

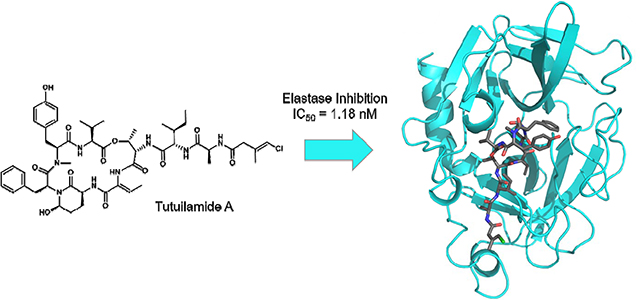
Abstract
Marine cyanobacteria (blue-green algae) have been shown to possess an enormous capacity to produce structurally diverse natural products that exhibit a broad spectrum of potent biological activities, including cytotoxic, antifungal, antiparasitic, antiviral and antibacterial activities. Using mass spectrometry-guided fractionation together with molecular networking, cyanobacterial field collections from American Samoa and Palmyra Atoll yielded three new cyclic peptides, tutuilamides A-C. Their structures were established by spectroscopic techniques including 1D and 2D NMR, HR-MS, and chemical derivatization. Structure elucidation was facilitated by employing advanced NMR techniques including non-linear sampling in combination with 1,1-ADEQUATE. These cyclic peptides are characterized by the presence of several unusual residues including 3-amino-6-hydroxy-2-piperidone and 2-amino-2-butenoic acid, together with a novel vinyl chloride-containing residue. Tutuilamides A-C show potent elastase inhibitory activity together with moderate potency in H-460 lung cancer cell cytotoxicity assays. The binding mode to elastase was analyzed by X-ray crystallography revealing a reversible binding mode similar to the natural product lyngbyastatin 7. The presence of an additional hydrogen bond with the amino acid backbone of the flexible side chain of tutuilamide A, compared to lyngbyastatin 7, facilitates its stabilization in the elastase binding pocket and possibly explains its enhanced inhibitory potency.
Keywords: Cyanobacteria, Cyclic depsipeptides, Elastase inhibitors, Molecular network, Natural products
Graphical Abstract
INTRODUCTION
Cyanobacteria produce a wide range of cyclic depsipeptides that contain an unusual 3-amino-6-hydroxy-2-piperidone (Ahp) moiety, generally referred to as cyanopeptolin-like peptides or Ahp-cyclodepsipeptides.1–3 The general structure includes an amino acid chain of variable length of which six amino acid residues form a macrocyclic ring. Most cyanopeptolin-like peptides contain the Ahp residue and an ester linkage between the β-OH group of L-threonine and the carboxyl group of the C-terminal amino acid. Additionally, among these Ahp containing peptides, there are more than 20 compounds that contain a highly conserved hexadepsipeptide core bearing an additional 2-amino-2-butenoic acid (Abu) moiety adjacent to the Ahp ring together with a highly variable side chain (Table 1).
Table 1.
Abu and Ahp-containing peptides isolated from Cyanobacteria.
| Compound name | Year | Collected from | Isolated organism |
|---|---|---|---|
| Dolastatin 135 | 1989 | East Africa, Indian Ocean | Dolabella auricularia (sea hare) |
| Symplostatin 26 | 1999 | Guam, Pago Bay | Symploca hydnoides |
| Somamide A+B7 | 2001 | Fiji, Somo Somo | Lyngbya majuscula / Schizothrix sp. assemblage |
| Lyngbyastatin 48 | 2007 | South Florida, Atlantic coast | Lyngbya confervoides |
| Lyngbyastatin 5–79 | 2007 | South Florida, Fort Lauderdale / Florida Keys, Summerland Key | Lyngbya confervoides |
| Lyngbyastatin 8–1010 / Bouillomide A+B11 | 2009 | Guam, Tumon Bay | Lyngbya semiplena |
| Molassamide12 | 2010 | Florida, Molasses Reef | Dichothrix utahensis |
| Stigonemapeptin4 | 2012 | Wisconsin, North Nokomis Lake | Stigonema sp. (freshwater) |
| Symplostatin 5–1013 | 2013 | Guam, Cetti Bay | red Symploca sp. |
| Kurahamide14 | 2014 | Japan, Kuraha | Lyngbya sp. assembly |
Ahp-containing peptides have frequently been observed from both marine and freshwater cyanobacteria as well as other bacteria, and over 200 are now known.3 However, peptides containing the Abu moiety are generally found in marine bacteria with the exception being stigonemapeptin which derives from a freshwater cyanobacterium (Table 1).4 Interestingly, the Abu moiety in stigonemapeptin is (E)-configured whereas the Abu moieties derived from marine sources are consistently (Z)-configured.
X-ray crystallography of the elastase complex formed with lyngbyastatin 7 (4) revealed that these Abu-cyclodepsipeptides act as substrate mimics.13 The Abu moiety was shown to occupy the S1 substrate binding pocket and engage in a non-covalent interaction. Additionally, a study of the binding mode of scyptolin showed that the Ahp moiety occupies a crucial part of the active site pocket and thereby prevents hydrolysis.15 It was suggested that the Abu moiety increases the potency of these elastase inhibitors and that the pendant side chain is responsible for modulating their activity and selectivity.13
In the current work, we report the isolation of three new members of this Ahp and Abu-containing family of peptides, named tutuilamides A (1) and B (2) based on the location of collection of the source cyanobacterium, Schizothrix sp., along with tutuilamide C (3) isolated from a Coleofasciculus sp. These structures were assembled by a combination of NMR, mass spectrometry (MS) and chiral chromatography analysis following acid hydrolysis, and feature an unusual vinyl chloride-containing residue never previously observed in this structure class. The new natural products, as well as two semisynthetic derivatives, were evaluated for serine protease inhibition and all of the cyclic species were found to be highly potent. The crystal structure of elastase in complex with tutuilamide A revealed extensive binding interactions in the substrate binding pocket, as has been shown previously with lyngbyastatin 7 (4). However, we identified additional hydrogen bond interactions between tutuilamide A and elastase that did not occur in the lyngbyastatin 7 co-crystal structure. These may be responsible for the increased potency of tutuilamide A compared to lyngbyastatin 7.
RESULTS AND DISCUSSION
Our discovery strategy to locate natural products with novel structural frameworks includes MS2-based metabolomics (Molecular Networking) for strain selection and dereplication as well as chromatographic methods for isolation driven by structural features. Cyanobacterial colonies of the genus Schizothrix sp. and Coleofasciculus sp. were collected by hand from the main island of Tutuila in American Samoa in 2016 and Palmyra Atoll in 2008, respectively, using SCUBA gear. The crude CH2Cl2-MeOH (2:1) extract was initially fractionated using vacuum-liquid chromatography (VLC) as well as solid phase extraction (SPE) for further analysis by MS and NMR. This approach revealed the presence of peptides with unusual features and led to the HPLC isolation of tutuilamides A and B from Schizothrix sp and tutuilamide C from Coleofasciculus sp. In addition, we identified related peptides such as symplostatin 2 together with several derivatives of dolastatin 10 that have yet to be characterized. Tutuilamide A (1) and B (2) share the same cyclic peptide core and possess the unusual Ahp and Abu moieties. They differ by a single side chain residue wherein isoleucine is replaced by valine (Figure 1). Meanwhile, tutuilamide C (3) is an analog of 2 that lacks an alanine moiety, but bears an additional methylene unite in the aliphatic side-chain. Moreover, they are the first cyanopeptolins to possess a vinyl chloride residue in the side chain. Vinyl chloride functionalities in cyanobacteria have been shown to biosynthetically result from a unique cassette of enzymes that involve polyketide synthase (PKS) beta branch formation along with radical-based chlorination of an intermediate, such as in jamaicamide A.16 Compounds 1-3 show moderate cytotoxic activity towards the H-460 lung cancer cell line with IC50’s of 0.53 ± 0.04 μM, 1.27 ± 0.21 μM and 4.78 ± 0.45 μM, respectively.
Figure 1.

Chemical structure of tutuilamide A (1), B (2) and C (3) together with the related lyngbyastatin 7 (4).9
High-resolution electrospray ionization mass spectrometry (HR-ESI-MS) of tutuilamide A (1) displayed an ion peak at m/z 1043.4616 [M + Na]+ (calculated for C51H69ClN8O12Na, 1043.4616, Δ = 0.0 ppm), consistent with the molecular formula C51H69ClN8O12 containing 21 double-bond equivalents. The isotope pattern for the molecular ion cluster indicated the clear presence of one chlorine atom.
The 1H NMR spectrum of 1 in DMSO-d6 exhibited signals characteristic of a peptide including seven α-proton signals at δ 4.65 (overlap), 4.89 (1H, dd, J = 11.5, 2.2 Hz), 4.73 (1H, dd, J = 11.4, 4.0 Hz), 3.79 (1H, m), 4.67 (overlap), 4.40 (1H, t, J = 7.8 Hz), and 4.36 (1H, p, J = 7.2 Hz), along with six amide NH signals at δ 7.53 (1H, t, J = 8.4 Hz), 7.22 (1H, broad), 9.23 (1H, broad), 7.91 (1H, broad), and 8.21 (1H, d, J = 7.4 Hz) (Table S1). Additionally, a downfield pair of triplets at δ 7.18 (2H, t, J = 7.2 Hz) and 7.15 (1H, t, J = 7.2 Hz) and a doublet at δ 6.83 (2H, d, J = 7.2 Hz) were characteristic of a phenyl group; similarly, two doublets at δ 7.00 (2H, d, J = 8.3 Hz) and 6.78 (2H, d, J = 8.3 Hz) were characteristic for a para-substituted phenol group. The proton NMR spectrum also showed a singlet at δ 2.77 (3H, s), indicative of an N-methyl amide, together with eight partially overlapping methyl signals at δ 0.75 (3H, d, J = 6.8 Hz), δ 0.81 (3H, t, J = 7.5 Hz), δ 0.84 (3H, d, J = 6.8 Hz), δ 0.88 (3H, d, J = 6.8 Hz), δ 1.19 (3H, d, J = 7.0 Hz), δ 1.21 (3H, d, J = 6.6 Hz), δ 1.48 (3H, d, J = 7.1 Hz), and δ 1.72 (3H, d, J = 1.2 Hz).
The HSQC spectrum revealed the presence of six methylene groups (δC-3Tyr 32.5, δH-3Tyr 3.11/2.69; δC-3Phe 35.2, δH-3Phe 2.88/1.83; δC-3Ahp 21.9, δH-3Ahp 2.42/1.58; δC-4Ahp 29.2, δH-4Ahp 1.72/1.58; δC-4Ile 23.9, δH-4Ile 1.43/1.08; δC-2 42.5, δH-2Cmb 2.97), two methines (δC-3Val 30.5, δH-3Val 2.07; δC-3Ile 36.5, δH-3Ile 1.81), two oxygenated methines (δC-5Ahp 73.7, δH-5Ahp 5.08; δC-3Thr 71.7, δH-3Thr 5.53), and two vinylic methines (δC-3Abu 131.5, δH-3Abu 6.52; δC-4Cmb 114.3, δH-4Cmb 6.10).
A detailed analysis of the 2D NMR data (HSQC, HMBC, and DQFCOSY) established the presence of valine, N-methyl-tyrosine, phenylalanine, threonine, isoleucine and alanine together with an Ahp, an Abu and a 4-chloro-3-methylbut-3-enoic acid (Cmb) residue. The Ahp residue was identified by COSY correlations following the sequential spin system starting at the α-proton at δ3.79 (H-2Ahp) followed by two methylene protons at δ2.42/1.58 (H-3Ahp), another set of methylene protons at δ1.72/1.58 (H-4Ahp), and an oxygenated methine at δ5.08 (H-5Ahp). The ring closure was based on HMBC correlations from the α-proton at δ3.79 (H-2Ahp), the methylene protons at δ2.42 and 1.58 (2H-3Ahp) and the oxygenated methine proton at δ5.08 (H-5Ahp) to the carbonyl carbon at δ168.5.
The Abu structure was based on a COSY correlation between the methyl protons at δ1.48 (Me-4Abu) and a vinylic methine proton at δ6.52 (H-3Abu) as well as HMBC correlations from both proton signals to carbon resonances at δ129.8 (C-2Abu) and 162.6 (C-1Abu). The structure of the remaining C5H6ClO residue was deduced as follows. Long-range COSY correlations between the vinylic methine proton at δ6.10 (H-4Cmb), the methyl protons at δ1.72 (Me-5Cmb) and the methylene protons at δ2.97 (2H-2Cmb) gave an unclear picture of the residue structure. Additionally, HMBC correlations from all three of these proton signals to a quaternary carbon at δ133.9 (C-3Cmb) and a carbonyl signal at δ168.4 (C-1Cmb) left the position of the double bond unresolved. To differentiate between a 4-chloro-3-methylbut-3-enoic acid and a 4-chloro-3-methyl-2-enoic acid residue, a 2-bond 1,1-ADEQUATE correlation with non-uniform sampling (NUS) was recorded from the methylene protons at δ2.97 (2H-2Cmb) to the carbonyl signal at δ168.4 (C-1Cmb) confirming this as a 4-chloro-3-methylbut-3-enoic acid residue; this assignment was also in better alignment with the predicted chemical shifts. Finally, an NOE correlation between the associated amide proton at δ8.21 (NHAla) and the methylene protons at δ 2.97 (2H-2Cmb) helped to confirm its identity.
The sequence of residues was established as Val-N-MeTyr-Phe-Ahp-Abu-Thr-Ile-Ala-Cmb through HMBC correlations between consecutive α-protons to carbonyl carbons of adjacent residues together with NOE correlations between α-protons and amide protons (Figure 2). The ring closure between the C-terminal valine and the β-hydroxy group of threonine was established based on NOE correlations from the valine α-proton at δ4.65 (H-2Val) to the threonine methine and methyl protons at δ5.53 (H-3Thr) and δ1.21 (Me-3Thr), respectively.
Figure 2.

Selected NOE correlations for tutuilamide A (1).
The second and considerably less abundant new compound isolated from this extract, tutuilamide B (2), showed a HR-ESI-MS molecular ion peak at m/z 1029.4445 [M + Na]+ (calcd for C50H67ClN8O12Na, 1029.4459, Δ = 1.36 ppm), thus having one less methylene unit than 1. Comparison of NMR features for 2 (Table S1) showed it to be highly similar in all regards to 1, with the primary difference being that the triplet methyl from the Ile residue at δ0.80 (Me-5Ile) in 1 was replaced by an additional doublet methyl at δ0.82 (Me-5Val2) in 2, thus revealing 2 to be the valine analog of 1.
The absolute configuration of the amino acid residues in both natural products were determined by LC-MS analysis of the 1-fluoro-2,4-dinitro-phenyl-5-D-alanineamide (D-FDAA) derivatives deriving from the acid hydrolyzate of 1 and 2 (Marfey’s method, Table S3).17 This revealed that all of the amino acid residues in both compounds were of L-configuration as is the case for all other cyanopeptolin-like peptides. In addition, PDC oxidation followed by acid hydrolysis liberated L-glutamic acid from the Ahp residue, as determined by Marfey’s analysis, therefore establishing the configuration of C-2Ahp as S. To determine the relative stereochemistry between C-2Ahp and C-5Ahp in 1, a combination of proton-proton coupling constants and NOE correlations was used (Figure 2). The relative conformation of the Ahp ring was determined based on the C-2Ahp to C-5Ahp coupling constant; a large value was observed (3JH-2,H-3a = 12.3 Hz), indicating that these two deshielded protons were axially oriented.18 NOE correlations between H-2Ahp, H-4aAhp and H-5Ahp as well as between H-3aAhp, OHAhp and NHAhp revealed the relative configuration of the two stereocenters, and therefore established the configuration of C-5Ahp as R. The geometry of the Abu olefinic bond was determined as Z based on an NOE correlation in DMSO-d6 between the methyl protons at δ1.48 (Me-4Abu) and the amide proton at δ9.23 (NHAbu). Finally, a 1D NOE experiment with selective eradiation of the vinylic proton at δ6.07 (in methanol-d4 to avoid overlapping signals with OHAhp) established the E geometry of the Cmb residue due to a correlation to the methylene protons at δ3.03 (2H-2Cmb).
Subsequently, a third tutuilamide was isolated from a Coleofasciculus sp. extract (3). The HR-ESI-MS of tutuilamide C showed a precursor ion at m/z 972.4252 [M + Na]+ (calcd for C48H64ClN7O11Na, 972.4255, Δ = 0.31 ppm) and the NMR data were highly similar to that of 2, except devoid of the alanine moiety resonances and possessing an additional methylene in the side-chain (Table S2). HMBC, TOCSY and ROESY correlations corroborated the same connectivities observed between the amino acid residues as for 1 and 2. Because the NMR data of 2 and 3 were highly similar, we propose that they are of the same relative configuration at comparable stereocenters. This hypothesis is additionally supported by the observation that all Abu and Ahp-containing peptides reported to date are composed only of L-amino acids.7–14
Inhibition of serine proteases with tutuilamide analogs
Studies have shown that Ahp-Abu-containing cyclic hexadepsipeptides inhibit serine proteases such as elastase by binding to the active site in a substrate-like manner. Therefore, 5 μM to 85 pM of 1, 2 and 3 were incubated with porcine pancreatic elastase and potency was directly compared to lyngbyastatin 7 (4) and symplostatin 2. Compounds 1 and 2 were found to be the most potent in this group, inhibiting elastase with IC50’s of 1–2 nM (Table 3, Figure S2 and S3). Compound 3 and symplostatin 2 had IC50 values of ~5 nM, while 4 was the weakest inhibitor with an IC50 of 11.5 nM.
Table 3:
Potency of compounds for select serine proteases following 30 minute incubations
| IC50 values (nM) with 95% Confidence Interval Values | ||||
|---|---|---|---|---|
| Name (Analog Number) | Elastase | Chymotrypsin | Trypsin | Proteinase K |
| Tutuilamide A (1) | 1.18 (0.33 – 1.97) | 1014 (853.0 –1227) | >20,000 | 103.7 (84.6 – 129.2) |
| Tutuilamide B (2) | 2.05 (1.68 – 2.46) | 576.6 (447.0 – 767.3) | >20,000 | 87.6 (65.2 – 122.2) |
| Tutuilamide C (3) | 4.93 (2.72 – 7.48) | 542.0 (445.0 – 671.2) | >20,000 | >5,000 |
| Lyngbyastatin 7 | 11.50 (9.25 – 14.28) | Not Done | Not Done | 1574 (806.0 to >5,000) |
| Symplostatin 2 | 5.41 (4.30 – 6.69) | Not Done | Not Done | 216.5 (155.2 – 326.0) |
| Tutuilamide A-linear (5) | 3,080 (2371 – 4248) | Not Done | Not Done | >5,000 |
| Tutuilamide A-methyl (6) | 1.83 (1.56 – 2.12) | Not Done | Not Done | 98.6 (94.4 – 100.1) |
Elastase is a member of the S1 family of serine proteases that forms a double β-barrel structure. Trypsin and chymotrypsin are also members of the S1 family and therefore we determined if tutuilamide A, B and C can also inhibit these structurally related enzymes. When bovine trypsin was incubated with up to 20 μM of these compounds, no inhibition was detected. This enzyme has a strong preference for binding to peptide inhibitors containing basic amino acids such as arginine and lysine, both of which are absent from these natural products. When incubated with chymotrypsin the IC50 values ranged from 542 nM to 1014 nM. Although the potency of these compounds is considerably weaker for chymotrypsin than elastase, the relative potency of the three tutuilamide analogs differs between enzymes. Chymotrypsin appears to have a preference for valine as found in 2 and 3 over isoleucine found in compound 1, resulting in a 2-fold reduction in potency. This inhibition profile is consistent with several other Ahp-Abu-containing cyclic hexadepsipeptides that preferentially inhibit elastase over chymotrypsin and trypsin.19
Outside of the S1 family, commonly studied serine proteases are from the S8 family. These enzymes have no sequence or structural homology with S1 family serine proteases and include the bacterial and fungal subtilisin enzymes commonly used in laundry detergents and the mammalian kexin family of enzymes.20 When 1 and 2 were incubated with proteinase K, a representative S8 protease isolated from the soil fungus Engyodontium album, we observed IC50s of 103.7 and 87.6 nM, respectively.
However, 3 had more than a 50-fold weaker inhibition for this enzyme (IC50 >5 μM) indicating that longer peptides have higher potency for this enzyme. When comparing the potency of symplostatin 2, an 8-mer cyclic hexadepsipeptides peptide to the 7-mer peptide 4, the IC50 value deceased 7.3-fold for the shorter peptide inhibitor. These data show that changes in peptide length and amino acid composition of these cyclic hexadepsipeptides result in differences in potency and selectivity for serine proteases.
In addition, we determined the cytotoxic activity of compounds 1-3 towards the H-460 lung cancer cell line. The compounds show moderate activity with IC50s of 0.53 ± 0.04 μM, 1.27 ± 0.21 μM and 4.78 ± 0.45 μM, respectively (Figure S1).
Synthesis of semi-synthetic analogues of 1
A limited structure-activity relationship study on tutuilamide A was performed to explore the structural features related to the elastase and proteinase K inhibitory activity (Scheme 1). In the first semi-synthetic analogue of 1, the depsipeptide ester was hydrolyzed using LiOH/H2O to obtain the acyclic analogue 5. This analogue failed to inhibit proteinase K activity at a concentration of 5 μM while potency for elastase was decreased 2,600-fold when compared to the cyclic tutuilamide A (1).
Scheme 1.

Synthesis of semi-synthetic analogues 5 and 6 of tutuilamide A (1)
These data demonstrate the importance of the cyclic structure to inhibit these serine proteases. In addition, the hemi-aminal moiety in 1 was methylated with methanol in the presence of pyridinium p-toluenesulfonate (PPTS) to generate analogue 6. This modification is predicted to improve the metabolic stability of the Abu moiety without affecting the potency of the compound; indeed, analog 6 showed excellent single digit nanomolar potency to elastase (Table 3).
Rationalizing the binding of tutuilamide A to elastase
We next determined the crystal structure of porcine pancreatic elastase in complex with 1 in order to compare the binding mode of the depsipeptide with related molecules. The complex crystallized in space group P 21 21 21, and data was collected to 2.2 Å. The structure was determined by molecular replacement using the published elastase apo structure (PDB ID: 1LVY) as a search model (Figure 3A). Full details of the data collection and refinement statistics can be found in Table S4. Compound 1 was found to occupy the same binding pocket as lyngbyastatin 7 (4), explaining the observed identical mode of action of tutuilamides compared to other Abu-bearing cyclic depsipeptides, with the Abu moiety and the N-terminal residues occupying the S1-S4 pockets (Figure 3B).13,15 However, compared to 4, the carbonyl group of the flexible Cmb moiety of 1 forms an additional hydrogen bond with the backbone amide of Arg217 (Figure 3B and Figure S4). This additional interaction could explain the slightly more potent inhibitory activity of tutuilamides compared to lyngbyastatin 7 in that this interaction would further stabilize the binding of tutuilamide A to the elastase binding pocket.
Figure 3.

A Cartoon representation of PPE (cyan) in complex with 1 (shown as grey sticks). B Comparison of 1 (grey) and 4 (PDB: 4GVU; orange) binding to PPE (cyan surface). The side chains of PPE residues Ser216 and Arg217 are shown as sticks. The additional hydrogen bond found in the PPE-1 complex structure is indicated by a dashed line.
The potency of semi-synthetic analogues 5 and 6 was evaluated in the context of our co-crystal structure. As shown for 4, the cyclic core of 1 also forms a network of direct and water mediated inter- and intramolecular hydrogen bonds (Figure S5). Therefore, one would expect any changes to the cyclic nature of these compounds to have a negative impact on their potency. This is indeed the case as shown by the observed 3,000-fold decrease in potency of acyclic analogue 5. The hemiaminal moiety of 1 is facing a hydrophobic pocket formed by Leu 63 and Phe 65 (Figure S5). This pocket is large enough to accommodate methylated analogue 6, without resulting in a steric clash with the binding pocket (Figure S5). Given the size of the pocket, even larger hydrophobic functional groups at this position may be accommodated. This would result in an enhancement of hydrophobic interactions, thereby further stabilizing the compound in the active site. However, further optimization of this group must be pursued cautiously, as the oxygen atom of the hydroxy or methoxy group is involved in intramolecular hydrogen bonding with the neighboring residues, and thus facilitates the formation of a cis peptide bond (Figure S5). In turn, this contributes to the rigidity of the cyclic core making these compounds less sensitive to protease activity.13,15,21,22
CONCLUSION
Tutuilamides A-C are new representatives of the family of Ahp-cyclodepsipeptides (cyanopeptolin-like peptides) isolated from Cyanobacteria. They share the cyclic peptide core including the distinctive 2-amino-2-butenoic acid (Abu) residue with a group of cyclic depsipeptides mainly isolated from marine cyanobacteria; however, they also contain an unprecedented 4-chloro-but-3-enoic acid moiety. The predicted biosynthesis of these new cyclic lipopeptides suggests initiation of the pathway by a polyketide synthase which produces an intermediate β-ketobutyrate moiety. This is expected to undergo transformation by a β-branch cassette of genes that also introduces a chlorine atom via radical chemistry, as shown in the case of jamaicamide A biosynthesis.16
The new compounds are potent inhibitors of porcine pancreatic elastase and fungal proteinase K. Direct comparison to the potent elastase inhibitor lyngbyastatin 7 revealed that the new compounds have 2 to 4-fold increased potency. Structural analysis of tutuilamide A in complex with the porcine pancreatic elastase confirms the same binding mechanism of lyngbyastatin 7 with an additional strong hydrogen bond (2.8 Å) between the Cmb carbonyl group and the backbone amide group of elastase residue R226; this additional hydrogen bond appears to stabilize the ligand in the binding pocket and may explain the increased potency of tutuilamides A over lyngbyastatin 7.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured on a JASCO P-2000 polarimeter, UV/Vis data were obtained using a Beckman DU800 spectrophotometer, and IR spectra were recorded on a Nicolet 100 FT-IR spectrometer. NMR data were obtained on a JEOL ECZ 500 NMR spectrometer equipped with a 3 mm inverse probe (H3X), the 1,1-ADEQUATE experiment was performed on a Bruker AVANCE III 600 MHz NMR with a 1.7 mm dual tune TCI cryoprobe. NMR data were recorded in either DMSO-d6 or methanol-d4 and adjusted to the residual solvent peak (DMSO-d6 δH 2.50, δC 39.52; methanol-d4 δH 3.31, δC 49.00). For HPLC-MS analysis, a Thermo Finnigan Surveyor HPLC System with a Phenomenex Kinetex 5 μm C18 100 × 4.6 mm column coupled to a Thermo-Finnigan LCQ Advantage Max Mass Spectrometer was used. Samples were analyzed using a linear gradient with (A) H2O + 0.1% FA to (B) CH3CN + 0.1% FA at a flow rate of 0.6 ml/min and UV detection at 220 nm, 254 nm and 280 nm. For HR-ESI-MS analysis, an Agilent 6530 Accurate Mass QTOF mass spectrometer was used in the positive ion mode. Semi-preparative HPLC was performed on a Thermo Fisher Scientific HPLC system with a Thermo Dionex UltiMate 3000 pump, RS autosampler, RS diode array detector, and automated fraction collector. All solvents were HPLC grade except for H2O, which was purified by a Millipore Milli-Q system before use.
Extraction and Isolation
A 1L packed volume of the purple Schizothrix sp. (voucher specimen available from W.H.G. as collection number ASG-23JUL14–2) was collected by SCUBA at 3–5 m in the Fagasa Bay, Tutuila, American Samoa. Samples were stored in 70% EtOH at −20°C prior to extraction. Approximately 40 g (dry wt) of cyanobacterial mat was extracted repeatedly with CH2Cl2/MeOH (2:1) to afford 1.8 g of crude extract. The material was fractionated by vacuum liquid chromatography (VLC) using a stepwise gradient of increasing polarity, starting with 10% EtOAc in hexanes and finishing with 100% MeOH, to produce nine fractions (B-J). Fraction 2241-H (eluted with 25% methanol in EtOAc, 447.6 mg) was subjected to fractionation via solid phase extraction using a 1 g SPE-C18 cartridge (35% to 100% MeOH in H2O) resulting in four fractions (H1-H4). Fraction 2241-H2 (eluted with 65% methanol) was subjected to semi-preparative HPLC purification using a linear gradient on a Phenomenex Kinetex C18 150 × 10.0 mm × 5 μm column from (A) H2O + 0.1% formic acid to (B) ACN + 0.1% formic acid at a flow rate of 4.7 mL/min, monitored at 220 nm. The gradient started with a 5 min isocratic step at 35% B followed by an increase to 39% B in 35 min, and yielded 46.5 mg of compound 1 (RT = 33.8 min) and 1.9 mg of compound 2 (RT = 26.6 min). This semi-preparative isolation also provided 4.2 mg of symplostatin 2 (RT = 16.0 min) (HR-ESI-MS displayed an ion peak at m/z 1051.5865 [M + H]+ (calcd for C52H75N8O13S, 1051.5169). Similar procedures were applied to a Coleofasciculus sp. sample collected in Palmyra Atoll (PAL22AUG08–1), yielding 2.1 mg of compound 3 (RT= 6.6 min) after HPLC fractionation of fraction H through the following elution gradient: 45% B to 85% B in 12 min at a flow rate of 2.5 mL/min.
Tutuilamide A (1)
White, amorphous solid. [α]25D −22.6 (c 0.80, MeOH); UV (MeOH) λmax 212 nm (log ε 3.82), 277 nm (log ε 3.18); IR (KBr) 3375, 2967, 1732, 1643, 1537, 1448, 1337, 1204, 1024, 997 cm−1; HR-ESI-MS [M + Na]+ m/z 1043.4616 (calcd for C51H69ClN8O12Na, 1043.4616).
Tutuilamide B (2)
White, amorphous solid. [α]25D − 21.3 (c 0.95, MeOH); UV (MeOH) λmax 209 nm (log ε 3.73), 277 nm (log ε 3.20); IR (KBr) 3379, 2964, 1733, 1644, 1538, 1441, 1337, 1203, 1019, 351 cm−1; HR-ESI-MS [M + Na]+ m/z 1029.4445 (calcd for C50H67ClN8O12Na, 1029.4459).
Tutuilamide C (3)
White, amorphous solid. [α]25D −7.6 (c 0.20, MeOH); UV (MeOH) λmax 221 and 275 nm; HR-ESI-MS [M + Na]+ m/z 972.4252 (calcd for C48H64ClN7O11Na, 972.4255).
Pyridinium dichromate (PDC) oxidation
An aliquot of tutuilamide A (1, 0.6 mg) was dissolved in CH2Cl2 (1.0 mL), to which was added 2 mg PDC and the reaction mixture was allowed to stand for 16 h at RT. The mixture was quenched by shaking with 2 × 1 mL portions of H2O, followed by separation of the CH2Cl2 phase and drying under N2. The residues were analyzed by Marfey’s method as described below.
Advanced Marfey’s Method
Small aliquots (0.6 mg) of both tutuilamide A (1) and B (2), as well as the residue of the PDC reaction, were hydrolyzed with 6 N HCl (0.6 mL) at 95°C for 16 h. The reaction mixtures were dried under N2 and lyophilized. The residues were dissolved in H2O (200 μL). Two 50 μL aliquots of each sample were mixed with 1 N NaHCO3 (20 μL) and 1% 1-fluoro-2,4-dinitrophenyl-5-alanine-amide (L-FDAA or D-FDAA solution in acetone, 100 μL) were added, respectively. Additionally, 1 to 2 mg samples of the respective L- and D-amino acid standards were dissolved in 200 μL of H2O and 1N NaHCO3 (80 μL) and 1% 1-fluoro-2,4-dinitrophenyl-5-alanine-amide (D-FDAA solution in acetone, 400 μL) were added. The mixtures were heated to 45 °C for 50 min and the reactions quenched by addition of 2 N HCl (10 μL for samples, 40 μL for standards). Solvents were evaporated under N2, and residues were redissolved in CH3CN and subsequently analyzed by HPLC-MS using a gradient of 5–55% solvent B (CH3CN + 0.1% formic acid at a flow rate of 4.7 mL/min) over a 60 min period.
Protease activity assays
Stocks of porcine pancreatic elastase, Type 1 (Sigma-Aldrich, P2308), human chymotrypsin (Sigma-Aldrich, 230900), bovine trypsin (Sigma-Aldrich, T-7409), and proteinase K (Sigma-Aldrich P-2308) were stored at −20°C and diluted to the appropriate concentration in assay buffer consisting of Dulbecco’s phosphate buffered saline, pH 7.4, 0.01% Tween-20 and 2 mM dithiothreitol. All inhibitors were stored in DMSO and diluted to 20 μM in assay buffer. Compounds were then serially diluted 3-fold in assay buffer and mixed with an equal volume of enzyme. After 30 minutes incubation at 22°C, 15 μL of the enzyme-inhibitor mixture was added to wells of black 384-well plates (Thermo Scientific, 262260) containing 15 μL of substrate diluted in assay buffer. Fluorescence was measured in 3-minute intervals at 22°C using a Synergy HTX Multi-Mode Microplate Reader (BioTek, Winooski, VT) with excitation and emission wavelengths of 360 and 460 nm, respectively. Protease activity was reported as the change in relative fluorescent units per second over a 30 minute time interval. The final concentration of inhibitor ranged from 20 μM to 339 pM for trypsin and chymotrypsin assays and from 5 μM to 85 pM for elastase and proteinase K assays. The final enzyme and substrate concentration in each assay were as follows: bovine trypsin (10 nM) and z-Leu-Arg-Arg-AMC (100 μM); human chymotrypsin (10 nM) and Suc-Ala-Ala-Pro-Phe-AMC (100 μM); porcine pancreatic elastase (10 nM) and MeOSuc-Ala-Ala-Pro-Val-AMC (223 μM), proteinase K (10 nM) and Suc-Ala-Ala-Pro-Phe-AMC (100 μM).
Semi-synthesis of tutuilamide analogue 5
To a solution of tutuilamide A (1.0 mg, 0.98 μmol, 1 equiv.) in THF/H2O (0.5 mL, 2:1) was added LiOH/H2O (0.1 mg, 2.4 mmol, 2.4 equiv.) and the reaction was stirred at 0 oC temperature for 4 h. After LC-MS indicated complete consumption of starting material, the reaction was evaporated and the resulting residue was suspended in H2O, acidified and extracted with ethyl acetate (3 × 2 mL). The combined organic layer was dried using anhydrous sodium sulfate, filtered and evaporated to obtain the free acid as a white solid. HPLC purification of the crude product afforded the pure product 5 as amorphous white solid (0.6 mg, 60% yield). 1H NMR (599 MHz, DMSO-d6) δ 9.51 (m, 1H), 9.28 (s, 1H), 8.22 (d, J = 7.4 Hz, 1H), 7.99 (bs, 1H), 7.84 (m, 1H), 7.19 (t, J = 7.4 Hz, 2H), 7.16 (t, J = 7.4 Hz, 1H), 7.06 (bs, 1H), 6.99 (d, J = 8.0 Hz, 2H), 6.84 (d, J = 7.1 Hz, 2H), 6.77 (d, J = 8.0 Hz, 2H), 6.73 (d, J = 9.4 Hz, 1H), 6.52 (q, J = 7.0 Hz, 1H), 6.11 (m, 1H), 5.01 (m, 1H), 4.93 (d, J = 11.1 Hz, 1H), 4.79 – 4.71 (m, 1H), 4.69 – 4.59 (m, 3H), 4.36 (m, 2H), 3.74 (q, J = 9.7 Hz, 1H), 3.06 (m, 2H), 2.95 (s, 2H), 2.76 (s, 3H), 2.73–2.69 (m, 2H), 2.22–2.20 (m, 2H), 2.04 (d, J = 13.9 Hz, 1H), 1.82 – 1.75 (m, 1H), 1.74–1.68 (m, 4H), 1.57 (t, J = 9.1 Hz, 1H), 1.46 (d, J = 7.1 Hz, 3H), 1.39 (t, J = 14.0 Hz, 2H), 1.25–1.15 (m, 6H), 1.06 (dp, J = 14.2, 7.2, 6.8 Hz, 1H), 0.89 (d, J = 6.6 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H), 0.84–0.77 (m, 6H). [α]25D −14.0 (c 0.12, MeOH); HR-ESI-MS [M + Na]+ m/z 1062.6120 (calcd for C51H71ClN8O13Na, 1062.6120).
Semi-synthesis of tutuilamide analogue 6
Tutuilamide A (4.0 mg, 3.92 μmol, 1 equiv.) was dissolved in MeOH and PPTS (5 mol%) was added. TLC indicated completion of the reaction after 1 h. The reaction was evaporated and purified using HPLC to afford the analytically pure product 6 as a colorless oil (1.0 mg, 23% yield). 1H NMR (599 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.20 (d, J = 7.4 Hz, 1H), 8.00 (bs, 1H), 7.82 – 7.78 (m, 1H), 7.20 (t, J = 7.4 Hz, 2H), 7.16 (t, J = 7.4 Hz, 1H), 7.06 (bs, 1H), 7.00 (d, J = 8.0 Hz, 2H), 6.84 (d, J = 7.1 Hz, 2H), 6.77 (d, J = 8.0 Hz, 2H), 6.73 (d, J = 9.4 Hz, 1H), 6.52 (q, J = 7.0 Hz, 1H), 6.05 – 6.01 (m, 1H), 5.55 (m, 1H), 5.01 (m, 1H), 4.93 (d, J = 11.1 Hz, 1H), 4.79 – 4.71 (m, 1H), 4.69 – 4.59 (m, 3H), 4.36 (m, 2H), 3.74 (q, J = 9.7 Hz, 1H), 3.06 (m, 2H), 2.95 (s, 2H), 2.76 (s, 3H), 2.73–2.69 (m, 2H), 2.22–2.20 (m, 2H), 2.04 (d, J = 13.9 Hz, 1H), 1.82 – 1.75 (m, 1H), 1.74–1.68 (m, 4H), 1.57 (t, J = 9.1 Hz, 1H), 1.46 (d, J = 7.1 Hz, 3H), 1.39 (t, J = 14.0 Hz, 2H), 1.25–1.15 (m, 6H), 1.06 (dp, J = 14.2, 7.2, 6.8 Hz, 1H), 0.89 (d, J = 6.6 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H), 0.84–0.77 (m, 6H). [α]25D +9.6 (c 0.08, MeOH); HR-ESI-MS [M + Na]+ m/z 1058.6247 (calcd for C52H71ClN8O12Na, 1058.6238).
Crystallization and structure determination
Porcine pancreatic elastase (PPE, E1250, Sigma-Aldrich) was passed over a desalting column (Desalt 16/10, GE healthcare) preequilibrated with 10 mM sodium acetate, pH 5.0. PPE was concentrated to 15 mg/mL and mixed with 1 at a final concentration of 1 mM. Complex crystals were grown with the sitting drop method using a reservoir solution containing 0.1 M tri-sodium citrate, pH 5.6, and 1.0 M ammonium phosphate. Crystals were cryoprotected in mother liquor supplemented with 30% glycerol and flash-frozen in liquid nitrogen. Diffraction data was collected at 100 °K at ESRF beamline ID30A-3. Data was processed using Xia2,23 and the structure was solved using PHASER24 molecular replacement with PPE (PDB ID: 1LVY) as the search model. The structure was manually rebuilt using COOT25 and refined using PHENIX.Refine.26 The structure was validated using MolProbity, and all images presented were created using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC). Interaction diagrams were created using Ligplot.27 Coordinates were deposited in the Protein Databank with accession number 6TH7.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the Territory of American Samoa for permitting the collection of this strain of Schizothrix sp. from Fagasa Bay, Tutuila in 2014, and the Department of Fish and Wildlife for permits to make collections in Palmyra Atoll. We acknowledge GM107550 and CA100851 for financial support of this project. L.K. would like to thank the German Research Foundation (DFG) for a Research Fellowship (KE 2172/3–1 and KE 2172/4–1). H.L. thanks R01 CA172310 for support of the chemical synthesis of lyngbyastatin 7. The authors are grateful to Y. Su for recording the HRMS spectra at the UCSD Molecular Mass Spectrometry Facility. We acknowledge use of ESRF beamline ID30A-3. J.K. thanks the German Research Foundation for an Emmy Noether Fellowship (KO 4116/3–2).
Footnotes
The authors declare no competing financial interest.
Supporting Information. Tabulated 1H and 13C NMR data for compound 1 in methanol-d4, 1H NMR, 13C NMR, 1H-1H COSY, 1H-1H NOESY, 1H-13C HSQC, 1H-13C HMBC and 1,1-ADEQUATE spectra in DMSO-d6 as well as 1D-NOE in methanol-d4 for compound 1, 1H NMR, 1H-1H COSY, 1H-13C HSQC and 1H-13C HMBC spectra in DMSO-d6 for compounds 2 and 3, dose response curves in elastase and H-460 cancer cell assays. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Weckesser J; Martin C; Jakobi C Cyanopeptolins, Depsipeptides from Cyanobacteria. Syst. Appl. Microbiol. 1996, 19 (2), 133–138. 10.1016/S0723-2020(96)80038-5. [DOI] [Google Scholar]
- (2).Welker M; Von Döhren H Cyanobacterial Peptides - Nature’s Own Combinatorial Biosynthesis. FEMS Microbiol. Rev. 2006, 30 (4), 530–563. 10.1111/j.1574-6976.2006.00022.x. [DOI] [PubMed] [Google Scholar]
- (3).Köcher S; Resch S; Kessenbrock T; Schrapp L; Ehrmann M; Kaiser M From Dolastatin 13 to Cyanopeptolins, Micropeptins, and Lyngbyastatins: The Chemical Biology of Ahp-Cyclodepsipeptides. Nat. Prod. Rep. 2019. 10.1039/c9np00033j. [DOI] [PubMed] [Google Scholar]
- (4).Kang HS; Krunic A; Orjala J Stigonemapeptin, an Ahp-Containing Depsipeptide with Elastase Inhibitory Activity from the Bloom-Forming Freshwater Cyanobacterium Stigonema Sp. J. Nat. Prod. 2012, 75 (4), 807–811. 10.1021/np300150h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pettit GR; Kamano Y; Herald CL; Dufresne C; Cerny RL; Herald DL; Schmidt JM; Kizu H Antineoplastic Agent. 174. Isolation and Structure of the Cytostatic Depsipeptide Dolastatin 13 from the Sea Hare Dolabella Auricularia. J. Am. Chem. Soc. 1989, 111 (13), 5015–5017. 10.1021/ja00195a084. [DOI] [Google Scholar]
- (6).Harrigan GG; Luesch H; Yoshida WY; Moore RE; Nagle DG; Paul VJ Symplostatin 2: A Dolastatin 13 Analogue from the Marine Cyanobacterium Symploca Hydnoides. J. Nat. Prod. 1999, 62 (4), 655–658. 10.1021/np980553b. [DOI] [PubMed] [Google Scholar]
- (7).Nogle LM; Williamson RT; Gerwick WH Somamides A and B, Two New Depsipeptide Analogues of Dolastatin 13 from a Fijian Cyanobacterial Assemblage of Lyngbya Majuscula and Schizothrix Species. J. Nat. Prod. 2001, 64 (6), 716–719. 10.1021/np010348n. [DOI] [PubMed] [Google Scholar]
- (8).Matthew S; Ross C; Rocca JR; Paul VJ; Luesch H Lyngbyastatin 4, a Dolastatin 13 Analogue with Elastase and Chymotrypsin Inhibitory Activity from the Marine Cyanobacterium Lyngbya Confervoides. J. Nat. Prod. 2007, 70 (1), 124–127. 10.1021/np060471k. [DOI] [PubMed] [Google Scholar]
- (9).Taori K; Matthew S; Rocca JR; Paul VJ; Luesch H Lyngbyastatins 5–7, Potent Elastase Inhibitors from Floridian Marine Cyanobacteria, Lyngbya Spp. J. Nat. Prod. 2007, 70, 1593–1600. 10.1021/np0702436. [DOI] [PubMed] [Google Scholar]
- (10).Kwan JC; Taori K; Paul VJ; Luesch H Lyngbyastatins 8–10, Elastase Inhibitors with Cyclic Depsipeptide Scaffolds Isolated from the Marine Cyanobacterium Lyngbya Semiplena. Mar. Drugs 2009, 7 (4), 528–538. 10.3390/md7040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rubio BK; Parrish SM; Yoshida W; Schupp PJ; Schils T; Williams PG Depsipeptides from a Guamanian Marine Cyanobacterium, Lyngbya Bouillonii, with Selective Inhibition of Serine Proteases. Tetrahedron Lett. 2010, 51 (51), 6718–6721. 10.1016/j.tetlet.2010.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gunasekera SP; Miller MW; Kwan JC; Luesch H; Paul VJ Molassamide, a Depsipeptide Serine Protease Inhibitor from the Marine Cyanobacterium. J. Nat. Prod. 2010, 13–16. [DOI] [PubMed] [Google Scholar]
- (13).Salvador LA; Taori K; Biggs JS; Jakoncic J; Ostrov DA; Paul VJ; Luesch H Potent Elastase Inhibitors from Cyanobacteria: Structural Basis and Mechanisms Mediating Cytoprotective and Anti-Inflammatory Effects in Bronchial Epithelial Cells. J. Med. Chem. 2013, 56 (3), 1276–1290. 10.1021/jm3017305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Iwasaki A; Sumimoto S; Ohno O; Suda S; Suenaga K Kurahamide, a Cyclic Depsipeptide Analog of Dolastatin 13 from a Marine Cyanobacterial Assemblage of Lyngbya Sp. Bull. Chem. Soc. Jpn. 2014, 87 (5), 609–613. 10.1246/bcsj.20140008. [DOI] [Google Scholar]
- (15).Matern U; Schleberger C; Jelakovic S; Weckesser J; Schulz GE Binding Structure of Elastase Inhibitor Scyptolin A. Chem. Biol. 2003, 10 (10), 997–1001. 10.1016/j.chembiol.2003.10.001. [DOI] [PubMed] [Google Scholar]
- (16).Gu L; Wang B; Kulkarni A; Geders TW; Grindberg RV; Gerwick L; Håkansson K; Wipf P; Smith JL; Gerwick WH; et al. Metamorphic Enzyme Assembly in Polyketide Diversification. Nature 2009, 459 (7247), 731–735. 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Harada K; Fujii K; Mayumi T; Hibino Y; Suzuki M; Ikai Y; Oka H A Method UsingL/CMS for Determination of Absolute Configuration of Constituent Amino Acids in Peptide --- Advanced Marfey’s Method -−-. Tetrahedron Lett. 1995, 36 (9), 1515–1518. 10.1016/0040-4039(95)00078-Q. [DOI] [Google Scholar]
- (18).Minch MJ Orientational Dependence of Vicinal Proton-Proton NMR Coupling Constants: The Karplus Relationship. Concepts Magn. Reson. 1994, 6 (1), 41–56. 10.1002/cmr.1820060104. [DOI] [Google Scholar]
- (19).Liu L; Rein KS New Peptides Isolated from Lyngbya Species: A Review. Mar. Drugs 2010, 8 (6), 1817–1837. 10.3390/md8061817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Siezen RJ; Leunissen JA Subtilases: The Superfamily of Subtilisin-like Serine Proteases. Protein Sci. 1997, 6 (3), 501–523. 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lee AY; Smitka TA; Bonjouklian R; Clardy J Atomic Structure of the Trypsin-A90720A Complex: A Unified Approach to Structure and Function. Chem. Biol. 1994, 1 (2), 113–117. 10.1016/1074-5521(94)90049-3. [DOI] [PubMed] [Google Scholar]
- (22).Pelay-Gimeno M; Tulla-Puche J; Albericio F “Head-to-Side-Chain” Cyclodepsipeptides of Marine Origin. Mar. Drugs 2013, 11 (5), 1693–1717. 10.3390/md11051693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Winter G Xia2 : An Expert System for Macromolecular Crystallography Data Reduction. J. Appl. Crystallogr. 2010, 43 (1), 186–190. 10.1107/S0021889809045701. [DOI] [Google Scholar]
- (24).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40 (Pt 4), 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr. D. Biol. Crystallogr 2010, 66 (Pt 4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D. Biol. Crystallogr. 2010, 66 (Pt 2), 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wallace AC; Laskowski RA; Thornton JM LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions. Protein Eng. 1995, 8 (2), 127–134. 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.