;56:894–897. [DOI] [PubMed] [Google Scholar]
- 41. Pavese N., Gerhard A., Tai Y.F., et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 2006;66:1638–1643. [DOI] [PubMed] [Google Scholar]
- 42. Iannaccone S., Cerami C., Alessio M., et al. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat Disord 2013;19:47–52. [DOI] [PubMed] [Google Scholar]
- 43. Iaccarino L., Presotto L., Bettinardi V., et al. An in vivo 11C‐PK PET study of microglia activation in Fatal Familial Insomnia. Ann Clin Transl Neurol 2018;5:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iaccarino L., Moresco R.M., Presotto L., et al. An In Vivo 11 C‐(R)‐PK11195 PET and in vitro pathology study of microglia activation in Creutzfeldt‐Jakob Disease. Mol Neurobiol 2018;55:2856–2868. [DOI] [PubMed] [Google Scholar]
- 45. Tondo G., Iaccarino L., Caminiti S.P., et al. The combined effects of microglia activation and brain glucose hypometabolism in early‐onset Alzheimer’s disease. Alzheimer's Research & Therapy 2020;12 10.1186/s13195-020-00619-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tai Y.F., Pavese N., Gerhard A., et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007;130:1759–1766. [DOI] [PubMed] [Google Scholar]
- 47. Bevan‐Jones W.R., Cope T.E., Jones P.S., et al. In vivo evidence for pre‐symptomatic neuroinflammation in a MAPT mutation carrier. Ann Clin Transl Neurol. 2019;6:373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ludolph A., Drory V., Hardiman O., et al. A revision of the El Escorial criteria‐2015. Amyotroph Lateral Scler Front Degener 2015;16:291–292. [DOI] [PubMed] [Google Scholar]
- 49. Presotto L., Iaccarino L., Bettinardi V., et al. IEEE nuclear science symposium and medical imaging conference (NSS/MIC). IEEE 2015;2015:1–3. [Google Scholar]
- 50. Matarrese M., Moresco R.M., Cappelli A., et al. Labeling and evaluation of N‐[11C] methylated quinoline‐2‐carboxamides as potential radioligands for visualization of peripheral benzodiazepine receptors. J Med Chem 2001;44:579–585. [DOI] [PubMed] [Google Scholar]
- 51. Turkheimer F.E., Rizzo G., Bloomfield P.S., et al. The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans 2015;43:586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Turkheimer F.E., Edison P., Pavese N., et al. Reference and target region modeling of [11C]‐(R)‐PK11195 brain studies. J Nucl Med 2007;48:158–167. [PubMed] [Google Scholar]
- 53. Gunn R.N., Lammertsma A.A., Hume S.P., Cunningham V.J.. Parametric imaging of ligand‐receptor binding in PET using a simplified reference region model. NeuroImage 1997;6:279–287. [DOI] [PubMed] [Google Scholar]
- 54. Lammertsma A.A., Hume S.P.. Simplified reference tissue model for PET receptor studies. NeuroImage 1996;4:153–158. [DOI] [PubMed] [Google Scholar]
- 55. Comi C., Tondo G.. Insights into the protective role of immunity in neurodegenerative disease. Neural Regen Res 2017;12:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sochocka M., Diniz B.S., Leszek J.. Inflammatory response in the CNS: friend or foe? Mol Neurobiol 2017;54:8071–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Motataianu A., Barcutean L., Balasa R.. Neuroimmunity in amyotrophic lateral sclerosis: focus on microglia. Amyotroph Lateral Scler Front Degener 2020;1–8. [DOI] [PubMed] [Google Scholar]
- 58. Gargiulo S., Anzilotti S., Coda ARD, et al. Imaging of brain TSPO expression in a mouse model of amyotrophic lateral sclerosis with 18 F‐DPA‐714 and micro‐PET/CT. Eur J Nucl Med Mol Imaging 2016;43:1348–1359. [DOI] [PubMed] [Google Scholar]
- 59. Keller A.F., Gravel M., Kriz J.. Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia 2009;57:1130–1142. [DOI] [PubMed] [Google Scholar]
- 60. Beers D.R., Henkel J.S., Xiao Q., et al. Wild‐type microglia extend survival in PU. 1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci 2006;103:16021–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gravel M., Béland L.‐C., Soucy G., et al. IL‐10 controls early microglial phenotypes and disease onset in ALS caused by misfolded superoxide dismutase 1. J Neurosci 2016;36:1031–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Corcia P., Tauber C., Vercoullie J., et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One 2012;7:e52941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zürcher N.R., Loggia M.L., Lawson R., et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: assessed with [11C]‐PBR28. NeuroImage Clin 2015;7:409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alshikho M.J., Zürcher N.R., Loggia M.L., et al. Glial activation colocalizes with structural abnormalities in amyotrophic lateral sclerosis. Neurology 2016;87:2554–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Alshikho M.J., Zürcher N.R., Loggia M.L., et al. Integrated magnetic resonance imaging and [11C]‐PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol 2018;83:1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cerami C., Iaccarino L., Perani D.. Molecular imaging of neuroinflammation in neurodegenerative dementias: the role of in vivo PET imaging. Int J Mol Sci 2017;18:993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brettschneider J., Del Tredici K., Toledo J.B., et al. Stages of pTDP‐43 pathology in amyotrophic lateral sclerosis. Ann Neurol 2013;74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Agosta F., Valsasina P., Riva N., et al. The cortical signature of amyotrophic lateral sclerosis. PLoS One 2012;7:e42816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Verstraete E., Veldink J.H., Hendrikse J., et al. Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2012;83:383–388. [DOI] [PubMed] [Google Scholar]
- 70. Walhout R., Westeneng H.‐J., Verstraete E., et al. Cortical thickness in ALS: towards a marker for upper motor neuron involvement. J Neurol Neurosurg Psychiatry 2015;86:288–294. [DOI] [PubMed] [Google Scholar]
- 71. Menke RAL, Körner S., Filippini N., et al. Widespread grey matter pathology dominates the longitudinal cerebral MRI and clinical landscape of amyotrophic lateral sclerosis. Brain 2014;137:2546–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tan R.H., Devenney E., Dobson‐Stone C., et al. Cerebellar integrity in the amyotrophic lateral sclerosis‐frontotemporal dementia continuum. PLoS One 2014;9:e105632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bede P., Chipika R.H., Finegan E., et al. Brainstem pathology in amyotrophic lateral sclerosis and primary lateral sclerosis: a longitudinal neuroimaging study. NeuroImage Clin 2019;24:102054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Agosta F., Spinelli E.G., Marjanovic I.V., et al. Unraveling ALS due to SOD1 mutation through the combination of brain and cervical cord MRI. Neurology 2018;90:e707–e716. [DOI] [PubMed] [Google Scholar]
- 75. Turner M.R., Hammers A., Allsop J., et al. Volumetric cortical loss in sporadic and familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2007;8:343–347. [DOI] [PubMed] [Google Scholar]
- 76. Turner M.R., Hammers A., Al‐Chalabi A., et al. Distinct cerebral lesions in sporadic and ‘D90A’SOD1 ALS: studies with [11C] flumazenil PET. Brain 2005;128:1323–1329. [DOI] [PubMed] [Google Scholar]
- 77. Cistaro A., Valentini M.C., Chiò A., et al. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging 2012;39:251–259. [DOI] [PubMed] [Google Scholar]
- 78. Canosa A., Pagani M., Cistaro A., et al. 18F‐FDG‐PET correlates of cognitive impairment in ALS. Neurology 2016;86:44–49. [DOI] [PubMed] [Google Scholar]
- 79. Sala A., Iaccarino L., Fania P., et al. Testing the diagnostic accuracy of [18F] FDG‐PET in discriminating spinal‐and bulbar‐onset amyotrophic lateral sclerosis. Eur J Nucl Med Mol Imaging 2019;46:1117–1131. [DOI] [PubMed] [Google Scholar]
- 80. Cistaro A., Pagani M., Montuschi A., et al. The metabolic signature of C9ORF72‐related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging 2014;41:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Castelnovo V., Caminiti S.P., Riva N., et al. Heterogeneous brain FDG‐PET metabolic patterns in patients with C9orf72 mutation. Neurol Sci 2019;40:515–521. [DOI] [PubMed] [Google Scholar]
- 82. Pagani M., Chiò A., Valentini M.C., et al. Functional pattern of brain FDG‐PET in amyotrophic lateral sclerosis. Neurology 2014;83:1067–1074. [DOI] [PubMed] [Google Scholar]
- 83. Van Laere K., Vanhee A., Verschueren J., et al. Value of 18fluorodeoxyglucose–positron‐emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol 2014;71:553–561. [DOI] [PubMed] [Google Scholar]
- 84. Yamanaka K., Chun S.J., Boillee S., et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 2008;11:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Visani M., De Biase D., Bartolomei I., et al. A novel T137A SOD1 mutation in an Italian family with two subjects affected by amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2011;12:385–388. [DOI] [PubMed] [Google Scholar]
- 86. Martinelli I., Zucchi E., Gessani A., et al. A novel p. N66T mutation in exon 3 of the SOD1 gene: report of two families of ALS patients with early cognitive impairment. Amyotroph Lateral Scler Front Degener 2020;21:296–300. [DOI] [PubMed] [Google Scholar]
- 87. Forsberg K., Graffmo K., Pakkenberg B., et al. Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD‐associated genes. J Neurol Neurosurg Psychiatry 2019;90:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gellera C.. Genetics of ALS in Italian families. Amyotroph Lateral Scler Other Mot Neuron Disord 2001;2:s43–s46. [DOI] [PubMed] [Google Scholar]