

Abstract
The CREB-binding protein (CBP) pathway plays an important role in transcription and activity of acetyltransferase that acetylates lysine residues of histones and non-histone proteins. In the present study, we hypothesized that genetic variants in the CBP pathway genes played a role in survival (OS) of non-small cell lung cancer (NSCLC). We tested this hypothesis using the genotyping data from the genome-wide association study (GWAS) dataset from the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. In the single locus analysis, we evaluated associations between 13 176 (1107 genotyped and 12 069 imputed) single-nucleotide polymorphisms (SNPs) in 72 genes and survival of 1185 NSCLC patients. The identified 106 significant SNPs in the discovery were further validated in additional genotyping data from another GWAS dataset of 984 NSCLC patients in the Harvard Lung Cancer Susceptibility Study. The combined results of two datasets showed that two independent, potentially functional SNPs (i.e., HDAC2 rs13213007G>A and PPARGC1A rs60571065T>A) were significantly associated with NSCLC overall survival, with a combined hazards ratio (HR) of 1.26 [95% confidence interval (CI) = 1.09–1.45 and P=0.002] and 1.23 (1.04–1.47 and P=0.017), respectively. Furthermore, we performed an expression quantitative trait loci analysis and found that the survival-associated HDAC2 rs13213007A allele (GA+AA), but not PPARGC1A rs60571065A allele (TA+AA), was significantly associated with increased mRNA expression levels of HDAC2 in 373 lymphoblastoid cell lines. These results indicate that HDAC2 rs13213007A allele is a potential predictor of NSCLC survival, likely by altering HDAC2 expression.
Keywords: Non-small cell lung cancer, Genetic susceptibility, CREB-binding protein pathway, Single Nucleotide Polymorphisms
1. INTRODUCTION
Lung cancer has been the leading cause of cancer-related mortality since 1985, with over a million deaths each year worldwide [1]. In 2018, it is estimated that there were approximately 234,030 new cases and 154,050 lung cancer deaths in the United States (https://seer.cancer.gov/statfacts/html/lungb.html). Non-small cell lung cancer (NSCLC) is the most common histological type of lung cancer, accounting for approximately 80–85% of all lung cancer diagnosis [2]. Over the past few decades, advances in surgical procedures, chemo-radiotherapy, targeted therapy, and more recently immunotherapy have contributed to modest improvements in the survival of lung cancer [3]; yet, the five-year survival rate is still at an underwhelming 15–20%. Previous studies have found that molecular and genetic factors may play an important role in lung cancer progression and outcomes [4]; thus, it is important to search for additional molecular markers that may predict the survival of NSCLC patients who would benefits from individualized therapies.
Single nucleotide polymorphisms (SNPs) can affect gene expression and functions and thus may be associated with susceptibility to cancer development and progression [5]. To date, several genome-wide association studies (GWASs) of lung cancer have discovered SNPs that are associated with cancer risk, but few have found markers predictive of outcomes of NSCLC patients. The vast majority of SNPs under GWAS investigation did not reach the stringent genome-wide level of significance, and most of those identified as significant SNPs are located within non-coding regions of the genome, making such discoveries fundamentally difficult to comprehend and harder to reveal the underlying mechanisms. As a result, it is recommended that research strategies in the post-GWAS era should include “discovery, expansion, and replication”, “biological studies”, and “epidemiologic studies” (https://epi.grants.cancer.gov/gameon/#funded) [6]. By using a hypothesis-driven approach of the pathway gene analysis pooling together GWAS datasets, one may have a better chance with an improved statistical power to identify novel loci with minor yet detectable effects, to examine functional consequences of such novel loci, and thus, to unravel possible mechanisms underlying the observed associations.
The CREB-binding protein (CBP) pathway regulates the post-translational modification with the activity of acetyltransferase that acetylates lysine residues of histones and non-histone proteins, such as p53 [7]. The CBP pathway is also involved in basic cellular functions, including cell growth, differentiation, DNA repair, and apoptosis [8, 9]. Studies have revealed oncogenic roles of the CBP pathway genes; for example, it has been shown that CBP is highly expressed in lung cancer cells and tumor tissues, upregulating hTERT expression and promoting tumor growth in human lung cancer cells [10]. Furthermore, CBP overexpression by members of the activator protein-1 (AP-1) family and downregulation of the retinoid acid receptor β might promote lung tumor proliferation [11]. Intriguingly, CBP exerts its actions mainly by acetylation of histones and other regulatory proteins [12], and could be fused to MOZ and several other gene products in acute myeloid leukemia by chromosomal translocations [13]. In addition, mice monoallelic for the CBP gene may induce multilineage defects in hematopoietic differentiation with an elevated chance of hematologic malignancies [14].
To date, the role of SNPs in the CBP pathway genes and their functionality related to tumor growth and progression are still unknown, and there are few reports about the SNPs in CBP pathway genes and its related gene-set in NSCLC prognosis. In the present study, by using two publicly available GWAS datasets, we performed a CBP pathway gene-set analysis to evaluate the association between genetic variants in this gene-set and the survival of NSCLC patients.
2. MATERIALS AND METHODS
2.1. Study populations
As shown in the study flowchart (Figure 1), the discovery phase included 1,185 NSCLC patients obtained from the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial, after application and access approval from the National Cancer Institute (NCI). The PLCO is an NCI funded multicenter randomized trial of screening for cancer from ten medical centers in the United States between 1993 and 2011 [15]. The screening trial enrolled 77,500 men and 77,500 women aged 55–74. All individuals were randomized to either the intervention arm with screening or the control arm with standard care [15]. The PLCO trial collected blood specimens from the first screening visit and gathered extensive information about each individual, including smoking history, family history of cancer and demographic information [16]. All participants were followed for at least 13 years after the enrollment. Genomic DNA extracted from the blood samples was genotyped in a genome-wide association study (GWAS) with Illumina HumanHap240Sv1.0, Human-Hap300v1.1 and HumanHap550v3.0 (dbGaP accession: phs000093.v2.p2 and phs000336.v1.p1) [17, 18]. In 1,187 Caucasian NSCLC patients from the PLCO trial, two with missing follow-up information were excluded. Therefore, the eligible subsets of the PLCO lung cancer dataset for survival analysis included 1185 NSCLC patients, whose clinicopathological variables and genotype data were available. Tumor staging was determined according to the fifth edition American Joint Committee on Cancer staging system. The institutional review boards of each participating institution approved the PLCO trial and the use of biological specimens for further research, and all subjects signed a written informed consent permitting the research represented here [15].
FIGURE 1.
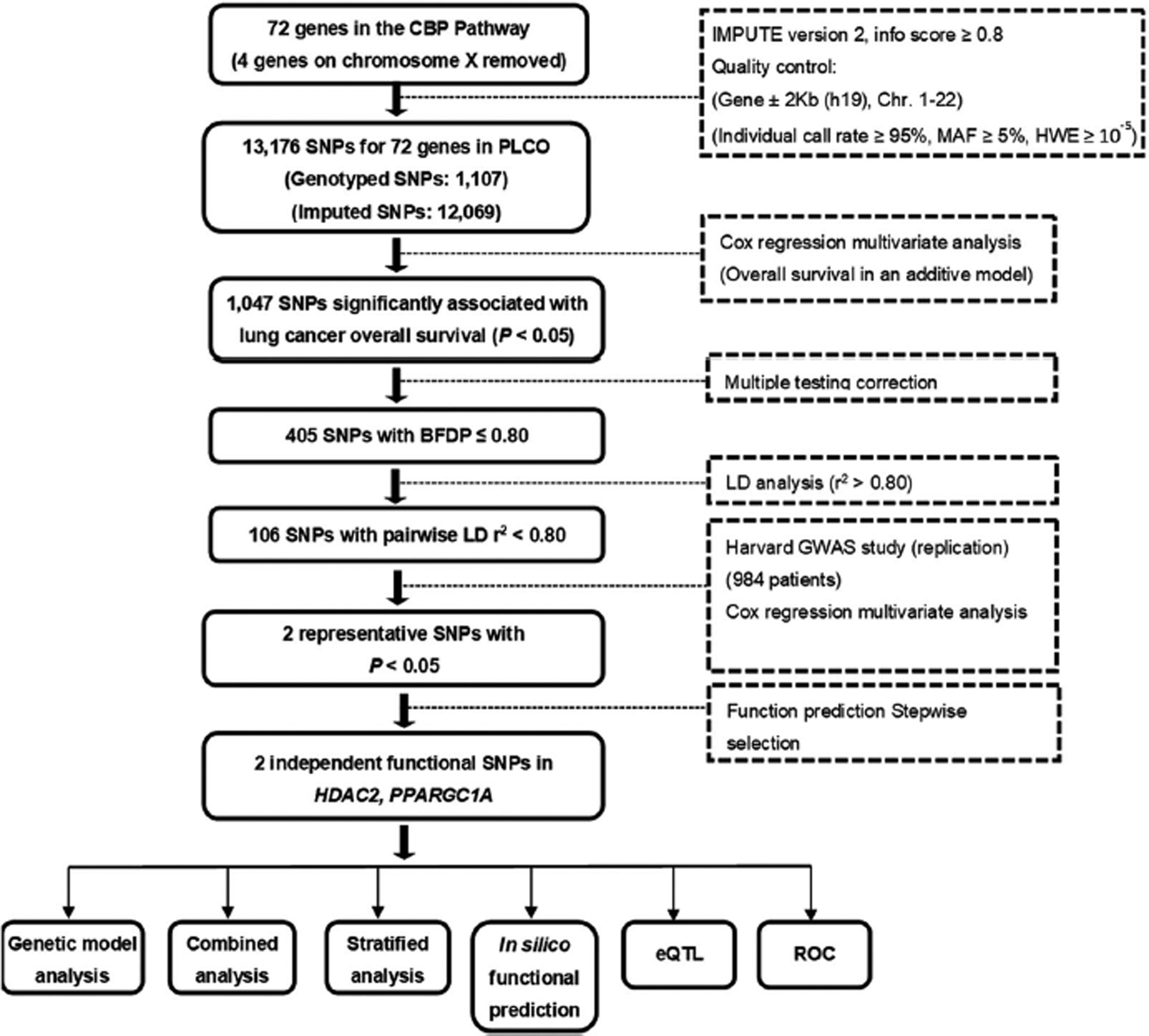
Study workflow chart. BFDP, Bayesian false-discovery probability; eQTL, expression quantitative trait loci; Harvard Study, Harvard Lung Cancer Susceptibility Study; PLCO, Prostate, Lung, Colorectal and Ovarian cancer trial; ROC, receiver operating characteristic; SNP, single-nucleotide polymorphism
The validation phase used the GWAS dataset from the Harvard Lung Cancer Susceptibility (HLCS) Study with 984 histology-confirmed Caucasian NSCLC patients. The histological classification of the tumors were recorded by two staff pulmonary pathologists at the Massachusetts General Hospital. The time of blood collection was within 1–4 weeks of the diagnosis for each patient. DNA was extracted from blood samples by using the Auto Pure Large Sample Nucleic Acid Purification System (QIAGEN Company, Venlo, Limburg, Netherlands). Genotype data were obtained by using Illumina Humanhap610-Quad arrays, and imputation was performed by using MaCH based on the 1000 Genomes project [19]. Details of the participants in the Harvard study were described previously [20]. The comparison of the characteristics between the PLCO and the Harvard study is presented in the Supplemental Table 1.
2.2. Gene and SNP selection
The genes or gene-set involved in the CBP pathway were selected through the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb/index.jsp), by the keyword “CBP”. After removing duplicated genes and excluding genes in the X chromosome, 72 genes remained as candidate genes for further analysis (Supplemental Table 2). We first performed imputation for the 72 genes plus the 500-kb flanking buffer regions by using IMPUTE2 and the 1000 Genomes Project data (phase 3) [19]. After imputation, we extracted all the SNPs in these genes and within their ±2 kb flanking regions according to the following criteria: a minor allele frequency ≥ 0.05, a genotyping rate ≥ 95%, and a Hardy-Weinberg equilibrium P value ≥ 1×10−5. As a result, 1107 genotyped SNPs were chosen from the PLCO GWAS dataset with an additional 12,069 SNPs as a result of imputation.
2.3. Statistical analyses
The follow-up time in both PLCO and HLCS datasets was from the diagnosis of lung cancer to the last follow-up or time of death. OS was the primary endpoint of the present study, and disease-specific survival (DSS) of lung cancer was also examined. In the single-locus analysis, multivariate Cox proportional hazards regression analysis was used to evaluate associations between each of the SNPs and survival (in an additive genetic model) with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery and the top four principal components of the PLCO dataset using the GenABEL package of R software [21]. We first used the false discovery rate (FDR) to correct multiple testing with a threshold of 0.2.
Since the majority of SNPs were imputed and there was a high level of linkage disequilibrium (LD), we used the Bayesian false discovery probability (BFDP) with a cutoff value of 0.8 for multiple test corrections as recommended [22]. We assigned a prior probability of 0.10 to detect an HR of 2.0 for an association with variant genotypes or minor alleles of the SNPs with P < 0.05. Then, we chose SNPs for the HLCS GWAS dataset validation, which satisfied the following conditions: SNPs passed the threshold of BFDP ≤ 0.8, and tagging SNPs based on their LD. To identify independent SNPs, we included the validated SNPs in a multivariate stepwise Cox model with adjustment for demographic characteristics, previously published SNPs, clinical variables and the top four principal components of the genotyping data in the PLCO dataset. Combined-analysis of discovery and validation datasets was also performed to provide summary results. The fixed-effects model was applied If the Cochran’s Q-test P value > 0.100 and the heterogeneity statistic (I2) < 50%; otherwise, the random-effects model was employed. Kaplan-Meier curves were used to estimate the survival associated with the genotypes. The combination of risk or unfavorable genotypes was used to estimate the cumulative effects of the identified SNPs.
Functions of the validated SNPs were further predicted by HaploReg23 , SNPinfo24 and RegulomeDB25. The criteria for functional SNPs were: 1) the SNPs were associated with survival in both the PLCO trial and HLCS study and 2) genotypes of the SNPs were associated with mRNA expression of their genes as shown in one of these databases. Expression quantitative trait loci (eQTL) analysis was further performed to assess correlations between SNPs and mRNA expression levels by using linear regression analysis with the R (version 3.5.0) software. mRNA expression data of genes were obtained from lymphoblastoid cell lines derived from the 373 European descendants included in the 1000 Genomes Project19, and from the whole blood and normal lung tissues in the genotype-tissue expression (GTEx) project26. Using the data from the Cancer Genome Atlas (TCGA) database (dbGaP Study Accession: phs000178.v9.p8), we examined the differences in mRNA expression levels between paired tumor tissues and adjacent normal tissues by the paired t test27. Next, we detected the association of mRNA expression and OS through Kaplan-Meier (KM) analysis (n=1,926) (http://kmplot.com/analysis/index.php?p=service&cancer=lung). Finally, receiver operating characteristic (ROC) curves were constructed and time-dependent ROC analysis were performed to examine the prediction accuracy of models integrating both clinical and genetic variables on NSCLC survival with the “timeROC” package in R (version 3.5.0)28. Unless specified, all statistical analyses were performed using the SAS software (version 9.4; SAS Institute, Cary, NC, USA).
3. RESULTS
3.1. Associations between SNPs in the CBP pathway gene-set and NSCLC survival in both PLCO and HLCS datasets
The study flowchart is shown in Figure 1, and basic characteristics of 1,185 NSCLC patients have been described previously29 (also see Supplemental Table 3). In the PLCO discovery dataset with an additive genetic model, the multivariate Cox model with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery and the first four principal components (Supplemental Table 4) identified 405 SNPs that were significantly associated with NSCLC survival after multiple test correction by BFDP ≤ 0.8 but not by False Discovery Rate ≤ 0.2 – the results are summarized in a Manhattan plot (Figure 2A). Potentially functional SNPs were further validated by the HLCS dataset. As a result, two SNPs in two different genes (i.e., rs13213007 in HDAC2 and rs60571065 in PPARGC1A) were validated (Table 1), both of which were imputed. Further combined analysis of these SNPs of the two datasets showed a worse survival associated with the rs13213007 A and rs60571065 A alleles (Padjusted = 0.0003 and 0.002, respectively), and no heterogeneity between the two studies were observed (Table 1).
FIGURE 2.
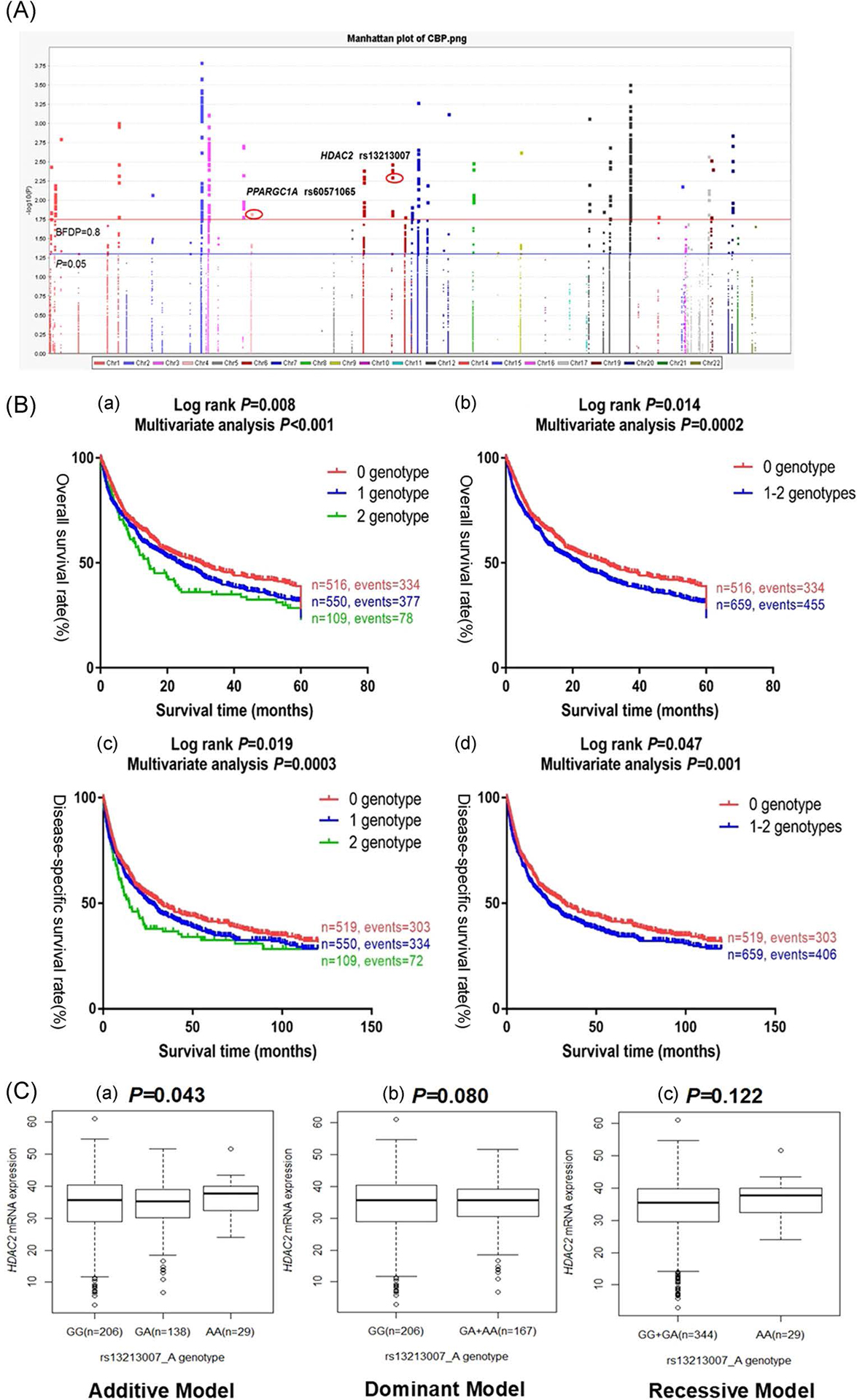
Functional and survival-associated SNPs. A. Manhattan plot of 13,176 SNPs of the CBP Pathway genes in the PLCO trial. The statistical values across the autosomes for associations between 13,176 SNPs and overall survival of patients with NSCLC are plotted as −log10 P values. The blue horizontal line indicates P = 0.05 and the red line indicates BFDP = 0.80. Abbreviations: NSCLC, non-small cell lung cancer; PLCO, Prostate, Lung, Colorectal and Ovarian cancer screening trial; BFDP, Bayesian false-discovery probability. B. Kaplan-Meier (KM) survival curves for NSCLC patients of two validated SNPs and combined unfavorable genotypes in the PLCO trial. (a) by 0, 1 and 2 unfavorable genotypes (log-rank test for trend: P) in OS, (b) by 0 and 1–2 unfavorable genotypes (log-rank test and multivariate analysis: P) in OS from the PLCO trial, (c) by 0, 1 and 2 unfavorable genotypes (log-rank test for trend: P) in DSS, and (d) by 0 and 1–2 unfavorable genotypes (log-rank test and multivariate analysis: P) in DSS from the PLCO trial. Abbreviations: NSCLC, non-small cell lung cancer; PLCO, Prostate, Lung, Colorectal and Ovarian cancer screening trial; OS, overall survival; DSS, disease-specific survival. C. eQTL analysis of HDAC2 rs13213007 and PPARGC1A rs60571065 genotypes and corresponding gene mRNA expression. All the data were from the 1000 Genome Project dataset. (a) rs13213007 additive model (P=0.043); (b) rs13213007 dominant model (P=0.080); and (c) rs13213007 recessive model (P=0.122) but no significant results for rs60571065 (P=0.556, 0.743 and 0.339, figures not shown). Abbreviations: eQTL, expression quantitative trait loci.
TABLE 1.
Combined analysis of the two unfavorable SNPs in PLCO and HLCS datasets
| SNP | Allelea | Gene | PLCO (n=1,185) | HLCS (n=984) | Combined analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EAF | HR (95% CI)b | Pb | BFDP | EAF | HR (95% CI)c | Pc | HR (95% CI)d | Pd | Phete | I 2 | |||
| rs13213007 | GG>A | HDAC2 | 0.26 | 1.18 (1.05–1.32) | 0.004 | 0.58 | 0.29 | 1.14 (1.02–1.29) | 0.022 | 1.16 (1.07–1.26) | 0.0003 | 0.758 | 0 |
| rs60571065 | TT>A | PPARGC1A | 0.11 | 1.22 (1.04–1.43) | 0.015 | 0.77 | 0.10 | 1.21 (1.00–1.46) | 0.050 | 1.22 (1.08–1.37) | 0.002 | 0.948 | 0 |
Abbreviations: PLCO, Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial; HLCS, Harvard Lung Cancer Susceptibility Study
Major > minor allele;
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, PC1, PC2, PC3 and PC4;
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, PC1, PC2 and PC3;
in fixed-effect model;
P value for heterogeneity by Cochrane’s Q test.
3.2. Identification of independent SNPs associated with survival of NSCLC in the PLCO dataset
Because the HLCS study provided only the summary data, we used only the PLCO dataset to identify independent SNPs after full adjustment for other co-variables. To identify potential functional SNPs associated with NSCLC survival with three online bioinformatics tools (i.e., RegulomeDB25, SNPinfo24 and HaploReg23), we found that both of the validated SNPs were located in the intron regions with some functions. In the RegulomeDB25, rs60571065 had a score of 5, indicating a potential transcriptional factor binding or DNase peak, while rs13213007 had no data (Supplemental Table 5); both of the validated SNPs had no obvious function based on SNPinfo24 or HaploReg23.
The two validated SNPs (i.e., HDAC2 rs13213007 and PPARGC1A rs60571065) were also included into a multivariate stepwise Cox model with adjustments for demographic and clinical variables as well as previously published SNPs and the first four principal components available in the PLCO dataset. As a result, both of the two SNPs remained independently associated with NSCLC survival (Table 2, also see Supplemental Table 6); a regional association plot of each SNP is shown in Supplemental Figure 1. In the PLCO dataset, patients with rs13213007 A or rs60571065 A alleles had an increased risk of death (Ptrend = 0.004 or 0.015, Table 3). Compared with the reference genotype in a dominant genetic model, HDAC2 rs13213007 GA+AA and PPARGC1A rs60571065 TA+AA were associated with a significantly increased risk of death (HR=1.26, 95% CI=1.09–1.45 and P=0.002 for rs13213007 GA+AA; and HR=1.23, 95% CI=1.04–1.47 and P=0.017 for rs60571065 TA+AA).
TABLE 2.
Predictors of OS obtained from stepwise Cox regression analysis in the PLCO dataset
| Variablesa | Category | Frequencyb | HR (95% CI) | P |
|---|---|---|---|---|
| Age | Continuous | 1,174 | 1.04 (1.02–1.05) | <0.001 |
| Sex | Male | 694 | 1.00 | |
| Female | 480 | 0.82 (0.70–0.95) | 0.010 | |
| Smoking status | Never | 114 | 1.00 | |
| Current | 416 | 1.78(1.32–2.39) | <0.001 | |
| Former | 644 | 1.78(1.35–2.35) | <0.001 | |
| Histology | Adenocarcinoma | 575 | 1.00 | |
| Squamous cell | 283 | 1.29 (1.07–1.56) | 0.008 | |
| others | 316 | 1.34 (1.13–1.59) | <0.001 | |
| Tumor stage | I-IIIA | 654 | 1.00 | |
| IIIB-IV | 520 | 2.97 (2.45–3.61) | <0.001 | |
| Chemotherapy | No | 638 | 1.00 | |
| Yes | 536 | 0.56 (0.46–0.66) | <0.001 | |
| Radiotherapy | No | 760 | 1.00 | |
| Yes | 414 | 0.96 (0.86–1.13) | 0.617 | |
| Surgery | No | 634 | 1.00 | |
| Yes | 540 | 0.19 (0.15–0.25) | <0.001 | |
| HDAC2 rs13213007 G>Ac | GG/GA/AA | 648/450/76 | 1.16 (1.03–1.30) | 0.012 |
| PPARGC1A rs60571065 T>Ac | TT/TA/AA | 925/231/18 | 1.21 (1.04–1.42) | 0.017 |
Abbreviations: CI = Confidence interval; HR = Hazards ratio; PLCO = Prostate, Lung, Colorectal and Ovarian cancer trial; OS = Overall survival; SNP, Single Nucleotide Polymorphism
Stepwise analysis included age, sex, smoking status, tumor stage, tumor histology, chemotherapy, radiotherapy, surgery, top four principal components and eight published SNPs and two new validated SNPs (HDAC2 rs13213007 and PPARGC1A rs60571065 in an additive model);
Ten observations missing of clinical variables (two of tumor stage and eight of chemotherapy/ radiotherapy/ surgery); one missing for TLE1 rs199731120 C>CA (published SNP); 1,174 patients remained for the stepwise analysis;
Two SNPs has been identified in the present study.
TABLE 3.
Associations of the two validated SNPs in the CBP pathway with OS of NSCLC in the PLCO dataset
| OS Univariate analysis | OS Multivariable analysisa | |||||||
|---|---|---|---|---|---|---|---|---|
| Frequency | Frequency | |||||||
| Genotype | (n=1,185) | Death (%) | HR (95% CI) | P | (n=1,175) | Death (%) | HR (95% CI) | P |
| HDAC2 rs13213007 G>A | ||||||||
| GG | 656 | 431 (65.7) | 1.00 | 648 | 424 (65.4) | 1.00 | ||
| GA | 453 | 311 (68.7) | 1.19 (1.03–1.38) | 0.018 | 451 | 309 (68.5) | 1.26(1.09–1.47) | 0.002 |
| AA | 76 | 56 (73.7) | 1.28(0.97–1.70) | 0.079 | 76 | 56 (73.7) | 1.24 (0.93–1.64) | 0.145 |
| Trend | 0.008 | 0.004 | ||||||
| GA+AA | 529 | 367 (69.4) | 1.21(1.05–1.39) | 0.008 | 527 | 365 (69.3) | 1.26 (1.09–1.45) | 0.002 |
| PPARGC1A rs60571065 T>A | ||||||||
| TT | 942 | 629 (66.8) | 1.00 | 934 | 621 (66.5) | 1.00 | ||
| TA | 225 | 159(70.7) | 1.11 (0.93–1.32) | 0.235 | 223 | 158 (70.9) | 1.22 (1.03–1.46) | 0.025 |
| AA | 18 | 10 (55.6) | 0.78 (0.42–1.46) | 0.441 | 18 | 10 (55.6) | 1.42 (0.76–2.67) | 0.276 |
| Trend | 0.555 | 0.015 | ||||||
| TA+AA | 243 | 169 (69.6) | 1.08 (0.91–1.29) | 0.351 | 241 | 168(69.7) | 1.23 (1.04–1.47) | 0.017 |
| NUGb | ||||||||
| 0 | 522 | 340 | 1.00 | 516 | 334 | 1.00 | ||
| 1 | 554 | 380 (68.6) | 1.17 (1.01–1.35) | 0.039 | 550 | 377 (68.6) | 1.28 (1.10–1.48) | 0.002 |
| 2 | 109 | 78 (71.6) | 1.32 (1.03–1.69) | 0.027 | 109 | 78 (71.6) | 1.50 (1.17–1.93) | 0.001 |
| Trend | 0.008 | <0.001 | ||||||
| 0 | 522 | 340 (65.1) | 1.00 | 516 | 334 (64.7) | 1.00 | ||
| 1–2 | 663 | 458 (69.1) | 1.19 (1.03–1.37) | 0.015 | 659 | 455 (69.0) | 1.31 (1.14–1.52) | 0.0002 |
| Phenotype missingc | N/A | 10 | ||||||
Abbreviations: SNPs = Single nucleotide polymorphisms; OS = Overall survival; NSCLC = Non-small cell lung cancer; PLCO = Prostate, Lung, Colorectal and Ovarian cancer trial; NPG = Number of protective genotypes; HR = Hazards ratio; 95%CI = 95%Confidence interval; N/A = Not applicable.
Multivariate Cox hazards regression analyses were adjusted for age, sex, smoking, stage, histology, chemotherapy, radiotherapy, surgery, and top four principal components;
NUG=number of unfavorable genotypes; NUG were HDAC2 rs11606640 GA+AA and PPARGC1A rs62053220 TA+AA;
Two observations missing of tumor stage and eight observations missing of chemotherapy/radiotherapy/surgery in PLCO dataset.
3.3. Combined and stratified analysis of the two independent and functional SNPs in the PLCO dataset
To provide a better estimation of the hazards for survival, we combined the risk genotypes (i.e., rs13213007 GA+AA and rs60571065 TA+AA) into a genetic score, which was used to divide all NSCLC patients into three groups. As shown in Table 3, in the multivariate analysis, an increased genetic score was associated with worse survival (trend test: P<0.001). After dichotomizing the genetic score, we re-grouped all the patients into a low-risk group (0 risk score) and a high-risk group (1–2 risk scores); patients in the high-risk group had significantly poorer survival compared with the low-risk group (HR=1.31, 95% CI=1.14–1.52 and P=0.0002). Kaplan-Meier survival curves were generated to depict the associations between risk genotypes and NSCLC OS (Figure 2Ba and 2Bb) and DSS (Figure 2Bc and 2Bd).
To assess the ability of risk genotypes to predict outcomes, we compared the area under the ROC curve (AUC) from the model with clinical variables to that from the model including both clinical variables and risk genotypes. The addition of risk genotypes to the prediction model of five-year OS increased the AUC from 87.0% to 87.27% (P=0.375, Supplemental Figure 2a); similarly, the addition of risk genotypes to the prediction model of five-year DSS increased the AUC from 86.7% to 87.01% (P=0.391, Supplemental Figure 2b). Finally, the time-dependent AUC curve was generated to quantify the ability of risk genotypes to predict NSCLC survival through the entire follow-up period (Supplemental Figure 2c and 2d).
We further analyzed the effect of combined risk genotypes on NSCLC OS, and whether it was modified by age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy and surgery (Table 4). The results showed no significant interactions (P>0.05) except with smoking status (P=0.002). Hence, as shown in Supplemental Tables 7 and 8, we further assessed the associations of HDAC2 rs13213007 and PPARGC1A rs60571065 with OS of NSCLC in three subgroups of never, current and former smokers and found that cigarette smoking and HDAC2 rs13213007 had an interactive effect on NSCLC OS, where the highest risk was observed amongst current smokers. Additionally, never smokers were at a higher risk than former smokers.
TABLE 4.
Stratified multivariate analyses for association between unfavorable genotypes and OS in NSCLC patients in the PLCO trial
| Characteristics | No. of 0 risk genotypes | No. of 1–2 risk genotypes | Multivariate analysisa | Pinterb | ||||
|---|---|---|---|---|---|---|---|---|
| All | Death (%) | All | Death (%) | HR (95% CI) | P | |||
| Age (years) | ||||||||
| ≤ 71 | 277 | 168 (60.7) | 357 | 251 (64.7) | 1.31 (1.07–1.61) | 0.009 | ||
| > 71 | 239 | 166 (69.5) | 302 | 224 (74.2) | 1.35 (1.10–1.65) | 0.005 | 0.697 | |
| Sex | ||||||||
| Male | 273 | 190 (69.6) | 422 | 315 (74.6) | 1.33 (1.11–1.60) | 0.002 | ||
| Female | 243 | 144 (59.3) | 237 | 140 (59.1) | 1.32 (1.03–1.69) | 0.029 | 0.906 | |
| Smoking status | ||||||||
| Never | 57 | 28 (49.1) | 57 | 34 (59.7) | 1.44 (0.81–2.57) | 0.212 | ||
| Current | 182 | 110 (60.4) | 235 | 156 (66.4) | 1.72 (1.33–2.23) | <0.001 | ||
| Former | 277 | 196 (70.8) | 367 | 265 (72.2) | 1.08 (0.90–1.31) | 0.409 | 0.002* | |
| Histology | ||||||||
| Adenocarcinoma | 259 | 149 (57.5) | 316 | 197 (62.3) | 1.36 (1.09–1.70) | 0.007 | ||
| Squamous | 129 | 87(67.4) | 155 | 104 (67.1) | 1.32 (0.98–1.79) | 0.069 | ||
| Others | 128 | 98 (76.6) | 188 | 154 (81.9) | 1.43 (1.10–1.87) | 0.009 | 0.780 | |
| Tumor stage | ||||||||
| I-IIB | 287 | 133 (46.3) | 367 | 181 (49.3) | 1.23 (0.98–1.55) | 0.077 | ||
| IIIA-IV | 229 | 201 (87.8) | 292 | 274 (93.8) | 1.36 (1.12–1.64) | 0.002 | 0.524 | |
| Chemotherapy | ||||||||
| No | 272 | 147 (54.0) | 366 | 219 (59.8) | 1.33 (1.07–1.65) | 0.009 | ||
| Yes | 244 | 187 (76.6) | 293 | 236 (80.6) | 1.22 (1.00–1.50) | 0.049 | 0.847 | |
| Radiotherapy | ||||||||
| No | 323 | 179 (55.4) | 438 | 270 (61.6) | 1.35 (1.11–1.64) | 0.002 | ||
| Yes | 193 | 155 (80.3) | 221 | 185 (83.7) | 1.25 (1.00–1.56) | 0.055 | 0.397 | |
| Surgery | ||||||||
| No | 283 | 242 (85.5) | 352 | 323 (91.8) | 1.34 (1.13–1.59) | 0.0008 | ||
| Yes | 233 | 92 (39.5) | 307 | 132 (43.0) | 1.33 (1.01–1.76) | 0.041 | 0.771 | |
| Phenotype missingc | 10 | |||||||
Abbreviations: CI = Confidence interval; HR = Hazards ratio; NSCLC = Non-small cell lung cancer; OS = Overall survival; PLCO = Prostate, Lung, Colorectal and Ovarian cancer trial.
Adjusted for age, sex, stage, histology, smoking status, chemotherapy, radiotherapy, surgery, PC1, PC2, PC3 and PC4;
Pinter: P-value for interaction analysis between characteristic and number of protective genotypes;
Two observations missing of tumor stage and eight observations missing of chemotherapy/radiotherapy/surgery in PLCO dataset.
3.4. in silico functional validation
Experimental data from the ENCODE project25 (Supplemental Figure 3a and 3b), revealed the two SNPs (i.e., HDAC2 rs13213007 and PPARGC1A rs60571065) to be located in a DNase I hypersensitive site, where the DNase hypersensitivity and histone modification H3K4 acetylation indicated some strong signals for active enhancer and promoter functions. The evidence from the DNase cluster and transcription factor CHIP-seq data25 predicted that rs60571065 is located in the Barhl2/ Msx-3/ Sox1 motif as shown by the position weight matrix (Supplemental Figure 3c, 3d and 3e), and that the minor allele may affect the binding activity to have an impact on the transcription factors.
In addition, we performed eQTL analysis to correlate genotypes of the SNPs and mRNA expression levels using data of the 373 European descendants available from the 1000 Genomes Project19. Only the HDAC2 rs13213007 A allele (GA+AA) showed a significant correlation with increased mRNA expression levels of the gene (P=0.043, Figure 2Ca), while this was not the case for the PPARGC1A rs60571065 A allele; however, in the whole blood and lung tissue data of the GTEx project, both SNPs were not associated with expression levels of HDAC2 and PPARGC1A (Supplemental Table 9). Taken together, these findings suggest that HDAC2 rs13213007A allele, but not PPARGC1A rs60571065A allele, may influence HDAC2 gene expression at the transcription level.
Finally, to find the expression of these genes in NSCLC, we compared the mRNA expression levels of these genes in 111 paired NSCLC tumor and adjacent normal tissue samples obtained from the TCGA database. Expression levels of HDAC2 were higher in the tumor tissues (P<0.001), compared with the adjacent normal tissues (Supplemental Figure 4a, 4b and 4c), and the higher expression levels were associated with a poorer NSCLC OS (Supplemental Figure 5a). On the other hand, the expression levels of PPARGC1A were lower in the tumor tissues (P<0.001), compared with the adjacent normal tissues (Supplemental Figure 4d, 4e and 4f), and the higher expression levels were associated with a better NSCLC OS30 (Supplemental Figure 5b).
3.5. Mutation analysis
We accessed the mutation status of HDAC2 and PPARGC1A in lung tumor tissues by using the public database of the cBioPortal for Cancer Genomics. As shown in Supplemental Figure 6a and 6b, HDAC2 had a low somatic mutation rate in NSCLC (1.05%, 12/1144) in the 2016 TCGA study31, LUAD (1.64%, 3/183 and 1.24%, 7/566) in the Broad32 and TCGA PanCan studies27, respectively; and LUSC (0.41%, 2/487) in the TCGA PanCan study. In contrast, PPARGC1A had a relative higher somatic mutation rate in NSCLC (3.06%, 35/1144) in the TCGA 2016 study31, LUAD (3.28%, 6/183; 3.89%, 22/566; and 3.89%, 22/566) in the Broad32 and TCGA PanCan studies27, respectively; and LUSC (3.37%, 6/178; 2.26%, 11/487; and 0.98%, 5/511) in the TCGA pub, TCGA PanCan and TCGA27 studies, respectively. Given the rarity of mutations in these two genes, our results suggest that the functional SNPs in HDAC2 may play a relatively important role in the dysregulation of mRNA expression in tumor tissues, whereas the mutations might also play a role in altered functions and expression of PPARGC1A in addition to the causal SNPs.
4. DISCUSSION
In the present study, we found two genetic variants (i.e., HDAC2 rs13213007 G>A and PPARGC1A rs60571065 T>A) in the CBP pathway gene-set to be significantly associated with survival of patients with NSCLC in both the PLCO trial and the HLCS GWAS datasets. The results suggest that patients with risk genotypes (i.e., HDAC2 rs13213007 GA+AA or PPARGC1A rs60571065 TA+AA) had a worse prognosis. Furthermore, HDAC2 rs13213007 G>A appears to influence HDAC2 mRNA expression, which makes this SNP-associated risk of death biologically plausible.
Histone deacetylases (HDACs) function to deacetylate the ε-amino group of lysine residues of both histone and non-histone substrates7. HDACs are grouped into class I–IV enzymes33, i.e., Class I contains HDAC1, 2, 3 and 8, class II HDAC4, 5, 6, 7, 9 and 10, and class IV HDAC, whereas Class III enzymes contains the SIRT deacetylases34. Studies suggest that HDAC2 is crucial for embryonic development, growth regulation, and affects cytokine signaling relevant for immune responses35 and is often overexpressed in lung cancer and other tumors36. Both published in vitro and in vivo experiments have indicated that the aberrant regulation of HDAC2 may confer an oncogenic potential for lung cancer cells and hepatocellular carcinoma cells by deregulating apoptosis and expression of cell cycle proteins37, while overexpression of HDAC2 has been found to be associated with a shorter relapse-free survival after radical prostatectomy in prostate cancer38. Furthermore, the upregulation of HDAC2 may enhance proliferation of gastric cancer cells by deregulating cell cycle proteins39, and HDAC2 may also play a fundamental role in the Myc-mediated oncogenesis40. These are consistent with our findings that HDAC2 expression was increased in lung cancer tissues, compared with normal tissues in the TCGA dataset. However, the detailed mechanisms of HDAC2 SNPs underlying the observed death-risk associations warrant further investigations.
Few studies have reported the role of HDAC2 SNPs in cancer outcomes. One study found that the rs11391 SNP in the 3’-UTR of HDAC2 was a potential genetic marker of poor prognosis for NSCLC41, while another study identified several functional SNPs in HDAC2 and evaluated their associations with clinical outcomes in hepatocellular carcinoma, but no significant associations were found42. In the present study, we showed that the HDAC2 rs13213007 A allele was associated with a poorer survival of NSCLC, likely due to its association with an increase in mRNA expression levels of the gene; moreover, HDAC2 mRNA expression levels were found to be higher in tumor tissues, and higher expression levels were also associated with a worse survival in NSCLC. According to the ENCODE project database25, HDAC2 rs13213007 is located in a DNase I hypersensitive site with considerable levels of histone modification H3K4 acetylation, which may lead to an enhanced transcriptional activity. Therefore, it is likely that that HDAC2 might act as an oncogene in NSCLC.
Similarly, there are also few studies that have reported the role of PPARGC1A SNPs in cancer outcomes. Previous studies have reported that abnormalities in cell metabolism are associated with tumorigenesis where mitochondria are key regulators43. PPARGC1A is a known regulator of mitochondrial biogenesis and is a multiple-function transcriptional coactivator that has been noted to be associated with various human diseases, including type II diabetes mellitus44, coronary disease45 and others. One study utilized ChIP-seq to obtain PPARGC1A binding sites across the genome in hepatoma cells HepG2 and the subsequent conserved-motif analysis showed that the majority of the PPARGC1A binding sites were located in multiple regulatory factor binding regions46. To date, however, the studies on the expression and function as well as oncogenic roles of PPARGC1A in lung cancer are inconsistent. For example, one report found that PPARGC1A might be an oncogene47, while the other demonstrated that it might be a suppressor gene48.
In the TCGA database, we found that mRNA expression levels of PPARGC1A were higher in normal lung tissues than in tumor tissues (n=111), while its lower expression levels were associated with a worse survival in NSCLC. However, PPARGC1A was also reported to act as an oncogene, because its expression levels were upregulated in tumor tissues (n=3), facilitating lung cancer metastasis49. It is possible that the expression levels of PPARGC1A in lung cancer tissues may be affected by other factors, such as an imbalanced activation by risk genotypes, which may cause abnormal expression of PPARGC1A, in addition to mutations. Based on the ENCODE Project data, PPARGC1A rs60571065 is located in a DNase I hypersensitive site, indicating its potential function to modify gene expression. Clearly, additional studies are warranted to investigate possible mechanisms underlying PPARGC1A rs60571065-associated death risk of NSCLC.
In the clinical setting for NSCLC patients with EGFR mutations, treatment with epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) has resulted in an increased survival rate in comparison with the standard chemotherapy50. Because EGFR-TKIs have been used as the first-line treatment in the application of concurrent or sequential chemo-radiotherapy regimens, different treatment options should be incorporated into the phenotype analysis51. Recently, the 3rd-generation EGFR-TKIs (EGFR-TKI3rd) have displayed remarkable efficacy against the T790M-related resistance in mutation-positive NSCLC52. Despite dramatic initial responses, a genetically driven resistance to the targeted therapy eventually emerged; thus, limiting their effectiveness in prolonged treatments52. Some reports suggest that it is due in part to differences in co-occurring genetic alterations, such as mutations of TP53 or PTEN53, but few reports have addressed the roles of SNPs in other oncogenes. In the present study, we found that risk genotypes of HDAC2 rs13213007 promoted its mRNA expression levels, suggesting that the risk genotypes may have an impact on T790M-related resistance in NSCLC, which may provide a clue for individualized therapy54.
While tobacco smoking is a major risk factor for lung cancer, smoking behavior could enhance the adverse genetic effect of the variants by increasing c-Jun expression in lung cancer55. In the present study, we found that smoking might increase the risk of death associated with HDAC2 rs13213007 SNP in patients with NSCLC. It is likely that smoking could affect the function of epigenetics genes or the H3K4 acetylation activity associated with HDAC2, a similar phenomenon seen on other cancers, including cancers of the pancreas, stomach, bladder, head and neck, and the colorectum56.
Although we observed associations between genetic variants in two CBP pathway genes and NSCLS survival along with some functional evidence, the exact molecular mechanisms are still unclear. Further functional experiments are required to corroborate the present findings. Additionally, since both the discovery and validation datasets used in the present study were from Caucasian populations, the results may not be generalizable to other ethnic populations. Although the sample size of PLCO was relatively large, the number of patients in each subgroup were still relatively small, which might have reduced the statistical power to detect a small effect in one particular subpopulation. Furthermore, since only a few clinical factors were available for additional analysis, other covariates, such as performance status and treatments, were not available for further adjustments. Additionally, it is worth noting that the detailed genotype and phenotype data from the HLCS study were not accessible for us to perform additional stratified analysis in a larger sample size.
In conclusion, two independent functional SNPs (i.e., HDAC2 rs13213007 G>A and PPARGC1A rs60571065 T>A) were found to be significantly associated with NSCLC survival in both the PLCO trial and the HLCS GWAS datasets. The combined analysis revealed that the two SNPs had a significant association with survival and that patients with more risk genotypes had a worse prognosis, possibly due to the progression acceleration and metastatic effects of overexpressed HDAC2. Knowing the characteristic overexpression of HDAC2 in cancer, the discovery of the novel genetic variant of HDAC2 rs13213007 may serve to showcase additional insights for potential therapeutic capabilities of HDAC inhibitors and in cancer treatment. The findings in the present study also provides a solid foundation for further functional studies to identify molecular mechanisms underlying the observed death-risk associations in the progression of NSCLC.
Supplementary Material
ACKNOWLEDGEMENTS
Carolyn Glass, Yu Chen Zhao, Danwen Qian, Hongliang Liu and Sipeng Shen participated in the data collection and analysis. Patricia G. Moorman, Li Su, and David C. Christiani participated in the design of the study. Dongfang Tang, Sheng Luo, Edward F. Patz, Wen Gao, and Qingyi Wei participated in the writing of the manuscript and the interpretation of the data. All authors read and approved the final manuscript. The authors thank all the participants of the PLCO Cancer Screening Trial. The authors also thank the National Cancer Institute for providing the access to the data collected by the PLCO trial. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by the National Cancer Institute. The authors would also like to acknowledge dbGaP repository for providing cancer genotyping datasets. The accession numbers for the datasets of lung cancer are phs000336.v1.p1 and phs000093.v2.p2. A list of contributing investigators and funding agencies for those studies can be found in the Supplemental Data. Qingyi Wei was supported by the V Foundation for Cancer Research (D2017-19) and partly supported by the Duke Cancer Institute as part of the P30 Cancer Center Support Grant (Grant ID: NIH/NCI CA014236). Dongfang Tang was supported by the short-term international training program for PhD from Fudan University, P.R. China. The Harvard Lung Cancer Susceptibility Study was supported by NIH grants 5U01CA209414, CA092824, CA074386 and CA090578 to David C. Christiani.
FUNDING
This work was supported by the National Institute of Health [CA090578, CA074386, CA092824, 5U01CA209414]; the short-term international training program for PhD training from Fudan University, P.R. China; the Duke Cancer Institute as part of the P30 Cancer Center Support Grant [NIH/NCI CA014236]; the V Foundation for Cancer Research [D2017-19].
ABBREVIATIONS
- NSCLC
Non-small cell lung cancer
- SNP
single nucleotide polymorphism
- CBP
CREB-binding protein
- GWAS
Genome-Wide Association Study
- PLCO
the Prostate, Lung, Colorectal and Ovarian cancer screening trial
- HLCS
Harvard Lung Cancer Susceptibility study
- OS
overall survival
- DSS
disease-specific survival
- LD
linkage disequilibrium
- BFDP
Bayesian false discovery probability
- eQTL
expression quantitative trait loci
- TCGA
the Cancer Genome Atlas
- ROC
receiver operating characteristic
- HR
hazards ratio
- CI
confidence interval
- AUC
area under the curve
Footnotes
CONFILICT OF INTERSET
The authors declare no conflict of interest.
SUPPLEMENTARY MATERIAL
Supplementary data can be found at the Molecular Carcinogenesis online.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during the present study are included in this article.
REFERENCES
- 1.Zarredar H, Ansarin K, Baradaran B, Ahdi Khosroshahi S, Farajnia S. Potential Molecular Targets in the Treatment of Lung Cancer Using siRNA Technology. Cancer Invest. 2018; 36: 37–58. [DOI] [PubMed] [Google Scholar]
- 2.Houston KA, Mitchell KA, King J, White A, Ryan BM. Histologic Lung Cancer Incidence Rates and Trends Vary by Race/Ethnicity and Residential County. J Thorac Oncol. 2018; 13: 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnfield PC, Ellis PM. Second-Line Treatment of Non-Small Cell Lung Cancer: New Developments for Tumours Not Harbouring Targetable Oncogenic Driver Mutations. Drugs. 2016; 76: 1321–36. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Sheu CC, Ye Y, et al. Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. The Lancet Oncology. 2010; 11: 321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosell R, Wei J. Single nucleotide polymorphisms (SNPs) in non-small cell lung cancer (NSCLC) patients. The oncologist. 2012; 17: 1484–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li MJ, Sham PC, Wang J. Genetic variant representation, annotation and prioritization in the post-GWAS era. Cell research. 2012; 22: 1505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scolnick DM, Chehab NH, Stavridi ES, et al. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer research. 1997; 57: 3693–6. [PubMed] [Google Scholar]
- 8.Du Y, Teng X, Wang N, et al. NF-kappaB and enhancer-binding CREB protein scaffolded by CREB-binding protein (CBP)/p300 proteins regulate CD59 protein expression to protect cells from complement attack. The Journal of biological chemistry. 2014; 289: 2711–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Z, Feng R, Li J, et al. Nuclear translocation of CBP/p300-interacting protein CITED1 induced by parathyroid hormone requires serine phosphorylation at position 79 in its 63–84 domain. Cellular signalling. 2014; 26: 2436–45. [DOI] [PubMed] [Google Scholar]
- 10.Guo W, Lu J, Dai M, et al. Transcriptional coactivator CBP upregulates hTERT expression and tumor growth and predicts poor prognosis in human lung cancers. Oncotarget. 2014; 5: 9349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu FF, Chiang MT, Li FA, et al. Acetylation is essential for nuclear heme oxygenase-1-enhanced tumor growth and invasiveness. Oncogene. 2017; 36: 6805–14. [DOI] [PubMed] [Google Scholar]
- 12.Levine AA, Guan Z, Barco A, et al. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci U S A. 2005; 102: 19186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serravalle S, Melchionda F, Astolfi A, et al. A novel specific signature of pediatric MOZ-CBP acute myeloid leukemia. Leukemia research. 2010; 34: e292–3. [DOI] [PubMed] [Google Scholar]
- 14.Kung AL, Rebel VI, Bronson RT, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes & development. 2000; 14: 272–7. [PMC free article] [PubMed] [Google Scholar]
- 15.Weissfeld JL, Schoen RE, Pinsky PF, et al. Flexible sigmoidoscopy in the randomized prostate, lung, colorectal, and ovarian (PLCO) cancer screening trial: added yield from a second screening examination. J Natl Cancer Inst. 2012; 104: 280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oken MM, Marcus PM, Hu P, et al. Baseline chest radiograph for lung cancer detection in the randomized Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. J Natl Cancer Inst. 2005; 97: 1832–9. [DOI] [PubMed] [Google Scholar]
- 17.Tryka KA, Hao L, Sturcke A, et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014; 42: D975–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mailman MD, Feolo M, Jin Y, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007; 39: 1181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lappalainen T, Sammeth M, Friedlander MR, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013; 501: 506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai R, Yu X, Wei Y, Su L, Christiani DC. Smoking and smoking cessation in relation to the development of co-existing non-small cell lung cancer with chronic obstructive pulmonary disease. Int J Cancer. 2014; 134: 961–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007; 23: 1294–6. [DOI] [PubMed] [Google Scholar]
- 22.Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004; 96: 434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012; 40: D930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009; 37: W600–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012; 22: 1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348: 648–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chambless LE, Diao G. Estimation of time-dependent area under the ROC curve for long-term risk prediction. Stat Med. 2006; 25: 3474–86. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Liu H, Ready NE, et al. Genetic variants in ABCG1 are associated with survival of nonsmall-cell lung cancer patients. Int J Cancer. 2016; 138: 2592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013; 8: e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016; 48: 607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012; 150: 1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neugebauer RC, Sippl W, Jung M. Inhibitors of NAD+ dependent histone deacetylases (sirtuins). Current pharmaceutical design. 2008; 14: 562–73. [DOI] [PubMed] [Google Scholar]
- 34.Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. Journal of biomedicine & biotechnology. 2011; 2011: 371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krämer OH. HDAC2: a critical factor in health and disease. Trends in Pharmacological Sciences. 2009; 30: 647–55. [DOI] [PubMed] [Google Scholar]
- 36.Noh JH, Jung KH, Kim JK, et al. Aberrant regulation of HDAC2 mediates proliferation of hepatocellular carcinoma cells by deregulating expression of G1/S cell cycle proteins. PLoS One. 2011; 6: e28103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung KH, Noh JH, Kim JK, et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. Journal of cellular biochemistry. 2012; 113: 2167–77. [DOI] [PubMed] [Google Scholar]
- 38.Weichert W, Roske A, Gekeler V, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. British journal of cancer. 2008; 98: 604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wisnieski F, Calcagno DQ, Leal MF, et al. Differential expression of histone deacetylase and acetyltransferase genes in gastric cancer and their modulation by trichostatin A. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014; 35: 6373–81. [DOI] [PubMed] [Google Scholar]
- 40.Stojanovic N, Hassan Z, Wirth M, et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene. 2017; 36: 1804–15. [DOI] [PubMed] [Google Scholar]
- 41.Xu J, Tian S, Yin Z, et al. MicroRNA-binding site SNPs in deregulated genes are associated with clinical outcome of non-small cell lung cancer. Lung cancer. 2014; 85: 442–8. [DOI] [PubMed] [Google Scholar]
- 42.Yang Z, Zhou L, Wu LM, et al. Combination of polymorphisms within the HDAC1 and HDAC3 gene predict tumor recurrence in hepatocellular carcinoma patients that have undergone transplant therapy. Clinical chemistry and laboratory medicine. 2010; 48: 1785–91. [DOI] [PubMed] [Google Scholar]
- 43.Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harbor perspectives in medicine. 2013; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ling C, Del Guerra S, Lupi R, et al. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia. 2008; 51: 615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Xu W, Li X, et al. Association between PPARGC1A gene polymorphisms and coronary artery disease in a Chinese population. Clinical and experimental pharmacology & physiology. 2008; 35: 1172–7. [DOI] [PubMed] [Google Scholar]
- 46.Charos AE, Reed BD, Raha D, et al. A highly integrated and complex PPARGC1A transcription factor binding network in HepG2 cells. Genome research. 2012; 22: 1668–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li JD, Feng QC, Qi Y, Cui G, Zhao S. PPARGC1A is Upregulated and Facilitates Lung Cancer Metastasis. Experimental cell research. 2017; 359: S0014482717304275. [DOI] [PubMed] [Google Scholar]
- 48.Luo C, Lim JH, Lee Y, et al. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature. 2016; 537: 422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li JD, Feng QC, Qi Y, Cui G, Zhao S. PPARGC1A is upregulated and facilitates lung cancer metastasis. Experimental cell research. 2017; 359: 356–60. [DOI] [PubMed] [Google Scholar]
- 50.Zhai H, Zhong W, Yang X, Wu YL. Neoadjuvant and adjuvant epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) therapy for lung cancer. Transl Lung Cancer Res. 2015; 4: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu C, Zhou Q, Wu YL. Can EGFR-TKIs be used in first line treatment for advanced non-small cell lung cancer based on selection according to clinical factors? - A literature-based meta-analysis. Journal of hematology & oncology. 2012; 5: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mountzios G Making progress in epidermal growth factor receptor (EGFR)-mutant non-small cell lung cancer by surpassing resistance: third-generation EGFR tyrosine kinase inhibitors (EGFR-TKIs). Annals of translational medicine. 2018; 6: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim Y, Lee B, Shim JH, et al. Concurrent genetic alterations predict the progression to target therapy in EGFR-mutated advanced non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2018. [DOI] [PubMed] [Google Scholar]
- 54.Yu Z, Boggon TJ, Kobayashi S, et al. Resistance to an irreversible epidermal growth factor receptor (EGFR) inhibitor in EGFR-mutant lung cancer reveals novel treatment strategies. Cancer Res. 2007; 67: 10417–27. [DOI] [PubMed] [Google Scholar]
- 55.Huang B, Liu B, Yang L, et al. Functional genetic variants of c-Jun and their interaction with smoking and drinking increase the susceptibility to lung cancer in southern and eastern Chinese. International journal of cancer. 2012; 131: E744–58. [DOI] [PubMed] [Google Scholar]
- 56.Cleary SP, Cotterchio M, Shi E, Gallinger S, Harper P. Cigarette smoking, genetic variants in carcinogen-metabolizing enzymes, and colorectal cancer risk. American journal of epidemiology. 2010; 172: 1000–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.