

Abstract
Erythroid cell formation critically depends upon signals transduced via EPO/EPOR/JAK2 complexes. This includes not only core response modules (e.g., JAK2/STAT5, RAS/MEK/ERK), but also specialized effectors (e.g., Erythroferrone, ASCT2 glutamine transport, Spi2A). By employing phospho-proteomics and a human erythroblastic cell model, we presently identify 121 new EPO target proteins, together with their EPO- modulated domains and phosphosites. Gene Ontology enrichment for ‘Molecular Function’ identified adaptor proteins as one top EPO target category. This includes a novel EPOR/JAK2-coupled network of actin assemblage modifiers, with adaptors DLG-1, DLG-3, WAS, WASL and CD2AP as prime components. ‘Cellular Component’ GO analysis further identified 19 new EPO- modulated cytoskeletal targets including the erythroid cytoskeletal targets SPECTRIN-A, SPECTRIN–B, ADDUCIN-2 and GLYCOPHORIN-C. In each, EPO-induced phosphorylation occurred at p-Y sites and subdomains that suggest coordinated regulation by EPO of the erythroid cytoskeleton. GO analysis of ‘Biological Processes’ further revealed metabolic regulators as a likewise unexpected EPO target set. Targets included ALDOLASE-A, PYRUVATE DEHYDROGENASE-A1 and THIOREDOXIN- INTERACTING PROTEIN (TXNIP), with EPO-modulated pY-sites in each occurring within functional subdomains. In TXNIP, EPO- induced phosphorylation occurred at novel p-T349 and p-S358 sites, and was paralleled by rapid increases in TXNIP levels. In UT7epo-E and primary human HSC-derived erythroid progenitor cells, lentivirus-mediated shRNA knockdown studies revealed novel pro-erythropoietic roles for TXNIP. Specifically, TXNIP’s knockdown sharply inhibited c-KIT expression; compromised EPO dose-dependent erythroblast proliferation and survival; and delayed late-stage erythroblast formation. Overall, new insight is provided into EPO’s diverse action mechanisms, and TXNIP’s contributions to EPO-dependent human erythropoiesis.
Keywords: EPO, EPOR, JAK2, phospho-proteomics, human erythropoiesis, signal transduction factors, TXNIP
Graphical abstract
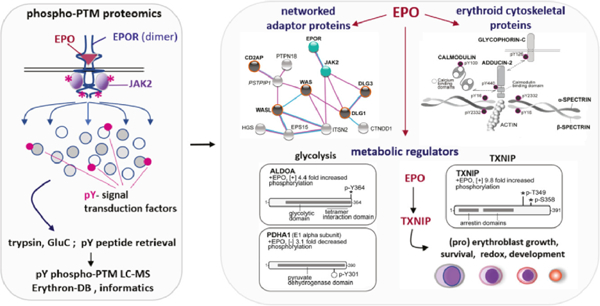
INTRODUCTION
The production of erythroid progenitors requires signals provided by EPO and its JAK2- coupled receptor, EPOR3. rh-EPO and related EPOR agonists are also important therapeutics for the anemia of chronic kidney disease4, myelodysplastic syndrome5 and chemotherapy6. While EPO’s actions have accordingly been intensely studied7,8, an advanced understanding of EPO/EPOR/JAK2 effects is important for several reasons. Clinically, and via poorly understood mechanisms, EPO also elicits hypertensive and thrombolytic side effects9. Additionally, strategies to counter JAK2 mutation effects on myeloproliferative neoplasms10,11 and myelofibrosis12 can be aided by a detailed understanding of JAK2 effectors. The EPO/EPOR/JAK2 system therefore continues to serve as an insightful paradigm for determining how hematopoietic growth factors (HGFs) trigger diverse responses within developing blood cell populations7,8,13.
Within erythroid progenitor cells (EPCs), EPO action studies have typically focused on anti-apoptotic responses, with early investigations describing Bcl-x mediated cytoprotection as perhaps a singular prime EPO effect14,15. The field further concluded that EPO (and HGFs) might essentially fail to provide instructive signals16. Recent investigations, however, have revealed several unexpected EPO actions. Lineage commitment studies describe EPO’s recruitment of pluripotent progenitors to an erythroid fate17. In proerythroblasts, a non-canonical EPOR/JAK2/STAT5 cytoprotective response involving serpin Spi2A inhibition of leached lysosomal executioner cathepsins has been defined18. In maturing erythroblasts, the EPO- induced expression and release of Erythroferrone19 can inhibit Hepcidin, derepress Ferroportin, and heighten systemic iron. These examples underscore unanticipated versatility in EPO’s signaling mechanisms, and functional effects.
For the above unexpected EPO signal transduction factors, each was discovered via gene profiling13,17–19. At the genomic level, 25,000 genes can generate up to 100,000 transcripts. Ten-fold greater diversity, however, can exist at the protein level due to dynamic post-translational modifications (PTMs). During HGF signaling, PTMs can play major regulatory roles7,8. We therefore presently have applied phospho-PTM proteomics20,21 together with an EPO-dependent human erythroid progenitor UT7epo-E cell model to interrogate EPO’s actions. Overall, we identify 121 novel EPO/EPOR target proteins, together with their specific EPO-regulated p-Y and/or p-TPP sites. Gene Ontology (GO)22 enrichment analysis of ‘Molecular Function’ terms first identified major cytokine signaling associated categories including receptor binding factors, kinases and phosphatases, Ras guanyl exchange proteins, and signal adaptors. Each EPO target set was substantial, including 18 newly discovered EPO-regulated adaptor proteins. Among enriched “Biological Processes”, novel EPO-modulated targets unexpectedly included the core metabolic regulators ALDOLASE-A and PYRUVATE DEHYDROGENASE-α1, and the redox and glycemic regulator THIOREDOXIN INTERACTING PROTEIN (TXNIP)23,24.
For the following reasons, TXNIP was selected as a new EPO target to investigate via shRNA knockdown experiments in primary (pro-erythroblasts). In at least certain cell types, TXNIP is known to modulate cellular oxidants by inhibiting Thioredoxin25. In transplanted hematopoietic stem cells, however, TXNIP unexpectedly has been demonstrated to exert important anti-oxidant effects24. Additionally, in a context of EPO- signaling, EPO has been shown to modulate ROS in (pro)erythroblasts, and in a global knockout mouse model, Txnip KO skews splenic basophilic erythroid cell formation and transferrin receptor levels26. Our present studies reveal that in human erythroblastic cells, EPO rapidly induces the phosphorylation of TXNIP at novel p-T349 and p-S358 sites, and heightens TXNIP levels. Knockdown studies further reveal novel roles for TXNIP in supporting c-Kit expression, EPO dose-dependent primary human erythroblast growth and survival, and the development of maturing erythroblasts.
Finally, among “Cellular Components”, EPO regulated the p-Y phosphorylation of 19 novel cytoskeletal targets, four of which are erythroid- specific, and required for late erythroblast differentiation. In silico analyses of their p-Y regulated functional subdomains suggest attenuating effects of EPO on erythroid cytoskeletal assembly. These overall findings are discussed in the context of unexpectedly expanded phospho-PTM EPO signaling networks, together with new experimentally defined pro-erythropoietic roles for TXNIP during human erythropoiesis.
METHODS
A detailed description of Methods is provided as a Supplemental to this report.
RESULTS
Phospho-PTM proteomic discovery of 121 novel EPO- modulated targets
PTM-directed proteomics can provide detailed insight into protein signaling networks20,21, but to date has not been stringently applied to analyze hematopoietic growth factor action. Presently, we have employed phospho-PTM profiling to explore EPO/EPOR transduced signals (Figure 1A). To generate sufficient material for effective PTM-based LC-MS, EPO-dependent human UT7epo-E cells were employed as a parental UT7epo cell subline27 generated via 50+ passages in EPO. To aid interpretations of phospho-PTM studies (and EPO target knockdown experiments), we first assessed UT7epo-E cells for possible complicating mutations among 18 myeloproliferative driver genes (including JAK2, MPL, and CALRETICULIN)28. No such mutations were detected (Figure 1B). Gene profiling further demonstrated an erythroid gene expression profile for UT7epo-E cells (Figure 1C), and globin chain HBA analysis showed levels in UT7epo-E cells to approximate those in basophilic erythroblasts (Figure 1D). Overall globin chain levels were also elevated compared to parental UT7epo, and K562 cells (LC-MS analyses, Supplemental Figure S-1). UT7epo-E cells also were shown to exhibit sharp EPO dose-dependent proliferation, survival and cell surface EPOR expression (Figures 1E, 1F).
Figure 1. Proteomic profiling of EPO/EPOR/JAK2 regulated phospho-PTM targets.

(A-F) Phospho-PTM workflow, and characterization of EPO-dependent human erythroid progenitor UT7epo-E cells. A: In the outlined phospho-PTM proteomic workflow, steps employed to profile EPO-regulated p-Y or p-TPP motif modified target proteins in UT7epo-E cells are defined. B: Analyses of mutations in 18 myeloproliferative driver genes were assessed, with no such mutations detected. C: Profiling of erythroid vs. megakaryocytic marker transcripts in UT7epo-E cells indicated a predominant erythroid phenotype. D: HBA globin levels in UT7epo-E cells (via western blotting) approximate levels in primary basophilic erythroblasts (western blot, right panel). E,F: UT7epo-E cells retain sharp EPO dose- dependency for growth and survival (E), and dynamic cell surface EPOR expression (F) (flow cytometry assays). (G,H) Validation data for known EPO- modulated phospho-PTM targets – G: To illustrate insight generated via this affinity LC-MS/MS approach, PTM-modulated signal transduction factors known to associate with EPO/EPOR/JAK2 complexes are first considered. Data illustrated for ten validation targets include: The fold-modulation of specific phospho-PTMs (“EPO, fold-modulation” column); parent target protein identities (“protein target” column); the use of trypsin (t) or Glu-C (g) to generate peptides (“study” column); EPO-regulated phospho-residues within the parent protein sequence (“site(s)” column); LC-MS/MS defined sequences of the EPO-regulated phospho-peptide (“peptide” column); and identifiers for parent proteins (“protein description” column). H: For comparison, western blot signals for select known EPO- regulated phospho-PTM targets are also shown including p-Y1007/p-Y1008-JAK2, p-Y570-JAK2, p-Y132/p-Y141-RHEX, and p-Y986/p-Y987 INPPL1.
Discovery proteomic studies were designed to capture early EPOR/JAK2 induced phospho-PTM events. Specifically, UT7epo-E cells were challenged with EPO (+/−) for 15 minutes. Total extracted proteins and derived tryptic or GluC peptides were then prepared, and peptides modified at p-Y or p-TPP motifs were retrieved using optimized immunoadsorption protocols. For eluted peptides, LC-MS and spectral analysis defined sequences, EPO-mediated phosphorylation levels, sites of regulated phosphorylation, and parent proteins. As validating phospho-PTM data, findings for ten known EPO targets previously associated with EPO’s actions are first summarized (Figure 1G). As illustrated for JAK2, validating EPO-regulated p-Y1007, p-Y1008, p-Y570 and p-Y813 sites occurred within unique, as well as confirming overlapping phospho-peptides.
Within known EPO targets, novel phospho-PTM sites additionally were discovered. As one example, PIK3’s p85 subunit is a known target for EPO induced p-Y607 phosphorylation29: Our analyses additionally revealed the 40.4-fold EPO-induced phosphorylation of PIK3’s p110 catalytic subunit at a novel Y508 site (Figure 1G). For select known EPO targets and p-Y motifs with available antibodies, western blot analyses are also shown for comparison (Figure 1H).
Novel EPO-modulated phospho-PTM targets, and ‘Molecular Function’ GO annotation enrichment of pY-modified signal adaptor networks
Bioinformatic approaches next were employed to identify novel EPO-regulated target phosphoproteins, and their networked associations. With the EPOR centrally positioned, connections among EPO-modulated targets were determined via STRINGdb analysis (Figure 2A), for three sets of phospho-PTM experiments (“pY-trypsin”, “pY-GluC”, “pTPP-GluC” – see Supplemental Methods and Supplemental Table S-1). Among 138 proteins revealed to be regulated by EPO ≥2-fold in defined phospho-PTMs, 121 were novel targets (orange chords, no mapped STRINGdb interactions, and not previously linked to EPO/EPOR signaling). Target protein identities, phospho-peptide sequences, and their fold-modulation due to EPO are also defined. In Supplemental Tables S-2,3,4, annotated phospho-PTM LC-MS/MS datasets are also provided via a dedicated ErythronDB platform (see Supplemental Methods, and https://www.cbil.upenn.edu/ErythronDB_alpha/app). For novel EPO- modulated phosphoproteins, annotation terms enriched (via GO analysis) for ‘Molecular Function’ strongly reflected cytokine receptor transduction events. These include ‘signaling adaptor activity’, ‘receptor binding’, ‘non-receptor binding’, and ‘RAS guanyl-nucleotide exchange factor activity’ (Figure 2B, and Supplemental Table 5). Based on their high GO representation (confirmed via UniProt and NCBI resource mining), adaptor proteins are a highlighted EPO target set with 24 such EPO- modulated adaptor protein targets identified, 18 as novel. When candidate associations were analyzed (using the full set of presently identified targets and STRINGdb) several specific new associations among novel EPO-regulated adaptor proteins were revealed within an EPOR/JAK2 signal transduction factor network (Figure 3A). The scaffold proteins (and tumor suppressors) DISCS LARGE-1 and −3 (ref.30) provide one example. Within each, EPO induced the phosphorylation of a novel p-Y site within their guanylate kinase domains (DLG1, p-Y760; DLG3, p-Y673) (Figure 3B). DLG-1 and −3 each also connected to EPOR/JAK2 signaling via EZRIN, CALM1 and FYN circuits (Figure 3A).
Figure 2. Discovery phospho-PTM proteomics identifies 121 novel EPO-regulated targets with diverse molecular functions.
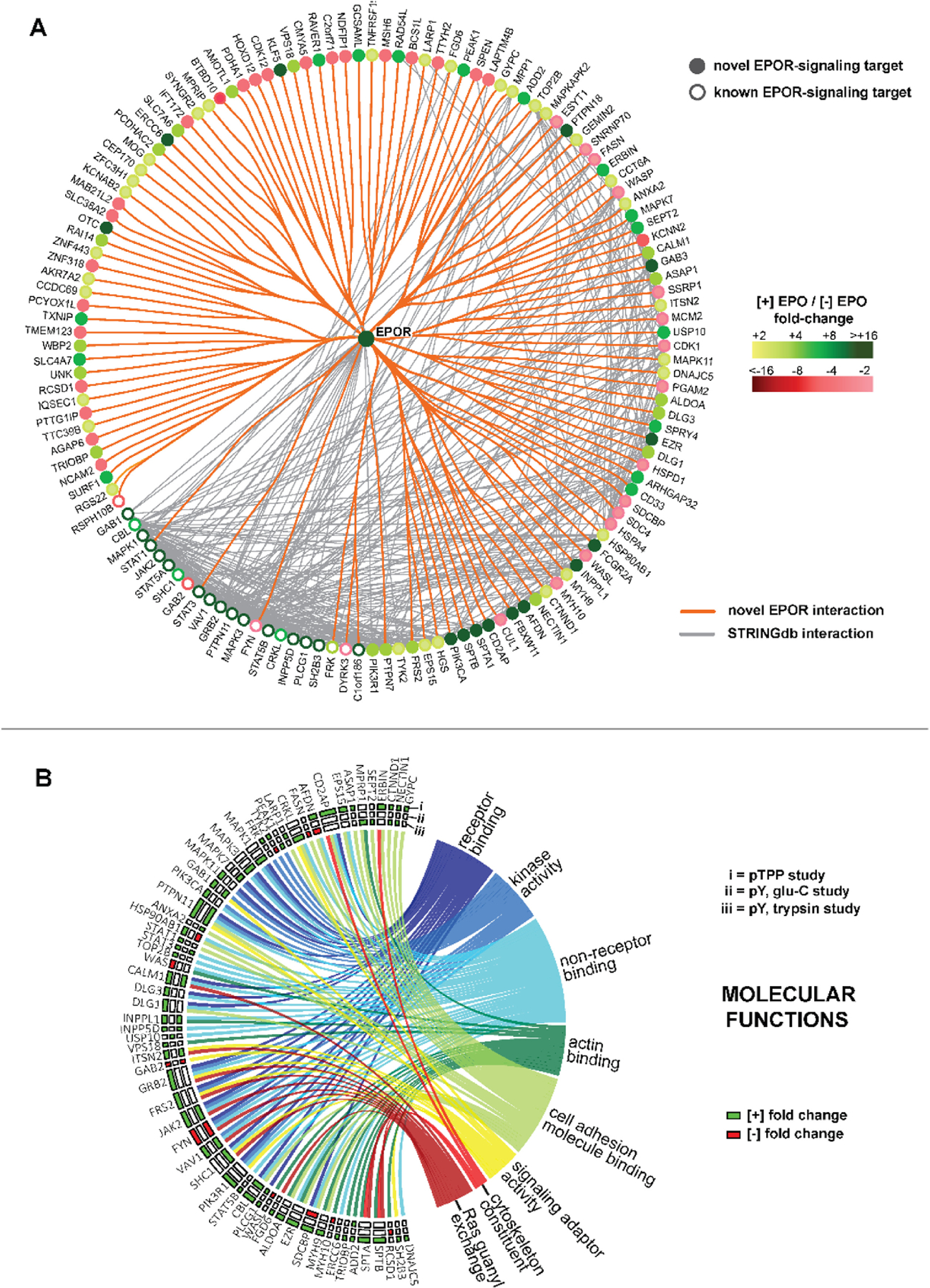
(A) Overall EPO-regulated phospho-PTM targets discovered in UT7epo-E cells via p-Y trypsin, p-Y GluC, and p-TPP GluC proteomic studies: Among 138 unique proteins identified, 121 proved to be novel EPO/EPOR regulated proteins (filled circles) that contain one or more p-Y and/or p-TPP site with ≥2-fold regulation due to EPO. For novel EPO targets, orange lines illustrate their new connections to the EPOR. As defined via STRINGdb, gray lines indicate existing associations among overall targets. Fold-change effects of EPO on phospho-PTM targets are coded by green intensities (induced by EPO), and red intensities (decreased due to EPO). (For p-TPP studies, fold changes are median-adjusted; see Methods). (B) GO enrichment analysis of ‘Molecular Function’ identifies eight principle sets of EPO-regulated phospho-PTM targets: EPO targets analyzed for ‘Molecular Function’ include those discovered in p-TPP(i), p-Y Glu-C (ii), and p-Y trypsin phospho-PTM studies (iii) (see insert key “i, ii, iii”). Target proteins are identified on the left border, and are connected via chords to associated ‘molecular function’ categories (as semantically similar sets of enriched GO terms). Enriched annotations include ‘receptor binding’ (n=15 proteins), ‘kinase activity’ (n=16), non-receptor binding terms (n=30), ‘actin binding’ (n=14), ‘cell adhesion molecule binding’ (n=23), ‘signaling adaptor activity’ (n=10), ‘structural constituent of cytoskeleton’ (n=4) and ‘Ras guanyl-nucleotide exchange factor activity’ (n=11) (also, see Supplemental Table S-5).
Figure 3. Analysis of 18 novel EPO- regulated phospho-PTM modified molecular adaptors defines an extended signaling network.
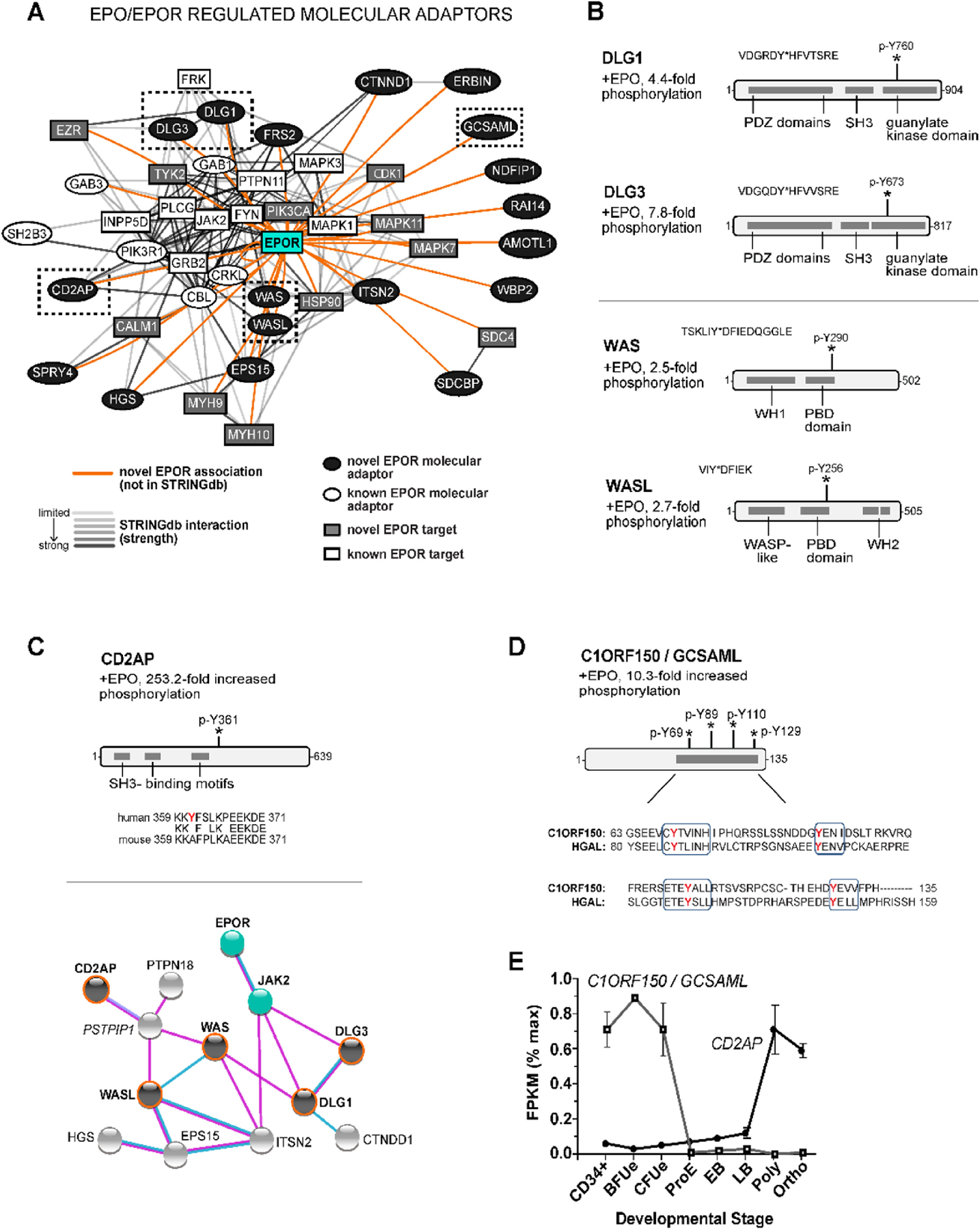
(A) EPO/EPOR/JAK2 molecular adaptor networks: As identified via gene ontology “signaling adaptor activity” assignments (together with NCBI UniProt, Gene Cards, and Phosphosite-Plus® database mining), eighteen novel EPO- and phospho- PTM regulated adaptor proteins were defined (closed circles). For twelve, STRINGdb analysis further established EPOR interactions among DLG1, DLG3, WAS, WASL, CD2AP, SPRY4, HGS, EPS15, FRS2, CTNND1, ERBIN, and EPS15. For C1ORF150/GCSAML, SDCBP, NDFIP1, RAI14, AMOTL1 and WBP2, associations (as presently established) were restricted to the EPOR. (B) Coordinated EPO- and phospho-PTM modulation of actin regulating scaffold proteins DLG1, DLG3, WAS and WASL – Upper panels: For the related scaffold proteins DLG-1 and −3, EPO-induced their phosphorylation at aligned novel p-Y760 (DLG-1) and p-Y673 (DLG-3) sites within guanylate kinase domains. Lower panels: WAS and WASL (co-regulators of actin, and DLG interacting proteins) likewise were coordinately regulated by EPO in their aligned p-Y290 (WAS) and p-Y256 (WASL) phosphorylations within central PBD domains. (C) CD2AP as a novel H sapiens EPO- and phospho-PTM regulated signal adaptor: For CD2AP (a direct actin binding scaffold protein), EPO induced its phosphorylation 253.2-fold at a novel p-Y361 site which is conserved in human and primate CD2AP (not shown), but not mouse, rat or lower vertebrates. CD2AP further exhibited connectivity (via PSTPIP1) to WAS, WASL, DLG1, DLG3 within a defined EPOR/JAK2 signaling network (lower subpanel, biochemical evidence; blue connectors). (D) C1ORF150/GSCAML as a novel EPO- regulated signal adaptor (and HGAL orthologue): In C1ORF150, EPO induced the phosphorylation of four C- terminal p-Y sites with singular homology to the B-cell receptor adaptor protein HGAL. (E) During human erythropoiesis, C1ORF150 and CD2AP exhibit stage-selective expression (lower panel) with maximal C1ORF150 expression in BFUe, and maximal CD2AP expression in polychromatic erythroblasts (abbreviations: proerythroblasts, ProE; early erythroblasts, EB; late erythroblasts, LB; polychromatic, poly; orthochromatic, ortho).
As a related second example, EPO stimulated the dephosphorylation of the Wiskott-Aldrich syndrome adaptor proteins WAS/WASP31 and WASL/N-WASP32, each at a conserved P21-Rho-binding domain (PBD) p-Y site (WAS, p-Y290; WASL, p-Y256) (Figure 3B, lower panel). For WASL, phosphorylation at Y290 is known to promote actin restructuring33, further implicating EPO effects on cytoskeletal assemblages. This focus on signaling adaptors also revealed species- restricted EPO-regulated PTMs. CD2AP (a cytoskeleton modulator34) was a top EPO-regulated target, with >250-fold induced phosphorylation at a novel p-Y361 site (Figure 3C). p-Y361 is conserved in humans and primates, but is not represented in mouse, rat, or lower vertebrate CD2APs. CD2AP likewise couples to a DLG1, DLG3, WAS, WASL signaling network (Figure 3C, lower panel). Finally, for C1ORF150 / GCSAML (Figure 3D), EPO induced its phosphorylation at four p-Y sites within a C-terminal region shared with HGAL, an important B-cell receptor adaptor protein35. Unlike HGAL, C1ORF150 is not represented in mouse, rat, or lower vertebrate genomes. During erythroid development, C1ORF150’s expression peaks at a BFUe stage, while CD2AP is elevated in maturing erythroblasts (Figure 3E).
Enriched ‘Biological Processes’ of EPO-modulated targets, including metabolic regulators and TXNIP
In GO analyses, enriched annotations for ‘Biological Process’ included major categories of EPO modulated phospho-targets as ‘signal transduction’, ‘phosphorylation’, ‘cell differentiation’, ‘cell migration’ and ‘response to stimulus’ (Figure 4A, and Supplemental Table S-6). For focus, and to further illustrate opportunities to frame data driven hypotheses, new EPO-modulated targets in the categories of ‘intrinsic apoptotic signaling pathway’ (under ‘signal transduction’), and ‘metabolic process’ are highlighted. In apoptotic pathways, two novel EPO targets, MSH6 and ERCC6 were discovered (Figure 4B) that play important (anti)apoptotic roles during DNA mismatch repair36. MSH6 heterodimerizes with MSH2 to form MutSα, a complex that binds mismatched base pairs, and recruits key repair enzymes36. EPO induced MSH6’s de-phosphorylation at novel p-Y977 and p-Y994 sites, each within a DNA interacting subdomain36. During mismatch repair, ERCC6 functions subsequently as a DNA helicase37,38. EPO stimulated ERCC6’s 52.7-fold phosphorylation at novel p-Y882 and p-Y901 sites, each within ERCC6’s helicase domain. As effectors of DNA repair and apoptotic signaling, MSH6 and ERCC6 (and their defined phospho-PTMs), may relate to anemia triggered by chemotherapeutic DNA alkylating agents39.
Figure 4. GO Biological processes implicated for novel EPO-and phospho-PTM modulated target proteins, including DNA repair and energy metabolism factors.
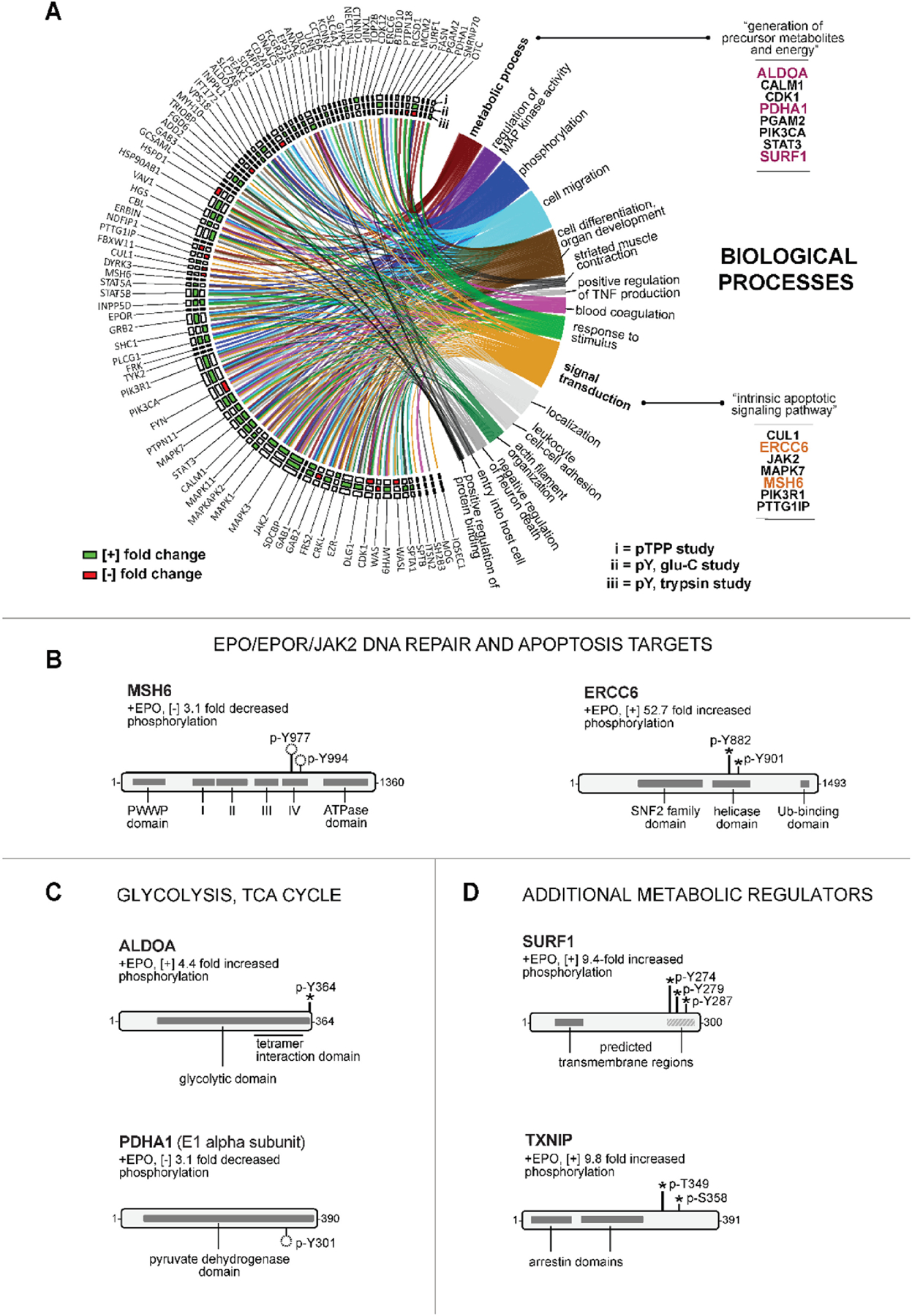
(A) Enrichment analysis for ‘biological process’ terms defines sixteen diverse EPO target sets: Anticipated validating enriched annotations included ‘phosphorylation’, ‘regulation of MAP kinase activity’, ‘response to stimulus’, and ‘signal transduction’. Under ‘signal transduction’, ‘intrinsic apoptotic signaling pathway’ included the anti-apoptotic DNA repair factors ERCC6 and MSH6. Within ‘metabolic process’ category, eight novel EPO- and phospho-PTM regulated targets were ‘generation of precursor metabolites and energy’ annotated proteins, including ALDOA,PDHA1; and SURF1. (B) EPO modulation of MSH6, and ERCC6: For these DNA mismatch repair factors, novel dual sites of EPO regulated p-Y (de)-phosphorylation were within MSH6’s DNA binding region (p-Y977 and p-Y994), and ERCC6’s helicase domain (p-Y882 and p-Y901). (C,D) EPO modulation of core metabolic regulators – C: EPO-modulated metabolic targets included ALDOLASE A (ALDOA; 4.4-fold EPO-induced p-Y364 phosphorylation, tetramerization domain) and PYRUVATE DEHYDROGENASE A1 (PDHA1; 3.1-fold decreased phosphorylation at Y301, catalytic domain). D: For the cytochrome oxidase C component, SURF1 (upper sub-panel) EPO induced the phosphorylation of clustered sites (p-Y274, p-Y279, and p-Y287) within a hydrophobic C-terminal domain. For THIOREDOXIN INTERACTING PROTEIN (TXNIP), EPO induced phosphorylation was at novel p-T349 and p-S358 sites (lower sub-panel).
For EPO’s actions, ‘metabolic process’ was an unanticipated category, with several new EPO targets specifically identified within a sub-category of ‘generation of precursor metabolites and energy’. Two are central glycolytic enzymes (Figure 4C). In ALDOA (mutated in hemolytic anemia)40, EPO stimulated p-Y364 phosphorylation within a C-terminal domain involved in ALDOA tetramerization and substrate binding41. In PDH-A1, EPO triggered its dephosphorylation at p-Y301, a site that when phosphorylated can inhibit PDH’s catalytic activity42. These EPO-induced p-Y events therefore implicate EPO’s heightening of triose and pyruvate- derived acetyl-CoA formation. EPO additionally induced the novel phosphorylation of SURF1, a cytochrome oxidase complex associated factor43 at three clustered pY sites within a C-terminal hydrophobic subdomain of this inner mitochondrial membrane protein (Figure 4D, upper panel). Finally, the redox and glycemic regulator THIOREDOXIN INTERACTING PROTEIN (TXNIP)23,24, was identified as a new EPO-modulated metabolic target (Figure 4D, lower panel), with EPO stimulating TXNIP’s phosphorylation at novel p-T349 and p-S358 sites (proximal to a p-S308 site that triggers TXNIP turnover)23.
TXNIP promotes EPO-dependent human erythroblast proliferation, survival and development
As a novel EPO- modulated phospho-PTM target, TXNIP was selected for direct functional investigations for three prime reasons: (1) TXNIP negatively regulates insulin expression in beta cells44 and glucose transport in HepG2 cells23, but in hematopoietic cells can support hematopoietic stem cell repopulation24. (2) TXNIP is an emerging drug target for diabetes25, but its inhibition might compromise hematopoiesis24. (3) In a global KO mouse model, TXNIP LOF has been observed to perturb splenic erythroblast formation26, raising important new questions concerning TXNIP’s potential erythroid cell intrinsic roles in human medullary progenitors, and candidate functional ties to EPO’s actions.
Using UT7epo-E cells as an initial model, several novel connections between TXNIP and EPO signaling were defined. EPO regulation of TXNIP phosphorylation at p-T349 and p-S358 first was validated in phospho-PTM studies using an EPOR agonist, Hematide (Figure 5A). Western blotting using an antibody prepared against a p-T349 peptide additionally specifically detected EPO- induced p-T349-TXNIP (Figure 5A, lower panel). TXNIP and EPO-induced phospho-T349 TXNIP were also shown to reside in cytoplasmic and membrane cell fractions, with apparent heightening effects of EPO on TXNIP levels. Repeated experiments (Figures 5B, and Supplemental Figure S-3) confirmed this effect. Using a shRNA lentivirus approach (Figure 5C), TXNIP knock-down (“KD”) was observed to attenuate EPO-dependent UT7epo-E cell proliferation (Figure 5D). Experiments employing a CDK4,6 inhibitor mediated G1 phase block- and-release design further implicated G1 to S phase effects of TXNIP KD (Figure 5E). Finally, in studies analyzing possible effects of TXNIP knockdown on KIT and GLYCOPHORIN A (GPA) marker transcripts, KIT expression proved to be substantially repressed, while GPA levels were modestly increased (Figure 5F).
Figure 5. EPO effects on TXNIP levels, and TXNIP effects on EPOR mediated UT7epo-E erythroid progenitor cell growth and c-KIT expression.
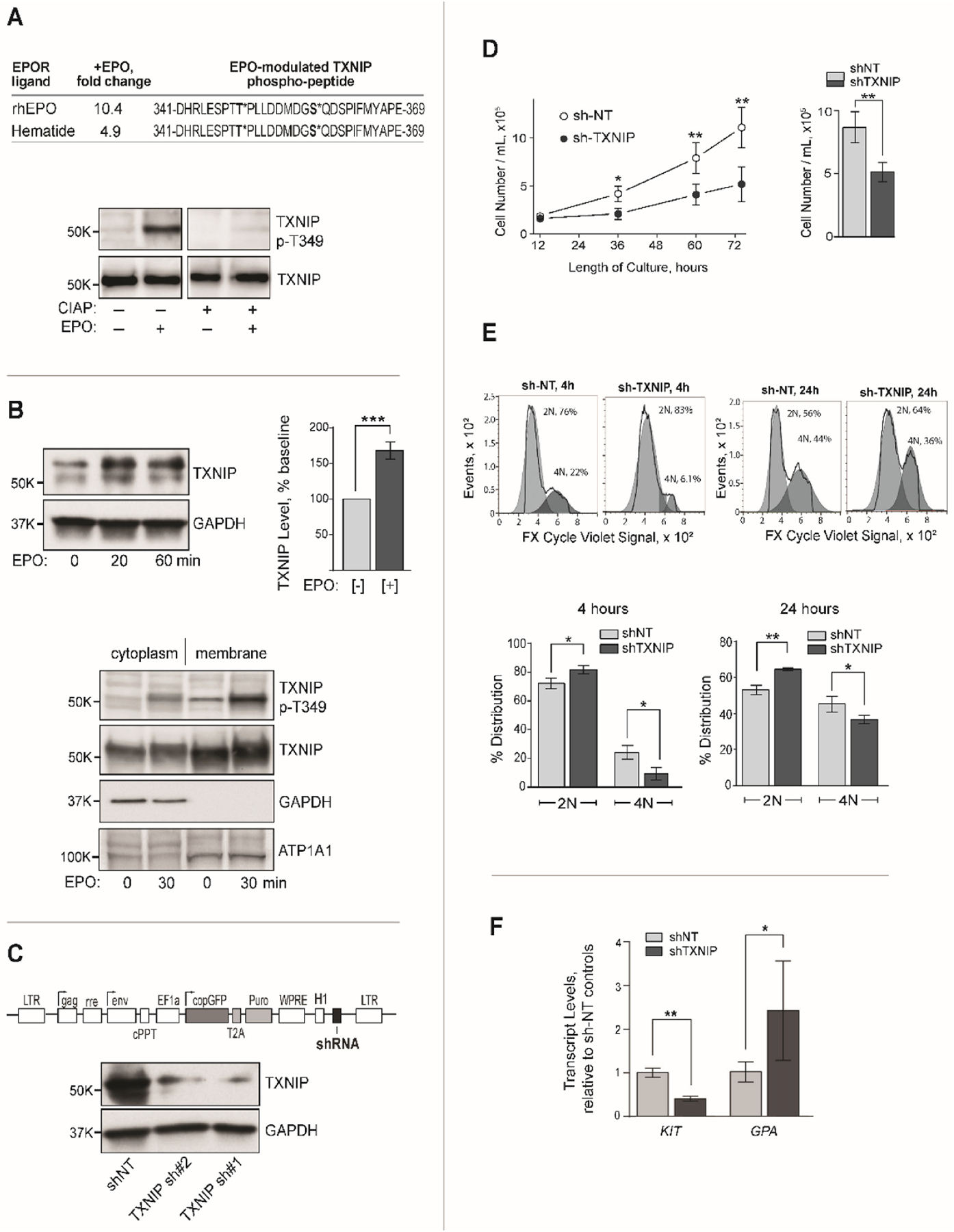
(A) Validation of TXNIP phosphorylation using an EPOR agonist (Hematide), and a TXNIP- p-T349 specific antibody – Upper panel: In phospho-PTM profiling of UT7epo-E cells, Hematide also induced TXNIP’s p-Y phosphorylation at p-T349 and S-358 sites. Lower panel: In EPO challenge experiments in UT7epo-E cells, EPO (+/− EPO, 10 min) induced TXNIP’s phosphorylation at p-T349 as assayed via western blotting. Upon blot pre-treatment with CIAP, this signal was abolished. (B) EPO rapidly increases TXNIP levels – Upper panels: In EPO challenge experiments, exposure of UT7epo-E cells (post EPO withdrawal) for 20 or 60 min rapidly increased TXNIP levels (western blot, total cell lysates). Bar graph data are normalized mean values +/− SE (n=3). Lower panel: Cell fractionation experiments demonstrated cytoplasmic and membrane localization of total and p-T349 TXNIP, with EPO-dependent increases in TXNIP again observed (also see Supplemental Figure S2). (C) Lentivirus mediated shRNA knockdown of TXNIP in UT7epo-E cells: Lentivirus pEF1aGreenPuro template pDNAs (upper panel) were used to generate lentiviruses expressing non-target (NT) or TXNIP transcript targeting shRNAs. In UT7epo-E cells transduced with these lentiviruses at low MOIs (and selected in puromycin), efficient knockdown (“KD”) of TXNIP was confirmed by western blotting. (D) Knockdown of TXNIP inhibits EPO-dependent growth of UT7epo-E cells: UT7epo-E cells stably expressing NT or 150#1 shRNAs were plated at 2x105 cells in 10% FBS, IMDM, P-S and EPO (2U/mL). At the indicated time points, viable cell numbers were determined (mean +/− SE, n=3). Under these assay conditions, no significant effects of TXNIP KD on cell viability were observed (data not shown). (E) G1 to G2 phase progression is attenuated due to TXNIP knockdown: UT7epo-E sh-NT and sh-TXNIP cells were cultured for 20h in the presence of PD-0332991 (0.2 µg/mL), with EPO at 0.5 U/mL to establish G1 phase block. Cells were then washed free of inhibitor, plated in EPO at 2 U/mL and assayed for 2N to 4N advancement (FX Cycle Violet, flow cytometry). Delays in progression to G2 were observed at 4h and 24h (means +/− SE). (F) TXNIP knockdown decreases c-KIT, and increases GPA levels: In UT7epo-E cells, shTXNIP compared to control UT7epo-E sh-NT cells, TXNIP KD effects on KIT and GPA transcripts were assessed by quantitative RT-PCR. Values are means +/− SE (n=3).
The above findings in UT7epo-E cells prompted TXNIP KD studies in primary human progenitors. Two erythroid culture systems initially were employed. One included SCF, EPO and dexamethasone. A second included SCF, IL3, EPO, human serum, and A/B plasma. In each, analyses of transcriptome profiling revealed late-stage specific spikes in TXNIP levels in developing human (but not mouse) medullary erythroblasts (Figure 6A – see next page). In each system, TXNIP KD repressed c-KIT expression multi-fold, while modestly increasing GPA levels (Figure 6B). Studies of KD effects on erythroid progenitor cell (EPC) growth next were performed, and revealed role roles for TXNIP in supporting EPO-dependent erythroid progenitor cell (EPC) proliferation, and survival (Figure 6C and Supplemental Figure S-4). When EPO was limited, TXNIP KD compromise of EPC growth and survival was further amplified (Figures 6C and Supplemental Figure S-5). Possible effects of TXNIP KD on EPC development also were assessed. As analyzed in phase-I cultures, late stage erythroblasts (d7) were under-represented several fold (Figures 6D). Ultimately, and at d10 of culture, TXNIP KD polychromatic erythroblasts did form at heightened frequencies of ~200% over shNT control levels (Figure 6D). TXNIP KD also visually attenuated the hemoglobinization of maturing erythroblasts (Figure 6E). This effect, however, did not include apparent effects on HBA or HBB globin levels (Figure 6E, lower panel). Mechanistically, possible effects of TXNIP knockdown on peroxide sensitivity, and ROS levels, were studied. This was prompted by reported TXNIP’s heightening of oxidants in beta cells45, and limiting of ROS in transplanted murine hematopoietic stem cells24. In EPCs, TXNIP KD sensitized erythroblasts to peroxide cytotoxicity (Figure 6F). Endogenous ROS levels, however, were not significantly affected. As summarized in Figure 6G, these overall studies define novel positive roles for TXNIP during EPO-dependent human (pro)erythroblast formation, including possible effects on late stage erythroid development.
Figure 6. TXNIP knockdown studies in primary human erythroid progenitor cells define positive roles for TXNIP in EPC growth, survival and development.
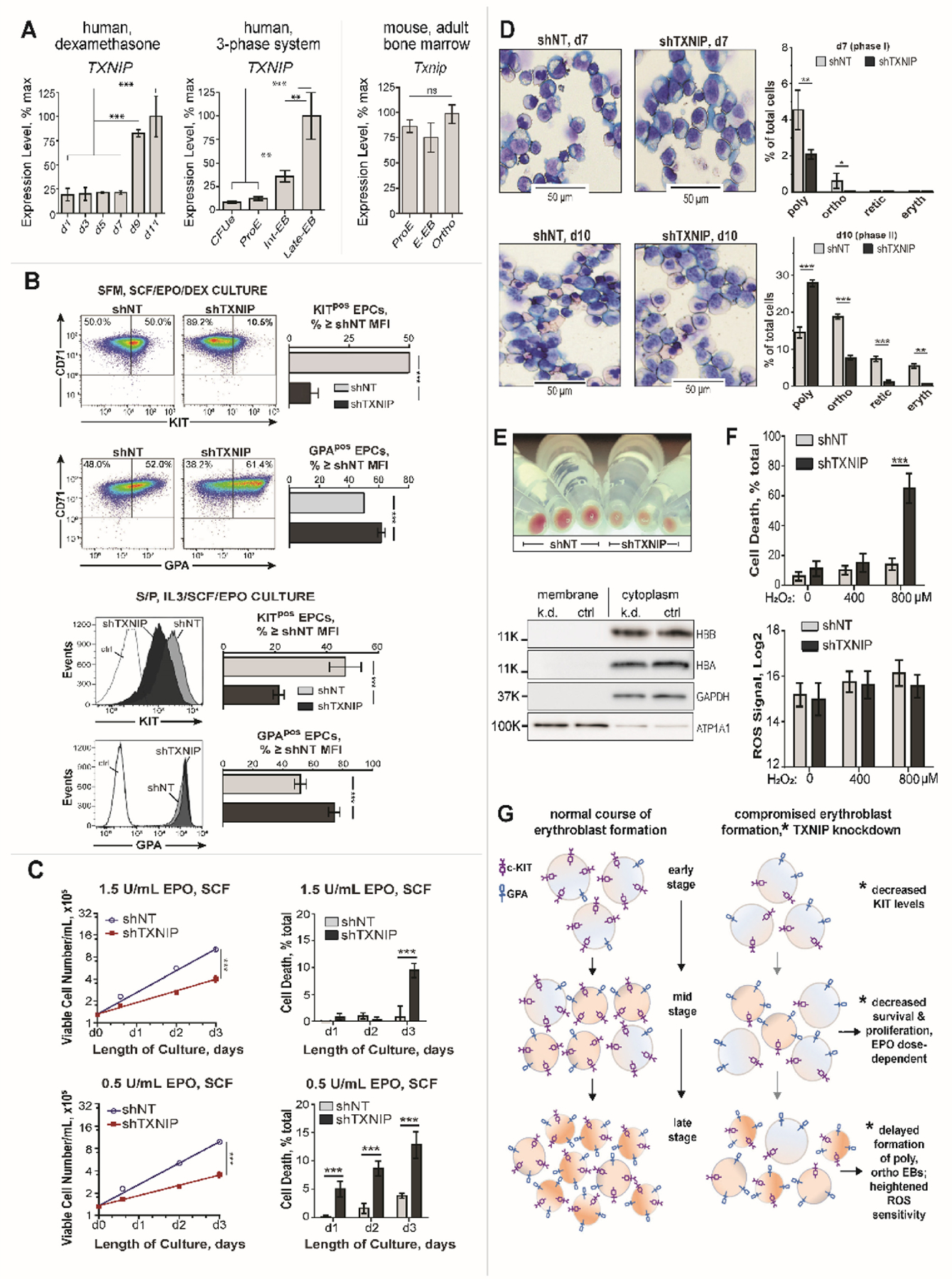
(A) TXNIP expression heightens during late stage erythroblast development: Analysis of transcriptome profiles for staged human EPCs from serum-free dexamethasone, and serum/plasma/IMDM cultures1 define 3− to 10− fold increases in TXNIP levels in late-stage poly- and ortho- chromatic erythroblasts (left and center panels). In developing murine EPCs, this late stage increase in Txnip is not observed (right panel). (B) In primary human EPCs, TXNIP KD sharply decreases c-KIT levels: CD34pos HSCs were pre-expanded (3.5 days; StemSpan, FLT3-L, TPO, IL3, SCF) and transduced with lentiviruses expressing shTXNIP or shNT (non-target shRNA). Lentiviruses were pre-calibrated for matched, ~50% transduction efficiencies (and single integrations). By d4 of erythroid culture in puromycin, ≥95% of cells were GFP positive (see Methods, and Supplemental Figure S-3). Transduced HSCs were cultured in dexamethasone serum-free medium (SFM, SCF/EPO/DEX; upper panels), or in IMDM medium with human serum and plasma (S/P, IL3/SCF/EPO; lower panels). By d7, and in each system, TXNIP KD led to marked decreases in cell surface KIT levels (flow cytometry), with modest increases in GPA also observed. Graphed data are median fluorescence intensities (MFIs) (mean values +/− SE, n=3). Results are representative of two independent experiments (for each culture system). (C) TXNIP supports EPO- dose dependent (pro)erythroblast growth, and survival: shTXNIP and shNT transduced and selected EPCs (S/P, IL3/SCF/EPO medium) were washed and replated at 1.5x105 cells/mL, with EPO at 1.5 U/mL or 0.5 U/mL, and SCF at 15 ng/mL. At the indicated time points, numbers of viable, and non-viable cells were determined (means +/− SE, n=3). shTXNIP KD significantly limited EPC growth, and survival (also, see Supplemental Figure S-4). (D) TXNIP knockdown limits the formation of late stage erythroblasts: EPCs transduced with shTXNIP or shNT lentiviruses were cultured in S/P, IL3/SCF/EPO medium (3 U/mL EPO). At d7 of culture (upper panels), late-stage sh-TXNIP erythroblasts (basophilic, polychromatic, orthochromatic) were observed in replicate cytospin preparations to be 3.3-fold under-represented vs sh-NT EPC controls (mean values +/− SD, n=3). At d10 of culture (lower panels), sh-TXNIP KD decreased numbers of orthochromatic erythroblasts, reticulocytes and terminal erythrocytes while increasing polychromatic erythroblast relative frequencies. (E) TXNIP KD delays erythroblast hemoglobinization: At d7 of culture, visual inspection of cells indicated decreased hemoglobinization of erythroblasts due to TXNIP KD (equal cell numbers collected). Levels of HBB and HBA globin chains, however, were closely matched in d7 shTXNIP and shNT erythroblasts (western blotting, lower panel). (F) TXNIP knockdown sensitizes erythroblasts to peroxide induced cell death: In shTXNIP erythroblasts at d7 of culture (vs control shNT cells), exposure to hydrogen peroxide (800 µM, 4h) resulted in a 4.6-fold increased cell death among TXNIP KD cells (EthD-1 assays). Graphed values are means +/− SD (n=3). As shown in the lower sub-panel, this was not associated with differences in endogenous ROS levels between shTXNIP vs shNT EPCs. (G) Summary model of the observed course of human primary erythroblast development in normal versus TXNIP knockdown conditions: Altered erythroblast formation due to TXNIP knockdown included decreased KIT levels, compromised survival and proliferation, delayed formation of late-stage erythroblasts, and increased ROS sensitivity.
‘Cellular Component’ annotation enrichment, with cytoskeletal proteins as a major new EPO- modulated target set
In assessing the subcellular localizations of EPO target phosphoproteins, GO enrichment analysis of ‘Cellular Component’ annotations identified nine significantly enriched categories, (Figure 7A, and Supplemental Table S-7), each consistent with cytokine receptor signaling. Within plasma membrane components (including ‘membrane raft’, ‘basolateral plasma membrane’, ‘cytoplasmic side of membrane’), 31 novel EPO targets were represented. Additional targets were associated with related annotation terms including ‘cell leading edge’, ‘axon’, and ‘synapse’, and more broadly cellular ‘midbody’, ‘nuclear part’, ‘vesicle’. Related to the above actin-modulating molecular adaptors, cytoskeletal proteins proved to be a major EPO- modulated cellular component EPO target set. Unexpectedly, this included the erythroid- specific cytoskeletal proteins alpha (SPTA) and beta (SPTB) spectrins, ADDUCIN-2 (ADD2) and GLYCOPHORIN-C (GYPC), together with the associated factors EZRIN (EZR) and CALMODULIN-1 (CALM1).
Figure 7. Coordinated EPO induced p-Y phosphorylation of networked erythroid cytoskeletal proteins.
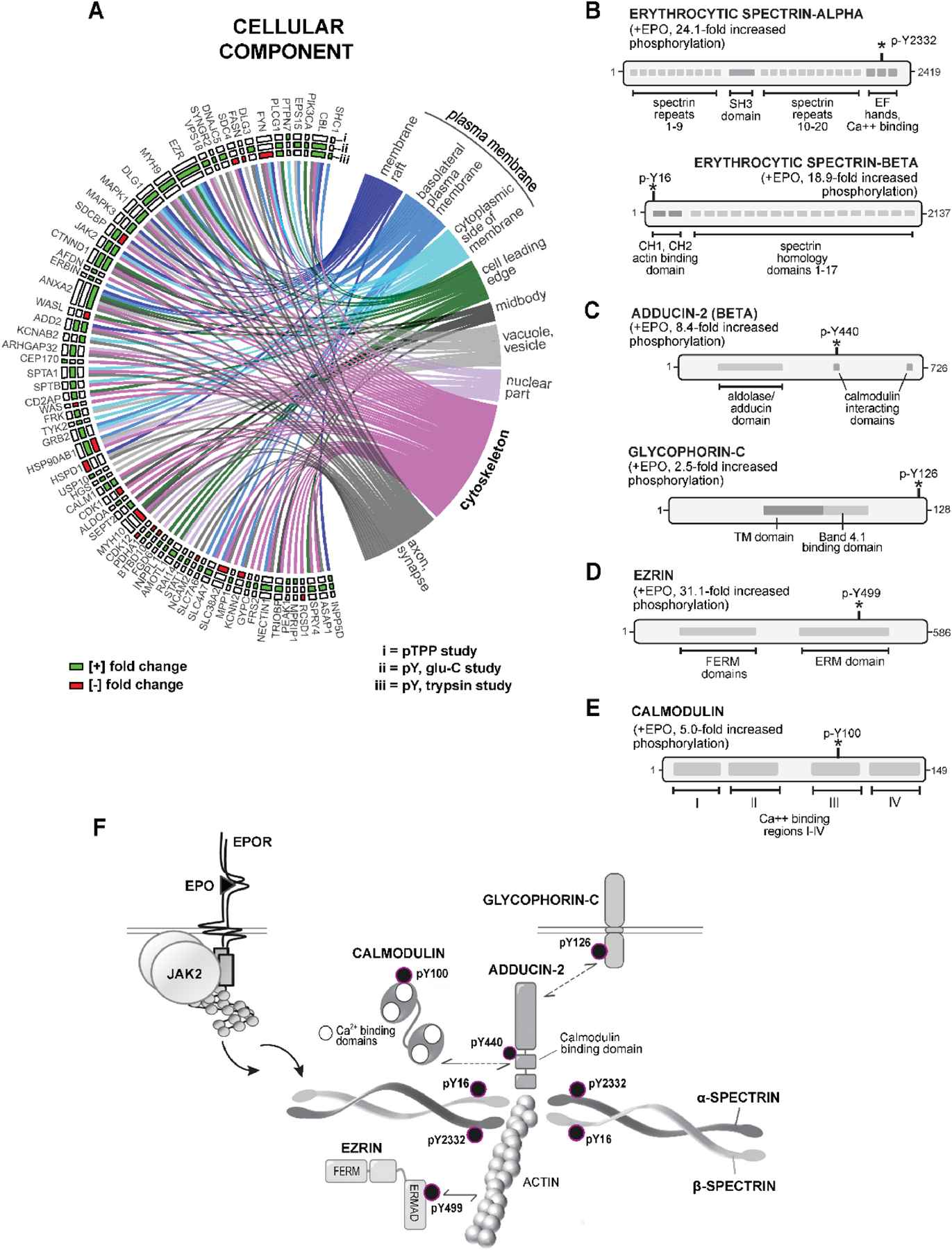
(A) Enrichment analysis of GO ‘cellular component’ terms highlights plasma membrane and cytoskeletal-associated EPO-regulated phospho-PTM targets: Chord plots illustrate high representation among enriched Cellular Component’ terms for EPO phospho-PTM targets including ‘plasma membrane’ and „membrane raft’ term targets (n=13), ‘basolateral plasma membrane’ term targets (n=9), ‘cytoplasmic side of membrane’ term targets (n=11) and ‘cytoskeleton’ term targets (n=19). (For overall ‘cellular component’ listings, see Supplemental Table S-6). (B-F) EPO induced phosphorylation of the inter-connected cytoskeletal proteins ALPHA and BETA ERYTHROCYTIC SPECTRIN (SPTA, SPTB), BETA-ADDUCIN (ADD2), and GLYCOPHORIN-C (GYPC) at novel phospho-sites within functional subdomains: B: In erythrocytic SPTA, EPO induced p-Y2332 phosphorylation within a C-terminal calcium binding and EF hand domain, and at SPTB p-Y16 in an N-terminal actin-binding and CH1,2 sub-domain. C: Within ADD2, EPO induced the phosphorylation of p-Y440 within a CALM interacting domain. In GYPC, EPO induced the phosphorylation of p-Y126 (C-terminal cytoplasmic domain). D,E: Within EZRIN (EZR), EPO regulated the phosphorylation of p-Y499 phosphorylation within an actin-interacting ERMAD subdomain. In CALM-1, EPO regulated the phosphorylation of p-Y100 within calcium binding domain III. F: Findings define a network of interacting cytoskeletal factors that are rapidly and coordinately regulated by EPO at unique novel p-Y sites within functionally important subdomains.
For these novel erythroid cytoskeletal EPO targets, EPO-induced phosphorylation occurred at novel p-Y sites (Figures 7B–7E). Each target and regulated p-Y site therefore were further characterized using Prosite, Phosphosite-Plus®, GeneCards, STRINGdb and NCBI mining resources. For erythroid spectrins, EPO induced the pY-2332 phosphorylation of SPTA within a distal C-terminal EF-hand domain, and of SPTB at p-Y16 within a proximal N-terminal region (Figure 7B) as domains that affect SPTA and SPTB heterodimerization46. For the SPTA/SPTB/actin complex partner, ADD247, EPO induced its phosphorylation at a p-Y440 site within a CALMODULIN interacting domain48 (Figure 7C), while also inducing the phosphorylation of GLYCOPHORIN-C at a distal cytoplasmic C-terminal p-Y126 site. For EZRIN and CALMODULIN, EPO induced EZRIN’s phosphorylation at Y499 within an ERM actin binding domain, and CALMODULIN’s phosphorylation at a Y100 residue essential for calcium binding, and CALM1’s activity49 (Figure 7D,E). EPO is therefore implicated in modulating the assembly of a late erythroid cytoskeleton through the coordinated p-Y phosphorylation of these new EPO targets at novel sites within interacting functional subdomains (Figure 7F).
DISCUSSION
Studies of hematopoietic growth factor signal transduction are often restricted in scope by limited collections of specific probes such as activation-state antibodies. PTM proteomic profiling via LC-MS/MS, however, can overcome this limitation, and uncover extended sets of signal transducers and modified phospho- residues and sequence motifs within target protein subdomains while also unambiguously identifying protein isoforms20,21. The present study sought to discover new mediators of EPO’s actions by analyzing target proteins modified in their phospho-PTMs following the ligation of EPOR. Our investigation further expanded target coverage by including three sets of phospho-PTM interrogations (p-Y trypsin, p-Y GluC, pTPP GluC), and by the use of EPO- dependent human proerythroblast UT7epo-E cells to provide sufficient cells (108 per sample) for effective phosphopeptide IPs, quantitative LC-MS/MS, and target validation. Overall, 121 novel EPO-modulated, phospho-modified target proteins are identified, with data attributes provided via a dedicated and upgraded Erythron-DB database [https://www.cbil.upenn.edu/ErythronDB_alpha/app].
Within a GO enrichment annotation category of ‘Molecular Function’, adaptor proteins were a major EPO signaling target set (24 targets, 18 novel) among which new connections to EPOR signaling were defined for the actin assemblage modifiers DLG1, DLG3, WAS, WASL and CD2AP. Scaffold proteins provide an example of how functional inferences can be generated via considerations of individual EPO target proteins and their PTM-modified subdomains. In both DLG-1 and −3, EPO induced the phosphorylation of a novel consensus pY motif with the C-terminal guanylate (pseudo) kinase domain (GKD). This includes p-Y760 in DLG1 and p-Y673 in DLG3, each of which map to a crystallographically defined subdomain that mediates the binding of DLG to interacting phosphoproteins (e.g., LGN, LGL, CASK)50. EPO regulation of these novel phosphosites therefore is implicated in modulating these functional interactions. In parallel, EPO regulated the phosphorylation of the actin nucleators WAS and WASL, each at a conserved motif within their PROFILIN binding domains51. PROFILIN directly binds G- and F- actin52, and WAS and WASL furthermore interact with DLGs53. Dynamic restructuring of actin networks therefore is implicated as a novel EPO effect that also may be exerted in non-hematopoietic EPO target cells potentially including EPORpos metastatic melanoma and breast carcinoma cells54,55.
Within a ‘Biological Process’ GO enrichment category for novel EPO- modulated target sets, attention focused primarily on metabolic regulators (see Figures 4–6). In erythroid progenitor cells (EPCs), EPO can stimulate glutamine transport56, ROS57, and iron transport58. The present studies are the first to reveal EPO modification of central metabolic regulators, including PDHA1 and ALDOA. PDHA1 (PDH E1a subunit)59 functions as a rate limiting decarboxylase to metabolize pyruvate to acetyl co-A for TCA utilization (and phosphorylation of PDHA1 at p-Y301 is known to inhibit pyruvate binding42). For ALDOA (mutated in hemolytic anemia)40, EPO induced its phosphorylation at p-Y364 within a C-terminal domain required for ALDOA tetramerization, and catalytic activity in converting fructose 1,6 bisphosphate to trioses41. EPO’s phospho-PTM modifications of PDHA1 and ALDOA therefore are predicted to enhance glycolysis and oxidative phosphorylation.
For TXNIP, the present focus on this redox and glycemic regulator in the context of EPO action were prompted by recent reports on EPO modulation of ROS in pro-erythroblasts60; unexpected actions of TXNIP in decreasing ROS damage in transplanted HSCs24; and effects of global Txnip KO on basophilic erythroblast formation in spleen26. In UT7epo-E cells, EPOR- mediated TXNIP phosphorylation was confirmed using Hematide, and a p-T349 site-specific antibody. EPO additionally rapidly heightened TXNIP levels (without affecting TXNIP transcript levels, data not shown). In HepG2 cells, 2-deoxyglucose induces AMPK- mediated TXNIP p-S308 phosphorylation and associated turnover, thereby suppressing glucose dependent TXNIP inhibition of GLUT-1 and −4(ref.23). Possible effects of novel EPO-induced p-T349 and p-S358 phosphorylation sites on TXNIP turnover therefore merit future investigation. Database analyses (e.g. PhosphoNET) point to JNK kinases as possible p-T349 and p-S358 phosphorylating kinases. EPO is known to activate JNK kinases, that can modulate EPC proliferation61.
In both UT7epo-E cells and primary EPC’s, TXNIP KD proved to markedly decrease c-KIT levels. c-KIT levels decrease during erythroid differentiation62, and attenuated SCF/KIT signaling can accelerate differentiation63. TXNIP KD, however, increased immature EPC representation. Mechanisms via which c-KIT expression levels depend upon TXNIP are presently undefined, but may contribute to reported effects of TXNIP KO on HSC transplantation24, which c-KIT is known to sharply affect64. TXNIP KD also compromised the EPO- dependent proliferation and survival of (pro)erythroblasts. The observed EPO dose- dependency of this effect additionally is consistent with a role for TXNIP as an EPO response mediator. Direct evidence, however, awaits studies of mutated EPO- regulated TXNIP p-T349 and p-S358 sites in a TXNIP-null background. In part, the limited formation of late stage erythroblasts due to TXNIP KD may reflect compromised survival, but this developmental effect was also observed at high EPO dosing. In addition, late stage erythroblasts did ultimately form at delayed time-points, consistent with delaying effects of TXNIP KD on their development. Despite this observed attenuated EPC development, globin levels were unaffected by TXNIP KD. TXNIP can also modulate ROS24, and in primary human EPCs, TXNIP KD was observed to significantly heighten damage due to ROS. A working model for TXNIP during EPO- dependent human erythropoiesis (see Figure 6G) therefore includes novel enforcement of c-KIT levels; supporting effects of EPO dose-dependent EPC proliferation and survival; and indicated roles in supporting late-stage medullary EPC development. In pancreatic beta cells, TXNIP hinders insulin expression via miRNA44 and ROS mechanisms65, and is an emerging therapeutic target for diabetics66. For TXNIP inhibitors, our findings therefore also serve to raise caution for possible anemia inducing effects.
Within a ‘Cellular Component’ GO enrichment category, cytoskeletal proteins were identified as a related major EPO- modulated phospho-PTM modified target set that unexpectedly includes the erythroid specific factors SPT-A, SPT-B, ADD2 and GYP-C. To our knowledge, this represents the first report of EPO targets directly involved in late erythroid differentiation. Based on emerging connections to Plasmodium invasion67–69, GYP-C and EZRIN are considered further. In particular, Plasmodium erythroid binding antigen EBA175 induces GYP-C phosphorylation70; EBA-140 directly binds to GYP-C71; and EZRIN is also a target of Plasmodium vivax induced phosphorylation69. Functionally, such phosphorylation events deform the erythroid cytoskeleton, and facilitate merozoite invasion69,70. For six related new EPO- modulated pY-modified cytoskeletal targets, including GYP-C and EZRIN, sites of EPO- induced phosphorylation occur within sub-domains involved in their co-assembly (see Figure 7). Their EPO induced site-specific p-Y phosphorylation therefore may similarly limit these interactions, and increase erythroblast deformability. During stress erythropoiesis, this may facilitate rapid erythroblast division69, and/or red cell egress from long bones72.
A final aspect of the present work that merits brief discussion is the insight into hematopoietin signaling and blood cell formation that PTM profiling can provide. This approach is being aggressively used in oncology73 and epigenetics74 and recently has been employed in certain hematological signal transduction systems75. In one example, reverse phase protein arrays were employed to monitor signatures for select phospho-STFs in polycythemia vera (PV) versus normal erythroblasts75. In PV cells, one interesting finding was a notable relative increase in activated p-STAT3 over p-STAT5. Presently, we similarly observed heightened activation of p-STAT3 over p-STAT5 in EPO-stimulated UT7epo-E cells, as originally derived from UT-7 megakaryoblastic leukemia cells27. As a second example, LC-MS/MS phospho-proteomics has been applied to gain new insight into thrombin effects on PAR1 signaling76. While LC-MS/MS is highly specific and informative, LC is a limiting bottleneck. Here, we update that we (and colleagues) are advancing a novel PTM affinity bead array which, as coupled to a scanning MALDI MS platform, promises to enable rapid multiplex profiling of customizable panels of PTM-target proteins (manuscript submitted).
Supplementary Material
Supplemental Table 1. EPO-regulated phospho-PTM target proteins, LC-MS/MS-identified, maximally EPO-modulated peptides, and specific phosphorylated sites.
Supplemental Table 2. p-Y Trypsin Study: EPO-regulated p-Y PTM targets, peptide sequences, and specific modulated phospho-sites.
Supplemental Table 3. p-Y GluC Study: EPO-regulated p-Y PTM targets, peptide sequences, and specific modulated phospho-sites.
Supplemental Table 4. p-TPP Glu-C Study: EPO-regulated p-TPP targets, peptide sequences, and specific modulated phospho-sites.
Supplemental Table 5. “Molecular Function” gene ontology categories significantly enriched among all EPOregulated phospho-PTM target proteins.
Supplemental Table 6. “Biological Process” gene ontology categories significantly enriched for all EPO-regulated phospho-PTM target proteins.
Supplemental Table 7. “Cellular Compartment” gene ontology categories significantly enriched among all EPO- regulated phospho-PTM target proteins.
HIGHLIGHTS.
Phospho-PTM proteomics reveals diverse new EPO- modulated signaling targets
Novel EPO-modulated adaptor proteins DLG-1,-3 & WAS-L,-P network as actin regulators
EPO modulates SPTA, SPTB, ADD2, GYPC at key cytoskeleton assembly domains
EPO modulates the metabolic factors ALDOA, PDH1, TXNIP at functional domains
TXNIP knockdown reveals proerythropoietic roles during erythroblast formation
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the following significant contributors: Dr. Boris Fehse laboratory (University of Hamburg) for contributing to exon sequencing and digital PCR analysis of possible leukemogenic driver mutations in UT7epo-E cells; Drs. Claudia Escher, Jakob Vowinckel and Karel Novy (Biognosys AG) for LC-MS/MS analysis of globin levels in UT7epo-E and parental UT7epo cells; Ashley Johnson and Ruth Asch (Maine Medical Center Research Institute) for western blot validations of phospho-PTM targets (AJ), and cell phenotype analyses of UT7epo-shTXNIP cells (RA); Drs. Jennifer Green (Pharmacyclics) and Peter Schatz (Companion Sciences, Inc.) for supporting efforts with phospho-PTM LC-MS/MS datasets; Mark Townley (UNH University Instrument Center) for confocal microscopy image analysis; Dr. Karen Miller (UNH, Durham) for contributions to data mining, illustrations and manuscript construction. Investigations were supported by NIH grant R01 HL044491 (DMW, PI), and by core facility support via University of New Hampshire’s Instrumentation Center (confocal imaging, EVOS cell imaging and flow cytometry; in association with NIH P20 GM113131); Maine Medical Center Research Institute’s Cell Phenotyping Core (in association with NIH P30 GM106391); and Fred Hutchinson Cooperative Center for Excellence in Hematology core facility for the provision of mobilized CD34pos human hematopoietic stem cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTERESTS: The authors declare no competing interests.
REFERENCES
- 1.Sathyanarayana P, Dev A, Pradeep A, Ufkin M, Licht JD, Wojchowski DM. Spry1 as a novel regulator of erythropoiesis, EPO/EPOR target, and suppressor of JAK2. Blood. 2012;119(23):5522–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gautier EF, Ducamp S, Leduc M, et al. Comprehensive Proteomic Analysis of Human Erythropoiesis. Cell Rep 2016;16(5):1470–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83(1):59–67. [DOI] [PubMed] [Google Scholar]
- 4.Biggar P, Kim GH. Treatment of renal anemia: Erythropoiesis stimulating agents and beyond. Kidney Res Clin Pract 2017;36(3):209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stauder R, Valent P, Theurl I. Anemia at older age: etiologies, clinical implications, and management. Blood. 2018;131(5):505–514. [DOI] [PubMed] [Google Scholar]
- 6.Aapro M, Beguin Y, Bokemeyer C, et al. Management of anaemia and iron deficiency in patients with cancer: ESMO Clinical Practice Guidelines. Ann Oncol 2018. [DOI] [PubMed] [Google Scholar]
- 7.Rainville N, Jachimowicz E, Wojchowski DM. Targeting EPO and EPO receptor pathways in anemia and dysregulated erythropoiesis. Expert Opin Ther Targets. 2016;20(3):287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koulnis M, Porpiglia E, Hidalgo D, Socolovsky M. Erythropoiesis: from molecular pathways to system properties. Adv Exp Med Biol 2014;844:37–58. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal R. Mechanisms and mediators of hypertension induced by erythropoietin and related molecules. Nephrol Dial Transplant. 2018;33(10):1690–1698. [DOI] [PubMed] [Google Scholar]
- 10.Vannucchi AM, Harrison CN. Emerging treatments for classical myeloproliferative neoplasms. Blood. 2017;129(6):693–703. [DOI] [PubMed] [Google Scholar]
- 11.Meyer SC. Mechanisms of Resistance to JAK2 Inhibitors in Myeloproliferative Neoplasms. Hematol Oncol Clin North Am 2017;31(4):627–642. [DOI] [PubMed] [Google Scholar]
- 12.Tefferi A Primary myelofibrosis: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol 2016;91(12):1262–1271. [DOI] [PubMed] [Google Scholar]
- 13.Perreault AA, Venters BJ. Integrative view on how erythropoietin signaling controls transcription patterns in erythroid cells. Curr Opin Hematol 2018;25(3):189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/−5b−/−mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98(2):181–191. [DOI] [PubMed] [Google Scholar]
- 15.Koulnis M, Porpiglia E, Porpiglia PA, et al. Contrasting dynamic responses in vivo of the Bcl-xL and Bim erythropoietic survival pathways. Blood. 2012;119(5):1228–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Socolovsky M, Lodish HF, Daley GQ. Control of hematopoietic differentiation: lack of specificity in signaling by cytokine receptors. Proc Natl Acad Sci U S A. 1998;95(12):6573–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grover A, Mancini E, Moore S, et al. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J Exp Med 2014;211(2):181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dev A, Byrne SM, Verma R, Ashton-Rickardt PG, Wojchowski DM. Erythropoietin-directed erythropoiesis depends on serpin inhibition of erythroblast lysosomal cathepsins. J Exp Med 2013;210(2):225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014;46(7):678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elia AE, Boardman AP, Wang DC, et al. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol Cell. 2015;59(5):867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aebersold R, Agar JN, Amster IJ, et al. How many human proteoforms are there? Nat Chem Biol 2018;14(3):206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Gene Ontology C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 2019;47(D1):D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu N, Zheng B, Shaywitz A, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49(6):1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung H, Kim MJ, Kim DO, et al. TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab 2013;18(1):75–85. [DOI] [PubMed] [Google Scholar]
- 25.Alhawiti NM, Al Mahri S, Aziz MA, Malik SS, Mohammad S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr Drug Targets. 2017;18(9):1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gasiorek JJ, Mikhael M, Garcia-Santos D, Hui ST, Ponka P, Blank V. Thioredoxin-interacting protein regulates the differentiation of murine erythroid precursors. Exp Hematol 2015;43(5):393–403 e392. [DOI] [PubMed] [Google Scholar]
- 27.Komatsu N, Yamamoto M, Fujita H, et al. Establishment and characterization of an erythropoietin-dependent subline, UT-7/Epo, derived from human leukemia cell line, UT-7. Blood. 1993;82(2):456–464. [PubMed] [Google Scholar]
- 28.Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667–679. [DOI] [PubMed] [Google Scholar]
- 29.He TC, Zhuang H, Jiang N, Waterfield MD, Wojchowski DM. Association of the p85 regulatory subunit of phosphatidylinositol 3-kinase with an essential erythropoietin receptor subdomain. Blood. 1993;82(12):3530–3538. [PubMed] [Google Scholar]
- 30.Stephens R, Lim K, Portela M, Kvansakul M, Humbert PO, Richardson HE. The Scribble Cell Polarity Module in the Regulation of Cell Signaling in Tissue Development and Tumorigenesis. J Mol Biol 2018. [DOI] [PubMed] [Google Scholar]
- 31.Biswas A, Shouval DS, Griffith A, et al. WASP-mediated regulation of anti-inflammatory macrophages is IL-10 dependent and is critical for intestinal homeostasis. Nat Commun 2018;9(1):1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu C, Bai X, Wu J, et al. N-wasp is essential for the negative regulation of B cell receptor signaling. PLoS Biol 2013;11(11):e1001704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alekhina O, Burstein E, Billadeau DD. Cellular functions of WASP family proteins at a glance. J Cell Sci 2017;130(14):2235–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaddanapudi S, Altintas MM, Kistler AD, et al. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J Clin Invest 2011;121(10):3965–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu X, Sicard R, Jiang X, et al. HGAL localization to cell membrane regulates B-cell receptor signaling. Blood. 2015;125(4):649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato N, Kawasoe Y, Williams H, et al. Sensing and Processing of DNA Interstrand Crosslinks by the Mismatch Repair Pathway. Cell Rep 2017;21(5):1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raney KD, Byrd AK, Aarattuthodiyil S. Structure and Mechanisms of SF1 DNA Helicases. Adv Exp Med Biol 2013;767:17–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diouf B, Cheng Q, Krynetskaia NF, et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med 2011;17(10):1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Konopleva M, Thall PF, Yi CA, et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 2015;100(7):927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao DC, Tolan DR, Murray MF, et al. Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase, ALDOA(Arg303X/Cys338Tyr). Blood. 2004;103(6):2401–2403. [DOI] [PubMed] [Google Scholar]
- 41.Esposito G, Vitagliano L, Costanzo P, et al. Human aldolase A natural mutants: relationship between flexibility of the C-terminal region and enzyme function. Biochem J 2004;380(Pt 1):51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan J, Kang HB, Shan C, et al. Tyr-301 phosphorylation inhibits pyruvate dehydrogenase by blocking substrate binding and promotes the Warburg effect. J Biol Chem 2014;289(38):26533–26541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kovarova N, Cizkova Vrbacka A, Pecina P, et al. Adaptation of respiratory chain biogenesis to cytochrome c oxidase deficiency caused by SURF1 gene mutations. Biochim Biophys Acta 2012;1822(7):1114–1124. [DOI] [PubMed] [Google Scholar]
- 44.Xu G, Chen J, Jing G, Shalev A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med 2013;19(9):1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shalev A Minireview: Thioredoxin-interacting protein: regulation and function in the pancreatic beta-cell. Mol Endocrinol 2014;28(8):1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Machnicka B, Czogalla A, Hryniewicz-Jankowska A, et al. Spectrins: a structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim Biophys Acta 2014;1838(2):620–634. [DOI] [PubMed] [Google Scholar]
- 47.Robledo RF, Ciciotte SL, Gwynn B, et al. Targeted deletion of alpha-adducin results in absent beta- and gamma-adducin, compensated hemolytic anemia, and lethal hydrocephalus in mice. Blood. 2008;112(10):4298–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scaramuzzino DA, Morrow JS. Calmodulin-binding domain of recombinant erythrocyte beta-adducin. Proc Natl Acad Sci U S A. 1993;90(8):3398–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stateva SR, Salas V, Benaim G, Menendez M, Solis D, Villalobo A. Characterization of phospho-(tyrosine)-mimetic calmodulin mutants. PLoS One. 2015;10(4):e0120798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mori S, Tezuka Y, Arakawa A, et al. Crystal structure of the guanylate kinase domain from discs large homolog 1 (DLG1/SAP97). Biochem Biophys Res Commun 2013;435(3):334–338. [DOI] [PubMed] [Google Scholar]
- 51.Gaucher JF, Mauge C, Didry D, Guichard B, Renault L, Carlier MF. Interactions of isolated C-terminal fragments of neural Wiskott-Aldrich syndrome protein (N-WASP) with actin and Arp2/3 complex. J Biol Chem 2012;287(41):34646–34659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alkam D, Feldman EZ, Singh A, Kiaei M. Profilin1 biology and its mutation, actin(g) in disease. Cell Mol Life Sci 2017;74(6):967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silva O, Crocetti J, Humphries LA, Burkhardt JK, Miceli MC. Discs Large Homolog 1 Splice Variants Regulate p38-Dependent and -Independent Effector Functions in CD8+ T Cells. PLoS One. 2015;10(7):e0133353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Julius A, Desai A, Yung RL. Recombinant human erythropoietin stimulates melanoma tumor growth through activation of initiation factor eIF4E. Oncotarget 2017;8(18):30317–30327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan KK, Matchett KB, Coulter JA, et al. Erythropoietin drives breast cancer progression by activation of its receptor EPOR. Oncotarget 2017;8(24):38251–38263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oburoglu L, Tardito S, Fritz V, et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell. 2014;15(2):169–184. [DOI] [PubMed] [Google Scholar]
- 57.Luo ST, Zhang DM, Qin Q, et al. The Promotion of Erythropoiesis via the Regulation of Reactive Oxygen Species by Lactic Acid. Sci Rep 2017;7:38105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pantopoulos K TfR2 links iron metabolism and erythropoiesis. Blood. 2015;125(7):1055–1056. [DOI] [PubMed] [Google Scholar]
- 59.Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 2009;389(2):157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao B, Mei Y, Yang J, Ji P. Erythropoietin-regulated oxidative stress negatively affects enucleation during terminal erythropoiesis. Exp Hematol 2016;44(10):975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jacobs-Helber SM, Sawyer ST. Jun N-terminal kinase promotes proliferation of immature erythroid cells and erythropoietin-dependent cell lines. Blood. 2004;104(3):696–703. [DOI] [PubMed] [Google Scholar]
- 62.Spinello I, Quaranta MT, Pasquini L, et al. PLZF-mediated control on c-kit expression in CD34(+) cells and early erythropoiesis. Oncogene 2009;28(23):2276–2288. [DOI] [PubMed] [Google Scholar]
- 63.Deshpande S, Bosbach B, Yozgat Y, Park CY, Moore MA, Besmer P. KIT receptor gain-of-function in hematopoiesis enhances stem cell self-renewal and promotes progenitor cell expansion. Stem Cells. 2013;31(8):1683–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Czechowicz A, Palchaudhuri R, Scheck A, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun 2019;10(1):617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rani S, Mehta JP, Barron N, et al. Decreasing Txnip mRNA and protein levels in pancreatic MIN6 cells reduces reactive oxygen species and restores glucose regulated insulin secretion. Cell Physiol Biochem 2010;25(6):667–674. [DOI] [PubMed] [Google Scholar]
- 66.Thielen L, Shalev A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr Opin Endocrinol Diabetes Obes 2018;25(2):75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilder JA, Hewett EK, Gansner ME. Molecular evolution of GYPC: evidence for recent structural innovation and positive selection in humans. Mol Biol Evol 2009;26(12):2679–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maier AG, Duraisingh MT, Reeder JC, et al. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat Med 2003;9(1):87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Panichakul T, Ponnikorn S, Roytrakul S, et al. Plasmodium vivax inhibits erythroid cell growth through altered phosphorylation of the cytoskeletal protein ezrin. Malar J 2015;14:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tolia NH, Enemark EJ, Sim BK, Joshua-Tor L. Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell. 2005;122(2):183–193. [DOI] [PubMed] [Google Scholar]
- 71.Lobo CA, Rodriguez M, Reid M, Lustigman S. Glycophorin C is the receptor for the Plasmodium falciparum erythrocyte binding ligand PfEBP-2 (baebl). Blood. 2003;101(11):4628–4631. [DOI] [PubMed] [Google Scholar]
- 72.Zeman K, Engelhard H, Sackmann E. Bending undulations and elasticity of the erythrocyte membrane: effects of cell shape and membrane organization. Eur Biophys J 1990;18(4):203–219. [DOI] [PubMed] [Google Scholar]
- 73.Radzisheuskaya A, Shliaha PV, Grinev V, et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol 2019;26(11):999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gilan O, Lam EY, Becher I, et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat Struct Mol Biol 2016;23(7):673–681. [DOI] [PubMed] [Google Scholar]
- 75.Hricik T, Federici G, Zeuner A, et al. Transcriptomic and phospho-proteomic analyzes of erythroblasts expanded in vitro from normal donors and from patients with polycythemia vera. Am J Hematol 2013;88(9):723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zimman A, Titz B, Komisopoulou E, Biswas S, Graeber TG, Podrez EA. Phosphoproteomic analysis of platelets activated by pro-thrombotic oxidized phospholipids and thrombin. PLoS One. 2014;9(1):e84488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. EPO-regulated phospho-PTM target proteins, LC-MS/MS-identified, maximally EPO-modulated peptides, and specific phosphorylated sites.
Supplemental Table 2. p-Y Trypsin Study: EPO-regulated p-Y PTM targets, peptide sequences, and specific modulated phospho-sites.
Supplemental Table 3. p-Y GluC Study: EPO-regulated p-Y PTM targets, peptide sequences, and specific modulated phospho-sites.
Supplemental Table 4. p-TPP Glu-C Study: EPO-regulated p-TPP targets, peptide sequences, and specific modulated phospho-sites.
Supplemental Table 5. “Molecular Function” gene ontology categories significantly enriched among all EPOregulated phospho-PTM target proteins.
Supplemental Table 6. “Biological Process” gene ontology categories significantly enriched for all EPO-regulated phospho-PTM target proteins.
Supplemental Table 7. “Cellular Compartment” gene ontology categories significantly enriched among all EPO- regulated phospho-PTM target proteins.