

Abstract
Nintedanib, a FDA -approved drug for the treatment of patients with idiopathic pulmonary fibrosis, inhibits both tyrosine kinase receptors and non-receptor kinases, and block activation of platelet-derived growth factor receptors, fibroblast growth factor receptor, vascular endothelial growth factor receptors and Src family kinases. Preclinical and clinical studies have revealed the potent anti-fibrotic effect of nintedanib in idiopathic pulmonary fibrosis in human and animal models. Recent preclinical studies have also demonstrated the inhibitory effect of nintedanib on the development and progression of tissue fibrosis in other organs, including liver, kidney, and skin. The anti-fibrotic actions of nintedanib occur through a number of mechanisms, including blocking differentiation of fibroblasts to myofibroblasts, inhibition of epithelial-mesenchymal transition, and suppression of inflammation and angiogenesis. In this article, we summarize the mechanisms and efficacy of nintedanib in the treatment of fibrotic diseases in animal models and clinical trials, provide an update on recent advances in the development of other novel antifibrotic agents in preclinical and clinical study, and offer our perspective about the possible clinical application of these agents in fibrotic diseases.
Keywords: Receptor tyrosine kinases, Src, Nintedanib, Antifibrotic agent, Fibrotic disease
Introduction
Nearly 45% of deaths in the developed world are attributable to some type of chronic fibro- proliferative disease, including idiopathic pulmonary fibrosis (IPF)[1], liver, kidney and cardiac fibrosis, that lead to organ dysfunction and failure [2]. Until recently, there have been no antifibrotic agents shown to be effective in decreasing the impact of fibrosis on mortality and morbidity rates, despite decades of research that have identified numerous potential targets to counter fibrosis in different organs [2]. Recent clinical trials have demonstrated that the receptor tyrosine kinases (RTKs) inhibitor nintedanib demonstrates a powerful anti-fibrotic effect in idiopathic pulmonary fibrosis (IPF), leading to its 2014 approval by the Food and Drug Administration (FDA) for treatment of IPF [3]. Inspired by this breakthrough, researchers conducting preclinical studies have shown that nintedanib has anti-fibrotic effects in other organs, including liver and kidney, and in systemic sclerosis. Preclinical and clinical studies are also underway on the effects of other antifibrotic agents [2]. This article reviews the use of nintedanib in treating IPF and non-pulmonary fibrotic diseases and summarizes scientific efforts to develop other novel antifibrotic agents to treat fibrotic diseases in humans (Table 1).
Table1.
Effects of Nintedanib on tissue fibrosis in various organs
| Objects | Tissue | Effects | Reference |
|---|---|---|---|
| Primary lung fibroblasts isolated from lungs of IPF patients; bleomycin-induced IPF mice model | lung | Inhibits pulmonary fibroblast activation, transdifferention, proliferation, ECM deposition and degradation, inflammatory cell infiltration, microangiopathy and TGF-β-induced fibrotic events | 12,13,14,15 |
| Cultured 3T3 fibroblasts and primary human hepatic stellate cells; CCl4-induced liver fibrogenesis in a mouse model | liver | Attenuates liver fibroblasts and HSC activation, collagen deposition, HSC differentiation, contractility and migration, intrahepatic inflammation and angiogenesis | 33 |
| Cultured renal interstitial fibroblasts; UUO and folic acid injection induced renal fibrosis mouse models | kidney | Inhibits renal fibroblasts activation, ECM overproduction and deposition, inflammatory cells infiltration and TGF-β-induced fibrotic events, may attenuate pre-established renal fibrosis | 36 |
| Dermal fibroblasts from patients with SSc; mouse model of bleomycin-induced skin fibrosis; model of murine sclerodermatous chronic graft-versus-host disease (cGvHD); murine Tight skin-1 (Tsk-1) model; Fos-related antigen-2 transgenic (Fra2 transgenic) mice | skin | Reduces PDGF-induced and TGF-β-induced dermal fibroblasts proliferation and migration, EMT and collagen release; attenuates dermal thickening, myofibroblast counts and hydroxyproline content of the skin, inflammatory cells infiltration ;regresses pre-established dermal fibrosis; | 37,42 |
| Vasoobliteration model; rodent model of neovascular age-related macular degeneration | eye | Interferes with retinal angiogenesis, retinal vaso-obliteration, reduces pathological neovascularization and promoted retinal vascularization; inhibits laser-induced choroidal neovascularization | 43,44 |
IPF: idiopathic pulmonary fibrosis; ECM: extra matrix; TGF: transforming growth factor; HSC: hepatic stellate cells; EMT: epithelial-mesenchymal transition; UUO: unilateral ureteral obstruction; SS: systemic sclerosis; PDGF: platelet-derived growth factor
Treatment of IPF with nintedanib
I. Nintedanib
Receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs) are the two subtypes of tyrosine kinases that regulate many biological processes, including metabolism, growth, differentiation and apoptosis [4]. RTKs are classified into a variety of superfamilies, including platelet-derived growth factor receptors (PDGFR), vascular endothelial growth factor receptors (VEGFR), fibroblast growth factor receptors (FGFR), epidermal growth factor receptors (EGFR), insulin-like growth factor receptors (IGFR); nRTKs, which lack extracellular and transmembrane domains, include Src, c-Abl, c-kit [4]. Increasing evidence indicates that RTKs and Src superfamily kinases are involved in the initiation and progression of tissue fibrosis [4].Tyrosine kinase Inhibitors (TKIs) are already known as the first-line therapies in various malignancies. Many basic science and clinical studies have also suggested the potential anti-fibrotic effects of TKIs in a variety of tissues.
Efforts to develop angiogenesis inhibitors for the treatment of cancer have been underway for 20 years [5]; nintedanib ethanesulfonate, a 6-methoxycarbonyl-substituted indolinone derivative identified during this time, has subsequently advanced to become a first-line targeted therapy against tumors. Nintedanib (BIBF 1120) is a potent, indolinone-derived small molecule, multiple-receptor TKI, that simultaneously blocks the intracellular ATP-binding pocket of RTKs, resulting in inhibition of the phosphorylation of specific tyrosine kinases, including PDFGR-α, PDFGR-β, FGFR-1, 2, 3, VEGFR-1, 2, VEGFR-3 and Src family kinases (Src, Lyn and Lck)[4]. As a first-line targeted cancer therapy, nintedanib has been shown to regulate tumor cell proliferation, differentiation, apoptosis, metastasis and angiogenesis, and has demonstrated broad clinical application in treating lung carcinoma, pancreatic carcinoma, colorectal carcinoma, breast carcinoma, leukemia [6–10].
RTKs as well as Src family kinases have all been implicated in fibrosis. Many TKIs that block specific RTKs have also demonstrated anti-fibrotic effects [4]. Researchers have come to regard combined inhibition of different profibrotic mediators as a promising approach for the treatment of fibrosis [11].Therefore, the broader spectrum of nintedanib-elicited inhibition of profibrotic signaling molecules distinguishes it from other single-receptor kinase inhibitors since nintedanib may offer additive effects with few side effects as compared with selective inhibition of individual profibrotic molecules. In the past decade, a number of preclinical studies and clinical trials have confirmed its antifibrotic effects in IPF through inhibition of pulmonary fibroblast activation, epithelial–mesenchymal transition (EMT), inflammatory cell infiltration, angiogenesis and TGF-β-induced fibrotic events [12–16]. In primary human lung fibroblasts from patients with IPF (IPF-HLF), nintedanib inhibited PDGF- and FGF-stimulated fibroblast motility in a concentration-dependent manner [12]. Nintedanib also inhibited TGF-β-induced fibroblast to myofibroblast transformation in primary human lung fibroblasts from IPF patients [13], while somewhat controversial results demonstrated that nintedanib at concentrations of ⩾300 nmol·L−1 did not change the morphology of primary alveolar type II epithelial cells derived from donors without IPF and showed no inhibitory activity on the TGF-β-induced EMT at 2 ng/mL [16]. Nintedanib reduced TGF-β-stimulated collagen secretion and deposition by primary human lung fibroblasts from patients with IPF cultured for 48 h and reduced secreted TIMP-2 levels [13]. At the same time it promoted secretion of pro-matrix metalloproteinase (MMP)-2, which induces degradation of collagen [12]. Nintedanib also induced apoptosis in human umbilical vascular endothelial cells (HUVEC) stimulated with VEGF or FGF-2, human umbilical artery smooth muscle cells (HUASMC) stimulated with PDGF-BB or FGF-2 and bovine retinal pericytes stimulated with PDGF-BB and FGF-2 [17]. In addition, nintedanib exerts an anti-angiogenic effect by inhibiting proliferation of three cell types that contribute to angiogenesis in the lung, including endothelial cells, pericytes and smooth muscle cells; it also reduces bleomycin-induced lung inflammation [17]. The latter was demonstrated by reduced lymphocyte counts in the BALF, diminished IL-1β levels and decreased percentage of myeloid dendritic cells in lung tissue [12]. Nintedanib can also reduce IL-1β, CXCL1/KC, TIMP-1 and total collagen levels in lung homogenates, and reduce lung inflammation, granuloma formation and fibrosis induced by silica [13].
II. Nintedanib treatment for IPF
In clinical trials, nintedanib was effective in improving lung function in patients with IPF, reducing acute attack frequency and improving patient quality of life [18, 19]. Based on those results, nintedanib was designated as a breakthrough therapy and approved by the FDA in October 2014 [3] and the European Commission Granted Marketing Authorisation for the treatment of patients with IPF in January 2015 [20–22].
The first clinical evidence for the efficacy of nintedanib in patients with IPF came from the TOMORROW trial, a multi-national, double-blinded, randomised, placebo-controlled, 52-week, phase II clinical trial [23]. In this study, researchers demonstrated that treatment with a 150-mg dose of nintedanib twice a day showed a promising numerical trend toward a reduction in the annual rate of decline in forced vital capacity (FVC) by approximately 68%. Nintedanib also reduced the incidence of acute exacerbations and improved the quality of life [23]. After completion of the 52-week period (period 1), patients could continue with nintedanib treatment for a further blinded period (period 2). The effect of nintedanib on slowing IPF progression persisted up to week 76.
The INPULSIS trials, two double-blind, randomized, placebo-controlled, 52-week, phase III clinical trials (INPULSIS-1 and INPULSIS-2)[19], enrolled 1061 patients from 24 countries and demonstrated that nintedanib significantly reduced the annual rate of decline in FVC. The researchers further showed that the beneficial effects of nintedanib were consistent irrespective of sex, age and race, and by FVC based on the 70% threshold [24, 25]. Some 734 patients out of 807 who completed the INPULSIS trials continued to be followed in INPULSIS-ON (430 continuing nintedanib, 304 initiating nintedanib). An interim analysis of a data snapshot in October 2016 suggests that the efficacy of nintedanib in slowing respiratory function decline persists beyond 144 weeks. The adjusted annual rates of decline in FVC of these patients during the 144-week period were similar to the decline in nintedanib recipients in the INPULSIS trials. The mean total exposure for patients treated with nintedanib in both INPULSIS and INPULSIS-ON was 40.7 months, with a maximum exposure of 63.1 months. Analyses of pooled data from two replicate phase III INPULSIS trials examining the effects on nintedanib on baseline FVC % predicted ≤90% or >90% showed that patients with IPF and preserved lung volume (FVC >90% predicted) have the same rate of FVC decline and gain the same benefit from nintedanib as patients with more impaired lung volume (baseline FVC ≤90% predicted)[25]. This also underscores the importance of prompt diagnosis of IPF to enable patients to receive nintedanib treatment as soon as possible and for as long as possible to obtain beneficial effects over the long term. These results suggest that nintedanib is effective in reducing the decline in FVC at least up to 3 years [22] and in improving the quality of life of patients with end stage of IPF [26].
Acute IPF exacerbations are associated with significantly increased morbidity and mortality. The TOMORROW and INPULSIS trials provided mixed results regarding the effect of nintedanib on acute exacerbations. Nintedanib significantly increased the time to the first acute exacerbation in INPULSIS-2, and reduced the incidence of acute exacerbations in TOMORROW [22].Treatment with nintedanib reduced the risk of experiencing a first investigator-reported acute exacerbation by 47% [27]. Acute exacerbations reported as serious adverse events occurred in 3.6% patients in the nintedanib group vs. 6.1% in the placebo group. The INPULSIS-ON trial also suggests that nintedanib is effective in reducing the number of acute exacerbations at least up to 3 years. These two trials showed that nintedanib reduces the risk of on-treatment mortality by 43% [27]. A recent post hoc analysis of pooled INPULSIS data showed that a smaller percentage of nintedanib recipients had a significant decline in FVC compared with placebo recipients.
Pirfenidone is another drug that has been approved for use in the treatment of IPF. In a real-world clinical setting, patients with IPF who were not eligible for pirfenidone treatment could be included in a compassionate use program for nintedanib [28]. Consistent with data from the INPULSIS-2 and TOMORROW trials, nintedanib reduced IPF progression in the majority of the patients who weren’t eligible for pirfenidone. After 6 months of nintedanib treatment, 63% of patients had a FVC decline of less than 5% from baseline [28].The authors of the study noted that nintedanib treatment stabilized disease progression in 62% of the patients who had not responded to pirfenidone in the previous 6 months [28].. These clinical trials demonstrated the effectiveness of nintedanib in slowing disease progression, and suggested that nintedanib treatment may ultimately reduce mortality in patients with IPF with good tolerability and safety profiles [22]. The beneficial effects of nintedanib in IPF patients are offset by adverse events and high cost. Some 50% of patients on the drug suffer an adverse event, leading to treatment discontinuation in 5% to 15% (depending on existing studies)[29]. The cost per quality-adjusted life year (QALY) exceeds 140,000 USD for nintedanib. This seems prohibitive as compared to the classical willingness to pay threshold of 50,000 USD per QALY under which an intervention is considered to be cost-effective [29].
Preclinical research of nintedanib in non-lung fibrotic diseases
The features of tissue fibrosis fibroblast activation, proliferation and transdifferention, inflammatory cells infiltration and extensive tissue remodeling are common to many organ systems. This has prompted several in vitro and in vivo investigations into the efficacy of nintedanib on fibrosis in liver, kidneys, skin and systemic disease, helping revealed nintedanib’s anti-fibrotic effect beyond the lungs (Figure 1).
Figure 1. The mechanism for antifibrotic actions of nintedanib.
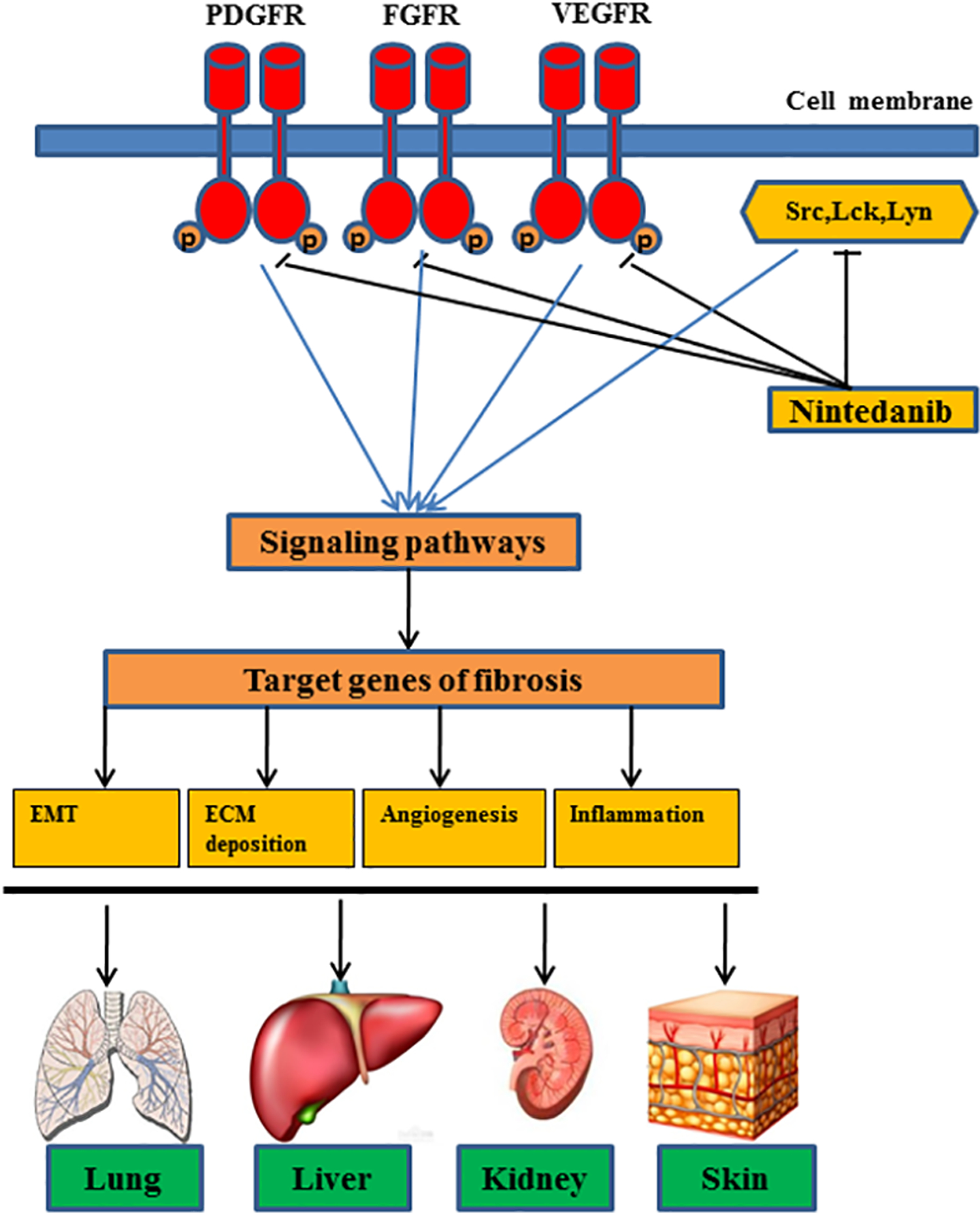
Nintedanib inhibits both tyrosine kinase receptors, including platelet-derived growth factor receptors, fibroblast growth factor receptor, vascular endothelial growth factor receptors and non-receptor kinases, such as Src, c-Abl, c-kit, leading to inactivation of their downstream stream signaling pathways and targeted profibrotic genes responsible for the epithelial–mesenchymal transition (EMT), extracellular matrix (ECM) deposition, angiogenesis and inflammation responses in multiple organs (i.e. lung, liver, kidney and skin).
I. Liver Fibrosis
Liver fibrosis is characterized by hepatocellular damage and inflammatory cell infiltration, which induces extensive accumulation of ECM and extensive tissue remodeling [30]. Activated hepatic stellate cells (HSCs or liver myofibroblasts) are the main effector cells of liver fibrosis initiation and progression [30]. Macrophages can also produce pro-fibrotic mediators and chemokines that activate fibroblasts and inflammatory cells, as well as control ECM turnover by regulating the balance of matrix metalloproteinases (MMPs) and tissue inhibitors of matrix metalloproteinases (TIMPs)[31]. Liver sinusoidal endothelial cells (LSECs) also play a critical role in liver fibrosis [32]. Recent studies have revealed that PDGF, VEGF and FGF are involved in liver fibrogenesis. In cultured HSCs and a mouse model of CCl4-induced liver fibrogenesis, Akcora et al found that nintedanib significantly inhibited liver fibrosis [33]. In cultured 3T3 fibroblasts and primary human hepatic stellate cells, nintedanib significantly blocked expression of collagen I, α –SMA, PDGFR and TIMP1 induced by PDGF and TGF-β. Nintedanib also inhibited expression of periostin, a TGF-β superfamily-responsive matricellular protein that is produced by fibroblasts and is involved in collagen fibrillogenesis and ECM organization. Moreover, nintedanib inhibited TGFβ1-stimulated integrin alpha 5 (ITGA5) expression in fibroblasts and promoted fibroblast motility and survival [34]. In a human hepatic stellate cell line (LX2), nintedanib significantly abrogated collagen I, TGF-β 1, TIMP1 and ITGA5 expression. All those results indicated that nintedanib may inhibit resident liver cell activation, differentiation, migration and contractility. In a CCl4-induced acute liver injury mouse model, nintedanib significantly down-regulated expression of collagen I, fibronectin, periostin, ITGA5, TIMP1, α -SMA and desmin (HSCs activation marker). Nintedanib administration also strongly attenuated expression of major inflammatory cytokines such as TNFα (tumor necrosis factor alpha) and CCL2 (C-C motif chemokine ligand 2), MCP1 (macrophage chemotactic protein) and IL-6 (Interleukin-6). Nintedanib induced no change in M1 (inflammatory) macrophages but induced M2 (restorative) macrophage polarization. Further, treatment with Nintedanib inhibited fibroblasts-driven angiogenesis, macrophage migration, inflammation, as well as primary human HSCs-driven LSEC and Kupffer cell activation in in vitro experiments.
In summary, nintedanib is able to attenuate liver fibroblast and HSC activation and suppress intrahepatic inflammation and angiogenesis[33], suggesting a potent inhibitory effect for nintedanib in liver fibrosis.
II. Renal Fibrosis
Renal fibrosis is characterized by activation and proliferation of renal interstitial fibroblasts and deposition of ECM components related to the progress of chronic kidney disease (CKD) [4]. Emerging evidence indicates that RTKs (PDGFR, FGFR, VEGFR) and non-receptor-RTKs (Src superfamily protein) play a critical role in the pathogenesis of renal fibrosis [4, 35]. In recent in vivo and in vitro experiments, our group found that nintedanib had both preventive and therapeutic effects on renal fibrosis [36]. In mouse models of renal fibrosis induced by unilateral ureteral obstruction (UUO) and folic acid injection, treatment with nintedanib significantly attenuated renal fibrosis when administered either immediately or starting 3 days after UUO injury or folic acid injection. In cultured renal interstitial fibroblasts, nintedanib also inhibited TGF-β1- or PDGF-BB-induced renal fibroblast activation and ECM protein expression. Interestingly, delayed application of nintedanib not only halted further deposition of ECM proteins, but also partially reduced their levels to below what had been accumulated before treatment. This suggests that nintedanib is also able to reverse established renal fibrosis in addition to attenuating development and progression of renal fibrosis. These finding are consistent with other reports that late treatment with nintedanib ameliorated fibrosis in animal models of lung and systemic sclerosis [13, 37]. The mechanism may be in part associated with its regulation of the imbalance between MMPs and TIMPs, which are involved in ECM metabolism [38]. Mechanistically, nintedanib can block activation of PDGFR, FGFR, VEGFR and Src family kinases (Src, Lyn, Lck) induced by UUO injury. Src can directly induce EGFR phosphorylation and also induce EGFR transactivation through cleavage of EGFR legends, releasing its soluble form [4]. Lck and Lyn kinase are proved to be involved in inflammation and fibrogenesis. Nintedanib reduced macrophage infiltration and proinflammatory cytokine/chemokine (i.e. MCP-1, TNF-α, IL-6, and IL-1β) production induced by UUO injury. It also blocked phosphorylation of RTK downstream signaling proteins, such as STAT3 and NF-κB in UUO-induced injuried kidneys. These signaling proteins play important roles in the signal transduction associated with inflammatory responses and renal fibrosis. Furthermore, nintedanib inhibits phosphorylation of Smad2/3, a key signaling molecule mediating biological actions of TGF-β1. It may accomplish that by specifically inhibiting tyrosine phosphorylation of the TGF-β1 receptor since phosphorylation of the five tyrosine residues within the cytoplasmic tail of the type II TGFβ receptor (TβRII) is indispensable for Smad-dependent fibrotic signaling in the kidney [39]. Collectively, nintedanib may protect against renal fibrosis through mechanisms associated with blockade of multiple RTKs, Src family kinases, TGF-β activation and their downstream signaling pathways involved in renal fibrogenesis.
III. Systemic Sclerosis
Systemic sclerosis (SSc) is characterized by vascular remodeling, through capillary and pulmonary arterial hypertension (PAH), and progressive fibrosis of the skin, lungs and heart [40]. Huang et al evaluated the antifibrotic effects of nintedanib in several preclinical studies and animal models of SSc [37]. They found that nintedanib dose-dependently reduced PDGF- and TGF-β-induced proliferation, migration and differentiation of myofibroblasts and collagen release from dermal fibroblasts in patients with SSc and healthy individuals [37]. Bleomycin-induced skin fibrosis in a mouse model is manifested by dermal thickening, deposition of collagen and myofibroblast differentiation [37]. Nintedanib ameliorated progression of fibrosis at all doses tested with significant decreases of dermal thickness, myofibroblast counts and hydroxyproline content [37]. Moreover, administration of nintedanib at doses of 50 mg/kg twice daily induced regression of pre-established fibrosis and reduced the dermal thickness, myofibroblast counts and hydroxyproline content to below the levels of vehicle treated mice injected with bleomycin [37]. These results suggest that nintedanib is able to prevent fibrosis and induce regression of pre-established fibrosis. In a murine model of sclerodermatous chronic graft-versus-host disease (cGvHD), administration of nintedanib to allogeneically transplanted mice ameliorated cGvHD-induced skin fibrosis as well [37]. Furthermore, nintedanib can reduce clinically detectable skin disease as evidenced by a reduced cGvHD score. In the murine Tight skin-1 (Tsk-1) model, treatment with nintedanib reduced hypodermal thickening, myofibroblast counts and hydroxyproline content of the skin [37].
Fos-related antigen-2 transgenic (Fra2 transgenic) mice display the core clinical fibrotic and vascular manifestations of SSc [41]. Fra2 transgenic mice develop a destructive microvascular disease with apoptosis of endothelial cells followed by systemic fibrotic manifestations and PAH [41]. By utilizing this model, Huang et al verified the antifibrotic and vasoprotective effects of nintedanib [42]. Nintedanib also decreased the number of apoptotic endothelial cells in the skin and lungs and normalized the levels of M-CSF1 and VEGF of Fra2 transgenic mice. Interestingly, nintedanib treatment completely prevented the increase in M2 macrophages, but M1 macrophage counts were not affected [42]. These inhibitory effects of nintedanib on macrophage polarization may be mediated by direct inhibition of CSF1R.
In summary, nintedanib inhibited proliferation and migration of fibroblasts, inhibited myofibroblast differentiation and decreased the synthesis of ECM proteins, reduced pulmonary, dermal and myocardial fibrosis of SSc. In addition to inhibition of cytokine-induced fibroblast activation, nintedanib also reduced the characteristic endogenous activation of SSc fibroblasts that may drive disease progression in later stages. Nintedanib may thus exert antifibrotic and vasoprotective effects on fibrosis and regression of pre-established fibrosis of SSc.
IV. Retinopathy
Retinopathy of prematurity (ROP) is characterized by an initial phase of retinal vascular constriction, followed by microvascular degeneration that results in ischemia and subsequent predisposition to an abnormal intravitreal neovascularization (NV). A large number of proangiogenic factors, such as PDGF, FGF and VEGF are involved in the progress [43, 44]. In aortic ring explants cultured in a basement membrane matrix containing growth factors, nintedanib significantly inhibited dose-dependently aortic vessel sprouting [44]. In the vasoobliteration model (VO), nintedanib interfered with retinal angiogenesis, retinal vaso-obliteration, reduced pathological neovascularization and promoted retinal vascularization, but did not compromise the established vascular network during development. The inhibitory effects of nintedanib on the oxygen-induced retinopathy (OIR) may be through blocking phosphorylation of MAPK (Erk1/2) and AKT. Nintedanib exerted suppression of OIR-induced expression of VEGF negative regulator Dll4, and repulsive factor EphrinB2 and its receptor EphB4, while attenuating the rise in VEGF and Netrin-1, resulting in enhanced normal retinal vascularization. Therefore, when nintedanib is injected at early stages of ROP, it can accelerate normal vascularization, which is essential to prevent pre-retinal neovascularization and long-term functional damage [44]. These properties could be of interest for the treatment of a variety of ischemic retinopathies associated with pathological neovascularization in addition to ROP, notably diabetic retinopathy.
Neovascular age-related macular degeneration (AMD) is one of the most common eye disorders that impair vision and is the leading cause of blindness in the elderly [45]. Nintedanib added to a light-sensitive polymer injected into in the posterior segment of the eye brown Norway rates was shown to inhibit angiogenesis significantly prior to laser induction of choroidal neovascularization [45], suggesting a role for nintedanib and this novel UV light-sensitive nanoparticle drug delivery system.
Other novel therapies for fibrotic disease
While nintedanib and pirfendone, discussed briefly above, have been approved as first-line therapies for IPF, other novel compounds have recently been identified that also hold promise as potential therapeutic agents in fibrotic disease. Those agents include PPARs agonist, pentoxifylline and bardoxolone methyl. We discuss them briefly here, noting that the different mechanisms of action could compliment that of nintedanin in IPF and may also have value as potential therapies in other fibrotic diseases, also complimenting nintedanib..
I. Pirfenidone
Pirfenidone is an antifibrotic, anti-inflammatory and antioxidant compound that was recently approved by the FDA for patients with IPF [20]. It inhibits production of TGF-α and free radical oxygen species (ROSs), as well as reduces IL-1, IL-6, IL-8, IL-12 and TNF-α levels [46]. Several preclinical studies and clinical trials have indicated that pirfenidone is effective in in treating IPF and renal fibrosis [20, 47]. The first clinical study of pirfenidone for IPF – a phase II, multicenter, randomized, double-blind trial – showed no significant therapeutic effect on the primary endpoint of a change from baseline in the lowest oxygen saturation by pulse oximetry [SpO2] during a 6-minute exercise test [48]. High-dose treatment (1800 mg/day) had a positive effect in reducing VC decline, a secondary endpoint, at 9 months [48]. A subsequent multicenter, randomized, double-blind phase III clinical trial of 275 patients with IPF randomly assigned to pirfenidone 1800 mg/day, pirfenidone 1200 mg/day, or placebo for 52 weeks, showed a significantly slower decline in FVC and better progression-free survival in the two pirfenidone groups compared with placebo [46].The placebo-controlled phase III CAPACITY 004 and 006 trials showed that the primary endpoint of a significant reduction in FVC declines at week 72 was only met in study 004, which assigned patients in a 2:1:2 ratio to pirfenidone 2403 mg/day, pirfenidone 1197 mg/day, or placebo [49]. In study 006, in which patients were assigned d in a 1:1 ratio to pirfenidone 2403 mg/day or placebo, the pirfenidone 2403 mg/day group had higher incidences of side-effects than the placebo group. The pooled analysis, however, revealed a significant reduction in the mean decline of absolute and percent predicted FVC; even patients with an FVC decline ≥10% at week 72 in the 2403 mg/day pirfenidone group still had longer progression-free survival and a lower rate of decline in mean 6-minute walk distance (6MWD) than patients in the placebo group [49]. Finally, the ASCEND trial looked at 555 patients from the USA, Europe, and Australia who were randomly assigned to receive 2403 mg/day of pirfenidone or placebo for 52 weeks [50]. It, too, showed that the treatment group had a significantly reduced absolute decline in FVC, fewer patients with an FVC decline ≥10%, better progression-free survival and fewer patients with a decrease of ≥50 m in distance walked during the 6MWD [50]. The CAPACITY and ASCEND trials showed a reduction in both all-cause and IPF-related mortality after one year [50].
Recent preclinical research has looked at the effect of pirfenidone on renal fibrosis. Rao et al. showed that pirfenidone inhibited mesangial matrix expansion and reduced levels of type I and IV collagen, and fibronectin gene expansion in kidneys of mice with diabetic nephropathy (DN). They suggest that the mechanism of action may be through inhibition of phosphorylation of eIF4E, thereby preventing mRNA processing and expression of fibrotic proteins [51]. Using a 5/6 nephrectomy rat model Sharma et al. found that pirfenidone inhibits M1 and M2 macrophage infiltration [52]. In UUO rats and cultured HK-2 cells, pirfenidone significantly attenuated TGF-β1-induced EMT and ECM synthesis, as determined by reducing expression of α-SMA, type I and III collagen, S100A4, fibronectin, and increased expression of E-cadherin, along with inhibiting TGF-β1-induced up-regulation of phosphorylation of ERK1/2, p38 and JNK [47]. Preclinical studies indicated a significant effect of pirfenidone on renal fibrosis. The only human trial to date so far has been a RCT in 77 patients with DN. The trial showed that after 1 year of therapy with pirfenidone, patients in the treatment arm with eGFR ranging from 20 mL/min/1.73 m2 to 75 mL/min/1.73 m2 experienced an average increase in eGFR of +8.5 mL/min/1.73 m2 compared with a mean eGFR decrease of 2.2 mL/min/1.73 m2 in the placebo group [52]. More experimental and clinical trials are needed to evaluate whether pirfenidone could be used for clinical treatment of fibrotic renal diseases.
II. PPARs agonist
Peroxisome proliferator-activated receptors (PPARs) are nuclear transcription factors that form obligate heterodimers with retinoid-X receptors to modulate transcription of target genes [53]. Several preclinical studies and clinical trials have identified three PPAR subtypes (PPARα, PPARβ/δ, and PPARγ) as potential targets for antifibrotic therapy.
PPARγ agonists, known as thiazolidinedione (TZD) drugs, are targeted primarily as a treatment for diabetes, disorder of lipid metabolism, inflammation and fibrosis [54]. Many preclinical experiments have indicated that PPARγ agonists prevent and inhibit tissue fibrosis in liver, kidneys, heart and lungs [2]. Clinical trials assessing the treatment of fibrotic diseases with PPARγ agonists have largely been limited to patients with non-alcoholic steatohepatitis (NASH) with mixed results [2]. Two early prospective studies of patients with NASH before and after treatment with either rosiglitazone or pioglitazone for 48 weeks and 12 months showed significant improvement in steatosis, inflammation and fibrosis [55]. A phase 3 randomized, multi-center placebo-controlled trial conducted to analyze the effect of 96 weeks treatment of pioglitazone, vitamin E or placebo on patients with NASH showed that pioglitazone had no significant effect on fibrosis, although it significantly improved liver function, steatosis, and inflammation [56]. A recent meta-analysis revealed that pioglitazone, but not rosiglitazone, significantly decreased fibrosis in NASH [57], suggesting differences in the relative effectiveness of TZDs. Moreover, TZDs have significant side effects, including weight gain, edema and bone density loss, increased risk of cardiovascular disease and bladder cancer that may limit their clinical usefulness [58].
A possible role for non-TZD PPARγ agonists on fibrotic diseases has been looked at in pre-clinical studies but in only limited clinic studies. Fenofibrate, a PPARα agonist, may prevent interstitial and perivascular cardiac fibrosis through a mechanism involving reduced endothelin-1 levels [59]. Fenofibrate also prevented pulmonary, hepatic and renal fibrosis in preclinical studies [2]. In addition, fenofibrate, gemfibrozil and BAY PP1 prevented interstitial renal fibrosis in a number of preclinical investigations using rodent models [2]. Similarly, fenofibrate and WY-14643 prevented hepatic fibrosis induced by several fibrotic substances [2]. Few clinical studies of PPARα agonists in fibrotic diseases have been conducted, and most of them were focused on primary biliary cirrhosis [60].
To date, there are only preclinical animal studies on the treatment of fibrotic diseases with PPARβ/δ agonists [2]. It has been reported that GW0742, a synthetic agonist of PPARβ/δ, prevented bleomycin-induced lung fibrosis, inflammation, pulmonary artery banding-induced cardiac hypertrophy and fibrosis in mice [61]. GW0742 also reduced markers of cardiac fibrosis in a rat model of type-1 diabetes-associated cardiac fibrosis [62]. Another PPARβ/δ agonist, GW610742, inhibited collagen deposition in a rat model of corneal wounding [63]. The PPARβ/δ agonist GW501516 reduced peritoneal fibrosis and inflammation in rats [64]. The PPARβ/δ agonist HPP593 may effectively abrogate renal fibrosis induced by chronic ischemia, through reduction in oxidative stress and preservation of mitochondrial function [65]. The PPARβ/δ agonist KD3010 has been reported to prevent liver fibrosis induced by CCl4 or chronic cholestasis [66]. More recent studies indicated that dual-, pan-, or mixed PPAR agonists have shown potential for treating fibrotic disease [2].
III. Pentoxifylline
Pentoxifylline, a methylxanthine derivative, has been approved recently for the treatment of vascular diseases. Its main clinical properties include improving the rheological properties of blood, anti-inflammatory effects, and antioxidative effects [67]. Several preclinical studies have shown an inhibitory effect of pentoxifylline on fibrotic diseases including radiation-induced fibrosis, schistosomiasis-induced hepatic granulomas, chronic pulmonary paracoccidioidomycosis, miscellaneous fibrotic conditions, peritoneal fibrosis and tubulointerstitial fibrosis. Pentoxifylline is thought to act through downregulation of the expression of TGF-β1 and reduction of proinflammatory and inflammatory factors [67].
The effect of pentoxifylline on radiation-induced fibrosis was largely spurred by the known antioxidant properties of pentoxifylline. In a observational study, 43 patients with 50 radiation-induced fibrotic lesions who had previously undergone radiotherapy for head and neck or breast cancers were orally administered pentoxifylline (800 mg/day) and tocopherol (1000 U/day). They showed a regression in the surface area of lesions and a reduction of Subjective Objective Medical Management and Analytic Evaluation (SOMA) grade [68]. A separate study involving 29 patients with 34 radiation-induced fibrotic lesions showed a 43% mean surface area regression of lesions and a 13% mean reduction of SOMA grade at 3 months. The improvements were sustained when treatment persisted for 6 months [69]. In a randomized, placebo-controlled, single-center study involving 22 patients with 27 radiation-induced fibrotic lesions who had previously undergone radiotherapy for breast cancer, the group that received combined pentoxifylline and tocopherol demonstrated significant mean surface area regressions of lesions after 3, 6 and even 48 months [70].
Pentoxifylline has been shown to be effective therapy for oral submucosal fibrosis. In a randomized, placebo-controlled clinical trial, 14 of 29 patients in the study were orally administered pentoxifylline (800 mg/day) for 1 month and then 1200 mg/day for the subsequent 6 months. The treatment arm demonstrated significant improvement of conditions associated with oral submucosal fibrosis, compared to the control group [71]. Similar results were observed in another randomized, placebo-controlled clinical trial involving a larger series in which 32 of 62 patients received the active drug [72]. A separate randomized, placebo-controlled clinical trial in which 106 patients were administered a smaller dose of pentoxifylline (800 mg/day) orally for a shorter duration (3 months) demonstrated significant improvement in all variables assessed, compared to the control group [73]. It will be of interest to investigate the potential effect of pentoxifylline on oral submucous fibrosis in other populations.
Pentoxifylline may have a role as a therapeutic agent in CKD through its anti-inflammatory and antiproteinuric properties. Perkins et al. documented slower progression of CKD in patients who took pentoxifylline versus placebo for 1 year, with the PTF treatment group showing an eGFR decline of −1.2 mL/min/1.73 m2 compared with −7.2 mL/min/1.73 m2 in the control group [74]. A prospective RCT in 91 patients with eGFR <60 mL/min/1.73 m2 randomized to receive pentoxifylline or placebo, Goicoechea et al. found that serum CRP, fibrinogen and TNF-α decreased significantly in the pentoxifylline group [75]. A single-center retrospective analysis conducted by Chen et al examined the combination of pentoxifylline with ACE inhibitors or ARBs in patients with advanced CKD. They found no change in overall mortality or risk of incident cardiovascular events [76]. The patients with the addition of pentoxifylline, however, had better renal outcomes, demonstrating a 40% lower risk of developing ESRD than treatment with an ACE inhibitor or ARB alone, especially in patients with heavy proteinuria [76].
IV. Bardoxolone methyl
Bardoxolone methyl is a promising antifibrotic compound with antioxidant and anti-inflammatory properties [77]. A phase 2, double-blinded RCT conducted by Pergola et al.showed that bardoxolone methyl was associated with improvement in eGFR in patients with advanced CKD and type 2 diabetes at 24 weeks of follow-up [78]. A larger RCT, in which more than 2000 patients with stage 4 CKD and diabetes were randomized to receive either bardoxolone methyl or placebo, failed to demonstrate a reduction in progression to ESRD or death and was also associated with a higher rate of cardiovascular events in the treatment arm [79]. In 2018, a post-hoc analysis of data from the Bardoxolone methyl Evaluation in patients with CKD and type 2 diabetes study (BEACON) suggested that bardoxolone methyl may preserve kidney function and delay the onset of ESRD in patients with T2D and stage 4 CKD. Conducted as a multinational, randomized, double-blind, placebo-controlled phase 3 trial, BEACON enrolled around 2,600 patients with T2D and CKD stage 4 [80]. Patients randomized to bardoxolone methyl experienced mean increases in eGFR that were sustained through study week 48 while increases in eGFR from baseline were sustained 4 weeks after cessation of treatment[80]. These patients were significantly less likely to experience the composite renal endpoint.
Conclusions and perspectives
The pathophysiologic process of tissue fibrosis is commonly characterized by fibroblast cell activation and proliferation, inflammatory cell infiltration and extensive tissue remodeling. There is substantial evidence to support a role for specific receptor tyrosine kinases and Src family kinases in the pathogenesis of tissue fibrosis. A variety of in vitro studies and animal studies have suggested that nintedanib, a multiple-receptors tyrosine kinase inhibitor, has a therapeutic potential in preventing and even reversing pre-established tissue fibrosis. In addition, based on the results from clinical trials of nitendanib on IPF and preclinical studies on several other non-pulmonary fibrotic diseases, it is likely that nintedanib may also have therapeutic value in the treatment of tissue fibrosis in other organs. While many cellular mechanisms responsible for fibrosis have been identified, and some anti-fibrotic agents, including nintedanib, show great promise in slowing down and even reversing fibrosis, translating that knowledge into effective antifibrotic therapies in humans has been limited due to lack of sufficient effects, off-target effects or both. As such, more clinical trials of nintedanib in other chronic fibrotic disease are warrantied.
Researchers are also encouraged by recent preclinical studies and clinical trials demonstrating the effectiveness, safety and tolerability of other novel antifibrotic agents such as perfenidone, PPARs agonist, pentoxifylline and bardoxolone methyl in slowing progression or even reversing renal fibrosis. But more clinical studies, in particular, large randomized-controlled clinical trials, are necessary to evaluate whether these promising antifibrotic agents will translate into actual therapies against chronic fibrotic diseases.
Acknowledgements
This work was supported by the grants from Economic Commission of Pudong New District of Shanghai (PKJ2017-Y19 to L.F.), the National Nature Science Foundation of China (81470920, 81670623, and 81830021 to S.Z) and the Branch grant of National key grants of Ministry of Science and Technology (2018YFA0108802 to S.Z.).
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117:524–9. doi: 10.1172/jci31487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McVicker BL and Bennett RG (2017) Novel Anti-fibrotic Therapies. Front Pharmacol 8:318. doi: 10.3389/fphar.2017.00318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCormack PL (2015) Nintedanib: first global approval. Drugs 75:129–39. doi: 10.1007/s40265-014-0335-0 [DOI] [PubMed] [Google Scholar]
- 4.Liu F and Zhuang S (2016) Role of Receptor Tyrosine Kinase Signaling in Renal Fibrosis. Int J Mol Sci 17. doi: 10.3390/ijms17060972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth GJ, Binder R, Colbatzky F, Dallinger C, Schlenker-Herceg R, Hilberg F, Wollin SL and Kaiser R (2015) Nintedanib: from discovery to the clinic. J Med Chem 58:1053–63. doi: 10.1021/jm501562a [DOI] [PubMed] [Google Scholar]
- 6.Otsubo K, Kishimoto J, Kenmotsu H, Minegishi Y, Ichihara E, Shiraki A, Kato T, Atagi S, Horinouchi H, Ando M, Kondoh Y, Kusumoto M, Ichikado K, Yamamoto N, Nakanishi Y and Okamoto I (2017) Treatment Rationale and Design for J-SONIC: A Randomized Study of Carboplatin Plus Nab-paclitaxel With or Without Nintedanib for Advanced Non-Small-cell Lung Cancer With Idiopathic Pulmonary Fibrosis. Clin Lung Cancer. doi: 10.1016/j.cllc.2017.06.003 [DOI] [PubMed] [Google Scholar]
- 7.Rossi A, Latiano TP, Parente P, Chiarazzo C, Limosani F, Di Maggio G and Maiello E (2017) The potential role of nintedanib in treating colorectal cancer. Expert Opin Pharmacother 18:1153–1162. doi: 10.1080/14656566.2017.1346086 [DOI] [PubMed] [Google Scholar]
- 8.Khalique S and Banerjee S (2017) Nintedanib in ovarian cancer. Expert Opin Investig Drugs 26:1073–1081. doi: 10.1080/13543784.2017.1353599 [DOI] [PubMed] [Google Scholar]
- 9.Liu CY, Huang TT, Chu PY, Huang CT, Lee CH, Wang WL, Lau KY, Tsai WC, Chao TI, Su JC, Chen MH, Shiau CW, Tseng LM and Chen KF (2017) The tyrosine kinase inhibitor nintedanib activates SHP-1 and induces apoptosis in triple-negative breast cancer cells. Exp Mol Med 49:e366. doi: 10.1038/emm.2017.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manzo A, Carillio G, Montanino A, Costanzo R, Sandomenico C, Rocco G and Morabito A (2016) Focus on Nintedanib in NSCLC and Other Tumors. Front Med (Lausanne) 3:68. doi: 10.3389/fmed.2016.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wuyts WA, Antoniou KM, Borensztajn K, Costabel U, Cottin V, Crestani B, Grutters JC, Maher TM, Poletti V, Richeldi L, Vancheri C and Wells AU (2014) Combination therapy: the future of management for idiopathic pulmonary fibrosis? Lancet Respir Med 2:933–942. doi: 10.1016/s2213-2600(14)70232-2 [DOI] [PubMed] [Google Scholar]
- 12.Hostettler KE, Zhong J, Papakonstantinou E, Karakiulakis G, Tamm M, Seidel P, Sun Q, Mandal J, Lardinois D, Lambers C and Roth M (2014) Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res 15:157. doi: 10.1186/s12931-014-0157-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wollin L, Maillet I, Quesniaux V, Holweg A and Ryffel B (2014) Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 349:209–20. doi: 10.1124/jpet.113.208223 [DOI] [PubMed] [Google Scholar]
- 14.Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, Antony VB, Zhang J, Zmijewski J and Thannickal VJ (2016) Novel Mechanisms for the Antifibrotic Action of Nintedanib. Am J Respir Cell Mol Biol 54:51–9. doi: 10.1165/rcmb.2014-0445OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knuppel L, Ishikawa Y, Aichler M, Heinzelmann K, Hatz R, Behr J, Walch A, Bachinger HP, Eickelberg O and Staab-Weijnitz CA (2017) A Novel Antifibrotic Mechanism of Nintedanib and Pirfenidone. Inhibition of Collagen Fibril Assembly. Am J Respir Cell Mol Biol 57:77–90. doi: 10.1165/rcmb.2016-0217OC [DOI] [PubMed] [Google Scholar]
- 16.Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S and Kolb M (2015) Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 45:1434–45. doi: 10.1183/09031936.00174914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A and Rettig WJ (2008) BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 68:4774–82. doi: 10.1158/0008-5472.can-07-6307 [DOI] [PubMed] [Google Scholar]
- 18.Woodcock HV, Molyneaux PL and Maher TM (2013) Reducing lung function decline in patients with idiopathic pulmonary fibrosis: potential of nintedanib. Drug Des Devel Ther 7:503–10. doi: 10.2147/dddt.s38833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B and Collard HR (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–82. doi: 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 20.Robalo-Cordeiro C, Campos P, Carvalho L, Borba A, Clemente S, Freitas S, Furtado S, Jesus JM, Leal C, Marques A, Melo N, Souto-Moura C, Neves S, Sousa V, Santos A and Morais A (2017) Idiopathic pulmonary fibrosis in the era of antifibrotic therapy: Searching for new opportunities grounded in evidence. Rev Port Pneumol (2006) 23:287–293. doi: 10.1016/j.rppnen.2017.05.005 [DOI] [PubMed] [Google Scholar]
- 21.Shaw J, Marshall T, Morris H, Hayton C and Chaudhuri N (2017) Idiopathic pulmonary fibrosis: a holistic approach to disease management in the antifibrotic age. J Thorac Dis 9:4700–4707. doi: 10.21037/jtd.2017.10.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-Portal JA (2018) Efficacy and Safety of Nintedanib for the Treatment of Idiopathic Pulmonary Fibrosis: An Update. Drugs R D 18:19–25. doi: 10.1007/s40268-017-0221-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Kluglich M and du Bois RM (2011) Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 365:1079–87. doi: 10.1056/NEJMoa1103690 [DOI] [PubMed] [Google Scholar]
- 24.Costabel U, Inoue Y, Richeldi L, Collard HR, Tschoepe I, Stowasser S and Azuma A (2016) Efficacy of Nintedanib in Idiopathic Pulmonary Fibrosis across Prespecified Subgroups in INPULSIS. Am J Respir Crit Care Med 193:178–85. doi: 10.1164/rccm.201503-0562OC [DOI] [PubMed] [Google Scholar]
- 25.Kolb M, Richeldi L, Behr J, Maher TM, Tang W, Stowasser S, Hallmann C and du Bois RM (2017) Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax 72:340–346. doi: 10.1136/thoraxjnl-2016-208710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crestani B, Huggins JT, Kaye M, Costabel U, Glaspole I, Ogura T, Song JW, Stansen W, Quaresma M, Stowasser S and Kreuter M (2019) Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study, INPULSIS-ON. Lancet Respir Med 7:60–68. doi: 10.1016/s2213-2600(18)30339-4 [DOI] [PubMed] [Google Scholar]
- 27.Richeldi L, Cottin V, du Bois RM, Selman M, Kimura T, Bailes Z, Schlenker-Herceg R, Stowasser S and Brown KK (2016) Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS((R)) trials. Respir Med 113:74–9. doi: 10.1016/j.rmed.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 28.Bonella F, Kreuter M, Hagmeyer L, Neurohr C, Keller C, Kohlhaeufl MJ, Muller-Quernheim J, Milger K and Prasse A (2016) Insights from the German Compassionate Use Program of Nintedanib for the Treatment of Idiopathic Pulmonary Fibrosis. Respiration 92:98–106. doi: 10.1159/000448288 [DOI] [PubMed] [Google Scholar]
- 29.Froidure Antoine J A, Mailleux Arnaud A., Crestani Bruno (2016) New targets in idiopathic pulmonary fibrosis: From inflammation and immunity to remodeling and repair. Expert Opinion on Orphan Drugs 4:511–520. doi: 10.1517/21678707.2016.1171140 [DOI] [Google Scholar]
- 30.Friedman SL (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655–69. doi: 10.1053/j.gastro.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wynn TA and Barron L (2010) Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 30:245–57. doi: 10.1055/s-0030-1255354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLeve LD (2015) Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 61:1740–6. doi: 10.1002/hep.27376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozturk Akcora B, Storm G, Prakash J and Bansal R (2017) Tyrosine kinase inhibitor BIBF1120 ameliorates inflammation, angiogenesis and fibrosis in CCl4-induced liver fibrogenesis mouse model. Sci Rep 7:44545. doi: 10.1038/srep44545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margadant C and Sonnenberg A (2010) Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep 11:97–105. doi: 10.1038/embor.2009.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan Y, Ma L, Zhou X, Ponnusamy M, Tang J, Zhuang MA, Tolbert E, Bayliss G, Bai J and Zhuang S (2016) Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int 89:68–81. doi: 10.1038/ki.2015.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu F, Wang L, Qi H, Wang J, Wang Y, Jiang W, Xu L, Liu N and Zhuang S (2017) Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin Sci (Lond) 131:2125–2143. doi: 10.1042/cs20170134 [DOI] [PubMed] [Google Scholar]
- 37.Huang J, Beyer C, Palumbo-Zerr K, Zhang Y, Ramming A, Distler A, Gelse K, Distler O, Schett G, Wollin L and Distler JH (2016) Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis 75:883–90. doi: 10.1136/annrheumdis-2014-207109 [DOI] [PubMed] [Google Scholar]
- 38.Arpino V, Brock M and Gill SE (2015) The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol 44–46:247–54. doi: 10.1016/j.matbio.2015.03.005 [DOI] [PubMed] [Google Scholar]
- 39.Chen X, Wang H, Liao HJ, Hu W, Gewin L, Mernaugh G, Zhang S, Zhang ZY, Vega-Montoto L, Vanacore RM, Fassler R, Zent R and Pozzi A (2014) Integrin-mediated type II TGF-beta receptor tyrosine dephosphorylation controls SMAD-dependent profibrotic signaling. J Clin Invest 124:3295–310. doi: 10.1172/jci71668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapoor R and Kapoor JR (2009) Scleroderma. N Engl J Med 361:826; author reply 826–7. [PubMed] [Google Scholar]
- 41.Reich N, Maurer B, Akhmetshina A, Venalis P, Dees C, Zerr P, Palumbo K, Zwerina J, Nevskaya T, Gay S, Distler O, Schett G and Distler JH (2010) The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum 62:280–90. doi: 10.1002/art.25056 [DOI] [PubMed] [Google Scholar]
- 42.Huang J, Maier C, Zhang Y, Soare A, Dees C, Beyer C, Harre U, Chen CW, Distler O, Schett G, Wollin L and Distler JHW (2017) Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. doi: 10.1136/annrheumdis-2016-210823 [DOI] [PubMed] [Google Scholar]
- 43.Virakul S, Heutz JW, Dalm VA, Peeters RP, Paridaens D, van den Bosch WA, Hirankarn N, van Hagen PM and Dik WA (2016) Basic FGF and PDGF-BB synergistically stimulate hyaluronan and IL-6 production by orbital fibroblasts. Mol Cell Endocrinol 433:94–104. doi: 10.1016/j.mce.2016.05.023 [DOI] [PubMed] [Google Scholar]
- 44.Rivera JC, Noueihed B, Omri S, Barrueco J, Hilberg F and Chemtob S (2015) BIBF1120 (Vargatef) Inhibits Preretinal Neovascularization and Enhances Normal Vascularization in a Model of Vasoproliferative Retinopathy. Invest Ophthalmol Vis Sci 56:7897–907. doi: 10.1167/iovs.15-17146 [DOI] [PubMed] [Google Scholar]
- 45.Huu VA, Luo J, Zhu J, Zhu J, Patel S, Boone A, Mahmoud E, McFearin C, Olejniczak J, de Gracia Lux C, Lux J, Fomina N, Huynh M, Zhang K and Almutairi A (2015) Light-responsive nanoparticle depot to control release of a small molecule angiogenesis inhibitor in the posterior segment of the eye. J Control Release 200:71–7. doi: 10.1016/j.jconrel.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, Taguchi Y, Takahashi H, Nakata K, Sato A, Takeuchi M, Raghu G, Kudoh S and Nukiwa T (2010) Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 35:821–9. doi: 10.1183/09031936.00005209 [DOI] [PubMed] [Google Scholar]
- 47.Li Z, Liu X, Wang B, Nie Y, Wen J, Wang Q and Gu C (2017) Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology (Carlton) 22:589–597. doi: 10.1111/nep.12831 [DOI] [PubMed] [Google Scholar]
- 48.Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, Sato A and Kudoh S (2005) Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 171:1040–7. doi: 10.1164/rccm.200404-571OC [DOI] [PubMed] [Google Scholar]
- 49.Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE Jr., Lancaster L, Sahn SA, Szwarcberg J, Valeyre D and du Bois RM (2011) Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 377:1760–9. doi: 10.1016/s0140-6736(11)60405-4 [DOI] [PubMed] [Google Scholar]
- 50.King TE Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ and Noble PW (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370:2083–92. doi: 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 51.RamachandraRao SP, Zhu Y, Ravasi T, McGowan TA, Toh I, Dunn SR, Okada S, Shaw MA and Sharma K (2009) Pirfenidone is renoprotective in diabetic kidney disease. J Am Soc Nephrol 20:1765–75. doi: 10.1681/asn.2008090931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma K, Ix JH, Mathew AV, Cho M, Pflueger A, Dunn SR, Francos B, Sharma S, Falkner B, McGowan TA, Donohue M, Ramachandrarao S, Xu R, Fervenza FC and Kopp JB (2011) Pirfenidone for diabetic nephropathy. J Am Soc Nephrol 22:1144–51. doi: 10.1681/asn.2010101049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu F, Mei X, Zhang Y, Qi H, Wang J, Wang Y, Jiang W, Zhang X, Yan H and Zhuang S (2014) Association of peroxisome proliferator-activated receptorgamma gene Pro12Ala and C161T polymorphisms with cardiovascular risk factors in maintenance hemodialysis patients. Mol Biol Rep 41:7555–65. doi: 10.1007/s11033-014-3645-0 [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Liu F and Chen N (2007) Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists attenuate the profibrotic response induced by TGF-beta1 in renal interstitial fibroblasts. Mediators Inflamm 2007:62641. doi: 10.1155/2007/62641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D and Bacon BR (2003) Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology 38:1008–17. doi: 10.1053/jhep.2003.50420 [DOI] [PubMed] [Google Scholar]
- 56.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH and Robuck PR (2010) Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 362:1675–85. doi: 10.1056/NEJMoa0907929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Musso G, Cassader M, Paschetta E and Gambino R (2017) Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern Med 177:633–640. doi: 10.1001/jamainternmed.2016.9607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tahrani AA, Barnett AH and Bailey CJ (2016) Pharmacology and therapeutic implications of current drugs for type 2 diabetes mellitus. Nat Rev Endocrinol 12:566–92. doi: 10.1038/nrendo.2016.86 [DOI] [PubMed] [Google Scholar]
- 59.Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, Tan Y, Feng W, Fu Y, Mellen N, Cheng R, Ma J, Zhang C, Li Z and Cai L (2016) Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin Sci (Lond) 130:625–41. doi: 10.1042/cs20150623 [DOI] [PubMed] [Google Scholar]
- 60.El-Haggar SM and Mostafa TM (2015) Comparative clinical study between the effect of fenofibrate alone and its combination with pentoxifylline on biochemical parameters and liver stiffness in patients with non-alcoholic fatty liver disease. Hepatol Int 9:471–9. doi: 10.1007/s12072-015-9633-1 [DOI] [PubMed] [Google Scholar]
- 61.Galuppo M, Di Paola R, Mazzon E, Esposito E, Paterniti I, Kapoor A, Thiemermann C and Cuzzocrea S (2010) GW0742, a high affinity PPAR-beta/delta agonist reduces lung inflammation induced by bleomycin instillation in mice. Int J Immunopathol Pharmacol 23:1033–46. doi: 10.1177/039463201002300408 [DOI] [PubMed] [Google Scholar]
- 62.Chang WT, Cheng JT and Chen ZC (2016) Telmisartan improves cardiac fibrosis in diabetes through peroxisome proliferator activated receptor delta (PPARdelta): from bedside to bench. Cardiovasc Diabetol 15:113. doi: 10.1186/s12933-016-0430-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gu Y, Li X, He T, Jiang Z, Hao P and Tang X (2014) The Antifibrosis Effects of Peroxisome Proliferator-Activated Receptor delta on Rat Corneal Wound Healing after Excimer Laser Keratectomy. PPAR Res 2014:464935. doi: 10.1155/2014/464935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su X, Zhou G, Wang Y, Yang X, Li L, Yu R and Li D (2014) The PPARbeta/delta agonist GW501516 attenuates peritonitis in peritoneal fibrosis via inhibition of TAK1-NFkappaB pathway in rats. Inflammation 37:729–37. doi: 10.1007/s10753-013-9791-z [DOI] [PubMed] [Google Scholar]
- 65.Fedorova LV, Sodhi K, Gatto-Weis C, Puri N, Hinds TD Jr., Shapiro JI and Malhotra D (2013) Peroxisome proliferator-activated receptor delta agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS One 8:e64436. doi: 10.1371/journal.pone.0064436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iwaisako K, Haimerl M, Paik YH, Taura K, Kodama Y, Sirlin C, Yu E, Yu RT, Downes M, Evans RM, Brenner DA and Schnabl B (2012) Protection from liver fibrosis by a peroxisome proliferator-activated receptor delta agonist. Proc Natl Acad Sci U S A 109:E1369–76. doi: 10.1073/pnas.1202464109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wen WX, Lee SY, Siang R and Koh RY (2017) Repurposing Pentoxifylline for the Treatment of Fibrosis: An Overview. Adv Ther 34:1245–1269. doi: 10.1007/s12325-017-0547-2 [DOI] [PubMed] [Google Scholar]
- 68.Delanian S (1998) Striking regression of radiation-induced fibrosis by a combination of pentoxifylline and tocopherol. Br J Radiol 71:892–4. doi: 10.1259/bjr.71.848.9828807 [DOI] [PubMed] [Google Scholar]
- 69.Haddad P, Kalaghchi B and Amouzegar-Hashemi F (2005) Pentoxifylline and vitamin E combination for superficial radiation-induced fibrosis: a phase II clinical trial. Radiother Oncol 77:324–6. doi: 10.1016/j.radonc.2005.09.014 [DOI] [PubMed] [Google Scholar]
- 70.Delanian S, Porcher R, Balla-Mekias S and Lefaix JL (2003) Randomized, placebo-controlled trial of combined pentoxifylline and tocopherol for regression of superficial radiation-induced fibrosis. J Clin Oncol 21:2545–50. doi: 10.1200/jco.2003.06.064 [DOI] [PubMed] [Google Scholar]
- 71.Rajendran R, Rani V and Shaikh S (2006) Pentoxifylline therapy: a new adjunct in the treatment of oral submucous fibrosis. Indian J Dent Res 17:190–8. [DOI] [PubMed] [Google Scholar]
- 72.Mehrotra R, Singh HP, Gupta SC, Singh M and Jain S (2011) Pentoxifylline therapy in the management of oral submucous fibrosis. Asian Pac J Cancer Prev 12:971–4. [PubMed] [Google Scholar]
- 73.Anuradha A, Patil B and Asha VR (2017) Evaluation of efficacy of aloe vera in the treatment of oral submucous fibrosis - a clinical study. J Oral Pathol Med 46:50–55. doi: 10.1111/jop.12463 [DOI] [PubMed] [Google Scholar]
- 74.Perkins RM, Aboudara MC, Uy AL, Olson SW, Cushner HM and Yuan CM (2009) Effect of pentoxifylline on GFR decline in CKD: a pilot, double-blind, randomized, placebo-controlled trial. Am J Kidney Dis 53:606–16. doi: 10.1053/j.ajkd.2008.11.026 [DOI] [PubMed] [Google Scholar]
- 75.Goicoechea M, Garcia de Vinuesa S, Quiroga B, Verdalles U, Barraca D, Yuste C, Panizo N, Verde E, Munoz MA and Luno J (2012) Effects of pentoxifylline on inflammatory parameters in chronic kidney disease patients: a randomized trial. J Nephrol 25:969–75. doi: 10.5301/jn.5000077 [DOI] [PubMed] [Google Scholar]
- 76.Chen PM, Lai TS, Chen PY, Lai CF, Wu V, Chiang WC, Chen YM, Wu KD and Tsai TJ (2014) Renoprotective effect of combining pentoxifylline with angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker in advanced chronic kidney disease. J Formos Med Assoc 113:219–26. doi: 10.1016/j.jfma.2014.01.002 [DOI] [PubMed] [Google Scholar]
- 77.Campbell D and Weir MR (2015) Defining, Treating, and Understanding Chronic Kidney Disease--A Complex Disorder. J Clin Hypertens (Greenwich) 17:514–27. doi: 10.1111/jch.12560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J and Warnock DG (2011) Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365:327–36. doi: 10.1056/NEJMoa1105351 [DOI] [PubMed] [Google Scholar]
- 79.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving HH, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D and Chertow GM (2013) Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369:2492–503. doi: 10.1056/NEJMoa1306033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chin MP, Bakris GL, Block GA, Chertow GM, Goldsberry A, Inker LA, Heerspink HJL, O’Grady M, Pergola PE, Wanner C, Warnock DG and Meyer CJ (2018) Bardoxolone Methyl Improves Kidney Function in Patients with Chronic Kidney Disease Stage 4 and Type 2 Diabetes: Post-Hoc Analyses from Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes Study. Am J Nephrol 47:40–47. doi: 10.1159/000486398 [DOI] [PMC free article] [PubMed] [Google Scholar]