

Abstract
Immunotherapies are becoming a promising strategy for malignant disease. Selectively directing host immune responses to target cancerous tissue is a milestone of human health care. The roles of the innate and adaptive immune systems in both cancer progression and elimination are now being realized. Defining the immune cell environment and identifying the contributions of each sub-population of these cells has lead to an understanding of the immunotherapeutic processes, and demonstrated the potential of the immune system to drive cancer shrinkage and sustained immunity against disease. Poorly treated diseases, such as high-grade glioma, suffer from lack of therapeutic efficacy and rapid progression. Immunotherapeutic success in other solid malignancies, such as melanoma, now provides the principals for which this treatment paradigm can be adapted for primary brain cancers. The central nervous system is complex, and relative contributions of immune sub-populations to high grade glioma progression are not fully characterized. Here, we summarize recent research in both animal and humans which add to the knowledge base of how innate and adaptive immune cells contribute to glioma progression, and outline work which has demonstrated their potential to elicit anti-tumorigenic responses. Additionally, we highlight Neuropilin 1, a cell surface receptor protein, describe its signaling functions in the context of immunity, and point to its potential to slow glioma progression.
Keywords: glioma TME, immunotherapy, T Cells, GAMs, neuropilin 1
1. Introduction
High grade gliomas (HGG), such as grade IV Glioblastoma Multiform (GBM), are the most common and lethal primary tumors arising in the CNS [1]. GBM are viciously invasive, present chemo- and radio-therapy resistance, are histologically heterogeneous, and more recently have been classified by molecular subtypes [1–4]. Standard therapy minimally increases median survival and involves maximal surgical debulking followed by Temozolomide and radiation treatment regiments [5]. GBM can arise from glial cells throughout the brain, and often results from malignant progression of grade II/III gliomas. The pathological hallmarks of glioma progression parallel those in other malignant solid tumor types, such as the dependency on vascular remodeling and angiogenesis, local tissue invasion, immune evasion, and resistance to standard of care therapies.
Increasing evidence supports the concept that the tumor microenvironment (TME) plays a modulatory role in glioma progression. The TME consists of non-cancerous stromal cell types, all of which ultimately contribute to maintenance and health of the bulk tumor [6]. While the CNS was once believed to be immune privileged, peripheral immune influences on CNS diseases is now a pragmatic subject. Here we discuss how major immune cell populations contribute to the progression and maintenance of HGG, and outline their potential to mitigate the advancement of disease. Lastly, we discuss Neuropillin 1 (NRP1), a prominent cell surface protein receptor with many distinct ligands, as a potential therapeutic target across immune cell populations, and suggest that NRP1 could be exploited in developing new treatments for GBM.
2. Discrete roles of immune cell populations in glioma progression, maintenance, and regression
2.1. Subpopulations of lymphocytes affect glioma progression
Although lymphoid populations vary across GBM cohorts, increased tumor infiltrating lymphocytes (TILs) correlate with glioma grade, but can also correlate with increased survival [7–9]. This paradox may be elucidated by considering the various lymphocytic populations present in the TME, and by identifying their contributions to tumor development. Naive CD4+ lymphocytes arise from hematopoietic thymic progenitors and are activated via MHC II antigen presentation on antigen presenting cells (APCs). Antigen exposure and humoral signaling initiate CD4+ T cells to expand into a variety of effector subsets. Polarized Th1/Th2 helper T cells (Th) are canonically derived from IL-12/INFγ or IL-4 exposure, respectively [10]. T regulatory cells (Treg) are another important subset of effector lymphocytes, which are potently induced following TGFβ exposure [11]. CD4+ Th cells have the ability to mount inflammatory responses, as well as activate processes of adaptive immunity. Alternatively, CD4+CD25+FOXP3+ Tregs are classically associated with immunosuppression, attenuation of autoimmunity, and the inhibition of CD4+ proliferation [11]. CD4+ cells are required for adaptive immune system activation, and thus, their presence would be expected to correlate with a stronger anti-tumoral adaptive immune response. However, the influences CD4+ Th2 and CD4+FOXP3+ Treg cells have within the TME have not been fully clarified in glioma.
Fecci et al. have previously reported that patients with GBM present with CD4+ lymphopenia, but noticeably also maintain higher proportions of CD4+CD25+FOXP3+ regulatory T cells (Tregs) [12]. Treg depletion studies in culture and in an orthotopic murine model of glioma suggested that the activity of the increased hematogenous populations of Tregs in GBM patients may be linked to Th2 responses and suppressed proliferation of CD4+ T cells [12]. The same group demonstrated that anti-CTLA-4 treatment in mice harboring malignant astrocytomas increased peripheral CD4+ cell numbers and conferred resistance to Treg immunosuppression [13]. Since this work, there has been an increased focus on the contributions of CD4+ Th cells and subset CD4+FOXP3+ Tregs to glioma progression at both the pre-clinical and clinical levels.
Following entry into the CNS/tumor compartment, lymphocytes downregulate CD28, and CD62L co-stimulatory molecule expression [8]. This may hint at a mechanism by which the existing immunosuppressive TME captures and represses TILs. Although CD4+ cell numbers alone do not directly correlate with clinical outcome in GBM patients [7,14], new evidence suggests that elevated CD4+ and/or CD4+FOXP3+ population ratios may be indicative of glioma disease severity and risk of recurrence [15,16].
The mechanisms underlying how Th and Treg populations aid glioma progression have not been fully characterized. Mu et al. conducted an elegant study which analyzed 44 paired samples from patients with recurrent HGG [17]. Elevated numbers of perivascular CD4+ TILs strongly correlated with CD34+ tumor vascularity in both primary and recurrent glioma [17]. In a subset of patients refractive to bevacizumab anti-angiogenic therapy, increased and activated CD4+ populations were found to be correlated with bevacizumab resistance, as such activation was not apparent in chemotherapy- naïve patient samples [17]. Elevated CD4+ and CD4+FOXP3+ populations were correlated with shorter recurrence-free survival, and the perivascular CD4+FOXP3+ Treg population in primary tumors was identified as an independent predictor of tumor recurrence in this cohort [17]. The close association to the perivascular region, and conspicuous relationship with tumor progression and recurrence may point to the angiogenic process which may contribute to grade III glioma progression to GBM, a mechanism which has long eluded glioma biology. Nevertheless, these data support negative roles of CD4+ population subsets in glioma progression.
Despite these supportive roles, selectively modulating CD4+ populations could be used to elicit tumor shrinkage. Anti-tumorigenic effector functions of these cells have been realized in mouse models of melanoma and pancreatic cancer [18–20]. In a syngeneic orthotopic murine model of glioma, CD4+ depletion completely nullified tumor lysate vaccine/Fc-OX40L treatment efficacy, and the survival effects were found to be driven in part by antibody-dependent cell mediated cytotoxicity (ADCC) and natural killer T cell (NKT) populations [21]. Similarly, CD4+ cell populations were found to be necessary for the complete efficacy of combined oncolytic herpes simplex virus (oHSV ΔG47-mIL12) and immune checkpoint inhibitor therapy in two distinct murine derived glioma models [22].
The anti-tumorigenic potential of alternate lymphocytic populations is also supported by a study, which expanded and differentiated glioma patient T cells (of mixed CD3+CD4−CD8−, CD4+, and CD8+ subsets) ex vivo using IL-2, IL-15, and IL-21. The study demonstrated preferential expansion of existing memory effector T cells populations, which were reactive against autologous tumor cells and shared tumor-associated antigens [23]. The authors also suggest that these expanded cell populations were resistant to TME immunosuppressive factors, and proposed this protocol for adoptive cell transfer therapy application [23]. While other specialized T cell subsets, such as γδ T cells, may lack roles in immune-mediated responses to HGG [24], CD4+ T cells and CD4+FOXP3+ Treg subsets cannot be ignored when considering HGG progression and treatment responses.
2.2. Challenges cytotoxic lymphocytes face during glioma rejection
Unlike CD4+ cells, there is appreciably more knowledge surrounding the mechanisms by which CD8+ cytotoxic lymphocyte (CTL) populations affect glioma progression. Stimulated by MHC I+ APCs, effector CD8+ T cells selectively target virus-infected, malfunctioning, and/or cancerous cells. CTL infiltrate is typically correlated with survival in GBM patients [25]. Consequently, ineffective tumor clearance arises when tumor cells express ligands, which directly inhibit CTL function. Programmed death ligand 1 (PD-L1) is a primary immunosuppressive molecule whose expression is correlated with glioma grade, and may be a prognostic marker of GBM survival [26]. However, it should be noted that PD-L1 expression among GBM subtypes is inconsistent [27]. Tumor cell expression of PD-L1 is a major mechanism by which the TME exerts immunosuppressive effects via ligation with PD-1 on CD8+ CTL effector immune cells. Success in phase I/II clinical trials for GBM patients using PD-1 checkpoint inhibitors Pembrolizumab and Nivolumab has demonstrated that PD-1 blockade may be a promising strategy to control glioma progression [28,29]. Animal models of GBM demonstrate that CTL effector function underlies PD-1/PDL-1 blockade responses [30]. However, efficacy of PD-1 blockade may be dependent on tumor PD-L1 expression levels, and the perquisite existence of PD-L1 subdued CTLs [31]. These caveats present interesting challenges when considering PD-1 blockade as a treatment strategy for GBM. As such, combinatorial treatments aim to more effectively stimulate, recruit and prime CTL populations. Supporting this theory, radiotherapy has shown to dramatically increase the efficacy of checkpoint inhibitor blockade in animal models of glioma, which is characterized by increased CTL infiltrate and diminished Treg populations [32,33]. This regimen can easily be applied to human subjects, as radiotherapy is already a component of standard of care for HGG.
Other methods designed to improve CTL effector function utilize adoptive cell transfer (ACT). ACT elicits potent anti-tumor responses via exogenously priming or genetically altering effector CD8+ T cells to recognize tumor specific antigens. This allows CTLs to enter the brain parenchyma, identify target cells, clonally expand, and elicit INFγ-dependent, cytotoxic anti-tumorigenic responses [34]. ACT has been successful in targeting primary melanoma and melanoma brain metastases [35,36], however they are still in initial phases of being developed for malignant gliomas. Intracranial and systemic delivery of autologous T cells expressing genetically engineered chimeric antigen receptor (CAR) against the tumor antigen IL13Rα2 has recently been demonstrated to be a tolerable platform to treat patients with advanced GBM [37]. Following this study, a recent individual case has reported compete regression of recurrent, multifocal tumors following modified IL13Rα2 CAR T-cell therapy [38]. Other modalities, such as the identification of a CD8-independent mechanism of tumor regression in a murine model of glioma via Fc-OX40L may shed light on how discrete lymphoid populations contribute to tumor control [21].
Perhaps the most striking advancement in GBM immunotherapy is the application of dendritic cell (DC) vaccines. Native CTLs are often insufficient to induce GBM disease regression, by virtue of the immunosuppressive TME, and due to the majority of unrecognizable surfaces within the bulk tumor. Exogenously introducing professional antigen presenting cells (APCs) which have been pre-educated to tumor antigen profiles can enhanced activation of the adaptive immune system, allowing for subsequent induction and recruitment of sufficient levels of host derived CTLs. This process may then tip the balance in favor of tumor rejection. Although it has been difficult to predict DC vaccination efficacy, the platform has proven to be a promising as well as tolerable approach to increase CTL infiltrate and GBM patient responses [39,40].
DC vaccines are faced with their own pitfalls including insufficient activation of the adaptive immune system, tumor heterogeneity, and limited migration of activated cells. An approach to circumvent the limited priming of the adaptive immune system is to combine DC vaccines with ACT. Tumor RNA-pulsed-DCs cocultured with autologous lymphocytes effectively expand tumor specific CTL populations, and in conjunction with DC vaccination, ACT significantly improves survival in animals with HGG [41]. Further protocols, which expand tumor specific CTLs, may be extended to humans. Tumor antigen-pulsed DCs from HLA-A*02-positive GBM patients can increase CD8+ T cell expansion and specificity ex vivo [42]. INFγ production is signature to functional CTL effector populations. CTL responses to tumor antigens can be measured by INFγ production as well as effector/target killing ratios, which could help identify potent CTL effector populations for ACT [42].
Deriving DC vaccination efficacy has also been demonstrated by altering the antigenic profile DCs present to the adaptive immune system. Immunogenic cell death (ICD) of glioma cells induced by photodynamic therapy elicited a significantly stronger DC vaccination response over typical DC priming techniques in a prophylactic animal model of glioma [43]. ICD generates reactive oxygen species and damage associated molecular patterns (DAMPs), which drives DCs to confer robust protection and inhibition of glioma progression [43]. Furthermore, ICD based DC vaccination increased brain Th1 and CTL infiltrates, INFγ levels, and reduced Treg population ratios following glioma induction [43]. This approach was also found to be synergistic with traditional DC priming techniques, such as glioma cell freeze/thaw necrosis, as well as standard Temozolomide treatment [43]. Thus, new efforts should consider where tumor antigens are derived and by which methods DCs are primed before vaccination, so that complete activation and specificity of CTLs may be produced.
Lack of DC activation and migration also presents an obstacle for DC vaccination. GBM patients receiving intracranial injection of recall antigen tetanus/diphtheria (Td) toxoid to precondition the vaccination site showed improved clinical outcomes following CMV-pp65 RNA pulsed DC vaccine treatment [44]. Patients displayed a robust increase in number of DCs draining into vaccine site lymph nodes. Further investigation identified a CD4+ T cell dependent mechanism, which mediated DC recall and Td precondition efficacy [44]. Prompting DC lymph node draining, cell maturation, and adaptive immune communication are all necessary to induce DC vaccine functionality.
2.3. Glioma associated microglia and macrophages
Glioma associated microglia and macrophages (GAMs) traffic to malignant lesions, where they become subverted by tumor cells to adopt a pro-tumorigenic phenotype. Although the paradigm of polarized “M1” or “M2” phenotypes has recently been challenged [45], it is widely accepted that GAMS are predominantly M2-like; and orchestrate tumor progression by secreting factors which promote chemoattraction, immune suppression, neoangiogenesis, tumor cell survival, and by influencing extracellular matrix (ECM) reorganization (reviewed in [6,46,47]). GAMs are considered to be an integral part of glioma pathology, and evidence suggests that modulation of this immune cell population could slow or cause regression of tumor growth.
HGG biopsies consistently show excessive GAM infiltrate [9,48,49], and GAM populations reportedly comprise up to 30% of tumor bulk [47]. GAM infiltrate has been correlated with poor prognosis, particularly those which express M2 markers [50–52]. In silico and transcriptional analyses of patient samples link excessive M2 GAM infiltrate to the aggressive mesenchymal GBM subtype, and suggest that alterations in the TME promote GAM recruitment and disease progression [53]. It has also been shown that GAMs from GBM patients express high levels of PDL1, and upregulate this immunosuppressive ligand in response to tumor secreted IL-10 [54]. By deeply infiltrating peritumoral and bulk lesions, pro-tumorigenic M2 GAMs largely exert negative influence over human glioma progression.
Consistent with pro-tumorigenic M2 GAMs found in human patient samples, dynamic characterization of immune cell populations and transcriptomic analysis of GAMs in C6 rat gliomas definitively identify accumulation of immunosuppressive CD4+ and Treg populations, and high expression of M2 markers [55]. The mechanisms by which GAMs contribute to glioma progression extend from regulating inflammatory responses to neoangiogenesis. Modulating immunosuppressive TGFβ activity through exogenous miRNA delivery can abrogate M2 GAM populations and prolong animal survival [56]. The anti-tumorigenic response in animal models, elicited by modulating GAM populations, is accompanied by significant downregulation of M2 associated genes, including Arginase-1 (Arg1), Adrenomedullin (Adm) and CD206 [57–61]. Suppressing the M2 phenotype is paramount for controlling GAM pro-tumorgenic functionality, however, shifting this innate immune cell population towards an M1 phenotype may be an additional mechanism to stifle glioma progression. GAM specific SOCS3 KO cells upregulate the JAK/STAT signaling pathway in conjunction with increased pro-inflammatory markers TNFα and CXCL10 [57]. M1 shifted GAMs were shown to induce anti-tumorgenic responses by altering the immune landscape through increasing CD8+ populations while simultaneously decreasing Treg populations, although, only mild improvements in animal survival and tumor burden were observed [57]. This presents the notion that GAMs within the TME may be persuaded to shed their subverted pro-tumorigenic phenotype and adopt more anti-tumorigenic behaviors.
Indeed, this idea has demonstrated effectiveness in the context of colony stimulating factor 1 receptor (CSF1R) inhibition, where GAMs can become “re-educated” within the glioma microenvironment to adopt an anti-tumorigenic phenotype [58]. Further, CSF1R inhibition using the experimental compound PLX3397 has been shown to reduce disease progression and M2 gene signatures in preclinical models of glioma, and has enhanced efficacy over the broader spectrum tyrosine kinase inhibitors Vatalanib and Dovitinib [59]. PLX3397 is currently in active clinical trials for malignant solid tumors including recurrent GBM. More recently, CSF1R inhibition has shown to prevent resistance to anti-VEGF therapy in orthotopic model using ovarian cancer cells [62]. These studies highlight CSF1R as a major target by which GAM contributions to glioma progression can be controlled.
Hypoxic tumor regions also induce potent angiogenic signaling in tumor associated macrophages (TAMs). This process regulates the expression of VEGF, contributes to vascular remodeling, and is reportedly dependent on the activity of hypoxia-inducible factor 1-alpha (Hif1α) [60,63]. Additionally, in vitro experiments demonstrate that M2 macrophages are responsible for inducing angiogenesis [64]. Thus, regulating GAM populations within the TME could provide a valid method to control immunosuppression and aberrant angiogenesis associated with HGG (Figure 1).
Figure 1.
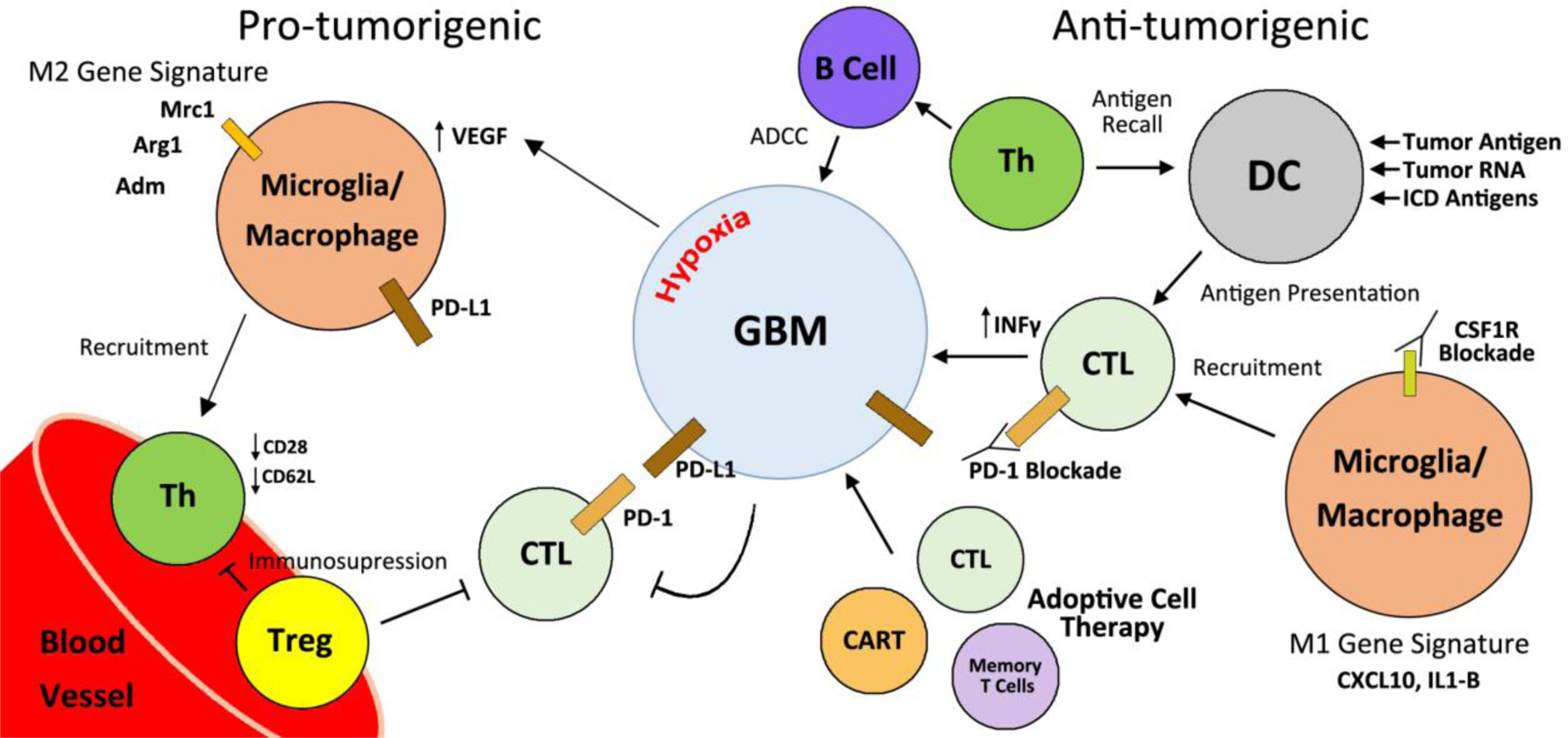
Contributions of immune cell populations in the maintenance, progression, and treatment options of glioma. Perivascular association of CD4+ Th and Treg correlates with glioma progression and recurrence. Treg populations are responsible for immunosuppressive effects within the TME and the periphery. CTLs are inhibited within the TME by relatively high Treg populations and PD-L1 ligation. M2 GAMs recruit immunosuppressive T Cells, express PD-L1, and contribute to the VEGF mediated angiogenesis feedback loop. Conversely, DC vaccines, adoptive cell therapy techniques, and GAM manipulation can modulate the immune landscape to exert anti-tumorigenic responses.
GAMs are potent sources of secreted chemokines, which drive immunosuppressive TIL recruitment. Chemokines, such as CCL2, have been correlated with TIL levels as well as decreased GBM patient survival [65]. To investigate this possible connection, Chang et al. utilized in vitro and in vivo systems in the GL261 murine glioma model and reported that soluble factors produced by tumor cells induce Arg1+ M2-like GAMs within the TME to secrete high levels of CCL2. CCL2 production correlated with distinct CCR4+ Treg and CCR2+ myeloid derived suppressor cell (MDSC) populations infiltrating the tumor, suggesting these molecular steps may be largely responsible for the recruitment of immunosuppressive cell types in glioma [65]. Other chemokines also play roles in local and peripheral immune recruitment during glioma progression. Activated microglia associated with NF1 low-grade optic gliomas were found to express significantly higher levels of CCL5 and CXCL13 RNA [66]. Antibody mediated CCL5 blockade reduced glioma growth and decreased microglia recruitment to tumor cells, indicating that this chemokine has local CNS effects and enhances the TME growth supporting functions [66].
Contribution to chemotaxis is not limited to GAMS; transplanted hematopoietic stem cell (HSC) prior to ACT in an animal model of glioma were shown to be necessary for lymphocyte recruitment and effective tumor rejection [41]. The chemoattracting properties of the HSCs, specifically the secretion of CLL3, was determined to be the governing factor for ACT efficacy [41], supporting the roles that immune signaling proteins have in glioma maintenance and progression. Thus, factors produced both by the tumor as well as immune cells of the TME contribute to the remodeling of the immune landscape. Cell signaling profiles may have important implications when considering how patients will respond to therapies.
Neuropilin 1 has recently been identified as a receptor involved in the activation of GAMs [67,68]. Our group has demonstrated that binding of the immunomodulatory tetrapeptide, tuftsin (TKPR), to Nrp1 in the setting of experimental autoimmune encephalomyelitis (EAE), a rodent model of Multiple Sclerosis, has the potential to polarize microglia to a more immunosuppressive phenotype via TGFβR1 and SMAD2/3 activation, thereby reducing the severity of the disease course [67]. This initial observation led to the examination of Neuropilin’s potential role in the immune regulation of glioma microenvironment.
3. Neuropilin 1: an immunotherapeutic, anti-proliferative, and anti-angiogenic target for glioma
Neuropilin 1 (Nrp1) is a cell surface receptor which was originally identified to contribute to signaling associated with axonal pathfinding and chemorepulsion via co-reception with one of its associated co-receptors, Plexin A1, in neurons [69]. Nrp1 has since been found by various groups to also have the potential to complex with other co-receptors including transforming growth factor β receptor I/II (TGFβRI/II), vascular endothelial growth factor receptor 2 (VEGFR2), hepatocyte growth factor receptor (cMET) and to amplify signaling pathways associated with these receptors [70–72].
Nrp1 is composed of an A1, A2, B1, B2, oligomerization, transmembrane, and cytoplasmic tail domain. The cytoplasmic tail domain is quite short and is considered to have no potential to elicit downstream signaling on its own (Figure 2). However, in complex with an associated receptor via interactions with its oligomerization domain, Nrp1 has the potential to amplify the associated receptor’s signaling pathway [73]. The A1 and A2 domain are semaphorin binding domains while the B1 and B2 domains are responsible for binding VEGF, TGFβ, PIGF, and HGF [74]. Additionally, it has been shown that Nrp1 can complex with ABL1 in endothelial cells and carry out angiogenic signaling independently of its association with VEGFR2 or VEGF [75].
Figure 2.
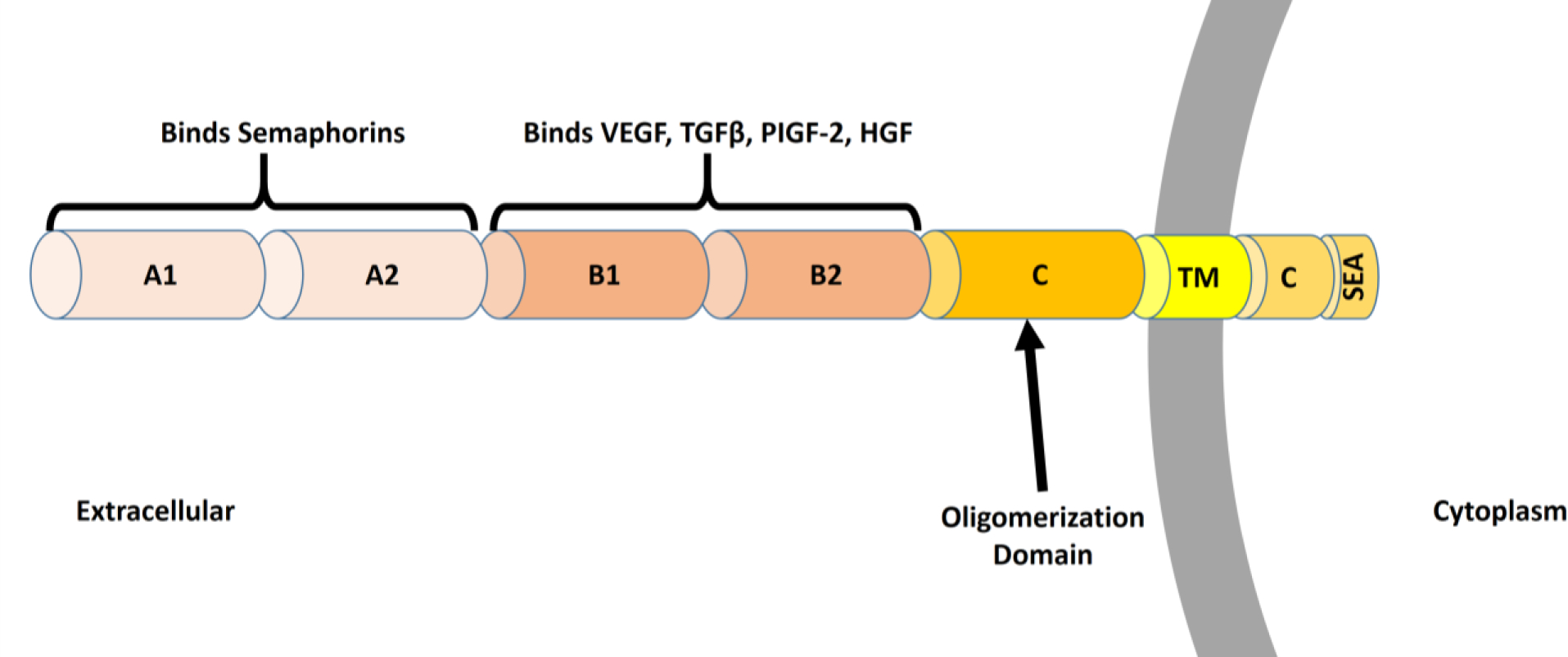
Structure and Function of Neuropilin 1. Neuropilin 1 (Nrp1) is a cell surface receptor composed of an A1, A2, B1, B2, C, transmembrane (TM), and a C-terminal (C) domain. The C-terminal domain contains a SEA motif which binds PDZ adaptor proteins. The A1 and A2 domains are responsible for binding semaphorins while the B1 and B2 domains have been characterized to bind VEGF, TGFβ, PlGF, and HGF. The C domain is an oligomerization domain, which allows Nrp1 to interact with its various co-receptors and amplify signaling via their associated ligands.
Nrp1’s expression is rather ubiquitous in terms of its tissue distribution in that it is expressed by endothelial cells, subsets of DCs, subsets of T cells, subsets of myeloid-derived cells, and microglia [74–76]. In mice and rats, the complete elimination of Nrp1 is lethal due to its crucial role in embryonic angiogenesis [77,78]. In a similar manner, mice which express Nrp1 with a point mutation in the B1 domain responsible for signaling via TGFβ, PlGF, HGF, and VEGF-A, survive to adulthood but exhibit abnormal vasculature [78]. These mice have been shown to have a resistance phenotype to the growth of xenograft tumors, attributed to poor neovascularization of the tumors [78].
3.1. Functional roles of NRP1 in T cells
Nrp1 is expressed by subsets of Tregs and plays a role in the suppression of adaptive immunity [76]. In skin allograft experiments in mice, it was seen that the survival of the tissue was partially dependent on the expression of Nrp1 by Treg populations. When this expression was lost, the allografts were rejected, hinting at an immunosuppressive role for Nrp1 in these cells [79]. NRP1 may also function to suppress autoimmunity, as CD4+ loss of NRP1 in a mouse model of multiple sclerosis skews inflammatory populations to a TH-17 phenotype and reduces Treg populations, worsening autoimmune disease progression [80]. It has also been shown in a murine melanoma model that Nrp1 expressing Tregs are attracted to tumors via the tumor’s secretion of VEGF, which is abrogated by the inhibition of Nrp1 signaling. Delgoffe et al. have shown that the stability of Nrp1+ Tregs in the tumor microenvironment is dependent on their activation by the Nrp1 ligand, semaphorin 4A (Sema4A). Antibody-mediated blockade of sema4A or genetic deletion of Nrp1 from these Treg populations using FOXP3-Cre resulted in enhanced anti-tumoral immunity in melanoma mouse models [81]. The accumulation of Nrp1 expressing Tregs in tumors is correlated with increased immunosuppression and the suppression of effector T cell functions [82].
Nrp1 expression in the majority of peripheral T cells is rather negligible in healthy people. However, in patients with certain advanced stage cancers, such as pancreatic adenocarcinoma and colorectal cancer, Nrp1 expressing T cells are significantly elevated in their blood and have been considered as potential biomarkers for their degree of immunosuppression in these patients [74]. Additionally, the expression of Nrp1 has also been consistently documented in naïve populations NKT cells in humans, but its expression is lost in mature populations of the cells [83].
3.2. Dendritic cells and innate immune cells
Immature plasmacytoid dendritic cells (iDCs) are another PBMC that has consistently been identified to express high levels of Nrp1 in humans. These cells have the potential to preferentially interact with Nrp1-expressing Tregs via the homotypic interaction of Nrp1 on both cell types, leading to the activation and expansion of these Treg populations, providing an explanation for how they can lead to increased immunosuppression. This “glue” between both cell types has also been postulated to contribute to greater sensitivity of these cells to antigen presentation [84]. Additionally, it has been shown that human Nrp1-expressing DCs have the potential to transfer Nrp1 to Tregs via trogocytosis along with VEGF and potentially other intracellular contents [85].
3.3. NRP1 functionality in monocytic populations
Macrophage-specific depletion of Nrp1 in mice via the use of LysM-Cre does not result in any apparent abnormalities in development or in adulthood [86]. However, it has been shown that mice with Nrp1 depletion from LysM-Cre expressing microglia and macrophages are resistant to pathological angiogenesis in a model of retinal sclerosis [87]. These same mice with Nrp1-deficient LysM expressing cells were shown to have slower disease courses in orthotopic breast and pancreatic cancer models, attributed to poorer vascularization of the tumors and increased infiltration of tumors by anti-tumorigenic macrophages and T cells [86]. Our group has reported the expression of Nrp1 by glioma associated microglia and macrophages (GAMs) associated with glioma biopsies of various grades [68]. Additionally, Zhang et al. reported that subsets of highly aggressive gliomas are populated by GAMs with significantly elevated Nrp1 expression [88].
As mentioned earlier, activation of Nrp1 by tuftsin during EAE resulted in polarization of microglia to a more M2-like phenotype via SMAD2/3 activation [67]. We have also observed that Nrp1 depletion from GAMs slows tumor progression and increases anti-tumoral immunity in a murine model of GBM [68]. The outcomes in these diseases models were partially attributable to the fact that Nrp1 complexes with TGFβRI/II to potentiate signaling via SMAD2/3, highlighting its potential as a therapeutic target in a similar fashion to the use of TGFβ inhibitors [89]. Additionally, using chimeric mouse models, we have shown that NRP1 ablation from either populations of peripheral monocytes or resident microglia can repress glioma progression, suggesting discriminant functionality of these cells [90].
3.4. Glioma-derived cancer cells
Overexpression of Nrp1 by cancerous cells in glioma biopsies has been directly correlated with poorer clinical outcome and worse progression free survival (PFS) [91]. Various studies have implicated most of the soluble factors known to signal via Nrp1 to be associated with poorer clinical outcomes and direct promotion of glioma growth in animal studies. Chen et al. demonstrated that blocking Nrp1 using a monoclonal antibody inhibited the proliferation and migration of the human-derived glioma cell line U87MG and slowed tumor progression in vivo when the cell line was xenografted in mice [92]. Additionally, the U87MG cell line has a highly invasive phenotype relative to glioma cell lines such as the LN18, T98, and U118 human glioma cell lines. In vitro analysis of the secretome of the U87MG cell line showed significant elevations in the amount of Nrp1 secreted by the cells relative to the less invasive lines [93] In the U373MG human glioma cell line, it was shown that siRNA-mediated knockdown of Nrp1 reduced proliferation and increased apoptosis of the cells, associated with reductions in Bcl-2 expression and ERK, JNK, and MAPK activation [94]. Additionally, Nasarre et al. showed that targeting the transmembrane domain of Nrp1 via a small peptide inhibitor has the potential to slow glioma progression due to reductions in angiogenesis and proliferation in pre-clinical human xenograft and rat models [95].
3.5. NRP1 signaling in glioma maintenance and progression
Semaphorin 3A (sema3A) has been implicated to promote the infiltration and spread of glioma-derived cells in an autocrine fashion and is overexpressed in a subset of gliomas in patients [96]. Sema3A is secreted by glioma-derived cells in vesicles, which have been shown to directly increase vascular permeability by interacting with Nrp1 on endothelial cells in xenograft mouse models. Blocking signaling via either sema3A or Nrp1 was shown to abrogate this. Additionally, these vesicles can be detected in the blood of patients, which may also hold some prognostic value [97]. The expression of the receptor for sema3A, PlexinA1, has been correlated with worse survival outcomes in patients with GBM. Additionally, in a murine xenograft model, it has been shown that a small peptide inhibitor that disrupts the oligomerization of Nrp1 and PlexinA1 reduces GBM proliferative potential and tumor angiogenesis in vivo [98]. This peptide inhibitor, interestingly, blocked VEGF-dependent angiogenesis in vitro as well, possibly by also blocking the oligomerization of Nrp1 with VEGFR2. Additionally, Casazza et al. have shown that Sema3A acts a chemoattractant for Nrp1-PlexinA1 expressing TAMs to infiltrate tumors where they downregulate Nrp1 expression once becoming entrapped in more hypoxic environments. Deletion of Nrp1 or mutating the sema3A-binding A1 domain of Nrp1 from TAMs was shown to prevent this [86]. As mentioned above, Nrp1 also binds and signal via Sema4A, playing an important role in the maintenance of immunosuppressive Treg populations.
VEGF-A and VEGF-B bind Nrp1 in complex to VEGFR2 and amplify pro-angiogenic signaling via the activation of AKT and p38 MAPK [99]. Glioma stem cells have also been shown to secrete VEGF-A, which not only serves to promote angiogenesis, but also enhances the proliferative index of glioma cells in an autocrine fashion via VEGFR2 in complex to Nrp1 [100]. VEGF-A overexpression is well documented in almost all cases of HGG and has received a great deal of attention as a therapeutic target in recent years. Phase III clinical trials were performed in 2014, evaluating concomitant Avastin (bevacizumab, an anti-VEGF antibody) with TMZ and RT as first line defense for newly diagnosed glioma. While increasing PFS significantly, the trials failed to meet pre-defined criteria for success and failed to show any increase in overall survival time (OST) for patients [101]. However, individual patients responded quite well, showing that some may serve to benefit from the adjuvant therapy. Bevacizumab is still under evaluation as a concomitant therapy in various clinical trials for HGG.
Nrp1 has been shown to bind TGFβ and LAP-TGFβ and amplify signaling associated with these ligands in cancer cells via co-reception with TGFβRI/II [89]. TGFβI and II overexpression, especially that of isoform II, has been correlated with poorer clinical outcomes in subsets of glioma [102,103]. Autocrine signaling within cancer cells serves to enhance epithelial to mesenchymal transition (EMT) and increases the invasive phenotype of tumor cells. TGFβ potentiates angiogenesis and is an immunosuppressive cytokine, which polarizes Tregs and attracts and polarizes immunosuppressive GAMs [104,105]. It can downregulate perforin, granzyme A/B, IFNγ, and FasL expression by CTLs, which are all mediators of CTL-mediated cytotoxicity [106]. Downregulation of the expression of TGFβRII in human xenograft-derived gliomas has been shown to reduce their tumorigenicity [107]. Inhibition of TGFβ-dependent pathways using TGFβRII inhibitors in GAMs has been shown to prevent their immunosuppressive polarization [108]. Blocking TGFβ-mediated signaling using systemically administered neutralizing antibodies was efficacious in slowing glioma progression in immunocompetent mice, partially by preventing the immunosuppressive polarization of GAMs [109]. For the treatment of glioma, clinical trials are ongoing, evaluating the TGFβRI small molecule inhibitor, LY2157299, for efficacy in combination with the standard of care. The drug is generally well tolerated and has shown efficacy in about 20% of patients [110].
Placental growth factor (PlGF) is an important angiogenic factor, which has been shown to bind the B1 domain of Nrp1 and act as a chemo-attractant for GAMs [111,112]. Clinical trials have been conducted using a monoclonal antibody against PIGF for recurrent glioma. The drug has shown acceptable safety profiles, but, unfortunately, the antibody was not shown to have any additive benefit for patients over bevacizumab alone [113]. In medulloblastoma, however, PlGF and Nrp1 are both highly expressed. Inhibition of signaling via either has been shown to slow tumor progression in murine xenograft models [114].
Hepatocyte Growth Factor/Scatter Factor (HGF/SF) is a ligand which binds Nrp1 in its complex to cMET and activates downstream signaling which promotes cell proliferation, angiogenesis, and survival [99]. The overexpression of HGF by glioma cells has been correlated with increased tumor microvascularity, increased tumor grade, and worse prognosis for patients. Downregulation of HGF in human-derived glioma cells was also shown to reduce their proliferative and migratory capacity [115]. Furthermore, Hu et al. demonstrated that Nrp1 expression by a subset of human glioma-derived xenografts potentiated their growth in an autocrine fashion via the amplification of pathways downstream of cMET and HGF [71]. A phase II clinical trial was conducted with rilotumumab, a HGF-blocking antibody, in patients with recurrent GBM, but the antibody showed little efficacy as a monotherapy [116]. However, cabozantinib, a small molecule inhibitor of both cMET and VEGFR2, underwent phase II clinical evaluation in patients with progressive and recurrent GBM, and was reported to result in modest improvements in PFS [117]. The drug is currently under evaluation in combination with standard of care RT and TMZ for newly diagnosed GBM [118]. The HGF-cMET signaling axis is a promising therapeutic target, but it would appear that targeting it in combination to other pathways is preferable. As Nrp1, serves to amplify cMET signaling, a similar rationale for targeting it in HGG is well substantiated.
3.6. NrpI as a PET tracer and for drug delivery
As Nrp1 is widely expressed in gliomas, the use of PET tracers that bind Nrp1 has been investigated in murine models. Using F-18 labelled peptides, Wu et al. were able to show that peptides targeting Nrp1 and integrin αvβ3 preferentially bound to glioma tissue [119]. This method may hold promise in monitoring glioma progression in patients. Additionally, more effective drug trafficking to gliomas has been proposed by packaging chemotherapeutics in liposomes coated by Nrp1-binding peptides [120–123]. In a similar fashion, targeting Nrp1 in order to deliver gadolinium oxide for MRI and chlorin for interstitial photodynamic therapy has been explored in rat xenograft studies. The nanoparticles were able to preferentially localize to peripheral tumoral vasculature and may hold some promise for translation to the clinic [124].
4. Conclusions
Identifying the contributions of immune cell populations within the TME will further the knowledge base by which we treat and develop therapies for GBM. The innate and adaptive immune systems are complex, multifaceted schemes. Providing protection from foreign pathogens, materializing sustained immunity, and regulating self/non-self-responses are immense tasks. Unfortunately, in scenarios of malignancy, the immune system often fails to protect the host. Modulating lymphocyte inflammatory responses may prove to be a method by which overall adaptive immunity can be coerced into rejection of bulk tumors. Additionally, with the advancements of ACT and DC vaccines, tools now exist to selectively activate effector cells of the adaptive immune system. Innate immune cells such as microglia and macrophages are now also recognized as pertinent players to glioma progression, and perhaps by invoking their phagocytic and pro-inflammatory functions, a greater foothold can be gained in controlling HGG disease (Figure 1). New targets which can modulate subsets, or entire arms, of the immune system need to be identified so that clinicians can combat GBM.
Directly targeting Nrp1 in the clinic has only been approached, thus far, for the treatment of advanced solid tumors via the use of a humanized monoclonal antibody, MNRP1685A, which blocks the binding of VEGF-A, VEGF-B, and PlGF-2 to the B1 domain of Nrp1. The antibody was hypothesized to benefit patients through a mechanism similar to bevacizumab, but was unfortunately poorly tolerated and associated with clinically significant levels of proteinuria in patients during phase I evaluation [125].
The efficacy and tolerability of other Nrp1-targeting drugs should be considered, and since Nrp1 plays so many roles in the glioma microenvironment (Figure 3), pursuing research in the development and implementation of Nrp1 antagonists in glioma therapy seems fruitful.
Figure 3.
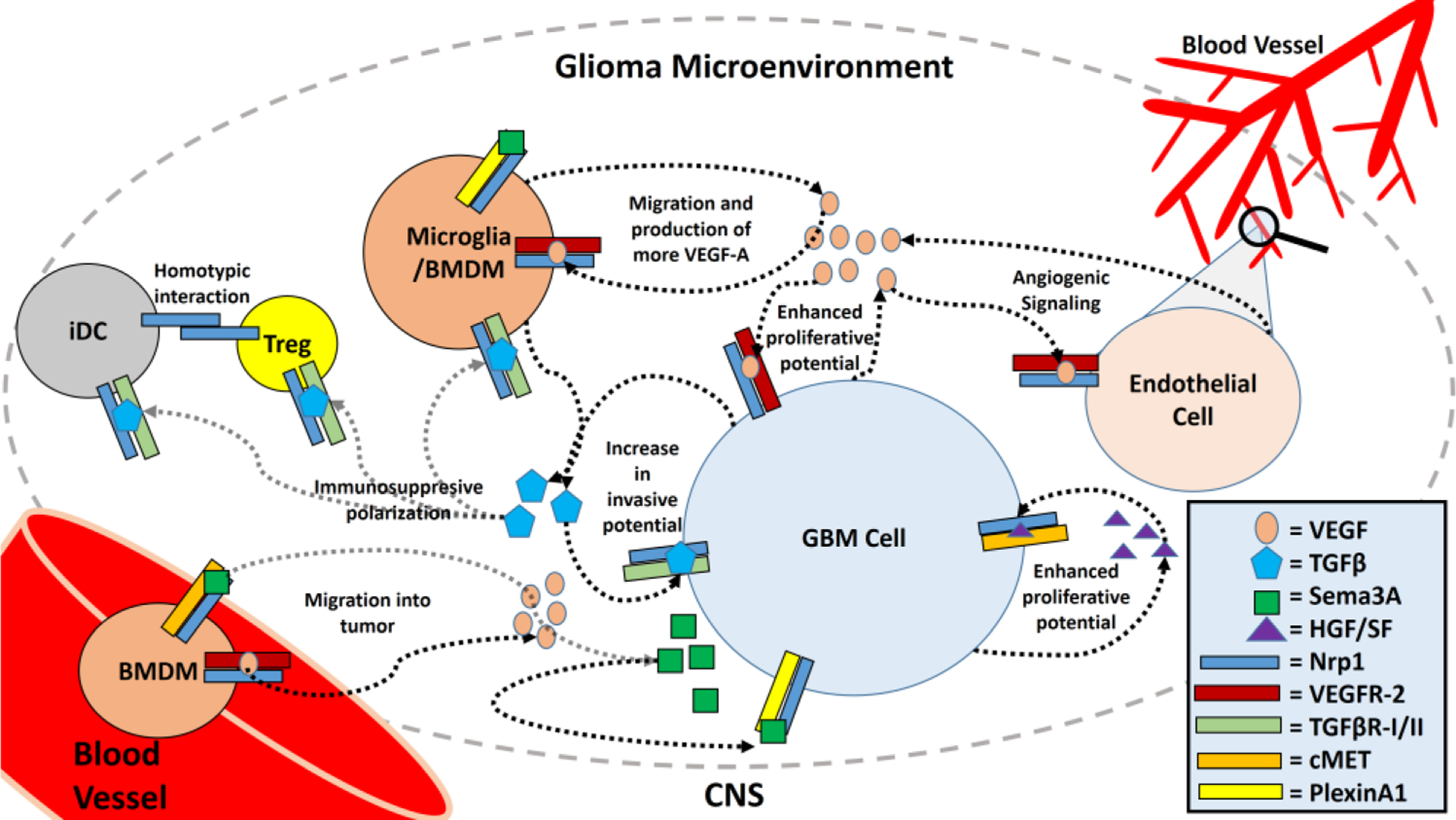
Roles of Neuropilin 1 in the Glioma Microenvironment. Neuropilin 1 (Nrp1) is expressed by various cell types which infiltrate the glioma microenvironment, including most glioma-derived cells (GBM cells), microglia, infiltrating BMDMs, endothelial cells, certain Treg subtypes, and certain dendritic cell subtypes (iDCs). GBM cells produce VEGF, TGFβ, and HGF/SF which increase the malignancy of the tumors by enhancing the proliferative and invasive potential of the GBM cells, mediated via Nrp1 and its associated co-receptors. Sema3A in the glioma microenvironment also causes microglial and BMDM migration into the tumor and enhances invasion by the GBM cells. VEGF produced by the tumors also serves to enhance angiogenesis and increase microglial and BMDM migration into the tumor. Microglia and BMDMs are also responsible for the production of VEGF and TGFβ. TGFβ can polarize microglia, BMDMs, Tregs, and iDCs to more immunosuppressive, tumor supporting phenotypes. Homotypic interactions between Nrp1 on iDCs and Tregs also enhances their contact times and allows for stronger stimulation of these immunosuppressive Treg subsets.
Acknowledgements
We thank the members of the Tsirka lab for helpful suggestions. This work was partially supported by the National Institutes of Health F30CA196110 and 2T32GM008444 (J.T. Miyauchi), R01NS42168 (S.E. Tsirka), and 5T32GM007518 and Scholars in BioMedical Sciences Program funds (M.D. Caponegro).
Footnotes
Conflict of interest
The authors declare no conflicts of interest in this paper.
References
- 1.Wen PY, Kesari S (2008) Malignant gliomas in adults. New Engl J Med 359: 492–507. [DOI] [PubMed] [Google Scholar]
- 2.Brennan C, Momota H, Hambardzumyan D, et al. (2009) Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One 4: e7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Louis DN, Perry A, Reifenberger G, et al. (2016) The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 131: 803–820. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, Mason WP, Van dBMJ, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med 352: 987–996. [DOI] [PubMed] [Google Scholar]
- 6.Quail DF, Joyce JA (2017) The microenvironmental landscape of brain tumors. Cancer Cell 31: 326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lohr J, Ratliff T, Huppertz A, et al. (2011) Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res 17: 4296–4308. [DOI] [PubMed] [Google Scholar]
- 8.Kmiecik J, Poli A, Brons NH, et al. (2013) Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol 264: 71–83. [DOI] [PubMed] [Google Scholar]
- 9.Yang I, Han SJ, Sughrue ME, et al. (2011a) Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J Neurosurg 115: 505–511. [DOI] [PubMed] [Google Scholar]
- 10.Swain SL (1995) T-cell subsets: Who does the polarizing? Curr Biol 5: 849–851. [DOI] [PubMed] [Google Scholar]
- 11.Mellanby RJ, Thomas DC, Lamb J (2009) Role of regulatory T-cells in autoimmunity. Clin Sci 116: 639–469. [DOI] [PubMed] [Google Scholar]
- 12.Fecci PE, Mitchell DA, Whitesides JF, et al. (2006) Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 66: 3294–3302. [DOI] [PubMed] [Google Scholar]
- 13.Fecci PE, Ochiai H, Mitchell DA, et al. (2007) Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 13: 2158–2167. [DOI] [PubMed] [Google Scholar]
- 14.Thomas AA, Fisher JL, Rahme GJ, et al. (2015) Regulatory T cells are not a strong predictor of survival for patients with glioblastoma. Neuro Oncol 17: 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han S, Zhang C, Li Q, et al. (2014) Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Brit J Cancer 110: 2560–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sayour EJ, McLendon P, McLendon R, et al. (2015) Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immun 64: 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mu L, Yang C, Gao Q, et al. (2017) CD4+ and perivascular Foxp3+ T cells in glioma correlate with angiogenesis and tumor progression. Front Immunol 8: 1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braumuller H, Wieder T, Brenner E, et al. (2013) T-helper-1-cell cytokines drive cancer into senescence. Nature 494: 361–365. [DOI] [PubMed] [Google Scholar]
- 19.Hirschhorn-Cymerman D, Budhu S, Kitano S, et al. (2012) Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 209: 2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez-Diez A, Joncker NT, Choi K, et al. (2007) CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 109: 5346–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy KA, Erickson JR, Johnson CS, et al. (2014) CD8+ T cell-independent tumor regression induced by Fc-OX40L and therapeutic vaccination in a mouse model of glioma. J Immunol 192: 224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha D, Martuza RL, Rabkin SD (2017) Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell 32: 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Meng Q, Bartek J, et al. (2017b) Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology 6: e1252894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck BH, Kim H, O’Brien R, et al. (2015) Dynamics of circulating gammadelta T cell activity in an immunocompetent mouse model of high-grade glioma. PLoS One 10: e0122387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang I, Tihan T, Han SJ, et al. (2010) CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci 17: 1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nduom EK, Wei J, Yaghi NK, et al. (2016) PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol 18: 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heiland DH, Haaker G, Delev D, et al. (2017) Comprehensive analysis of PD-L1 expression in glioblastoma multiforme. Oncotarget 8: 42214–42225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reiss SN, Yerram P, Modelevsky L, et al. (2017) Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J Immunother Cancer 5:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyauchi JT, Tsirka SE (2017) Advances in immunotherapeutic research for glioma therapy. J Neurol, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakashima H, Alayo QA, Penaloza-MacMaster P, et al. (2018) Modeling tumor immunity of mouse glioblastoma by exhausted CD8+ T cells. Sci Rep 8: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haanen J (2017) Converting cold into hot tumors by combining immunotherapies. Cell 170: 1055–1056. [DOI] [PubMed] [Google Scholar]
- 32.Zeng J, See AP, Phallen J, et al. (2013) Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol 86: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waitz R, Solomon SB, Petre EN, et al. (2012) Potent induction of tumor immunity by combining tumor cryoablation with anti-CTLA-4 therapy. Cancer Res 72: 430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masson F, Calzascia T, Di BBW, et al. (2007) Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol 179: 845–853. [DOI] [PubMed] [Google Scholar]
- 35.Hong JJ, Rosenberg SA, Dudley ME, et al. (2010) Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res 16: 4892–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenberg SA, Yang JC, Sherry RM, et al. (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17: 4550–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown CE, Badie B, Barish ME, et al. (2015) Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 21: 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown CE, Alizadeh D, Starr R, et al. (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med 375: 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liau LM, Prins RM, Kiertscher SM, et al. (2005) Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 11: 5515–5525. [DOI] [PubMed] [Google Scholar]
- 40.Polyzoidis S, Ashkan K (2014) DCVax(R)-L--developed by Northwest Biotherapeutics. Hum Vacc Immunother 10: 3139–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flores C, Pham C, Snyder D, et al. (2015) Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 4: e994374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller I, Altherr D, Eyrich M, et al. (2016) Tumor antigen-specific T cells for immune monitoring of dendritic cell-treated glioblastoma patients. Cytotherapy 18: 1146–1161. [DOI] [PubMed] [Google Scholar]
- 43.Garg AD, Vandenberk L, Koks C, et al. (2016) Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 8: 328ra27. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell DA, Batich KA, Gunn MD, et al. (2015) Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519: 366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19: 987–991. [DOI] [PubMed] [Google Scholar]
- 46.Charles NA, Holland EC, Gilbertson R, et al. (2011) The brain tumor microenvironment. Glia 59: 1169–1180. [DOI] [PubMed] [Google Scholar]
- 47.Da FA, Badie B (2013) Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol 2013: 264124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hewedi IH, Radwan NA, Shash LS, et al. (2013) Perspectives on the immunologic microenvironment of astrocytomas. Cancer Manag Res 5: 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Held-Feindt J, Hattermann K, Muerkoster SS, et al. (2010) CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res 316: 1553–1566. [DOI] [PubMed] [Google Scholar]
- 50.Nishie A, Ono M, Shono T, et al. (1999) Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res 5: 1107–1113. [PubMed] [Google Scholar]
- 51.Sorensen MD, Dahlrot RH, Boldt HB, et al. (2017) Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropath Appl Neuro 44: 185–206. [DOI] [PubMed] [Google Scholar]
- 52.Okada M, Saio M, Kito Y, et al. (2009) Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int J Oncol 34: 1621–1627. [DOI] [PubMed] [Google Scholar]
- 53.Wang Q, Hu B, Hu X, et al. (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32: 4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bloch O, Crane CA, Kaur R, et al. (2013) Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 19: 31653175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gieryng A, Pszczolkowska D, Bocian K, et al. (2017) Immune microenvironment of experimental rat C6 gliomas resembles human glioblastomas. Sci Rep 7: 17556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu S, Wei J, Wang F, et al. (2014) Effect of miR-142–3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J Natl Cancer Inst 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McFarland BC, Marks MP, Rowse AL, et al. (2016) Loss of SOCS3 in myeloid cells prolongs survival in a syngeneic model of glioma. Oncotarget 7: 20621–20635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pyonteck SM, Akkari L, Schuhmacher AJ, et al. (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19: 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan D, Kowal J, Akkari L, et al. (2017) Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 36: 60496058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colegio OR, Chu NQ, Szabo AL, et al. (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang I, Alizadeh D, Liang J, et al. (2016) Characterization of arginase expression in glioma-associated microglia and macrophages. PLoS One 11: e0165118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lyons YA, Pradeep S, Wu SY, et al. (2017) Macrophage depletion through colony stimulating factor 1 receptor pathway blockade overcomes adaptive resistance to anti-VEGF therapy. Oncotarget 8: 96496–96505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakayama T, Kurobe H, Sugasawa N, et al. (2013) Role of macrophage-derived hypoxia-inducible factor (HIF)-1alpha as a mediator of vascular remodelling. Cardiovasc Res 99: 705715. [DOI] [PubMed] [Google Scholar]
- 64.Jetten N, Verbruggen S, Gijbels MJ, et al. (2014) Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 17: 109–118. [DOI] [PubMed] [Google Scholar]
- 65.Chang AL, Miska J, Wainwright DA, et al. (2016) CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory t cells and myeloid-derived suppressor cells. Cancer Res 76: 5671–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Solga AC, Pong WW, Kim KY, et al. (2015) RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for neurofibromatosis-1 glioma growth. Neoplasia 17: 776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nissen JC, Selwood DL, Tsirka SE (2013) Tuftsin signals through its receptor neuropilin-1 via the transforming growth factor beta pathway. J Neurochem 127: 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miyauchi JT, Chen D, Choi M, et al. (2016) Ablation of neuropilin 1 from glioma-associated microglia and macrophages slows tumor progression. Oncotarget 7: 9801–9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koncina E, Roth L, Gonthier B, et al. (2007) Role of semaphorins during axon growth and guidance. Adv Exp Med Biol 621: 50–64. [DOI] [PubMed] [Google Scholar]
- 70.Glinka Y, Prud’homme GJ (2008) Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J Leukoc Biol 84: 302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu B, Guo P, Bar-Joseph I, et al. (2007) Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene 26: 5577–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gelfand MV, Hagan N, Tata A, et al. (2014) Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. Elife 3: e03720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakamura F, Goshima Y (2002) Structural and functional relation of neuropilins. Adv Exp Med Biol 515: 55–69. [DOI] [PubMed] [Google Scholar]
- 74.Chaudhary B, Khaled YS, Ammori BJ, et al. (2014) Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immun 63: 81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raimondi C, Fantin A, Lampropoulou A, et al. (2014) Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. J Exp Med 211: 1167–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Campos-Mora M, Morales RA, Gajardo T, et al. (2013) Neuropilin-1 in transplantation tolerance. Front Immunol 4: 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klagsbrun M, Takashima S, Mamluk R (2002) The role of neuropilin in vascular and tumor biology. Adv Exp Med Biol 515: 33–48. [DOI] [PubMed] [Google Scholar]
- 78.Zachary I, Fantin A, Herzog B, et al. (2014) Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF-A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development 141: 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Campos-Mora M, Morales RA, Perez F, et al. (2015) Neuropilin-1(+) regulatory T cells promote skin allograft survival and modulate effector CD4(+) T cells phenotypic signature. Immunol Cell Biol 93: 113–119. [DOI] [PubMed] [Google Scholar]
- 80.Solomon BD, Mueller C, Chae WJ, et al. (2011) Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 108: 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delgoffe GM, Woo SR, Turnis ME, et al. (2013) Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 501: 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hansen W, Hutzler M, Abel S, et al. (2012) Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med 209: 2001–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Milpied P, Massot B, Renand A, et al. (2011) IL-17-producing invariant NKT cells in lymphoid organs are recent thymic emigrants identified by neuropilin-1 expression. Blood 118: 2993–3002. [DOI] [PubMed] [Google Scholar]
- 84.Sarris M, Andersen KG, Randow F, et al. (2008) Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity 28: 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bourbie-Vaudaine S, Blanchard N, Hivroz C, et al. (2006) Dendritic cells can turn CD4+ T lymphocytes into vascular endothelial growth factor-carrying cells by intercellular neuropilin-1 transfer. J Immunol 177: 1460–1469. [DOI] [PubMed] [Google Scholar]
- 86.Casazza A, Laoui D, Wenes M, et al. (2013) Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24: 695–709. [DOI] [PubMed] [Google Scholar]
- 87.Dejda A, Mawambo G, Daudelin JF, et al. (2016) Neuropilin-1-expressing microglia are associated with nascent retinal vasculature yet dispensable for developmental angiogenesis. Invest Ophth Vis Sci 57: 1530–1536. [DOI] [PubMed] [Google Scholar]
- 88.Zhang W, Lv Y, Xue Y, et al. (2016b) Co-expression modules of NF1, PTEN and sprouty enable distinction of adult diffuse gliomas according to pathway activities of receptor tyrosine kinases. Oncotarget 7: 59098–59114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Glinka Y, Stoilova S, Mohammed N, et al. (2011) Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 32: 613–621. [DOI] [PubMed] [Google Scholar]
- 90.Miyauchi JT, Caponegro MD, Chen D, et al. (2017) Deletion of neuropilin 1 from microglia or bone marrow-derived macrophages slows glioma progression. Cancer Res 78: 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Osada H, Tokunaga T, Nishi M, et al. (2004) Overexpression of the neuropilin 1 (NRP1) gene correlated with poor prognosis in human glioma. Anticancer Res 24: 547–552. [PubMed] [Google Scholar]
- 92.Chen L, Miao W, Tang X, et al. (2013) Inhibitory effect of neuropilin-1 monoclonal antibody (NRP-1 MAb) on glioma tumor in mice. J Biomed Nanotechnol 9: 551–558. [DOI] [PubMed] [Google Scholar]
- 93.Formolo CA, Williams R, Gordish-Dressman H, et al. (2011) Secretome signature of invasive glioblastoma multiforme. J Proteome Res 10: 3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li X, Tang T, Lu X, et al. (2011) RNA interference targeting NRP-1 inhibits human glioma cell proliferation and enhances cell apoptosis. Mol Med Rep 4: 1261–1266. [DOI] [PubMed] [Google Scholar]
- 95.Nasarre C, Roth M, Jacob L, et al. (2010) Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene 29: 2381–2392. [DOI] [PubMed] [Google Scholar]
- 96.Bagci T, Wu JK, Pfannl R, et al. (2009) Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene 28: 3537–3550. [DOI] [PubMed] [Google Scholar]
- 97.Treps L, Edmond S, Harford-Wright E, et al. (2016) Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 35: 2615–2623. [DOI] [PubMed] [Google Scholar]
- 98.Jacob L, Sawma P, Garnier N, et al. (2016) Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget 7: 57851–57865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sulpice E, Plouet J, Berge M, et al. (2008) Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood 111: 2036–2045. [DOI] [PubMed] [Google Scholar]
- 100.Hamerlik P, Lathia JD, Rasmussen R, et al. (2012) Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med 209: 507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gilbert MR, Dignam JJ, Armstrong TS, et al. (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. New Engl J Med 370: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Frei K, Gramatzki D, Tritschler I, et al. (2015) Transforming growth factor-beta pathway activity in glioblastoma. Oncotarget 6: 5963–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maxwell M, Galanopoulos T, Neville-Golden J, et al. (1992) Effect of the expression of transforming growth factor-beta 2 in primary human glioblastomas on immunosuppression and loss of immune surveillance. J Neurosurg 76: 799–804. [DOI] [PubMed] [Google Scholar]
- 104.Platten M, Wick W, Weller M (2001) Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech 52: 401–410. [DOI] [PubMed] [Google Scholar]
- 105.Chen ML, Pittet MJ, Gorelik L, et al. (2005) Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci USA 102: 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thomas DA, Massague J (2005) TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8: 369–380. [DOI] [PubMed] [Google Scholar]
- 107.Wesolowska A, Kwiatkowska A, Slomnicki L, et al. (2008) Microglia-derived TGF-beta as an important regulator of glioblastoma invasion--an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 27: 918–930. [DOI] [PubMed] [Google Scholar]
- 108.Uhl M, Aulwurm S, Wischhusen J, et al. (2004) SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res 64: 7954–7961. [DOI] [PubMed] [Google Scholar]
- 109.Ueda R, Fujita M, Zhu X, et al. (2009) Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res 15: 6551–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rodon J, Carducci MA, Sepulveda-Sanchez JM, et al. (2015) First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res 21: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Falco SD (2012) The discovery of placenta growth factor and its biological activity. Exp Mol Med 44: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dewerchin M, Carmeliet P (2012) PlGF: a multitasking cytokine with disease-restricted activity. Csh Perspect Med 2: 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lassen U, Chinot OL, McBain C, et al. (2015) Phase 1 dose-escalation study of the antiplacental growth factor monoclonal antibody RO5323441 combined with bevacizumab in patients with recurrent glioblastoma. Neuro Oncol 17: 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Snuderl M, Batista A, Kirkpatrick ND, et al. (2013) Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 152: 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guo Y, Wang X, Tian X, et al. (2012) Tumor-derived hepatocyte growth factor is associated with poor prognosis of patients with glioma and influences the chemosensitivity of glioma cell line to cisplatin in vitro. World J Surg Oncol 10: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wen PY, Schiff D, Cloughesy TF, et al. (2011) A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol 13: 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.De Groot JF, Prados M, Urquhart T, et al. (2009) A phase II study of XL184 in patients (pts) with progressive glioblastoma multiforme (GBM) in first or second relapse. J Clin Oncol 27: 2047. [Google Scholar]
- 118.Schiff D, Desjardins A, Cloughesy T, et al. (2016) Phase 1 dose escalation trial of the safety and pharmacokinetics of cabozantinib concurrent with temozolomide and radiotherapy or temozolomide after radiotherapy in newly diagnosed patients with high-grade gliomas. Cancer 122: 582–587. [DOI] [PubMed] [Google Scholar]
- 119.Wu HB, Wang Z, Wang QS, et al. (2015) Use of labelled tLyP-1 as a novel ligand targeting the NRP receptor to image glioma. PLoS One 10: e0137676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ying M, Shen Q, Zhan C, et al. (2016) A stabilized peptide ligand for multifunctional glioma targeted drug delivery. J Control Release 243: 86–98. [DOI] [PubMed] [Google Scholar]
- 121.Ying M, Zhan C, Wang S, et al. (2016) Liposome-based systemic glioma-targeted drug delivery enabled by all-d peptides. ACS Appl Mater Inter 8: 29977–29985. [DOI] [PubMed] [Google Scholar]
- 122.Liu C, Yao S, Li X, et al. (2017) iRGD-mediated core-shell nanoparticles loading carmustine and O6-benzylguanine for glioma therapy. J Drug Target 25: 235–246. [DOI] [PubMed] [Google Scholar]
- 123.Liu Y, Mei L, Xu C, et al. (2016) Dual receptor recognizing cell penetrating peptide for selective targeting, efficient intratumoral diffusion and synthesized anti-glioma therapy. Theranostics 6: 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bechet D, Auger F, Couleaud P, et al. (2015) Multifunctional ultrasmall nanoplatforms for vascular-targeted interstitial photodynamic therapy of brain tumors guided by real-time MRI. Nanomed-Nanotechnol 11: 657–670. [DOI] [PubMed] [Google Scholar]
- 125.Patnaik A, Lorusso PM, Messersmith WA, et al. (2014) A Phase Ib study evaluating MNRP1685A, a fully human anti-NRP1 monoclonal antibody, in combination with bevacizumab and paclitaxel in patients with advanced solid tumors. Cancer Chemoth Pharm 73: 951–960. [DOI] [PubMed] [Google Scholar]