

Abstract
The curative potential of non-autologous cellular therapy is hindered by the requirement of anti-rejection therapy. Cellular encapsulation within nondegradable biomaterials has the potential to inhibit immune rejection, but the efficacy of this approach in robust preclinical and clinical models remains poor. While the responses of innate immune cells to the encapsulating material have been characterized, little attention has been paid to the contributions of adaptive immunity in encapsulated graft destabilization. Avoiding the limitations of animal models, we established an efficient, antigen-specific in vitro platform capable of delineating direct and indirect host T cell recognition to microencapsulated cellular grafts and evaluated their consequential impacts. Using ovalbumin (OVA) as a model antigen, we determined that alginate microencapsulation abrogates direct CD8+ T cell activation by interrupting donor-host interaction; however, indirect T cell activation, mediated by host antigen presenting cells (APCs) primed with shed donor antigens, still occurs. These activated T cells imparted cytotoxicity on the encapsulated cells, likely via diffusion of cytotoxic solutes. Overall, this platform delivers unique mechanistic insight into the impacts of hydrogel encapsulation on host adaptive immune responses, comprehensively addressing a long-standing hypothesis of the field. Furthermore, it provides an efficient benchtop screening tool for the investigation of new encapsulation methods and/or synergistic immunomodulatory agents.
Keywords: cell replacement therapy, adaptive immunity, islet transplantation, immunomodulation, alginate encapsulation
1. Introduction
Cell replacement therapy shows great promise in treating diseases caused by the absence or malfunction of specialized cells, such as the replacement of insulin-producing beta cells in Type 1 Diabetes (T1D) or parathyroid cells for parathyroid hormone deficiencies [1-4]. A substantial impediment to the success of any non-autologous cellular transplant, however, is the need for systemic immunosuppressive drugs to prevent host rejection of the graft [5, 6]. The encapsulation of cells within biocompatible and durable polymers has been proposed as a means to prevent or reduce this pharmaceutical load [7, 8]. The permeability of the encapsulating material is modulated to support the interchange of oxygen, nutrients, and cellular products, while also preventing direct cell interactions between the host and the transplant [9]. Numerous materials have been studied for this application, from nanoporous membranes to synthetic hydrogels [8]. Among the various choices of materials, alginate, a natural anionic polysaccharide that is easily gelled via exposure to divalent ions at physiological conditions, is one of the most widely used [10]. The encapsulation of various cell types within alginate has resulted in decreased immune rejection and improved long-term graft survival in rodent models [10-12]. However, the robustness of this protection declines when translated to larger preclinical models and clinical trials. For example, the implantation of encapsulated pancreatic islets for the treatment of T1D into non-human primates and humans has been plagued by inconsistent results, with modest, transient graft efficacy and evidence of immune-driven rejection [13-16].
The failure of alginate encapsulated cells in clinical trials has been attributed to multiple factors, from impurities in the material to inadequate nutrient delivery [7, 17, 18]. Evidence from transplant models also indicates that the host's adaptive immune cells contribute to the loss of graft function, despite retention of the polymeric barrier. For example, xeno-antibodies have been detected following the implantation of alginate encapsulated porcine islets in a non-human primate model, indicating xenoantigen recognition across the polymeric barrier [14]. Furthermore, others have observed that the addition of T cell co-stimulatory blocking agents (i.e., CTLA4-Ig and anti-CD154 mAb) improves the efficacy of alginate encapsulated cellular grafts in disparate murine and non-human primate transplant models, implying that T cells contribute to the loss of function in these encapsulated implants [13, 19, 20]. Collectively, these findings imply that adaptive immune responses are activated and impart detrimental impacts on encapsulated cells; however, due to the multi-factorial nature of transplant models, their specific roles and pathways have not been clarified.
It has been postulated that the host adaptive immune system can detect encapsulated cells via the indirect antigen recognition pathway [7, 21, 22]. For context, host immune cells recognize foreign antigens via two pathways: direct and indirect. For direct recognition, host T cells directly recognize donor antigens presented on the surface of foreign cells via donor MHC molecules [23]. Alternatively, indirect antigen recognition occurs when the host APCs collect and process donor shed antigens, and present them to the host T cells in a host MHC-restricted manner [23-25]. Regardless of the antigen presentation pathway, the final consequence is the clonal expansion and activation of graft-specific effector immune cells (e.g., cytotoxic CD8+ T cells, helper CD4+ T cells, and effector B cells) that destroy the foreign grafts via both cellular (i.e., cytotoxic CD8+ T cells) and humoral (i.e., graft-specific antibodies) responses [23]. Published reports have concluded that complete polymeric encapsulation prevents direct antigen recognition by blocking direct cell-cell contact between donor and host cells [9, 10, 12, 13, 16, 20, 26-28]. It is postulated, however, that host immune recognition may still occur via indirect antigen recognition, as foreign shed antigens may diffuse through the biomaterial and into the peri-transplant site, as summarized in Figure 1.
Figure 1. Summary of Antigen Recognition Pathways of Unencapsulated and Microencapsulated Cells by the Host Adaptive Immune System.
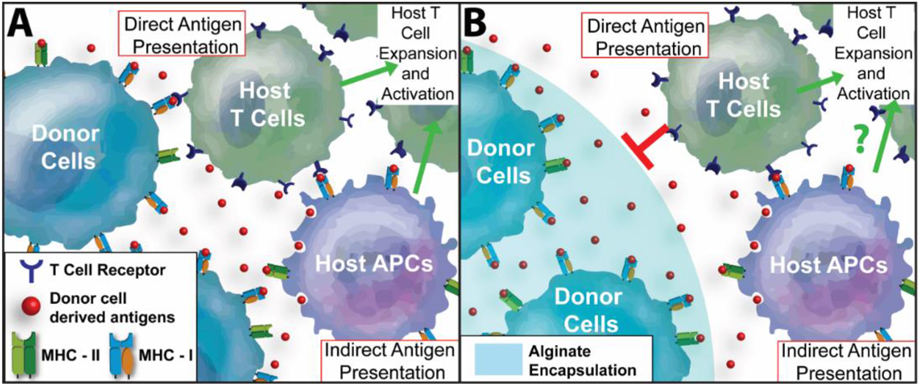
(A) Unencapsulated cells activate both direct and indirect antigen recognition pathways. For direct antigen recognition, host CD4+ and/or CD8+ T cells recognize foreign antigens presented on cells, via donor MHC-II and MHC-I molecules, respectively. Indirect antigen recognition is mediated by host antigen-presenting cells (APCs), where donor antigens are presented to host CD4+ and/or CD8+ T cells via host MHC molecules. Following antigen presentation, T cells are activated to proliferate and mature to graft-specific cytotoxic T cell effectors. (B) For encapsulated cells, e.g. alginate microencapsulation, direct cell-cell contact of donor cells to host T cells is blocked by the polymer barrier. However, antigens shed by the donor cells can diffuse out of the hydrogel for collection and presentation by host APCs, leading to indirect antigen recognition and subsequent antigen-specific T cell expansion and activation.
Despite this long-standing hypothesis, the immune crosstalk between host T cells and the encapsulated graft has remained unclear. Without a more comprehensive understanding of the pathways of host T cell activation and its subsequent role, if any, in damaging the underlying encapsulated cells, the capacity to generate immunoprotective biomaterial approaches for long-term clinical efficacy will remain elusive. While animal transplant models can provide physiologically relevant insight into the adaptive immune response to encapsulated grafts, a multitude of other factors that contribute to graft failure, such as the transplant environment, insufficient nutrient delivery, and foreign body responses, overlay and prevent clear delineation of their respective roles. Furthermore, reliance on animal models to characterize these responses is expensive and time-consuming.
Herein, we developed a reproducible, efficient, and antigen-specific in vitro platform to conduct mechanistic studies into the role of polymeric microencapsulation on host adaptive immune responses. To impart antigen specificity, encapsulated cells were sourced from membrane-bound ovalbumin (mOVA) transgenic mice. These mOVA stimulator cells express ovalbumin on their cell membrane and present OVA peptide derivatives as self-antigen in MHC-I [29]. To examine OVA-specific adaptive immune activation, responder cells were isolated from OT1 mice, a transgenic murine strain with a dominant CD8+ T cell population expressing an engineered Vα2/Vβ5 T cell receptor recognizing OVA257-264 peptide (i.e., SIINFEKL). Combining alginate encapsulation and these antigen-specific immunoreactive cells in an in vitro coculture platform permitted the exploration and delineation of the roles of direct versus indirect antigen recognition and their downstream impacts on effector T cell generation. This platform was then validated using microencapsulated mOVA pancreatic islets for potential application in diabetes therapy, where corresponding OT1 T cell activation and its immune impacts on the encapsulated islets were investigated. The contribution of adaptive immune cell recognition and activation on the destabilization of encapsulated cells is also explored.
2. Materials and Methods
2.1. Animals
All animal procedures were conducted under IACUC approved protocols at the University of Florida. Responder cells were sourced from OT1/GFP mice, 8-15 weeks of age, where the CD8+ T cells are clonally specific to SIINFEKL peptide via transgenic T cell receptors. Stimulator cells (splenocytes and islets) were sourced from mOVA mice (C57BL/6J (CAG-OVA)916Jen/J; Jackson laboratories), where all beta-actin expressing cells express both membrane-bound OVA and OVA peptide derivatives as self-antigen in MHC-I [29].
2.2. Splenocyte isolation and islet isolation
Spleens were collected from donors in cold HBSS buffer (Corning). A single-cell suspension was prepared by mechanically rupturing the spleen and filtering through a 40-μm cell strainer (Corning). Splenocytes were harvested by spinning down at 500×g for 5 min and erythrocytes were removed by 5 min treatment with ACK lysing buffer. Islets were isolated from mOVA or C57BL/6J mice as previously described[30, 31]. Isolated islets were cultured at 37°C, 5% CO2 in a humidified incubator in CMRL 1066 media (Mediatech) supplemented with 10% FBS (Hyclone, GE Healthcare), 20 mM HEPES buffer, 100 U/mL penicillin- streptomycin, and 2 mM L- glutamine until used for experiments.
2.3. Encapsulation materials and cell encapsulation
Sterile 1.6% (w/v%) UP MVG (cGMP grade, Pronova, NovaMatrix) alginate in saline was used for encapsulation. mOVA cells were encapsulated using a parallel air flow bead generator (Biorep, Inc.) with an average air rate of 3300 mL/min and 50 mM BaCl-MOPS cross-linking buffer, as previously reported [32]. Encapsulation density was 5x107 or 1x107 cells/mL alginate, depending on the experimental design. After three PBS washes, mOVA splenocyte microbeads were maintained in complete T cell media (RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin-streptomycin, 2 mM L- glutamine, 20 mM HEPES buffer and 55 μM β-mercaptoethanol). For islet encapsulation, islets were counted 24 h post-isolation using a standard algorithm for the calculation of 150 μm diameter islet equivalent (IEQ) number [33] and encapsulated at a density of 1000 IEQ/mL alginate. Encapsulated islets were maintained in complete CMRL media overnight prior to coculture. Cell-free alginate microbeads were generated as a control. Bead size was measured (n ≥ 10) with Carl Zeiss™ Primo Vert™ Inverted Microscope.
2.4. OTI/GFP splenic cell purification by cell sorting
Fresh OTI/GFP splenocytes were labelled with anti-mCD3-PacificBlue; anti-mCD8a-APC; anti-mCD4-PE/Cy5 and anti-mCD11c-PE antibodies at 4 °C for 20 min (Table S1). Labeled cells at the concentration of 25×106/mL were sorted using BD FACSAria™ sorter with over 98% efficiency. i) GFP+CD3+CD11c−CD8a+CD4− cells were sorted out as viable OTI splenic CD8+ T cells; and ii) GFP+CD3−CD11c+CD8a+CD4− cells were harvested as the viable OTI splenic cross-presenting CD8a+ DCs. Sorting was conducted by the Interdisciplinary Center for Biotechnology Research Cytometry Core and the Center for Immunology and Transplantation at the UF. A representative sorting scheme is shown in Figure S1.
2.5. mOVA-OTI in vitro coculture platform
To delineate direct and indirect antigen recognition pathways, 1) 100,000 purified OTI CD8+ T cells; 2) 75,000 sorted OTI CD8+ T cells with 25,000 pre-primed cross-presenting CD8a+ DCs (with or without inhibition); or 3) 100,000 unsorted OT1 splenocytes were used as immune responders and co-incubated with 100,000 mitomycin C (50 μg/mL) treated unencapsulated or encapsulated mOVA cells for 48 hrs. All immune responders were labeled with CellTrace™ violet dye for proliferation tracking. The frequency of proliferating granzyme B expressing OTI CD8+ T effector cells was quantified as the readout of the coculture system. Unstimulated control (T cell media), anti-CD3/CD28 Dynabeads® (ThermoFisher); 0.1μM SIINFEKL (InvivoGen) peptide, 0.1μM soluble ovalbumin (InvivoGen) protein, and cell-free alginate microbeads were used as control stimulators for OTI T cell activation. The stimulator/responder (S/R) ratio of the coculture system was adjusted, as outlined in the experimental design. For mOVA islet-OTI responder coculture, 50 mOVA or C57BL/6J islets, in unencapsulated or alginate encapsulated form, were hand-picked and cocultured with 100,000 CellTrace™ violet-labeled OTI/GFP splenocytes at 37°C for 72 h before downstream analysis. The same control groups were applied as mentioned above. For cross-presentation inhibition, 25,000 fresh sorted viable CD8a+ DCs were primed with 100,000 unencapsulated or encapsulated mOVA cells for 6 h with or without 5μg/mL brefeldin A (Sigma)[34]. 75,000 Cell Trace™ Violet labeled purified OTI CD8+ T cells were then added for 48 h stimulation.
2.6. Flow cytometry
To evaluate the OVA-specific CD8+ T cell activation, OTI responders post coculture were collected for flow cytometry analysis. OTI responders were sequentially stained with Live/Dead® Fixable Near IR dye (Invitrogen), anti-mCD8a-PE, anti-mCD25-PE/Cy7, and anti-mGranzymeB-APC (Table S1) for viability and immune phenotyping. Background signals were identified and excluded by isotype-matched and fluorescence-minus-one controls. The level of OTI CD8+ T cell activation was quantified as the percentage of proliferating granzyme B+ CD8+ T effector cells (Figure S2). Data were acquired using BD LSRII or FACSCelesta analyzer. Data analysis was performed using FCS Express 6.05 software (DeNovo software). Proliferation index (PI, the total number of cell divisions) and division index (DI, the average number of cell divisions) were calculated by fold dilution of CellTrace™ Violet signal and proliferation modeling using the embedded proliferation module of FCS Express 6.05 software.
2.7. In vitro islet assessments
To evaluate mOVA islet viability and function after coculture experiment, Live/Dead® imaging and static glucose-stimulated-insulin-release (GSIR) assay were used. For Live/Dead® assay, the encapsulated islets post coculture were stained with 26.67 μM calcein AM and ethidium homodimer-1 at 37°C for 30 min. Confocal images were then acquired using Leica TCS SP8 microscope. Fluorescent signals from calcein AM and ethidium homodimer-1 were quantified as the area of calcein AM+ (or ethidium homodimer-1+) normalized by the area of an islet using ImageJ software (n ≥ 10 images per group per test). For the GSIR assay, samples with 50 encapsulated islets were immobilized in chromatography columns using Sephadex G10 resin beads (GE Health) after coculture, as previously described[35]. The columns were then sequentially stimulated with 1 h of low (3 mM) glucose (L1), followed by 1 h of high (16.7 mM) glucose (H), and lastly an additional 1 h of low (3 mM) glucose (L2). Samples (1 mL) collected after each 1-hr stimulation were analyzed for insulin content via ELISA (Mercodia) and normalized by the islet number (50) of each sample.
2.8. Statistical analysis
Data are expressed as the mean ± standard deviation for each group, with individual data points shown as scattered points. For all experiments, a minimum of three independent biological replicates were included in each group. Outlier screening was performed for all data sets using Robust Fit with Cauchy estimate with multiplier K=2, using SAS JMP Pro v13.1.0. software. Statistical assessments were performed using one-way ANOVA with Tukey's multiple comparison analysis using GraphPad Prism Software. Statistical differences were considered significant when the probability value (p) was <0.05. Statistical difference is shown as — *p<0.05; **p<0.01; ***p<0.001; **** p < 0.0001 and n.s. indicates not significant.
3. Results
3.1. Alginate Encapsulation Blocks the Contact-dependent Direct Antigen Recognition
To provide an efficient means to evaluate host adaptive immune responses to encapsulated cells, an OVA-based antigen-specific platform was designed. For biomaterial encapsulation, ultra-pure, cGMP-grade, medium viscosity, high guluronic alginate (UP-MVG) cross-linked via barium was used. This formulation was selected due to its high stability, predictable and controlled porosity, and use in preclinical and clinical trials [9, 15, 16, 26, 36, 37]. mOVA stimulators were sourced from mOVA spleens, mitomycin C treated to prevent cellular proliferation within the microbeads, and encapsulated. Responder cells were sourced from OT1 splenocytes. To characterize CD8+ T cell activation, the frequency of viable, proliferating, and granzyme B+ CD8+ cells was quantified via flow cytometry, as these markers classically define an effector cytolytic T cell (Figure S2) [38, 39]. For select experiments, proliferation analysis of CD8+ T cells was also conducted.
To examine the role of direct antigen recognition in microencapsulation systems, CD8+ T cells were sorted from OTI splenocytes (Figure S1). Resulting sorted OTI CD8+ T cells were incubated with either unencapsulated or encapsulated mOVA cells and the activation of OTI CD8+ T cells was subsequently measured (Figure 2A). The response of purified OTI CD8+ T cells to unencapsulated mOVA cells was efficient and robust, with high activation (90.5 ± 1.3%) and proliferation (PI = 3.12 ± 0.23) (Figure 2B&D). This CD8+ T cell activation profile was statistically equivalent to anti-CD3/28 activator beads controls (p=0.05, Tukey post-hoc), demonstrating vigorous stimulation. Contrarily, OT1 CD8+ T cell proliferation and activation was completely ablated when cocultured with alginate encapsulated mOVA cells; T cell activation (2.7 ± 2.2%) and proliferation (PI = 1.35 ± 0.31) was statistically comparable to unstimulated controls (p=0.96, Tukey post-hoc; Figure 2C&D). While in vitro cultures of primary splenic T cells typically become apoptotic in the absence of stimulation [40], T cells harvested from cocultures of both unencapsulated and encapsulated mOVA cells were highly viable (p<0.0001 vs unstimulated control respectively; Tukey post-hoc) albeit statistically distinct (p = 0.005; t-test; Figure 2D inset & S3A).
Figure 2. Alginate Encapsulation Blocks Cell-Contact Dependent Direct CD8+ T Cell Antigen Recognition and Activation.
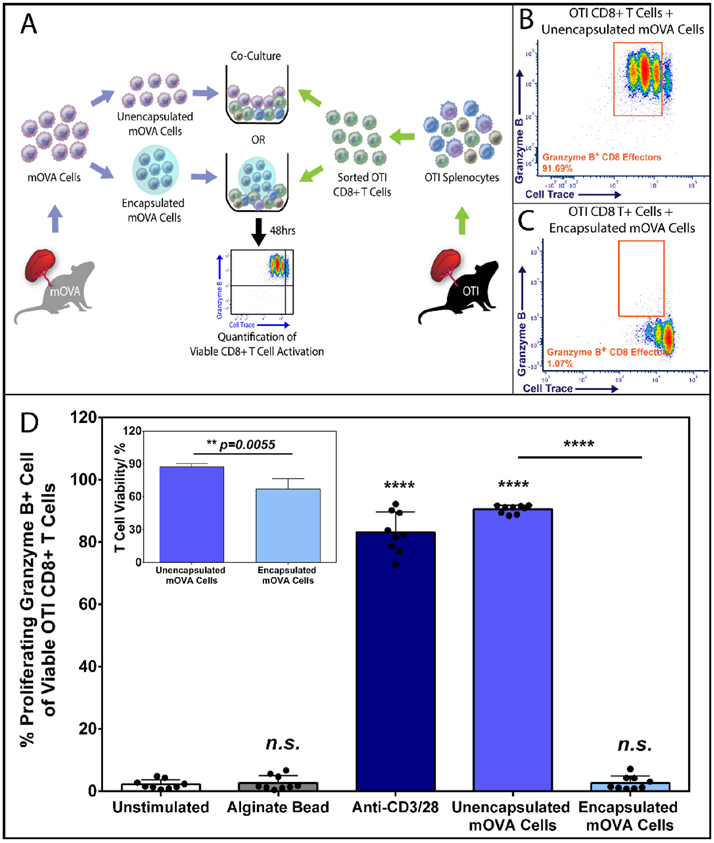
(A) Schematic overview of the 48hr coculture experiment co-incubating sorted OTI CD8+ T cells (see Figure S1 for sorting) with unencapsulated or encapsulated mOVA stimulatory splenocytes at 1:1 ratio. Antigen-specific OTI CD8+ T cell activation was quantified by the % of proliferating (Cell Trace® Violet labeled) and granzyme B + CD8+ T effector cells (see Figure S2 for gating) via flow cytometry analysis. Representative FCM data of OTI CD8+ T cell activation by unencapsulated (B) or alginate encapsulated (C) mOVA cells after a 48-hour stimulation. (D) Summary of the frequency of proliferating granzyme B+ CD8+ T effectors in response to designated stimuli (x-axis). Inset: CD8+ T cell viability after 48 h incubation with unencapsulated or encapsulated mOVA cells. Bars indicate the average of individual data points (N=3; n=9) with standard deviation. Statistical significance was determined as ****p < 0.0001 and n.s. = not significant via Tukey’s test when compared with the unstimulated control group.
To validate the inert nature of the base alginate biomaterial, cell-free alginate microbeads were included as a control group. CD8+ T cell responses to this group were statistically equivalent to unstimulated controls (p=0.87; Tukey post-hoc; Figure 2D). Of note, endotoxin levels from alginate eluates were below assay detection limits (sensitivity 0.1EU/mL, Table S2).
To confirm the antigen specificity of the coculture system, cells sourced from C57BL/6J mice were screened as non-specific stimulators (Figure S3A). OTI CD8+ T cells were nonresponsive to either unencapsulated or encapsulated C57BL/6J splenic cells; the percentage of effector CD8+ T cells were statistically equivalent to unstimulated controls (p = 0.54 and 0.97, respectively; Tukey post-hoc). As OTI mice are syngeneic with the C57BL/6J strain [29], the lack of a T cell response within the time frame of this assay validates that the observed OTI CD8+ T cell activation in response to mOVA stimulators was instigated by OVA antigens.
3.2. Indirect Antigen Recognition is Preserved in Alginate Microencapsulation.
Following the conclusion that alginate encapsulation suppresses direct antigen recognition, we sought to develop methods to assess host T cell activation via indirect antigen recognition pathways. While the CD8+ T cell is the dominant responsive population of lymphocytes in OTI splenocytes due to the nature of its transgenic development and MHC-I restriction [41], the splenocytes of OTI mice also contain CD4+ T cells, CD11c+ dendritic cells (DC), F4/80+ macrophages, and B cells (Figure S4). These immune populations can contribute to activating cytolytic CD8+ T cells via indirect antigen presentation pathways, e.g., DC cross-presentation and CD4+ T cell licensing [42, 43]. Thus, it was postulated that the inclusion of the full OTI splenic repertoire in this coculture platform could permit the study of CD8+ T cell activation via indirect antigen mechanisms (Figure 3A).
Figure 3. Alginate Encapsulated Cells Effectively Activate Antigen-Specific CD8+ T cells via the Indirect Antigen Recognition Pathway.
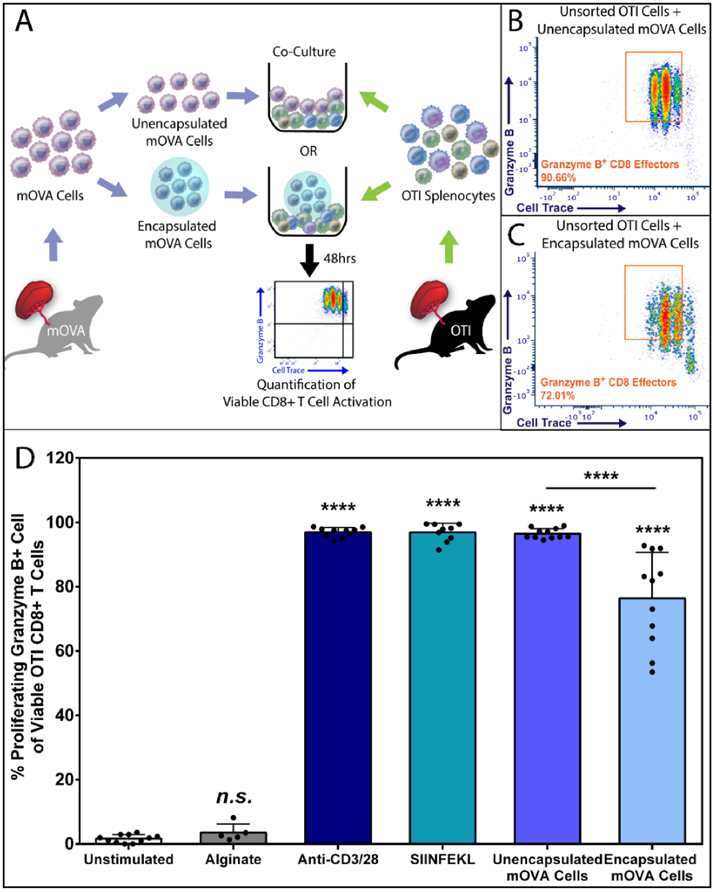
(A) Schematics of the 48hr coculture experiment co-incubating mOVA stimulator cells, either unencapsulated or encapsulated form, with the unsorted OTI splenic immune responder cells. OVA-specific OTI CD8+ T cell activation was quantified by the % of proliferating and granzyme B + CD8+ T effectors via flow cytometry analysis (see Figure S2). Representative FCM data of OVA-specific CD8+ T cell activation within the unsorted OT1 responder pool in response to the equivalent amount of unencapsulated (B) or alginate encapsulated (C) mOVA cells. (D) Summary of the frequency of proliferating granzyme B+ CD8+ effector T cells, gated from the whole OTI splenocyte population, in response to designated stimuli (x-axis). Bars indicate the average with individual data points (N=4; n=6-11) shown with standard deviation. Statistical difference was determined as ****p < 0.0001; n.s. = not significant via Tukey’s test when compared with the unstimulated control group.
As expected, the coculture of OTI splenocytes with unencapsulated mOVA cells resulted in significant CD8+ T cell activation (96.5 ± 1.5%) and proliferation (PI = 3.63 ± 0.52); values were statistically equivalent to cells stimulated via polyclonal activator beads (p>0.999, Tukey post-hoc) or soluble SIINFEKL peptide (p>0.999, Tukey post-hoc) (Figure 3B&D). Distinct from purified CD8 T cell cultures, when whole OTI splenocytes were incubated with encapsulated mOVA cells, the resulting CD8+ T cell activation and expansion was surprisingly vigorous, with significant activation (76.4 ± 14.3%; p < 0.0001 vs unstimulated control) and proliferation (PI = 2.30 ± 0.60), albeit statistically lower than those measured in response to unencapsulated mOVA cells (p<0.0001; Figure 3C&D).
To investigate the role of biomaterial porosity in the activation of indirect antigen recognition pathways, alginate microbeads containing mOVA cells were further coated with poly-L-lysine (PLL) and alginate (Figure S5). These classic alginate/PLL/alginate (APA) beads exhibit a more restrictive porosity (molecular weight cutoff (MWCO) of ~70 kDa versus ~125 kDa for this alginate) [32]. This outer PLL/alginate coating also ensures complete cellular encapsulation (Figure S5B) [44]. The co-incubation of mOVA APA beads with whole OT1 splenocytes resulted in OTI CD8+ T cell activation statistically identical to alginate microbead results (66.9±6.8% vs 68.1±6.1%; p=0.9998, Figure S5C-D). This further illustrates that indirect OT1 CD8+ T cell activation is independent of host-donor contact and also that it is unhindered by a more restrictive MWCO at ~70kDa.
This robust CD8+ T cell activation to alginate encapsulated mOVA cells, particularly when compared to the complete inhibition of T cell activation for purified CD8+ T cell responders, indicates that the inclusion of endogenous splenic lymphocytes alters T cell activation pathways. Of note, similar to the results from purified CD8+ T cell studies, stimulators incubated with either cell-free alginate microbead (Figure 3D), APA microbead (Figure S5C) or C57BL/6J cells (Figure S3B) exhibited T cell activation equivalent to unstimulated controls.
3.3. Professional APCs are Mediators of Indirect Antigen Recognition to Alginate Encapsulated Cells
The robust activation of OTI CD8+ T cells in response to alginate encapsulated mOVA cells when the entire splenocyte repertoire is present implicates that non-CD8+ T cells facilitate antigen recognition in this platform. It is known that CD8a+ dendritic cells (DC) uptake, process, and cross-present extracellular antigens to CD8+ T cells via the exogenous MHC-I pathway, resulting in antigen-specific CD8+ effector T cells [43]. To investigate their potential role in this benchtop model, OTI CD8a+ cross-presenting DCs were sorted from bulk OTI splenic responders via flow cytometry, as outlined in Figure S1. Sorted DCs were then primed for six hours with mOVA stimulating cells, followed by the addition of responding OTI CD8+ T cells (Figure 4A). To further delineate the role of cross-presentation for indirect T cell activation in this model, brefeldin A, an inhibitor of cross-presentation [34], was added to select groups during antigen priming.
Figure 4. Cross Presenting CD8+ Dendritic Cells Mediate the Indirect CD8+ T cell Activation by Alginate Encapsulated Cells.
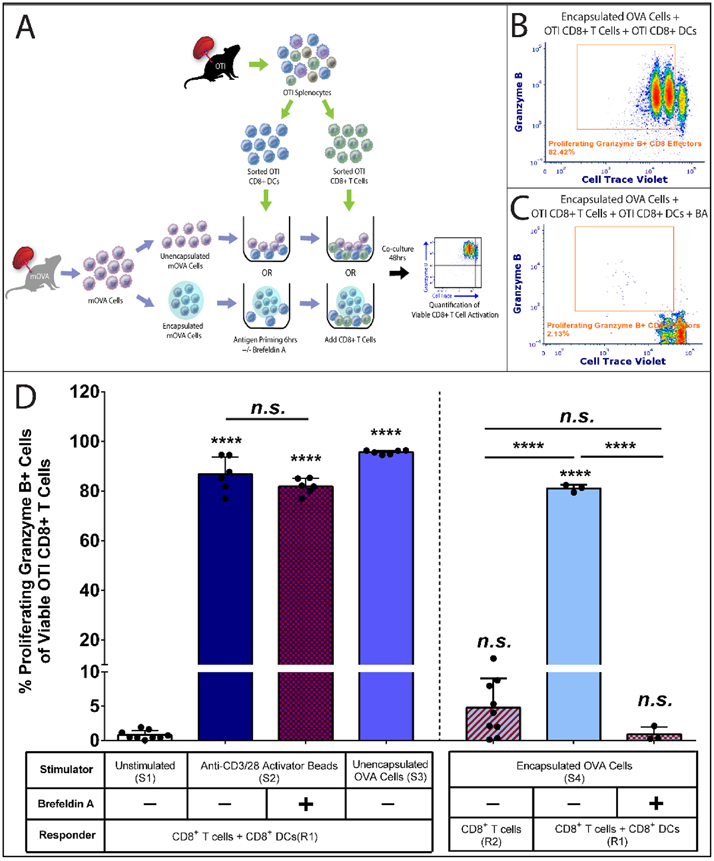
(A) Schematics of the coculture experiment. Firstly, 15,000 purified cross-presenting CD8+ DCs were primed by unencapsulated or encapsulated mOVA cells for 6hrs with or without the cross-presentation inhibitor brefeldin A (BA, 5μg/mL). Then 85,000 purified CD8+ T cells with Cell Trace® Violet labeling were added to the system. After 48h coculture. OVA-specific OTI CD8+ T cell activation was quantified by the % of proliferating and granzyme B + CD8+ T effectors via flow cytometry analysis. Representative FCM data of OTI CD8+ T cell activation within the CD8+ T cell and CD8+ DC responder pool stimulated by encapsulated mOVA cells (B) with or (C) without brefeldin A treatment. (D) Summary of the frequency of OT1 CD8+ effector T cells in response to designated stimuli (table at the bottom). Stimulator groups are annotated as unstimulated control (S1); anti-CD3/28 activator beads (S2); unencapsulated mOVA cells (S3) and encapsulated mOVA cells (S4). Responder groups are annotated as CD8+ T cells + CD8a+ DCs (R1) and purified CD8+ T cells (R2). Cross-presentation is labeled as untreated (BA−) or inhibited (BA+). Bars indicate the average with individual data points (n=3-9) shown with standard deviation. Statistical difference was determined as ****p < 0.0001; n.s. = not significant via Tukey’s test when compared with the unstimulated control group or in between groups.
As summarized in Figure 4D, the inclusion of purified CD8a+ DCs into CD8+ T cell cultures generated targeted effects. Consistent with our previous findings, robust CD8+ T cell activation was observed within the coculture of CD8+ T cells and CD8a+ DCs (responder group R1) with activator beads (anti-CD3/28, group S2 + R1) (86.8 ± 7.0%) or unencapsulated mOVA cells (group S3 + R1) (95.7 ± 0.8%).
When encapsulated mOVA cells were used as stimulators (S4), CD8+ T cell response was altered depending on the composition of the OT1 responder pool. Specifically, if purified CD8+ T cells were applied as immune responders, minimal activation was observed (4.8 ± 4.2%; group S4 + R2), consistent with the results reported in Section 3.1. However, the inclusion of CD8a+ DCs into the coculture system substantially altered this response, resulting in robust CD8+ T cell activation (81.0 ± 1.2%; group S4 + R1; Figure 4B&D) statistically equivalent to that observed in response to anti-CD3/28 activator beads (p = 0.27; Tukey post-hoc). To delineate the role of cross-presenting CD8a+ DCs in indirect CD8+ T cell activation, brefeldin A (BA) was added to the coculture system. The inclusion of this inhibitor during DC antigen priming resulted in the potent loss of the downstream indirect OT1 CD8+ T cell activation, with a granzyme B negative, nonresponsive CD8+ T cells population (1.2 ± 0.9%; group S4 + R1 + BA; Figure 4C&D); the resulting activation level was comparable to purified CD8+ T cell controls (p = 0.74; group S4 + R2, no BA; Tukey post-hoc). Of note, the inclusion of brefeldin A inhibition for control anti-CD3/28 activator bead cultures (group S2 + R1 + BA) did not impair CD8+ T cell activation (p = 0.08; Tukey post-hoc) or viability (Figure S6C, p=0.56; Tukey post-hoc), validating that the cross-presentation inhibition treatment does not compromise the activation capacity of the CD8+ T cells.
3.4. Indirect T cell Activation Level Correlates to Encapsulated Cell Number and Density
To further characterize the dynamics of T cell activation in response to encapsulated cells, a two-parameter antigen titration was performed. Specifically, the overall encapsulated stimulator cell number and the encapsulation density were independently varied and its impact on subsequent T cell activation was quantified. Whole OTI splenic responders and experimental parameters used for the titration were identical to those defined in Figure 3A.
As summarized in Figure 5A, antigen dose dependency was observed for CD8+ T cells stimulated by either unencapsulated or alginate encapsulated mOVA cells. The trend of increased OTI CD8+ T cell activation in response to unencapsulated mOVA cells was less evident, with robust stimulation observed using a modest stimulating cell dose (i.e., 10,000 mOVA). Contrarily, antigen dosage of the encapsulated mOVA stimulators strongly influenced the response of CD8+ T cells, with activation values ranging from insignificant to high (e.g., 2.8 ± 0.1% and 48.5 ± 0.8% in response to 10,000 and 50,000 mOVA encapsulated cells, respectively). Comparing CD8+ T cell responses to titrated levels of unencapsulated or encapsulated mOVA cells, it can be concluded that the presence of the alginate barrier plays a suppressive role in T cell activation. As shown, the level of CD8+ T cell activation in response to 50,000 encapsulated mOVA cells was almost half (51.8%) of its unencapsulated counterpart. To further dissect the difference in direct and indirect CD8+ T cell responses, proliferation modeling was employed (Figure 5B). A decrease in proliferating CD8+ T cell number (5260 ± 781) and a delay in T cell proliferation (PI = 1.78 ± 0.04) was measured for CD8+ T cells responding to encapsulated mOVA cells, when compared to its matched unencapsulated counterpart (T cell number: 10614 ± 1321, p< 0.0001 and PI: 3.22 ± 0.05, p < 0.0001, respectively; Tukey post-hoc). However, the immunosuppressive effect of encapsulation is incomplete, with increased indirect CD8+ T cell activation measured when the antigen dose increased (Figure 5A).
Figure 5. Distinct Antigen Dosage Impacts for Unencapsulated versus Alginate Encapsulated.
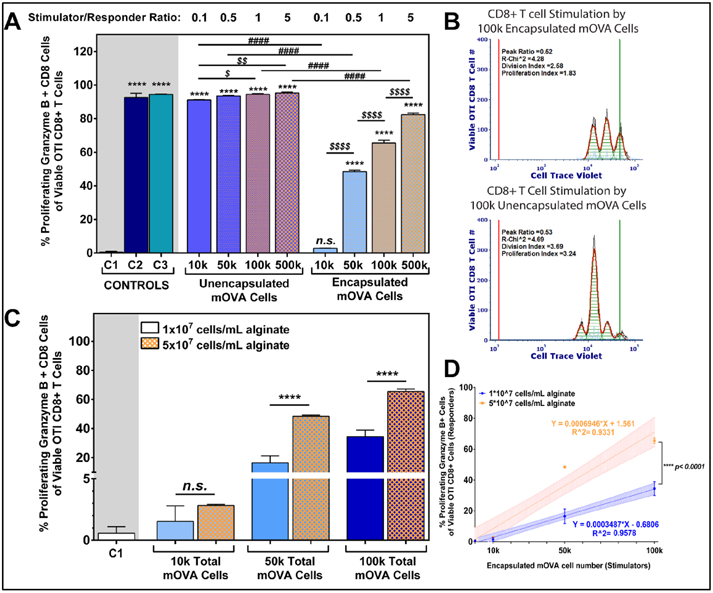
(A) OTI CD8+ T cell activation in response to titration of unencapsulated and encapsulated mOVA stimulator cells in a 48hr coculture (n=3); S/R ratio ranged from 0.1 to 5 (top label). Encapsulation density was 5x107 mOVA cells/mL alginate. Controls: C1: unstimulated; C2: anti-CD3/28 activator beads; C3: 0.1μM SIINFKEL peptide. Mean comparisons to unstimulated control C1 (statistics shown as *); within unencapsulated or encapsulated mOVA stimulators (statistics shown as $); and between unencapsulated and encapsulated stimulators (statistics shown as #) using Tukey's test. (B) Representative proliferation modeling of OTI CD8+ T cells in response to 100,000 unencapsulated or encapsulated mOVA cells, with the black lines represent histogram contour of raw data, green lines marked the undivided population and red lines represent basal signal. For proliferating modeling, the orange lines are the fitted histogram data with the light blue points represent noise events and the green shades as area under the fitted curve. (C) OTI CD8+ T cell activation in response to titration of alginate encapsulated mOVA cells with two different cell densities (1x107 or 5x107 cells/mL alginate; top legend; n=3). Means were compared using Tukey's test. Error = standard deviation. (D) Linear correlation between OTI CD8+ T cells activation levels and encapsulated mOVA cell number with two different densities (1x107 in blue or 5x107 in orange cells/mL alginate; top legend), with the regression equations shown and coefficient of determination labeled as R^2. Shaded area = standard deviation. Slope comparison analysis was performed between the regression curves. Statistical difference was determined as * p<0.05; ** p< 0.01; ****p < 0.0001; n.s. = not significant.
Of interest, cellular density within the microcapsules also played a role in modulating the level of CD8+ T cell activation. Specifically, increasing the cell density within the encapsulation platform consistently resulted in amplified responder cell activation, despite an equivalent total cell dosage (Figure 5C). Figure 5D summarizes this phenomenon, where the two cell loading densities resulted in distinct activation trends (p<0.0001, slope comparison, t-test).
3.5. Alginate encapsulated pancreatic islets activate T cells via indirect antigen presentation pathway
To examine the host adaptive immune responses to encapsulated cells relevant to a disease model, pancreatic islets were screened. In this approach, pancreatic islets of mOVA mice were encapsulated within alginate microbeads and cocultured with whole OTI splenic responders to investigate their response. Antigen-specific CD8+ T cell activation in response to mOVA islets was evaluated (Figure 6A). For stringent capture of indirect T cell activation, the complete encapsulation of the antigenic islets was visually validated by hand-picking the microbeads for all experiments (Figure S7A-B).
Figure 6. Alginate Encapsulated Islets Effectively Activate Antigen-Specific T cells via the Indirect Antigen Recognition Pathway.
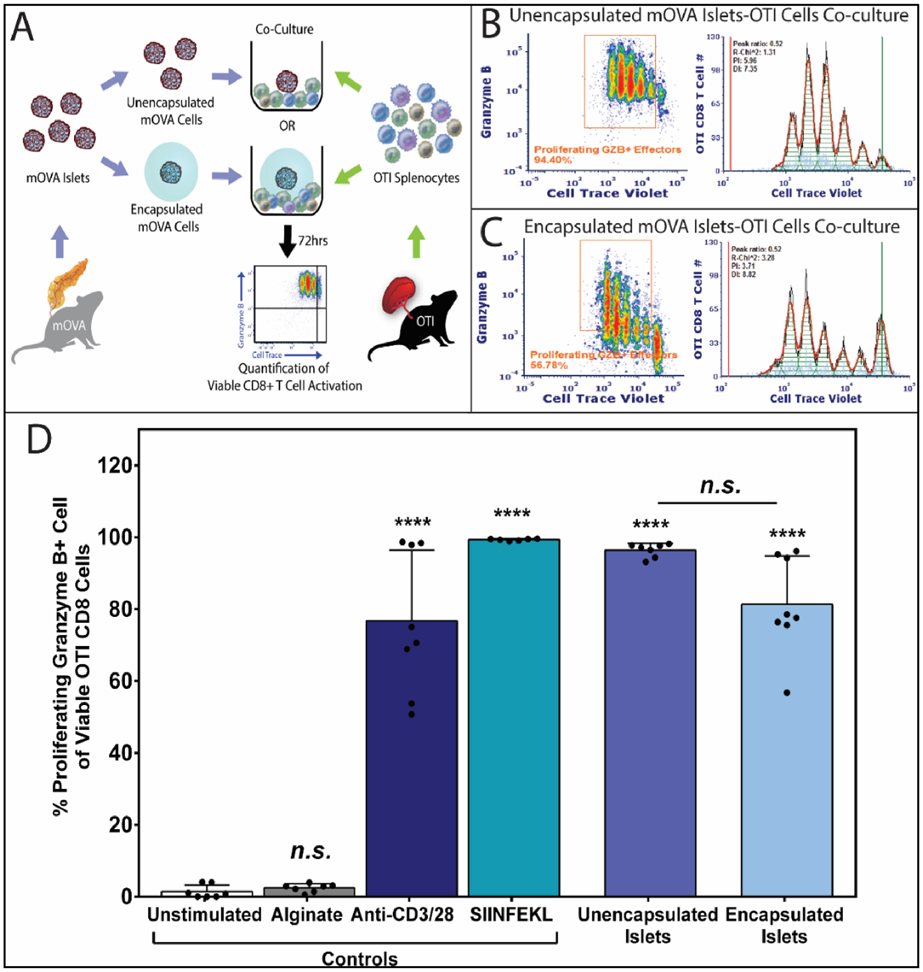
(A) Schematics of OTI splenocytes stimulated by unencapsulated or encapsulated mOVA islets for 72hrs. Antigen-specific CD8+ T cell activation was quantified using the method mentioned above. Representative FCM data of OVA-specific proliferating granzyme B+ OTI CD8+ T cells effector and the respective proliferation fitting when 100,000 OTI responders stimulated by (B) 50 unencapsulated islets or (C) 50 complete encapsulated islets. Proliferation modeling was performed using FCS Express 6 software, with the black lines represent histogram contour of raw data, green lines marked the undivided population and red lines represent basal signal. For proliferating modeling, the orange lines are the fitted histogram data with the light blue points represent noise events and the green shades as area under the fitted curve. (D) Quantification of OVA-specific activated OTI CD8+ T cells, characterized as the percentage of proliferating and granzyme B+ CD8+ T effector cells. Data were shown as mean ± standard deviation of individual data points (N=3; n=9). Outliers were identified using Robust Fit with Cauchy estimate with multiplier K=2, using SAS JMP Pro v13.1.0. software. Tukey’s test was used for mean comparison. Statistical significance was determined as ****p < 0.0001 or n.s. = not significant, with outliers removed for statistical analysis.
As the antigenicity of islets versus splenocytes may be disparate, the cell dosage and culture duration for islet experiments were evaluated (Figure S7C-D). From these trials, an antigen dose of 50 islets, and an extended incubation of 72 hours were identified as optimal (Figure 6A). Similar to the splenocyte coculture setting, polyclonal (anti-CD3/28 activator beads, 76.8 ± 19.7%) and monoclonal (SIINFEKL, 99.3 ± 0.3%) stimulators were used as positive controls, while unstimulated cell culture media (1.4 ± 1.8%) and cell-free alginate microbeads (2.4 ± 1.1%, Figure 6D) served as negative controls. Robust OT1 CD8+ T cell activation in response to both unencapsulated (96.4 ± 1.9%; Figure 6B&D) and encapsulated islets (81.3 ± 13.4%; Figure 6C&D) were observed, indicating strong adaptive indirect immune recognition to the alginate encapsulated islet grafts. While the percentage of viable, granzyme B+ CD8+ T cells responding to unencapsulated or encapsulated mOVA islets was statistically equivalent (p = 0.08; post-hoc Tukey, Figure 6D), proliferation analysis demonstrated a delayed T cell response to the encapsulated islets, characterized by a lowered CD8+ T cell proliferation (PI = 6.06 ± 1.35) when compared to its unencapsulated counterpart (PI = 8.15 ± 1.72, p = 0.04, t-test; Figure 6B-C). These results imply that the hydrogel membrane delays immune recognition; however, once initiated, the resulting graft-specific T cell response is comparable to unencapsulated islets.
3.6. Indirectly activated T cells impair encapsulated islets in a contact-independent manner
With evidence of robust indirect adaptive immune T cell activation to alginate encapsulated cells, it was of interest to evaluate the impact of this immune cell activation on the encased islets. While the alginate barrier impedes direct interactions with cytolytic CD8+ T cells, it is feasible that their activated state may produce an unfavorable microenvironment. To address this hypothesis, islet microbeads cocultured with naïve OT1 splenocytes for 72 h were subsequently evaluated via live/dead viability imaging and glucose-stimulated-insulin-release (GSIR) functional assays.
Aggressive T cell attack to the unencapsulated islets were observed by the co-incubated OTI splenocytes, resulting in completely dissociated islets unamendable to subsequent assessments. For encapsulated mOVA islets, comparative assessments were made between islet co-incubated with OTI splenocytes or cultured alone for the same time period. As shown in Figure 7A&B, the exposure of encapsulated islets to OTI splenocytes for 72 h resulted in detrimental impacts on peripheral islet cells, with a significant decline in viability when compared to untreated islets (p<0.0001 respectively; t-test; Figure 7B).
Figure 7. Indirectly Activated CD8+ T cells Impair the Viability and Function of Alginate Encapsulated Islets.
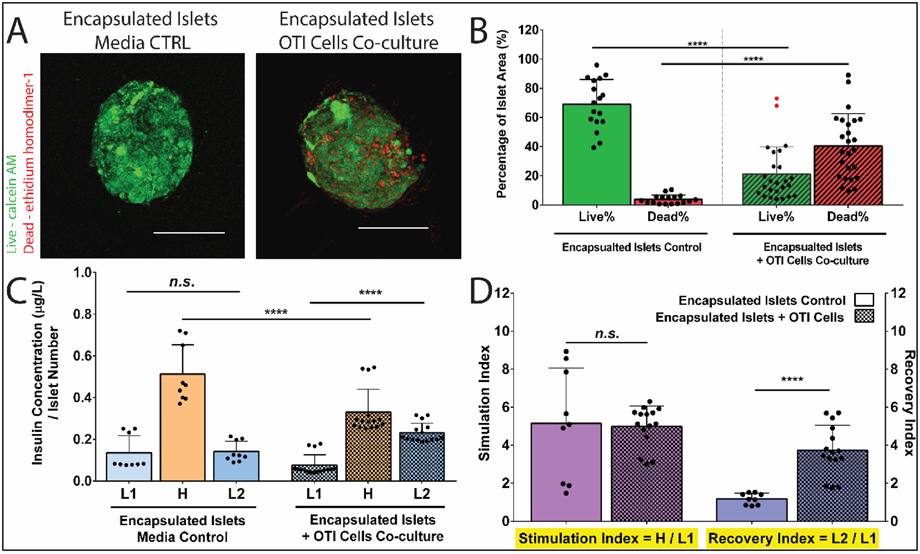
(A) Representative Live/Dead images of alginate encapsulated mOVA islets following the 72hr coculture with OTI splenocytes. Green= viable cells, Red = dead cells. Scale bars = 100 μm. Images analysis (B) quantified the % live cells and % dead cells of islets area (n≥18). Outliers were identified using Robust Fit with Cauchy estimate with multiplier K=2, via SAS JMP Pro v13.1.0. software. Tukey’s test was used for mean comparison for viability quantification including the outliers. (C) Representative GSIR data of alginate encapsulated islets following coculture with OTI splenocytes or media control (n≥9). Encapsulated islets were sequentially stimulated in 3 mM (Low1, L1); 16.7 mM (High, H), and another 3 mM (Low2, L2) glucose for 1hr respectively. Samples were collected after each hour stimulation, and the corresponding insulin level was measured by Elisa. Insulin secretion was normalized by the number of the encapsulated islets for each sample. (D) Stimulation index (SI, the ratio of H/L1) and recovery index (RI, the ratio of L2/L1) of encapsulated islets after the 72hrs coculture (n≥9). Data were shown as mean ± standard deviation. Mean comparison was conducted using Tukey’s test. Statistical significance was determined as **p < 0.01; ****p < 0.0001 or n.s. = not significant.
Islet function, following exposure to the various experimental conditions, was assessed via a glucose-stimulated-insulin response assay. In this test, islets were sequentially incubated in low (3 mM, L1), high (16.7 mM, H), and low (3 mM, L2) glucose-containing buffer for one hour respectively. This assay permitted evaluation of insulin secretion in response to a high glucose challenge, followed by the timely termination of insulin release when returned to basal glucose levels. A healthy islet response demonstrates a classic low-high-low insulin release profile, with a stimulation index (SI: H insulin release / L1 insulin release) > 1 and a recovery index (RI: L2 insulin release / L1 insulin release) ≈ 1 [45, 46]. For control islet microbeads, insulin release in response to glucose challenge was normal, with an SI = 5.15 ± 2.9 and an RI = 1.19 ± 0.3. When incubated with activated OTI CD8+ T cells, the function of encapsulated islets was negatively impacted, with decreased insulin production in response to high glucose stimulation (Hcontrol = 0.51 ± 0.14 vs. HT cell = 0.33 ± 0.10 μg/(L*IN); p<0.0001, t-test, Figure 7C). Impaired recovery of insulin release was also observed for islet microbeads challenged by extracapsular OTI CD8+ T cells, with an aberrantly high insulin release for the second low glucose incubation and a significant increase in RI (3.73 ± 1.32; p<0.0001, compared to control islet microbeads, t-test, Figure 7D).
To further delineate if the cytotoxicity observed in the encapsulated islet was attributed to soluble factors generated by activated T cells, culture media was procured from activated OTI CD8+ T cells and added to encapsulated islet cultures. As summarized in Figure S8, viability and functional assessments illustrate substantial cell death and dysfunction imparted by the activated T cell media. Comparing the responses of unencapsulated and encapsulated islets, the alginate polymer dampens the negative impacts of the activated T cell-conditioned media, with decreased cell death (Figure S8A-E) and improved GSIR (Figure S8F-G) compared to the unencapsulated islets. Nevertheless, the negative impacts of the activated T cell-derived cytotoxic factors on the underlying islets was profound, when compared to standard media controls (Figure S8E&H).
4. Discussion
The concept of using polymeric encapsulating materials, particularly alginate, to prevent host recognition and rejection of foreign cells has been explored for decades. Despite numerous company ventures and clinical trials, the clinical translation of cellular encapsulation-based devices for the treatment of diseases, specifically for autoimmune diabetes, has resulted in disappointing outcomes, with limited functional efficacy and retrieved encapsulated implants containing significantly distressed cells [47, 48]. There are multiple hypotheses as to what features contribute to the clinical failure of alginate encapsulated transplants, e.g., foreign body response, islet stress, and poor engraftment site [49, 50]. Analysis of explants, however, suggests that host immune cells play a role in graft destabilization [47, 48, 51, 52].
As the encapsulating material serves as the interface between the implant and the host, most investigations into host immune responses have focused on the "biocompatibility" of the materials via characterization of host innate immune cells and their corresponding responses. It has been shown that common contaminants in alginate can activate macrophages via NF-κB pathway and promote DC maturation, as measured by the up-regulation of CD86 and HLA-DQ [49, 53, 54]. Consequently, these activated innate cells direct classic foreign body responses that lead to fibrotic encapsulation [55, 56]. Furthermore, incomplete cellular encapsulation or biomaterial instability can contribute to their failure [44]. Modifications in alginate composition, purification, stability, and the final 3-D format have reduced innate immune activation, resulting in decreased fibrotic responses to empty hydrogels and subsequently improving cell transplant outcomes in preclinical models [17, 26, 57-60]. These mechanistic investigations, while important, focus primarily on one facet of the host immune response. As an indispensable factor to enable long-term efficacy of encapsulated grafts, a comprehensive understanding of the pathways of host T cell activation and its immune impacts on the grafts is particularly important.
Animal studies of encapsulated transplants indicate that host adaptive immune cells can sense, respond, and impact encapsulated cellular implants in an antigen-driven manner, supporting the theory that immune cells migrating to the implant site are not just reacting to the biomaterial [61-63]. For example, in rodent studies, the functional duration of encapsulated grafts is significantly reduced if the degree of antigen diversity between the donor and the recipient is elevated (i.e., allografts function longer than xenografts) or if T cells specific to the transplanted cell antigen are present (i.e., native murine recipients accept grafts longer than primed/pre-exposed recipients)[47, 61, 64]. T cell depletion and adoptive transfer studies implicate T cells in the rejection of encapsulated xenograft transplants [61]. The addition of pharmaceutical interventions targeted at host T cell suppression, such as CTLA-Ig and anti-CD154 mAb, have resulted in significant improvements in encapsulated xenograft survival [19, 20, 27]. B cells must also sense the encapsulated graft, as de novo anti-xenograft antibodies emerge following the implantation of encapsulated xenogeneic cells [61, 62, 65]. Altogether, these transplant studies prove the participation of host adaptive immunity in foreign graft recognition and highlight the potentially deleterious impacts of these responses to the encapsulated transplants.
The reliance on animal models to study immune activation pathways and screen new pharmaceutical targets for cellular encapsulation, however, is not ideal, as they cannot delineate host responses initiated by non-specific factors (e.g., the implantation procedure, material compatibility, and the transplant site) from antigen-specific responses. Furthermore, characterization of the distinct roles of different immune cell components and antigen recognition pathways (e.g., direct versus indirect) towards microencapsulated cells is difficult to ascertain in vivo. Finally, the substantial time and expense required for screening different encapsulation and pharmaceutical approaches in rodent models hinder their efficient identification. Developing a validated and effective in vitro platform that permits the clear investigation of host-donor immune interactions could alleviate many of these challenges and potentially advance the efficacy of cell-based therapies by identifying potent immunoprotective targets.
The in vitro platform developed herein was inspired by the classic mixed lymphocyte reaction (MLR), a simple in vitro assay that examines antigen-specific immune reactivity and histocompatibility. In the conventional MLR approach, immune cells from two donor sources are co-cultured, where T cell activation of one strain in response to the stimulator strain is quantified as the readout. Since a successful MLR screen requires robust phylogenetic disparity of the tested strains and an elevated frequency of reactive T cell precursors [28, 66], results from a standard MLR assay exploring naïve T cell reactivity to encapsulated cell grafts are typically weak and poorly reproducible [52, 67, 68]. For studying islets, this limitation can be further exacerbated by the poor proficiency of healthy, unstressed pancreatic beta cells at MHC-I or MHC-II antigen presentation [28, 69, 70].
To overcome these challenges, an mOVA-OTI single-antigen model was employed. mOVA stimulator cells provide the proficient surface presentation of OVA protein, as well as OVA peptide derivatives, such as SIINFEKL [71]. OTI T cells, which carry Vα2/Vβ5 receptors specific for OVA-derived antigen (SIINFEKL), render efficient T cell activation in response to encapsulated mOVA stimulators within a brief culture window (48 to 72 hours), permitting efficient screening [41]. In this study, alginate encapsulation completely inhibited the activation of purified OTI CD8+ T cells in response to mOVA cells. These results highlight that alginate encapsulation, using this specific alginate source and crosslinking method, completely blocks direct donor-host contact to prevent direct T cell activation. Conversely, when the entire repertoire of OTI splenocytes were used as responder cells, robust T cell proliferation and effector function in response to the encapsulated mOVA cells were observed. These results indicate that CD8+ T cells can be activated by alginate encapsulated cells, when other host immune cells, such as APCs, are present. The findings of this platform are the first reported evidence, to our knowledge, that clearly demonstrate the impact of encapsulation on the activation of T cells via indirect recognition pathway in vitro.
To delve into the hypothesis that indirect T cell activation in response to encapsulated cells is facilitated by host antigen presenting cells (APC), mechanistic studies using cross-presenting OTI CD8a+ DCs were conducted. CD8a+ DCs were selected as the APC population of interest due to the MHC-I restricted features of the mOVA-OTI model, which limits antigen-specific T cell activation solely to CD8+ T cells (in lieu of the classic MHC- II APC and CD4+ T cell activation pathway) [34, 72]. CD8a+ DCs also have an established role in apoptotic antigen uptake and presentation triggered by apoptotic and/or necrotic shed antigens, which is a likely pathway for indirect CD8+ T cell activation to encapsulated grafts [73, 74]. Our results found that the combination of CD8a+ DCs with purified CD8+ T cell responders caused vigorous indirect CD8+ T cell activation in response to encapsulated mOVA cells. This activation, meanwhile, was fully suppressed once DC cross-presentation was inhibited, further validating this host-donor contact-independent T cell activation pathway and its dependency on host APC function. Overall, this platform provides definitive evidence that CD8+ T cell activation in response to encapsulated cells persists via donor antigen cross-presentation by CD8a+ DCs. Of note, other immune cells, such as CD4+ T cells, macrophages, and B cells, likely also play a role in the collective adaptive immune responses to encapsulated grafts and are the subject of future studies.
With the observation of both direct and indirect antigen activation pathways, this benchtop assay can be subsequently leveraged to study rejection pathways and optimize new biomaterials for encapsulated cells, as well as to screen complementary immunomodulatory therapies. As an example of this utility, the role of antigen dosage on antigen-specific T cell activation was characterized. OTI CD8+ T cell activation in response to mOVA cell-derived antigens was generally depressed by the encapsulating hydrogels, both in terms of proliferation index and generation of effector T cells. Elevation of antigen dosage via increased total cell number, however, resulted in increased stimulation. Of interest, increasing the encapsulation cell density, while maintaining a constant total number of mOVA cells, resulted in increased T cell activation, when compared to its same cell dose counterpart. These results indicate that the number of cells and the manner by which they are loaded into an encapsulation system may impact downstream adaptive immune responses. These correlations also implicate additional activator factors distinct from the total cell number. Increasing the cell loading density within the microbead can result in an increased percentage of stressed and/or nutritionally deficient cells. Distressed cells are known to release instigative agents, such as damage-associated molecular patterns (known as DAMPS, e.g., high mobility group box-1) and inflammatory cytokines (e.g., IL-8, TNF-α), which can easily permeate out of the hydrogel [44, 75]. Co-delivery of these agents with antigen may exacerbate adaptive immune cell activation. As such, efforts that seek to improve nutrient availability to the grafts, via improved oxygenation, vascularization, and/or transplant sites[50, 76], as well as mitigating islets stress and inflammation by drug delivery[59, 77], can be leveraged to improve the efficacy of the encapsulated cell grafts.
To explore the role of hydrogel permeability in indirect antigen recognition and stimulation, our alginate microbeads were coated with PLL and alginate. These classic APA microbeads exhibit a more restrictive permselectivity (MWCO of ~ 70 kDa vs 125 kDa for alginate only) [32], which has been postulated to reduce immune recognition [44, 47]. For this permeability range and microbead size, no change in downstream CD8+ T cell activation was observed, indicating that the degree of antigen shedding was unaffected. This is likely due to the wide range of OVA-based antigens that can still be released from the cells, with variable structures and formats (e.g. SIINFEKL peptide, 0.94 kDa; ovalbumin protein, 43 kDa; and MHC bound OVA antigens, 20 – 45 kDa)[78]. Exploration of a more permselective coating or different alginate composition could potentially reduce antigen shedding [79, 80]. However, it is likely extremely challenging, if not impossible, to customize a material permselectivity that would completely prohibit antigen release without also restricting nutritional delivery and/or secretagogue exchange. This hypothesis, however, requires further investigation, which can be facilitated by the screening potential of this platform.
To apply this in vitro platform to a disease model, encapsulated pancreatic mOVA islets were investigated. Hand-picked mOVA islets were used as stimulators to OTI splenic responders. While fully encapsulated mOVA islets restricted T cell activation via direct antigen pathways, the coculture of these same encapsulated organoids with whole naïve OT1 splenocytes resulted in the robust generation of cytotoxic CD8+ T cells via the indirect antigen presentation pathway. The coculture time was increased from 2 to 3 days, likely due to the decreased immunogenicity of pancreatic islets compared to splenocytes [28, 69, 70]. Despite this modest delay, T cell activation was robust for both direct and indirect pathways.
After establishing the inability of encapsulation to prevent adaptive immune cell recognition and activation, a subsequent key question is the downstream impact of these activated cells on the encapsulated cells. It is postulated that the modulation of material permselectivity can exclude detrimental agents (e.g., effector T cells, antibodies, complement, and cytokines), with recommended targets of an exclusion criteria of 12 nm hydrodynamic radius (Rh) (e.g., MW ≥ 150 kDa) to prevent the influx of undesired immune agents (e.g., antibodies and large complement), while supporting the free diffusion of desired molecules (e.g., insulin, glucose, transferrin) that are typically Rh < 4 nm (e.g., MW < 80 kDa) [44]. While the UP-MVG alginate microbead used in this study was within this desired range (i.e., MWCO between 125-155 kDa)) [81-83] and prevented direct cell contact between the T effector cells and the encapsulated cells, indirectly activated T cells imparted detrimental effects to the encapsulated islets. Specifically, cell death at the periphery of the encapsulated organoid was observed, implicating an external cytotoxic stimulus, as opposed to insufficient nutritional availability. Islet function was also impaired, with decreased insulin release under a high glucose challenge and a reduced capacity to shut down insulin release following a return to non-stimulatory glucose levels. While the mechanism of persisting high insulin release following exposure to stimulatory glucose is not fully understood, it has been attributed to elevated oxidative stress or ionic channel leakage, which can be induced by exposure to inflammatory cytokines [84, 85]. Supplemental experiments using only activated T cell-conditioned media resulted in similar impairment to the encapsulated islets, further identifying T cell-derived diffusible solutes as the cytotoxic agents. These in vitro observations of immune-mediated cytotoxicity despite retention of the biomaterial barrier correlate with xenograft transplantation studies, which observed downstream impacts of delayed-type xenoantigen hypersensitivity and inflammatory cytokine infiltration that contributed to host immune infiltration and the subsequent graft failure [86-88].
Comparing the cytotoxicity observed for encapsulated versus unencapsulated islets in response to the T cell-condition media, however, clearly illustrate the capacity of the alginate biomaterial to significantly dampen the toxic impacts of these immune-derived solutes. This phenomenon may be related to inhibited and/or delayed diffusion of the agents [9, 89, 90]. As alginate permeability can be modified via alginate properties (e.g., concentration and monomer composition) and/or the fabrication process (e.g. crosslinker selection and encapsulation geometry) [91], this benchtop platform provides ease in investigating the role of porosity in both antigen recognition and downstream cytotoxicity to further optimize these materials. However, as with antigen shedding, the small size of most inflammatory cytokines (e.g. TNF-α: 17.5kDa, Rh ~2.3nm, and IFN-γ: 16.9kDa, Rh ~3.0nm) makes it likely impossible to redesign material permeability to completely prevent their diffusion into the material [92, 93]. However, as the variable features of the diffusing agent (e.g. MW, charge, shape) can impact their mobility through a hydrogel matrix, the identification of key cytokines responsible for this cytotoxicity can inform material design to target the selected restriction of these agents; this area of exploration could also be efficiently screened using this benchtop platform.
While this assay provides a powerful in vitro platform for immune screening, it is fully recognized that the efficiency of this platform is skewed by the high affinity of the transgenic Vα2/Vβ5 T cell receptors to OVA peptides, as well as the elevated antigen presence associated with the mOVA cells [94]. In vivo, the timeline of T cell activation would be extended, given the need for antigen uptake and processing by APCs, trafficking, antigen matching, and clonal expansion. However, the ease by which APC-mediated T cell activation occurs in our clonally specific platform permits clear evidence that the indirect antigen pathway is fully functional for encapsulated grafts. In addition, this clonal specificity serves to mimic clinical scenarios where indirect antigen presentation and T cell activation are dominant, such as in established autoimmunity [23, 28, 95].
Using this in vitro screening approach, future work will focus on exploring the roles of non-cross-presenting APCs and CD4+ T cells in indirect T cell activation, e.g., by adjusting the responder cells population or using MHC-II restricted OVA-specific responders [96]. Moreover, the system will be leveraged to screen immunomodulatory biomaterials targeted to dampen the unique adaptive immune cell pathways activated by encapsulated cells. In this manner, this in vitro platform could provide efficient and inexpensive screening and optimization of agents with validation of results using animal transplant models.
5. Conclusions
In this study, we successfully developed an efficient, antigen-specific in vitro coculture platform with robust reproducibility to study host adaptive immune responses to hydrogel encapsulated cell grafts. Leveraging our platform, we conclude, with detailed experimental evidence, that alginate microencapsulation effectively blocks the host-to-donor, contact-dependent, direct T cell activation, while host APCs-mediated indirect T cell activation to encapsulated cell grafts is preserved. Subsequently, indirect T cell activation imparts detrimental effects on the encapsulated cell grafts via the diffusion of soluble cytotoxic agents. Overall, this reported mOVA-OTI in vitro platform can inform the design of bioactive encapsulating materials that target the suppression of adaptive immune recognition for improved clinical outcomes for cell replacement therapy.
Supplementary Material
Highlights.
An efficient single-antigen benchtop model capable of characterizing adaptive immune responses to biomaterial encapsulated cells was developed.
The platform determined that encapsulated cells/organoids activate host T cells via an APCs-mediated indirect recognition pathway.
The amount and density of the encapsulated cells direct the magnitude of indirect T cell activation.
The resulting T effector cells were cytotoxic to the encaged organoids across the biomaterial barrier.
This benchtop platform could be used in future studies to screen immune interventions for therapeutic applications.
Acknowledgments
This work was supported by NIH grants DK100654 and DK122638. We thank Drs. Craig Moneypenny and Andria Doty of the University of Florida ICBR Cytometry Core and Howie Seay of UF Center for Immunology and Transplantation for cell sort assistance; Chad Rancourt of Animal Care Services of the UF for the assistance of animal breeding; and Cecilia Cabello of the University of Miami for the assistance of animal genotyping. We also thank all members of the Stabler lab for their collective assistance in animal care and monitoring, as well as islet isolations.
Appendix
Summary of Abbreviation
- APA
Alginate/poly-L-lysine/alginate
- APC
Antigen Presenting Cell
- DC
Dendritic cell
- DI
Division index
- DAMPS
Damage-associated molecular patterns
- ELISA
Enzyme-linked immunosorbent assay
- FCM
Flow cytometry
- GFP
Green Fluorescent Protein
- GSIR
Glucose stimulated insulin response
- HBSS
Hank’s Balanced Salt solution
- mAb
Monoclonal antibody
- MHC
major histocompatibility complex
- MLR
Mixed lymphocyte reaction
- mOVA
membrane-bound ovalbumin
- MWCO
Molecular weight cutoff
- OVA
Ovalbumin
- PBS
Phosphate buffered saline
- PI
Proliferation index
- RI
Recovery index
- ROS
Reactive oxygen species
- SI
Stimulation index
- T1D
Type 1 Diabetes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
Data Availability
The raw data required to reproduce these findings are available upon request to the corresponding author
References
- [1].Shapiro AM, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD, Berney T, Brennan DC, Cagliero E, Alejandro R, Ryan EA, DiMercurio B, Morel P, Polonsky KS, Reems JA, Bretzel RG, Bertuzzi F, Froud T, Kandaswamy R, Sutherland DE, Eisenbarth G, Segal M, Preiksaitis J, Korbutt GS, Barton FB, Viviano L, Seyfert-Margolis V, Bluestone J, Lakey JR, International trial of the Edmonton protocol for islet transplantation, N Engl J Med 355(13) (2006) 1318–30. [DOI] [PubMed] [Google Scholar]
- [2].Hering BJ, Clarke WR, Bridges ND, Eggerman TL, Alejandro R, Bellin MD, Chaloner K, Czarniecki CW, Goldstein JS, Hunsicker LG, Kaufman DB, Korsgren O, Larsen CP, Luo X, Markmann JF, Naji A, Oberholzer J, Posselt AM, Rickels MR, Ricordi C, Robien MA, Senior PA, Shapiro AM, Stock PG, Turgeon NA, C. Clinical Islet Transplantation, Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia, Diabetes Care 39(7) (2016) 1230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Toledo PC, Rossi RL, Caviedes P, Microencapsulation of Parathyroid Cells for the Treatment of Hypoparathyroidism, Methods Mol Biol 1479 (2017) 357–363. [DOI] [PubMed] [Google Scholar]
- [4].Eyjolfsdottir H, Eriksdotter M, Linderoth B, Lind G, Juliusson B, Kusk P, Almkvist O, Andreasen N, Blennow K, Ferreira D, Westman E, Nennesmo I, Karami A, Darreh-Shori T, Kadir A, Nordberg A, Sundstrom E, Wahlund LO, Wall A, Wiberg M, Winblad B, Seiger A, Wahlberg L, Almqvist P, Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer's disease patients: application of a second-generation encapsulated cell biodelivery device, Alzheimers Res Ther 8(1) (2016) 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chatenoud L, Chemical immunosuppression in islet transplantation--friend or foe?, N Engl J Med 358(11) (2008) 1192–3. [DOI] [PubMed] [Google Scholar]
- [6].Van Belle T, von Herrath M, Immunosuppression in islet transplantation, J Clin Invest 118(5) (2008) 1625–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stabler CL, Li Y, Stewart JM, Keselowsky BG, Engineering immunomodulatory biomaterials for type 1 diabetes, Nature Reviews Materials 4(6) (2019) 429–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Desai T, Shea LD, Advances in islet encapsulation technologies, Nat Rev Drug Discov 16 (2016) 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Olabisi RM, Cell microencapsulation with synthetic polymers, J Biomed Mater Res A 103(2) (2015) 846–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].de Vos P, van Hoogmoed CG, van Zanten J, Netter S, Strubbe JH, Busscher HJ, Long-term biocompatibility, chemistry, and function of microencapsulated pancreatic islets, Biomaterials 24(2) (2003) 305–12. [DOI] [PubMed] [Google Scholar]
- [11].Hasse C, Bohrer T, Barth P, Stinner B, Cohen R, Cramer H, Zimmermann U, Rothmund M, Parathyroid xenotransplantation without immunosuppression in experimental hypoparathyroidism: long-term in vivo function following microencapsulation with a clinically suitable alginate, World J Surg 24(11) (2000) 1361–6. [DOI] [PubMed] [Google Scholar]
- [12].Qi M, Morch Y, Lacik I, Formo K, Marchese E, Wang Y, Danielson KK, Kinzer K, Wang S, Barbaro B, Kollarikova G, Chorvat D Jr., Hunkeler D, Skjak-Braek G, Oberholzer J, Strand BL, Survival of human islets in microbeads containing high guluronic acid alginate crosslinked with Ca2+ and Ba2+, Xenotransplantation 19(6) (2012) 355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Safley SA, Kenyon NS, Berman DM, Barber GF, Willman M, Duncanson S, Iwakoshi N, Holdcraft R, Gazda L, Thompson P, Badell IR, Sambanis A, Ricordi C, Weber CJ, Microencapsulated adult porcine islets transplanted intraperitoneally in streptozotocin-diabetic non-human primates, Xenotransplantation 25(6) (2018) e12450. [DOI] [PubMed] [Google Scholar]
- [14].Kirchhof N, Shibata S, Wijkstrom M, Kulick DM, Salerno CT, Clemmings SM, Heremans Y, Galili U, Sutherland DE, Dalmasso AP, Hering BJ, Reversal of diabetes in non-immunosuppressed rhesus macaques by intraportal porcine islet xenografts precedes acute cellular rejection, Xenotransplantation 11(5) (2004) 396–407. [DOI] [PubMed] [Google Scholar]
- [15].Kollmer M, Appel AA, Somo SI, Brey EM, Long-Term Function of Alginate-Encapsulated Islets, Tissue Eng Part B Rev 22(1) (2016) 34–46. [DOI] [PubMed] [Google Scholar]
- [16].Tuch BE, Keogh GW, Williams LJ, Wu W, Foster JL, Vaithilingam V, Philips R, Safety and viability of microencapsulated human islets transplanted into diabetic humans, Diabetes Care 32(10) (2009) 1887–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Paredes Juarez GA, Spasojevic M, Faas MM, de Vos P, Immunological and technical considerations in application of alginate-based microencapsulation systems, Front Bioeng Biotechnol 2 (2014) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Korsgren O, Islet Encapsulation: Physiological Possibilities and Limitations, Diabetes 66(7) (2017) 1748–1754. [DOI] [PubMed] [Google Scholar]
- [19].Weber CJ, Hagler MK, Chryssochoos JT, Kapp JA, Korbutt GS, Rajotte RV, Linsley PS, CTLA4-Ig prolongs survival of microencapsulated neonatal porcine islet xenografts in diabetic NOD mice, Cell Transplant 6(5) (1997) 505–8. [DOI] [PubMed] [Google Scholar]
- [20].Kobayashi T, Harb G, Rayat GR, Prolonged survival of microencapsulated neonatal porcine islets in mice treated with a combination of anti-CD154 and anti-LFA-1 monoclonal antibodies, Transplantation 80(6) (2005) 821–7. [DOI] [PubMed] [Google Scholar]
- [21].Wilson JT, Chaikof EL, Challenges and emerging technologies in the immunoisolation of cells and tissues, Advanced Drug Delivery Reviews 60(2) (2008) 124–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Babensee JE, Anderson JM, McIntire LV, Mikos AG, Host response to tissue engineered devices, Adv Drug Deliv Rev 33(1) (1998) 111–139. [DOI] [PubMed] [Google Scholar]
- [23].Lin CM, Gill RG, Direct and indirect allograft recognition: pathways dictating graft rejection mechanisms, Curr Opin Organ Transplant 21(1) (2016) 40–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ali JM, Bolton EM, Bradley JA, Pettigrew GJ, Allorecognition pathways in transplant rejection and tolerance, Transplantation 96(8) (2013) 681–8. [DOI] [PubMed] [Google Scholar]
- [25].Kupfer T, Beilke JN, Pham K, Buhrman J, Gill RG, "Indirect" acute islet allograft destruction in nonobese diabetic mice is independent of donor major histocompatibility complex and requires host B lymphocytes, Transplant Proc 40(2) (2008) 462–3. [DOI] [PubMed] [Google Scholar]
- [26].Bochenek MA, Veiseh O, Vegas AJ, McGarrigle JJ, Qi M, Marchese E, Omami M, Doloff JC, Mendoza-Elias J, Nourmohammadzadeh M, Khan A, Yeh CC, Xing Y, Isa D, Ghani S, Li J, Landry C, Bader AR, Olejnik K, Chen M, Hollister-Lock J, Wang Y, Greiner DL, Weir GC, Strand BL, Rokstad AMA, Lacik I, Langer R, Anderson DG, Oberholzer J, Alginate encapsulation as long-term immune protection of allogeneic pancreatic islet cells transplanted into the omental bursa of macaques, Nat Biomed Eng 2(11) (2018) 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Safley SA, Cui H, Cauffiel SM, Xu BY, Wright JR Jr., Weber CJ, Encapsulated piscine (tilapia) islets for diabetes therapy: studies in diabetic NOD and NOD-SCID mice, Xenotransplantation 21(2) (2014) 127–39. [DOI] [PubMed] [Google Scholar]
- [28].Rayat GR, Johnson ZA, Beilke JN, Korbutt GS, Rajotte RV, Gill RG, The degree of phylogenetic disparity of islet grafts dictates the reliance on indirect CD4 T-cell antigen recognition for rejection, Diabetes 52(6) (2003) 1433–40. [DOI] [PubMed] [Google Scholar]
- [29].Ehst BD, Ingulli E, Jenkins MK, Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection, Am J Transplant 3(11) (2003) 1355–62. [DOI] [PubMed] [Google Scholar]
- [30].Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ, A protocol for islet isolation from mouse pancreas, Nat Protoc 4(11) (2009) 1649–52. [DOI] [PubMed] [Google Scholar]
- [31].Pedraza E, Brady AC, Fraker CA, Molano RD, Sukert S, Berman DM, Kenyon NS, Pileggi A, Ricordi C, Stabler CL, Macroporous three-dimensional PDMS scaffolds for extrahepatic islet transplantation, Cell Transplant 22(7) (2013) 1123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gattas-Asfura K, Valdes M, Celik E, Stabler C, Covalent layer-by-layer assembly of hyperbranched polymers on alginate microcapsulesto impart stability and permselectivity, J Mater Chem B 2(46) (2014) 8208–8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ricordi C, Gray DW, Hering BJ, Kaufman DB, Warnock GL, Kneteman NM, Lake SP, London NJ, Socci C, Alejandro R, et al. , Islet isolation assessment in man and large animals, Acta Diabetol Lat 27(3) (1990) 185–95. [DOI] [PubMed] [Google Scholar]
- [34].Thacker RI, Janssen EM, Cross-presentation of cell-associated antigens by mouse splenic dendritic cell populations, Front Immunol 3 (2012) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Frei AW, Li Y, Jiang K, Buchwald P, Stabler CL, Local delivery of fingolimod from three-dimensional scaffolds impacts islet graft efficacy and microenvironment in a murine diabetic model, J Tissue Eng Regen Med 12(2) (2018) 393–404. [DOI] [PubMed] [Google Scholar]
- [36].Qi M, Strand BL, Morch Y, Lacik I, Wang Y, Salehi P, Barbaro B, Gangemi A, Kuechle J, Romagnoli T, Hansen MA, Rodriguez LA, Benedetti E, Hunkeler D, Skjak-Braek G, Oberholzer J, Encapsulation of human islets in novel inhomogeneous alginate-ca2+/ba2+ microbeads: in vitro and in vivo function, Artif Cells Blood Substit Immobil Biotechnol 36(5) (2008) 403–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Darrabie MD, Kendall WF, Opara EC, Effect of alginate composition and gelling cation on micro-bead swelling, J Microencapsul 23(1) (2006) 29–37. [DOI] [PubMed] [Google Scholar]
- [38].Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC, Granzyme B: a natural born killer, Immunological Reviews 193(1) (2003) 31–38. [DOI] [PubMed] [Google Scholar]
- [39].Harty JT, Badovinac VP, Shaping and reshaping CD8+ T-cell memory, Nat Rev Immunol 8 (2008) 107. [DOI] [PubMed] [Google Scholar]
- [40].Klein AB, Witonsky SG, Ahmed SA, Holladay SD, Gogal RM Jr., Link L, Reilly CM, Impact of different cell isolation techniques on lymphocyte viability and function, J Immunoassay Immunochem 27(1) (2006) 61–76. [DOI] [PubMed] [Google Scholar]
- [41].Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR, Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection, Immunol Cell Biol 78(2) (2000) 110–7. [DOI] [PubMed] [Google Scholar]
- [42].Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, Belz GT, Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity, Nat Immunol 5(11) (2004) 1143–8. [DOI] [PubMed] [Google Scholar]
- [43].Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, Davey G, Kupresanin F, Li M, Maraskovsky E, Belz GT, Carbone FR, Shortman K, Heath WR, Villadangos JA, The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture, Proc Natl Acad Sci U S A 103(28) (2006) 10729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Barkai U, Rotem A, de Vos P, Survival of encapsulated islets: More than a membrane story, World Journal of Transplantation 6(1) (2016) 69–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pizarro-Delgado J, Hernandez-Fisac I, Martin-Del-Rio R, Tamarit-Rodriguez J, Branched-chain 2-oxoacid transamination increases GABA-shunt metabolism and insulin secretion in isolated islets, Biochem J 419(2) (2009) 359–68. [DOI] [PubMed] [Google Scholar]
- [46].Buchwald P, Tamayo-Garcia A, Manzoli V, Tomei AA, Stabler CL, Glucose-stimulated insulin release: Parallel perifusion studies of free and hydrogel encapsulated human pancreatic islets, Biotechnol Bioeng 115(1) (2018) 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Scharp DW, Marchetti P, Encapsulated islets for diabetes therapy: History, current progress, and critical issues requiring solution, Advanced Drug Delivery Reviews 67-68 (2014) 35–73. [DOI] [PubMed] [Google Scholar]
- [48].Matsumoto S, Abalovich A, Wechsler C, Wynyard S, Elliott RB, Clinical Benefit of Islet Xenotransplantation for the Treatment of Type 1 Diabetes, EBioMedicine 12 (2016) 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Paredes-Juarez GA, de Haan BJ, Faas MM, de Vos P, The role of pathogen-associated molecular patterns in inflammatory responses against alginate based microcapsules, J Control Release 172(3) (2013) 983–92. [DOI] [PubMed] [Google Scholar]
- [50].Coronel MM, Stabler CL, Engineering a local microenvironment for pancreatic islet replacement, Current Opinion in Biotechnology 24(5) (2013) 900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Omer A, Duvivier-Kali V, Fernandes J, Tchipashvili V, Colton CK, Weir GC, Long-term normoglycemia in rats receiving transplants with encapsulated islets, Transplantation 79(1) (2005) 52–8. [DOI] [PubMed] [Google Scholar]
- [52].Duvivier-Kali VF, Omer A, Parent RJ, O’Neil JJ, Weir GC, Complete protection of islets against allorejection and autoimmunity by a simple barium-alginate membrane, Diabetes 50(8) (2001) 1698–705. [DOI] [PubMed] [Google Scholar]
- [53].Yang D, Jones KS, Effect of alginate on innate immune activation of macrophages, J Biomed Mater Res A 90(2) (2009) 411–8. [DOI] [PubMed] [Google Scholar]
- [54].Park J, Babensee JE, Differential functional effects of biomaterials on dendritic cell maturation, Acta Biomater 8(10) (2012) 3606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].de Vos P, Smedema I, van Goor H, Moes H, van Zanten J, Netters S, de Leij LF, de Haan A, de Haan BJ, Association between macrophage activation and function of micro-encapsulated rat islets, Diabetologia 46(5) (2003) 666–73. [DOI] [PubMed] [Google Scholar]
- [56].Krishnan R, Ko D, Foster CE 3rd, Liu W, Smink AM, de Haan B, De Vos P, Lakey JR, Immunological Challenges Facing Translation of Alginate Encapsulated Porcine Islet Xenotransplantation to Human Clinical Trials, Methods Mol Biol 1479 (2017) 305–333. [DOI] [PubMed] [Google Scholar]
- [57].De Vos P, De Haan BJ, Wolters GH, Strubbe JH, Van Schilfgaarde R, Improved biocompatibility but limited graft survival after purification of alginate for microencapsulation of pancreatic islets, Diabetologia 40(3) (1997) 262–70. [DOI] [PubMed] [Google Scholar]
- [58].Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, Fenton P, Kang JW, Hollister-Locke J, Bochenek MA, Chiu A, Siebert S, Tang K, Jhunjhunwala S, Aresta-Dasilva S, Dholakia N, Thakrar R, Vietti T, Chen M, Cohen J, Siniakowicz K, Qi M, McGarrigle J, Graham AC, Lyle S, Harlan DM, Greiner DL, Oberholzer J, Weir GC, Langer R, Anderson DG, Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates, Nat Biotechnol 34(3) (2016) 345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mooranian A, Negrulj R, Arfuso F, Al-Salami H, Characterization of a novel bile acid-based delivery platform for microencapsulated pancreatic beta-cells, Artif Cells Nanomed Biotechnol 44(1) (2016) 194–200. [DOI] [PubMed] [Google Scholar]
- [60].Mooranian A, Negrulj R, Al-Salami H, Morahan G, Jamieson E, Designing anti-diabetic beta-cells microcapsules using polystyrenic sulfonate, polyallylamine, and a tertiary bile acid: Morphology, bioenergetics, and cytokine analysis, Biotechnol Prog 32(2) (2016) 501–9. [DOI] [PubMed] [Google Scholar]
- [61].Kobayashi T, Harb G, Rajotte RV, Korbutt GS, Mallett AG, Arefanian H, Mok D, Rayat GR, Immune mechanisms associated with the rejection of encapsulated neonatal porcine islet xenografts, Xenotransplantation 13(6) (2006) 547–59. [DOI] [PubMed] [Google Scholar]
- [62].Duvivier-Kali VF, Omer A, Lopez-Avalos MD, O'Neil JJ, Weir GC, Survival of Microencapsulated Adult Pig Islets in Mice In Spite of an Antibody Response, American Journal of Transplantation 4(12) (2004) 1991–2000. [DOI] [PubMed] [Google Scholar]
- [63].Siebers U, Horcher A, Brandhorst H, Brandhorst D, Hering B, Federlin K, Bretzel RG, Zekorn T, Analysis of the cellular reaction towards microencapsulated xenogeneic islets after intraperitoneal transplantation, J Mol Med (Berl) 77(1) (1999) 215–8. [DOI] [PubMed] [Google Scholar]
- [64].Weaver JD, Headen DM, Coronel MM, Hunckler MD, Shirwan H, García AJ, Synthetic poly(ethylene glycol)-based microfluidic islet encapsulation reduces graft volume for delivery to highly vascularized and retrievable transplant site, American Journal of Transplantation 19(5) (2019) 1315–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jones KS, Sefton MV, Gorczynski RM, In Vivo Recognition by the Host Adaptive Immune System of Microencapsulated Xenogeneic Cells, Transplantation 78(10) (2004) 1454–1462. [DOI] [PubMed] [Google Scholar]
- [66].Sherman LA, Chattopadhyay S, The molecular basis of allorecognition, Annu Rev Immunol 11 (1993) 385–402. [DOI] [PubMed] [Google Scholar]
- [67].Lee DY, Nam JH, Byun Y, Effect of polyethylene glycol grafted onto islet capsules on prevention of splenocyte and cytokine attacks, Journal of Biomaterials Science, Polymer Edition 15(6) (2004) 753–766. [DOI] [PubMed] [Google Scholar]
- [68].Godlewska E, Sitarek E, Iwahńska M, Orlowski T, In vitro activation of mice splenocytes by free and encapsulated rat islets and by components of capsular wall, Transplantation Proceedings 34(2) (2002) 659–660. [DOI] [PubMed] [Google Scholar]
- [69].Calderon B, Carrero JA, Unanue ER, The central role of antigen presentation in islets of Langerhans in autoimmune diabetes, Curr Opin Immunol 26 (2014) 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hamilton-Williams EE, Palmer SE, Charlton B, Slattery RM, Beta cell MHC class I is a late requirement for diabetes, Proc Natl Acad Sci U S A 100(11) (2003) 6688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Miyagawa F, Gutermuth J, Zhang H, Katz SI, The use of mouse models to better understand mechanisms of autoimmunity and tolerance, J Autoimmun 35(3) (2010) 192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tan JK, Quah BJ, Griffiths KL, Periasamy P, Hey YY, O’Neill HC, Identification of a novel antigen cross-presenting cell type in spleen, J Cell Mol Med 15(5) (2011) 1189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Blachere NE, Darnell RB, Albert ML, Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation, PLoS Biol 3(6) (2005) e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nierkens S, Tel J, Janssen E, Adema GJ, Antigen cross-presentation by dendritic cell subsets: one general or all sergeants?, Trends Immunol 34(8) (2013) 361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Cheng Y, Xiong J, Chen Q, Xia J, Zhang Y, Yang X, Tao K, Zhang S, He S, Hypoxia/reoxygenation-induced HMGB1 translocation and release promotes islet proinflammatory cytokine production and early islet graft failure through TLRs signaling, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1863(2) (2017) 354–364. [DOI] [PubMed] [Google Scholar]
- [76].Smink AM, de Haan BJ, Lakey JRT, de Vos P, Polymer scaffolds for pancreatic islet transplantation — Progress and challenges, American Journal of Transplantation 18(9) (2018) 2113–2119. [DOI] [PubMed] [Google Scholar]
- [77].Mooranian A, Negrulj R, Al-Salami H, The incorporation of water-soluble gel matrix into bile acid-based microcapsules for the delivery of viable beta-cells of the pancreas, in diabetes treatment: biocompatibility and functionality studies, Drug Deliv Transl Res 6(1) (2016) 17–23. [DOI] [PubMed] [Google Scholar]
- [78].Fremont DH, Stura EA, Matsumura M, Peterson PA, Wilson IA, Crystal structure of an H-2Kb-ovalbumin peptide complex reveals the interplay of primary and secondary anchor positions in the major histocompatibility complex binding groove, Proc Natl Acad Sci U S A 92(7) (1995) 2479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Thu B, Bruheim P, Espevik T, Smidsrød O, Soon-Shiong P, Skjåk-Bræk G, Alginate polycation microcapsules: I. Interaction between alginate and polycation, Biomaterials 17(10) (1996) 1031–1040. [DOI] [PubMed] [Google Scholar]
- [80].De Vos P, De Haan B, Van Schilfgaarde R, Effect of the alginate composition on the biocompatibility of alginate-polylysine microcapsules, Biomaterials 18(3) (1997) 273–278. [DOI] [PubMed] [Google Scholar]
- [81].Wee S, Gombotz WR, Protein release from alginate matrices, Adv Drug Deliv Rev 31(3) (1998) 267–285. [DOI] [PubMed] [Google Scholar]
- [82].Andresen I-L, Skipnes O, Smidsrod O, Ostgaard K, Hemmer PC, Some biological functions of matrix components in benthic algae in relation to their chemistry and the composition of seawater, ACS Publications; (1977). [Google Scholar]
- [83].Shoichet MS, Li RH, White ML, Winn SR, Stability of hydrogels used in cell encapsulation: An in vitro comparison of alginate and agarose, Biotechnol Bioeng 50(4) (1996) 374–81. [DOI] [PubMed] [Google Scholar]
- [84].Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, Collins S, Andersen ME, ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function, Toxicol Appl Pharmacol 244(1) (2010) 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Garcia-Contreras M, Tamayo-Garcia A, Pappan KL, Michelotti GA, Stabler CL, Ricordi C, Buchwald P, Metabolomics Study of the Effects of Inflammation, Hypoxia, and High Glucose on Isolated Human Pancreatic Islets, J Proteome Res 16(6) (2017) 2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dufrane D, Gianello P, Macro- or microencapsulation of pig islets to cure type 1 diabetes, World J Gastroenterol 18(47) (2012) 6885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Soderlund J, Wennberg L, Castanos-Velez E, Biberfeld P, Zhu S, Tibell A, Groth CG, Korsgren O, Fetal porcine islet-like cell clusters transplanted to cynomolgus monkeys: an immunohistochemical study, Transplantation 67(6) (1999) 784–91. [DOI] [PubMed] [Google Scholar]
- [88].Cole DR, Waterfall M, McIntyre M, Baird JD, Microencapsulated islet grafts in the BB/E rat: a possible role for cytokines in graft failure, Diabetologia 35(3) (1992) 231–7. [DOI] [PubMed] [Google Scholar]
- [89].Olabisi RM, Cell microencapsulation with synthetic polymers, Journal of Biomedical Materials Research Part A 103(2) (2015) 846–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kulseng B, Thu B, Espevik T, Skjak-Braek G, Alginate polylysine microcapsules as immune barrier: permeability of cytokines and immunoglobulins over the capsule membrane, Cell Transplant 6(4) (1997) 387–94. [DOI] [PubMed] [Google Scholar]
- [91].Mørch YA, Novel Alginate Microcapsules for Cell Therapy–A study of the structure-function relationships in native and structurally engineered alginates, (2008).
- [92].Narhi LO, Arakawa T, Dissociation of recombinant tumor necrosis factor-alpha studied by gel permeation chromatography, Biochem Biophys Res Commun 147(2) (1987) 740–6. [DOI] [PubMed] [Google Scholar]
- [93].Perez Sanchez H, Tatarenko K, Nigen M, Pavlov G, Imberty A, Lortat-Jacob H, Garcia de la Torre J, Ebel C, Organization of human interferon gamma-heparin complexes from solution properties and hydrodynamics, Biochemistry 45(44) (2006) 13227–38. [DOI] [PubMed] [Google Scholar]
- [94].Richard AC, Lun ATL, Lau WWY, Gottgens B, Marioni JC, Griffiths GM, T cell cytolytic capacity is independent of initial stimulation strength, Nat Immunol 19(8) (2018) 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Vendrame F, Pileggi A, Laughlin E, Allende G, Martin-Pagola A, Molano RD, Diamantopoulos S, Standifer N, Geubtner K, Falk BA, Ichii H, Takahashi H, Snowhite I, Chen Z, Mendez A, Chen L, Sageshima J, Ruiz P, Ciancio G, Ricordi C, Reijonen H, Nepom GT, Burke GW 3rd, Pugliese A, Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T-cells, Diabetes 59(4) (2010) 947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Barnden MJ, Allison J, Heath WR, Carbone FR, Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements, Immunol Cell Biol 76(1) (1998) 34–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.