

Abstract
Carboxy-terminus of Hsc70-interacting protein (CHIP) functions both as a molecular co-chaperone and ubiquitin E3 ligase playing a critical role in modulating the degradation of numerous chaperone-bound proteins. To date, it has been implicated in the regulation of numerous biological functions, including misfolded-protein refolding, autophagy, immunity, and necroptosis. Moreover, the ubiquitous expression of CHIP in the central nervous system suggests that it may be implicated in a wide range of functions in neurological diseases. Several recent studies of our laboratory and other groups have highlighted the beneficial role of CHIP in the pathogenesis of several neurological diseases. The objective of this review is to discuss the possible molecular mechanisms that contribute to the pathogenesis of neurological diseases in which CHIP has a pivotal role, such as stroke, intracerebral hemorrhage, Alzheimer’s disease, Parkinson’s disease, and polyglutamine diseases; furthermore, CHIP mutations could also cause neurodegenerative diseases. Based on the available literature, CHIP overexpression could serve as a promising therapeutic target for several neurological diseases.
Subject terms: Cerebrovascular disorders, Neurodegenerative diseases
Facts
CHIP has dual function both as co-chaperone and ubiquitin ligase.
CHIP can participate in inspecting and facilitating the refolding of misfolded proteins; moreover, it can also promote the degradation of ubiquitin-tagged proteins via the ubiquitin-proteasome pathway.
CHIP overexpression can function neuroprotectively in neurological diseases, suggesting that it may constitute a promising therapeutic strategy.
CHIP mutations can lead to spinocerebellar ataxia autosomal recessive 16 (SCAR16) or spinocerebellar ataxia 48 (SCA48).
Open Questions
What is the relationship between the location of the mutations and changes in the biological function of CHIP within the clinical spectrum of SCAR16 or SCA48?
Why can CHIP mutations lead to coexistence of autosomal recessive and dominant inheritance?
How can the most effective and practical new CHIP-based drugs for neurological disease treatment be developed?
Introduction
It is established that proteins fundamentally contribute to the maintenance of cellular function and that protein stability is a major mechanism underlying human disease, such as cancer and neurological diseases. The organism has developed meticulous mechanisms to monitor and maintain the health of its proteome, which comprise the protein quality control (PQC) system. The most important means employed by the PQC system is the network of molecular chaperones, which can inspect and facilitate the refolding of misfolded proteins; moreover, it can also promote the degradation of ubiquitin-tagged proteins via the ubiquitin-proteasome pathway1,2.
Carboxy-terminus of Hsc70-interacting protein (CHIP), also termed STUB1, is a 35 kDa protein with the dual function of co-chaperone and E3 ubiquitin ligase activity3,4. CHIP is ubiquitously expressed and, in particular, highly expressed in hypermetabolic tissues with rapid protein turnover such as in skeletal muscle, heart, and brain tissues3. Moreover, CHIP is an evolutionarily ancient protein with extensive conservation across species; for instance, human CHIP exhibits 98% amino acid similarity with mouse CHIP3. CHIP comprises triple tandem tetratricopeptide repeat (TPR) domains, which can mediate interactions with chaperones such as HSP70 and HSP90 in the N-terminal end, an U-box domain displaying E3 ubiquitin ligase activity in the C-terminal end, and a central coiled coil (CC) domain with a largely unknown function that may be required for dimerization5. Due to its unique structure, CHIP can combine with chaperones via the TPR domain and mediate the ubiquitination and degradation of various chaperone-bound proteins via its E3 ubiquitin ligase activity6–8. Hence, CHIP can act as a connecting link between molecular chaperones and proteasomes and is widely regarded as a vital player in the cellular PQC system.
In recent years, growing evidence has shown that CHIP can also regulate the activity of non-misfolded proteins, such as INSR9, RIPK310, TFEB11, and PKA12; in these pathways, CHIP does not degrade toxic proteins and even does not require chaperone partners, suggesting that CHIP can modulate the pathological process of human diseases through multiple pathways. Furthermore, according to the studies of our team and those of other groups13–15, some but not all CHIP mutants destabilize the structure and show defects in CHIP chaperone binding, which suggests that some CHIP mutations may cause diseases by influencing the non-canonical CHIP functions.
Emerging evidence indicates that CHIP is involved in multiple fundamental cellular processes relevant to the pathogenesis of neurological diseases. Herein, we will discuss in detail the available evidence of the physiological role of CHIP in the context of neurological diseases, such as intracerebral hemorrhage (ICH), ischemic stroke, Alzheimer’s disease (AD), Parkinson’s disease (PD), polyglutamine (PolyQ) diseases, and spinocerebellar ataxia autosomal recessive 16 (SCAR16) and spinocerebellar ataxia 48 (SCA48) caused by CHIP mutation (Fig. 1).
Fig. 1. CHIP involves in various of nervous system diseases.
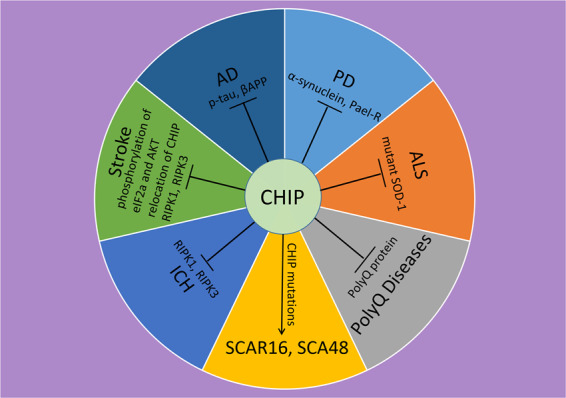
Schematic diagram of the substrates of CHIP in the context of various neurological disorders. Please refer to the main text for details.
Implication of CHIP in neurological diseases
Intracerebral hemorrhage
ICH, which accounts for ~15% of all stroke-related events16,17, is a severe condition associated with high mortality and morbidity18. Mechanical damage caused by hematoma19 and secondary brain injury can cause neuronal death and neurological deficits20. Accumulating evidence has shown that strategies designed to limit neuron death after ICH are therapeutically valid to reduce hematoma expansion and improve outcomes. Recently, studies indicated that necroptosis21–23 is an important mechanism influencing brain injury after ICH and that inhibiting necroptosis can markedly ameliorate the outcomes24,25. As reported, CHIP can regulate necroptosis through mediating the degradation of RIPK310, intimating the possible therapeutic function of CHIP after ICH.
Our team explored the role of CHIP following ICH26 and provided the first evidence that CHIP expression is upregulated after ICH in rat models. We constructed a CHIP overexpression rat model using AAVPHP.B viral particles, CHIP overexpression could remarkably reduce neurological scores and significantly decrease hemorrhagic-lesion volume after ICH26. Notably, CHIP overexpression observably decreased the number of necroptotic cells and the expression levels of RIPK1, RIPK3, and MLKL, three major regulators of necroptosis, around the hematomal region after ICH compared with those in the control groups. Furthermore, CHIP-deficient rats had significantly aggravated neurological impairments and markedly increased levels of RIPK1, RIPK3, and MLKL compared with wildtype (WT) rats; and these effects could be recovered through the reintroduction of CHIP. Moreover, CHIP overexpression ameliorated neuroinflammation in rats after ICH26.
However, the introduction of CHIP overexpression in our study was achieved through treatment with AAVPHP.B vectors prior to ICH induction, which is unattainable in clinical practice. Thus, it is necessary to develop effective and practical drugs that can rapidly induce CHIP overexpression. Moreover, the generation of neutralizing antibodies (NAbs) in peripheral circulation may be a major obstacle that impedes the repeated use of AAVPHP.B27. Therefore, establishing methods to circumvent NAbs should be a critical crux in the use of AAVPHP.B.
Ischemic stroke
Stroke is a devastating disease threatening ~30 million humans annually worldwide, and ~80% of cases of stroke are of the ischemic type28. In addition to immediate neuron death after stroke, neuronal loss also occurs in the tissues surrounding the affected area due to multiple destructive mechanisms, such as excitotoxicity, necroptosis29, and inflammation. Therefore, therapeutic strategies that can rescue damaged neurons are very important, but approaches that can alleviate neurological impairment are limited. Hence, developing new therapeutic agents is urgently needed. Recent studies have demonstrated that CHIP participates in the pathological process of ischemic stroke; interestingly, the implicated mechanisms vary30–34. Anderson et al. reported that CHIP expression is upregulated in the postmortem brain tissues of patients after stroke31, implying that CHIP may play an important role in the pathological mechanism after stroke and may be a potential therapeutic target for neuronal injury.
In our study, CHIP protected N2a cells following oxygen glucose deprivation (OGD) through inhibiting necroptosis30. In detail, we detected upregulation of RIPK1, RIPK3, and MLKL in N2a cells exposed to OGD accompanied by increased cell death, and Nec-1, a RIPK1 inhibitor, could reverse these effects30. These results suggested that necroptosis was activated under OGD challenging. Moreover, anisomycin can reduce OGD-induced necroptosis through upregulation of CHIP expression in N2a cells and primary hippocampal neurons. As expected, CHIP overexpression could reduce the RIPK3 level by post-translational regulation under OGD-challenging. Moreover, while other functions of anisomycin may contribute to the benefit of OGD, the role of CHIP may be much more important30. Neither the TPR nor the U-box domain of CHIP alone possesses the protective function of inhibiting necroptosis, suggesting that both the co-chaperone and ubiquitin E3 ligase functions of CHIP are necessary for anti-necroptosis following OGD30. Similarly, Cabral-Miranda et al. reported that CHIP overexpression protected against hippocampal neuronal death through decreasing the phosphorylation of eIF2a and AKT in experimental brain ischemia34. Conversely, another study reported that CHIP overexpression exerted harmful effects and that downregulation of CHIP led to improved survival following OGD33; this distinction could likely result from the use of dissociated cell cultures, as Cabral-Miranda et al. suspected in their study34.
The relocation of CHIP subcellularly also plays an important role in ischemia31. In a cell model, the nuclear levels of CHIP increased in response to OGD, which can significantly delayed cell death, suggesting that there is an intimate relationship between CHIP nuclear localization and neuronal survival31. Furthermore, in an OGD neuronal model, CHIP and PINK1 expressions increased after stress, and CHIP was observed to relocate from cytosolic and perinuclear sites to the mitochondria following OGD32. Given that PINK1 changes synchronously with CHIP after OGD, we speculate that the re-localization of CHIP may be promoted by PINK1 in a similar manner to PINK1 recruitment of parkin, a E3 ligase35–37. Furthermore, CHIP-deficient neurons have lower cell viability compared to WT ones following OGD, which underscores the important role of CHIP in maintaining neuronal survival32.
As previously described, although the CHIP-related mechanisms in neuronal degradation or neuronal death vary, these studies have suggested that CHIP could be a promising therapeutic target to prevent brain injury after ischemic stroke.
Parkinson’s disease
PD is the second most common neurodegenerative disorder and affects ~2% of the population over 60 years old. Although the pathogenesis of PD remains unclear, α-Synuclein (α-Syn), a major component of Lewy bodies, has long been associated with PD. α-Syn can form prefibrillar and fibrillar cellular aggregates, and these oligomeric assemblies can mediate α-Syn neurotoxicity38,39. Therefore, reducing α-Syn oligomerization or clearing existing a-Syn oligomers may provide a potential therapeutic target38,39. Shin et al. reported that CHIP plays a role in α-Syn aggregation and degradation40. They showed that CHIP co-localizes with α-Syn and Hsp70 in Lewy bodies and in α-Syn inclusions, and CHIP overexpression reduces α-Syn aggregation and increases α-Syn degradation. Moreover, CHIP can mediate α-Syn degradation by two discrete mechanisms, i.e., the TPR domain is critical for proteasomal degradation, but the U-box domain directs α-Syn toward the lysosomal degradation pathway40. Moreover, CHIP preferentially recognizes and mediates degradation of toxic oligomeric forms of α-Syn41, and that CHIP is an E3 ubiquitin ligase of α-Syn42. These results suggest that CHIP may be a target for PD treatment.
Although most cases of PD are sporadic, a Mendelian pattern of inheritance has also been recognized, such as cases of leucine-rich repeat kinase-2 (LRRK2)43, PTEN-induced putative kinase 1 (PINK1)44, and Parkin45. Mutation of LRRK2 is the most frequent genetic cause of familial PD and has also been identified in individuals with sporadic PD46. Studies reported that CHIP binds, ubiquitinates, and promotes the degradation of LRRK247,48. Hsp90 can attenuate CHIP-mediated LRRK2 degradation, and this can be blocked by a Hsp90 inhibitor47,48. Moreover, there are some variants that are not located in the central ROC-COR-kinase triple domain, including G2385R49. Rudenko et al. reported that G2385R LRRK2 has decreased kinase activity and affinity to the LRRK2 interactor50. Furthermore, G2385R LRRK2 has a lower steady state and can increase LRRK2 protein turnover compared to WT LRRK251. CHIP can directly recognize different regions of LRRK2, including the WD40 domain where G2385R is located, and G2385R LRRK2 has a higher affinity for CHIP compared to WT LRRK251. CHIP overexpression decreased both G2385R mutant and WT LRRK2, while CHIP knockdown had the opposite effect and induced neuronal death51. These results suggest that CHIP may be a potentially valid candidate for the treatment of LRRK2-related PD.
Mutation of Parkin is also a genetic cause of PD45. Parkin is a protein with a ubiquitin-like domain, and two RING finger motifs52. Pael receptor (Pael-R) is a substrate of parkin. Accumulation of Pael-R in the endoplasmic reticulum (ER) of dopaminergic neurons can induce ER stress, eventually leading to neurodegeneration53. CHIP, Hsp70, parkin, and Pael-R formed a complex both in vitro and in vivo54. The expression of CHIP in the complex was increased under ER stress. CHIP enhanced the dissociation of Hsp70 from parkin and Pael-R, then accelerating Pael-R ubiquitination mediated by parkin54. Moreover, CHIP enhanced parkin-mediated ubiquitination of Pael-R, and CHIP strengthened the ability of parkin to inhibit neuronal death induced by Pael-R54. Taken together, these data indicate that CHIP positively regulates parkin E3 activity and might be an excellent therapeutic target for the treatment of parkin-related PD.
PINK1 is the second most frequent cause of autosomal recessive PD. Via its kinase activity, PINK1 regulates mitochondrial quality control and neuronal cell survival. CHIP is a novel ubiquitin E3 ligase that targets PINK1, promoting its ubiquitination and degradation, but this effect was not seen with mutants CHIP-H260Q and CHIP-ΔU55. CHIP overexpression suppressed PINK1 mutant phenotypes in flies, such as locomotion defects and loss of dopaminergic neurons56. Moreover, CHIP, but not its ligase-dead mutants, could rescue mitochondrial defects in PINK1 mutants56, suggesting that E3 ubiquitin ligase activity is required for CHIP to protect against mitochondrial dysfunction in PINK1-mutant flies.
Alzheimer’s disease
AD is the most common neurodegenerative disease worldwide and is characterized by gradual onset of dementia, cognitive decline, even inability to live independently57. Although numerous causal hypotheses have been recently proposed, the presence of beta-amyloid (Aβ) deposits and hyperphosphorylated tau (p-tau) have gained most of the attention58. Thus, eliminating Aβ and p-tau may be promising therapeutic strategies.
Tau is a protein that plays a key role in the dynamics of microtubules. In the pathology of AD, tau becomes hyperphosphorylated, separates from the microtubules, misfolds, and aggregates into neurofibrillary tangles in the intracellular space, playing a critical role in AD59,60. Several studies have reported that CHIP can directly ubiquitinate and eliminate p-tau61–66. Moreover, CHIP strongly interacts with caspase cleavaged tau (tauΔC) compared to full-length tau (FL-tau), leading to increased ubiquitylation and rapid degradation of tauΔC61. Both the U-box and TPR domains are necessary for CHIP binding to tau; binding of Hsp70 to the TPR domain is necessary for reducing tau levels61,63,64. CHIP overexpression can also rescue the mitochondrial deficit caused by tau overexpression63. CHIP expression was increased in human brains with AD and tauopathy mouse models67, and CHIP deficiency could induce increase of insoluble tau67. Furthermore, tau expression was upregulated in the brain of CHIP-T246M rat model13, implying that tauopathy may be importantly implicated in the pathogenesis of CHIP mutation-related diseases. In an AD mouse model, overexpressing CHIP could reduce tau phosphorylation68. There results indicate that CHIP plays a critical role in mediated tau and p-tau degradation, and CHIP overexpression may protect against p-tau aggregation and neurofibrillary-tangle formation.
A large body of evidence supports that the accumulation of intracellular Aβ42, a proteolytic cleavage of amyloid precursor protein (APP) by β-site APP-cleaving enzyme 1 (BACE1), which plays a central role in AD pathogenesis by processing APP to Aβ, is a critical event in the pathogenic mechanism of AD, and reducing the accumulation of Aβ can mitigate the progression of AD69–71. Several studies have suggested that CHIP can act to lessen the toxicity of Aβ4272–74, and CHIP overexpression can directly interact with holo-βAPP and stabilize steady-state holo-βAPP levels73. In addition, a complex with several HSPs and CHIP can hasten the degradation of Aβ42 levels and protect against toxicity in neurons73. CHIP overexpression can decrease BACE1 levels by promoting its ubiquitination and degradation, thus reducing APP processing to Aβ74, and both the U-box and TPR domains are essential for ubiquitination and degradation of BACE174. Furthermore, CHIP can also negatively regulate BACE1 through the regulation of p53-mediated transrepression74.
Moreover, emerging evidence has suggested that Aβ and tau may be mechanistically linked75,76, and exploration of the possible mechanistic links between Aβ and tau pathology may greatly aid the understanding of AD. One of the molecular mechanisms underlying the effects of Aβ on tau is mediated by CHIP77. Aβ42 plays a major role in the mechanism of tau pathology; reducing Aβ42 levels or preventing Aβ42 accumulation markedly delays the onset of tau pathology77. Moreover, Aβ42, not full-length APP, can reduce CHIP levels, which is sufficient to interfere with tau turnover and degradation, thereby facilitating the buildup of tau aggregates. More importantly, Aβ42-induced tau pathology can be rescued by restoring CHIP levels77. These results suggest CHIP as a valid therapeutic target, and increasing its activity may reduce Aβ42-induced tau pathology.
Several small-molecule agonists that can upregulate CHIP expression have been reported recently (Table 1)30,68,78,79. Sulforaphane, a herbal isothiocyanate draw from cruciferous vegetables, can clear the accumulation of Aβ and tau and then ameliorate learning and memory defaults in 3×Tg-AD mice through upregulating CHIP expression in the cortex and hippocampus68. Moreover, several traditional Chinese medications can also upregulate CHIP expression in the cerebral cortex and hippocampus of AD disease models, highlighting the role of CHIP in AD80,81. These results indicate that small-molecule agonists of CHIP such as sulforaphane may constitute promising drugs that could be used for the treatment of neurological diseases.
Table 1.
Small-molecule agonists that can induce CHIP overexpression.
| # | Small-molecule agonists | Disease model | References |
|---|---|---|---|
| 1 | Sulforaphane | 3×Tg-AD mouse model | 68 |
| 2 | Anisomycin | Oxygen-glucose deprivation (OGD) cell model | 30 |
| 3 | Peptidoglycan (PGN) | RAW264.7 cells, peritoneal macrophage isolated from WT and TLR2 KO mice | 78 |
| 4 | 2-(4-hydroxy-3-methoxyphenyl)-benzothiazole (YL-109) | MDA-MB-231 cells (breast cancer) | 79 |
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a late-onset fatal neurodegenerative disease selectively affecting motor neurons and other neuronal cells in the spinal cord, brainstem, and cortex, leading to severe disability and eventually death. The common pathological features of ALS include the accumulation of misfolded protein inclusions in motor neurons and other neurons in the cortex and other neuroanatomical regions. Approximately 10% of ALS cases are inherited familial cases (fALS), and genetic mutations in SOD1 are shared by 20% of patients with fALS82,83. Lines of evidence have indicated that the formation of aggregates is a common feature due to the misfolding of mutant SOD1 in over 100 SOD1 mutants84, and that mutant SOD1 is conjugated to a multi-ubiquitin chain and degraded in a chaperone-dependent manner, which suggests that UPS may play a critical role in the pathological mechanism of ALS.
CHIP can promote the degradation of mutant SOD1 but not of WT SOD1 at the proteasome85. Interestingly, CHIP does not directly target mutant SOD1, which differs from the mechanism of other CHIP substrates85. Moreover, CHIP immunoreactivity and high Hsc70-immunoreactivity are observed in inclusions of motor neurons of an ALS mouse model that contains SOD1 mutations, denoting the important role of the Hsp/Hsc70-CHIP machinery in the degradation of mutant SOD185.
Dorfin is the first identified E3 protein that can specifically ubiquitylate mutant SOD1 and attenuate the cytotoxicity of mutant SOD1 in ALS cell models86. However, dorfin’s neuroprotective effect has been shown to be modest in dorfin/mutant SOD1 double transgenic mice87; this limited effect might be due to its short half-life88. Several studies have reported that engineered chimera E3s can degrade certain substrates with high efficiency89–91; Ishigaki et al. engineered a series of dorfin-CHIP chimeric proteins combined with the hydrophobic region of dorfin and the U-box domain of CHIP87. Some engineered proteins such as dorfin-CHIPD, J, and L have longer half-lives and stronger binding activity than WT dorfin. Moreover, dorfin-CHIPL presents stronger E3 activity when bound to mutant SOD1 compared to dorfin or CHIP alone, and dorfin-CHIPL showed a greater therapeutic effect against mutant SOD1. It is noteworthy that dorfin-CHIPL specifically ubiquitylates mutant SOD1 but not WT SOD1, just as dorfin87. Dorfin can be found in both fALS and sALS; and proteasomal abnormalities also occur in sALS. Thus, the neuroprotective function of dorfin-CHIPL could render it an appropriate therapeutic candidate to be tested in clinical trials87. Furthermore, developing new chimera E3 ligases such as dorfin-CHIPL may be a promising therapeutic concept for ALS and other neurodegenerative disorders.
PolyQ diseases
PolyQ diseases are a group of inherited neurodegenerative diseases caused by a genetic mutation of the cytosine–adenine–guanine (CAG) triplet repeat expansion, which encodes for polyQ tracts in the causative proteins92–94. To date, nine disorders have been reported, including spinal and bulbar muscular atrophy (SBMA), Huntington’s disease (HD), several spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17), and dentatorubral pallidoluysian atrophy (DRPLA)95. A common pathological feature of polyQ diseases is the degeneration of neurons in the brain. Misfolding and aggregation of the expanded polyQ proteins constitute the most upstream events in the most common pathogenic cascade of polyQ diseases; thus, approaches targeting misfolding and aggregation of expanded polyQ proteins are likely therapeutically important.
Several studies have reported that CHIP plays a critical role in regulating the degradation of polyQ96–102. CHIP can reduce the aggregation and increase the solubility of mutant huntingtin, which depends on the function of the TPR domain, and thus rescued inclusion formation and toxicity in disease models of HD99. Furthermore, CHIP deficiency can exacerbate and accelerate neuronal dysfunction and phenotype in an HD model99. Together, these results identified the beneficial effects of CHIP in HD, strengthening its potential as a therapeutic target for polyQ diseases. CHIP can interact with expanded huntingtin or ataxin-3, and that CHIP overexpression increased the ubiquitination and rate of degradation of expanded huntingtin or ataxin-3100. In a SCA3 mouse model, CHIP deficiency accelerated the disease and markedly increased the level of ataxin-3 micro-aggregates100. Moreover, CHIP overexpression suppressed the aggregation and neuronal death mediated by expanded polyQ proteins, and this function was more prominent when CHIP was overexpressed along with Hsc70100.
The role of CHIP in SCA1 has also been explored. CHIP and ataxin-1 directly interact and co-localize in nuclear inclusions (NIs) both in cell-model and patient postmortem neurons98. CHIP promotes the ubiquitination of expanded ataxin-1, and this effect involves the TPR domain98, Hsp70 can increase CHIP-mediated ubiquitination of ataxin-198. Interestingly, CHIP also interacts with and ubiquitinates unexpanded ataxin-198. Moreover, CHIP overexpression decreased the protein steady-state levels of both expanded and unexpanded ataxin-1 and suppressed their toxicity in a drosophila SCA1 model98. It is worth noting that the effect of CHIP is impaired by the mutation of Ser776 of ataxin-1 whose phosphorylation is critical for ataxin-1 aggregation101, suggesting that the role of CHIP in polyQ-protein aggregation greatly depends on the context of full-length polyQ proteins.
In addition, the role of CHIP has also been studied in SBMA. CHIP overexpression can improve motor disorders and inhibited nuclear accumulation of mutant AR in an SBMA animal model97. Moreover, mutant AR was preferentially degraded over WT AR under CHIP overexpression97, suggesting that mutant AR is more sensitive to CHIP than WT AR. These results demonstrate that CHIP overexpression mitigates SBMA phenotypes by reducing mutant AR by enhancing its degradation. Thus, we conclude that CHIP is a vital factor in response to misfolded polyQ proteins and represents a potential therapeutic target for polyQ diseases.
Neurological diseases caused by CHIP mutations
Recently, there have been emerging reports of diseases caused by CHIP mutations. Gordon Holmes syndrome (GHS) is a rare neurodegenerative disease characterized by ataxia and hypogonadism103. In 2014, our team reported the first GHS family in China mainland and identified the third GHS-related pathogenic gene, i.e., CHIP with p.Thr246Met mutation in the U-box domain, which abolishes the ubiquitin ligase activity of CHIP104. Interestingly, the first and second pathogenicity genes of GHS, E3 ligase RNF216 and deubiquitinase OTUD4, also suggest that ubiquitination plays an essential role in the pathophysiology of GHS, further highlighting the role of deficiency of ubiquitin ligase activity in the pathogenesis of GHS105. Moreover, we found that CHIP knockout (CHIP−/−) mice also showed ataxia, hypogonadism, and cognitive impairment, closely reflecting the clinical manifestations of GHS patients. Histological examination of the CHIP−/− mice cerebellum revealed cellular loss throughout the cerebellum specifically in the Purkinje cells compared with WT mice104. This strong similarity between our GHS patients and the CHIP−/− rodent model suggests a vital role of CHIP in maintaining cerebellar function and the reproductive–endocrine axis104.
In further study, we found that CHIP p.Thr246Met mutation disrupted the structure of the U-box and promoted the formation of soluble oligomers, while still maintaining chaperone-interaction activity, i.e., interactions between CHIP and known interactors were not affected13. However, CHIP-T246M could change solubility and increase turnover. Moreover, we prepared mouse and rat models with the CHIP-T246M mutation, and both showed decreased expression of CHIP in the brain and cerebellum and decreased steady-state protein expression13. The CHIP-T246M rodent animal models recapitulated the key features of GHS13, which support our previous findings that CHIP plays a critical role in cerebellar maintenance. It is especially worth mentioning that some of the behavioral deficits in the CHIP-T246M rodent models were not shared by CHIP-/- mice, implying that disease-causing mutations in CHIP and total loss of CHIP are not functionally equivalent; we suspect that while CHIP-T246M no longer functions as an E3 ubiquitin ligase, other CHIP functions remain intact13.
To date, more than 20 new pedigrees have been reported worldwide, and the autosomal recessive cerebellar ataxia caused by CHIP mutation was classified as SCAR16104,106–116 (Fig. 2 and Table 2). The pathogenic gene sites in CHIP of these patients were distributed in the U-box and TPR domains and even in the CC domain, suggesting that all three domains are indispensable in maintaining CHIP function. The clinical symptoms of SCAR16 are complex; in addition to early-onset progressive cerebellar ataxia, cerebellar atrophy, dementia, and hypogonadism, the patients also present with hyperkinetic movement disorder114 and significant pyramidal tract damage113,114, and in some patients, ataxia was not the first symptom114. Some patients with CHIP mutation also show action-verb impairment117. These findings imply that the phenotype of SCAR16 is not limited to ataxia syndrome, but rather involves almost the entire central nervous system. These results also show that CHIP not only plays a vital role in maintaining the cerebellum function but is also critically implicated in preserving the function of other areas of the brain.
Fig. 2. CHIP protein domains are diagramed.

The locations (arrows) of the various mutations and respective nucleic acid and amino acid changes associated with SCAR16 and SCA48 are indicated in Table 2.
Table 2.
A summary of the various mutations and respective nucleic acid and amino acid changes associated with SCAR16 and SCA48.
| # | Genotype | Amino Acid | Inheritance Mode | Disease | References |
|---|---|---|---|---|---|
| 1 | c.737C>T211 | p.Thr246Met | ARa | SCAR16 | 104 |
| 2 | c.612+1G>C, c.823C>Gb | p.Leu275Val | AR | SCAR16 | 106 |
| 3 | c.433A>C; c.687_690delCTACb | p.Lys145Gln; p.Asn230Cysfs*8 | AR | SCAR16 | 107 |
| 4 | c.194A>G | p.Asn65Ser | AR | SCAR16 | 116 |
| c.82G>A; c.430A>Tb | p.Glu28Lys; p.Lys144Ter | ||||
| 5 | c.433A>C; 721C>Tb | p.Lys145Gln; Arg241Trp | AR | SCAR16 | 108 |
| c.433A>C; 694T>Gb | p. Lys145Gln; Cys232Gly | ||||
| 6 | c.358+1G>A; c.566A>Cb | p.Asp189Ala | AR | SCAR16 | 109 |
| 7 | c.633G>A; c.712G>Tb | p.Met211Ile; p.Glu238Ter | AR | SCAR16 | 115 |
| 8 | c.389AT; c.441GTb | p.Asn130Ile; p.Trp147Cys | AR | SCAR16 | 110 |
| c.621CG; c.707GCb | p.Tyr207*; p.Ser236Thr | ||||
| c.493CT | p.Leu165Phe | ||||
| 9 | c.103C>A; c.678_679delb | p.Arg35Ser; p.Ile227Profs*11 | AR | SCAR16 | 111 |
| 10 | c.367C>G, | p.Leu123Val, | AR | SCAR16 | 112 |
| c.719T>C, | p.Met240Thr, | ||||
| c.235G>A; c.236C>Ab | p.Ala79Thr; p.Ala79Asp | ||||
| 11 | c.*240T>C | Alteration the polyadenylation signal from AATAAA to AACAAA | AR | SCAR16 | 113 |
| 12 | c.355C>T; c.880A>Tb | p.Arg119*; p.Ile294Phe | AR | SCAR16 | 114 |
| c.433A>C; c.728C>Tb | p.Lys145Gln; p.Pro243Leu | ||||
| 13 | c.823_824delCT | p.Leu275Aspfs*16 | ADc | SCA48 | 119,122 |
| 14 | c.731_732delGC | p.Cys244Tyrfs*24 | AD | SCA48 | 123 |
| 15 | c.158T>C | p.Ile53Thr | AD | SCA48 | 124 |
| c.111C>G | p.Phe37Leu | ||||
| 16 | c.97G>A, | p.Gly33Ser, | AD | SCA48 | 125 |
| c.682C>T | p.Pro228Ser | ||||
| 17 | c.170C>T, c.199G>A, | p.Pro57Leu, p.Ala67Thr, | AD | SCA48 | 126 |
| c.721C>T, c.673C>T, | p.Arg241Trp, p.Arg225*, | ||||
| c.791_792delTG, | p.Val264Glyfs*4, | ||||
| c.818_819dupGC, | p.Pro274Alafs*3, | ||||
| c.689_692delACCT, | p.Tyr230Cysfs*9, | ||||
| c.823_824delCT | p.Leu275Aspfs*16 |
aAutosomal recessive.
bCompound heterozygous mutation.
cAutosomal dominant.
Consistent with our results13, studies of other CHIP mutations also found that some SCAR16 mutations destabilize CHIP, causing defects in CHIP function, including decreased interactions with chaperones, diminished substrate ubiquitination, increased formation of soluble oligomers, and reduced steady-state levels14. Astonishingly, two mutants, N65S and L123V, located in the TPR domain can also decrease the formation of free ubiquitin chains, implying that the co-chaperone function of the TPR domain may be important for activating the ability of E3 ligase activity15. However, it remains unclear whether there is a relationship between the location of the mutation and changes in the biological function of the corresponding mutated protein with the clinical spectrum of SCAR16. Madrigal et al. reported that the mutations located in the U-box domain strongly induce loss of CHIP function and are intensely related to cognitive impairment in patients with SCAR16118; TPR and CC mutations mildly affect CHIP function and are associated with an increased tendon reflex. They also reported that inhibiting the interaction between mutant CHIP and HSP70 can lead to later age of onset and less severe ataxia118.
In 2018, researchers reported a family with autosomal dominant cerebellar ataxia caused by a frameshift heterozygous STUB1 pathogenic variant, c.823_824delCT STUB1 (p.L275Dfs*16), which is distinct from SCAR16119. The same phenomena also occurred to SPTBN2, causing both SCA5 and SCAR15 due to dominant and recessive mutations, respectively120,121. The researchers named this disease SCA48119,122–126 (Fig. 2 and Table 2). The earliest and most prominent clinical manifestation of the patients was cognitive impairment, and cerebellar ataxia appeared years later119. Moreover, p.L275Dfs*16 STUB1 heterozygous carriers show no pyramidal signs or seizures, which can often be seen in patients with SCAR16, and extra-cerebellar symptoms can only be observed at the end stage119. Furthermore, c.823.824delCT of STUB1 leads to selective atrophy in cognitive- and emotion-related cerebellar areas that precedes the appearance of ataxia by years before motor-related cerebellar areas become involved119. It is worth noting that patients with SCA48 can also show cognitive impairment and motor cerebellar signs119. These results suggest that CHIP is a critical element in maintaining cognitive cerebellar function, but the mechanism requires further research.
Recently, two other SCA48 pedigrees respectively caused by p.Gly33Ser and p.Pro228Ser in CHIP were reported, and the phenotype of the patients appeared more complex125. In these two families with SCA48, the patients presented adult-onset ataxia associated with cognitive impairment and psychiatric disorders and a combination of movement disorders that have also been described in SCAR16 such as parkinsonism and dystonia; they also showed cerebellar atrophy and a widespread degeneration pattern in areas including the cerebellum, basal ganglia, and cerebral cortex125. The researchers also found a characteristic T2-weighted hyperintense signal extending from the dentate nuclei bilaterally to the middle cerebellar peduncles, which may be a useful imaging biomarker of SCA48125. Nowadays, patients with SCA48 with new mutations continue to be reported and their clinical features are more complex, including hypogonadism and extrapyramidal features122, which have also been observed in patients with SCAR16. Moreover, the extrapolated frequency in Lieto’s reference cohort reached 3.4% overall and ~23% among familial cases; the lack of evidence of founder mutations suggests that SCA48 may not be uncommon among the SCAs worldwide126. In these SCA48 families, p.Gly33Ser occurred in the TRP domain, whereas p.Pro228Ser and p.L275Dfs*16 were located in the ubiquitin ligase region, similar to findings pertaining to more than 10 SCAR16 families, and it can be seen that both protein regions are critical in maintaining CHIP function.
Several studies regarding the function of mutated CHIP have reported that some STUB1 mutations mediate disease by affecting the CHIP E3 ubiquitin ligase interactions and function through modification of its oligomeric states and structural stability, which indicates that change in structure may lead to gain- or loss-of-function and thus cause disease. However, we should also note that numerous mutations have limited effects on the structure and ubiquitination activity of CHIP, suggesting that these mutations may affect CHIP function though other mechanisms13–15,118. The most intensively studied mutation site in vitro and in vivo was T246M, and these studies could serve as grounds for further research of other CHIP mutation sites.
According to these studies, an interesting question remains regarding why CHIP mutations can lead to the coexistence of autosomal recessive and dominant inheritance. It is evident that carrier parents in SCAR16 families are healthy, although they bear mutation protein products in their brains. The type and location of the mutations cannot explain this phenomenon, as missense, frameshift, or nonsense mutations occurring in SCAR16 are scattered in the U-box and TPR domains and even in the adjacent regions without hotspots. Therefore, the pathogenetic mechanisms of SCA48 and its relationship with SCAR16 should be further explored.
Besides the use of rodents or cells, other model systems, such as worms and drosophilas, are also widely used in studying the role of CHIP in neurodegenerative diseases9,56,98. For example, Tawo et al. reported a competitive relationship between proteostasis and longevity regulation through CHIP-assisted proteolysis9, and almost all the experiments were performed using worms and flies, denoting that worms and flies can serve as good model animals for exploring the role of CHIP.
As almost all CHIP mutations in SCAR16 and SCA48 are associated with abnormal function, gene therapy delivering functional gene copies of CHIP may be the obvious beneficial solution; alternatively, gene-editing approaches or antisense oligonucleotide therapy may also prove to be effective therapeutic avenues.
Conclusions
In this review, we highlighted the neuroprotective role of CHIP in neurological diseases such as stroke, AD, and PD, and diseases caused by CHIP mutations. These results suggest that CHIP overexpression could be a potential therapeutic target. However, almost all these studies introduced CHIP overexpression via gene transfection, which may be of limited clinical use, and thus further studies are required to determine the most effective and practical regimens for CHIP treatment in neurological diseases. Furthermore, the combination of whole-exome sequencing analysis with traditional linkage analysis is a fruitful approach that can further extend the phenotypical spectrum of rare Mendelian disorders.
Acknowledgements
This work was funded by the National Natural Science Foundation of China to Dr Yu-ming Xu [81530037; 91849115]; the National Natural Science Foundation of China to Dr Chang-he Shi [81771290; 81974211].
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by B. Joseph
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chang-he Shi, Email: shichanghe@gmail.com.
Yu-ming Xu, Email: xuyuming@zzu.edu.cn.
References
- 1.Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta. 2014;1843:182–196. doi: 10.1016/j.bbamcr.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 2.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger CA, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connell P, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 5.Zhang M, et al. Chaperoned ubiquitylation-crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. cell. 2005;20:525–538. doi: 10.1016/j.molcel.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 6.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cyr DM, Hohfeld J, Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem. Sci. 2002;27:368–375. doi: 10.1016/s0968-0004(02)02125-4. [DOI] [PubMed] [Google Scholar]
- 8.Esser C, Alberti S, Hohfeld J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim. Biophys. Acta. 2004;1695:171–188. doi: 10.1016/j.bbamcr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 9.Tawo R, et al. The Ubiquitin ligase CHIP integrates proteostasis and aging by regulation of insulin receptor turnover. Cell. 2017;169:470–482.e413. doi: 10.1016/j.cell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo J, et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 2016;18:291–302. doi: 10.1038/ncb3314. [DOI] [PubMed] [Google Scholar]
- 11.Sha Y, Rao L, Settembre C, Ballabio A, Eissa NT. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 2017;36:2544–2552. doi: 10.15252/embj.201796699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rinaldi L, et al. Feedback inhibition of cAMP effector signaling by a chaperone-assisted ubiquitin system. Nat. Commun. 2019;10:2572. doi: 10.1038/s41467-019-10037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi CH, et al. Disrupted structure and aberrant function of CHIP mediates the loss of motor and cognitive function in preclinical models of SCAR16. PloS Genet. 2018;14:e1007664. doi: 10.1371/journal.pgen.1007664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pakdaman, Y. et al. In vitro characterization of six STUB1 variants in spinocerebellar ataxia 16 reveals altered structural properties for the encoded CHIP proteins. Biosci. Rep.37, BSR20170251 (2017). [DOI] [PMC free article] [PubMed]
- 15.Kanack AJ, Newsom OJ, Scaglione KM. Most mutations that cause spinocerebellar ataxia autosomal recessive type 16 (SCAR16) destabilize the protein quality-control E3 ligase CHIP. J. Biol. Chem. 2018;293:2735–2743. doi: 10.1074/jbc.RA117.000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. 2009;8:355–369. doi: 10.1016/S1474-4422(09)70025-0. [DOI] [PubMed] [Google Scholar]
- 17.Sacco S, Marini C, Toni D, Olivieri L, Carolei A. Incidence and 10-year survival of intracerebral hemorrhage in a population-based registry. Stroke. 2009;40:394–399. doi: 10.1161/STROKEAHA.108.523209. [DOI] [PubMed] [Google Scholar]
- 18.Balami JS, Buchan AM. Complications of intracerebral haemorrhage. Lancet Neurol. 2012;11:101–118. doi: 10.1016/S1474-4422(11)70264-2. [DOI] [PubMed] [Google Scholar]
- 19.Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373:1632–1644. doi: 10.1016/S0140-6736(09)60371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. doi: 10.1161/STROKEAHA.110.596718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang X, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to renal epithelial cell damage in an ATP-depleted renal ischemia model. Mol. Med. Rep. 2014;10:719–724. doi: 10.3892/mmr.2014.2234. [DOI] [PubMed] [Google Scholar]
- 22.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 23.Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017;24:1184–1195. doi: 10.1038/cdd.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su X, et al. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem. Res. 2015;40:643–650. doi: 10.1007/s11064-014-1510-0. [DOI] [PubMed] [Google Scholar]
- 25.Shen H, et al. Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. 2017;8:e2641. doi: 10.1038/cddis.2017.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S, et al. AAV/BBB-mediated gene transfer of CHIP attenuates brain injury following experimental intracerebral hemorrhage. Transl. stroke Res. 2020;11:296–309. doi: 10.1007/s12975-019-00715-w. [DOI] [PubMed] [Google Scholar]
- 27.Shinohara Y, et al. Effects of neutralizing antibody production on AAV-PHP.B-mediated transduction of the mouse central nervous system. Mol. Neurobiol. 2019;56:4203–4214. doi: 10.1007/s12035-018-1366-4. [DOI] [PubMed] [Google Scholar]
- 28.Cheng, X. et al. Exploring the potential value of miR-148b-3p, miR-151b and miR-27b-3p as biomarkers in acute ischemic stroke. Biosci. Rep.38, BSR20181033 (2018). [DOI] [PMC free article] [PubMed]
- 29.Vieira M, et al. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol. Dis. 2014;68:26–36. doi: 10.1016/j.nbd.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Tang MB, et al. Anisomycin prevents OGD-induced necroptosis by regulating the E3 ligase CHIP. Sci. Rep. 2018;8:6379. doi: 10.1038/s41598-018-24414-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson LG, Meeker RB, Poulton WE, Huang DY. Brain distribution of carboxy terminus of Hsc70-interacting protein (CHIP) and its nuclear translocation in cultured cortical neurons following heat stress or oxygen-glucose deprivation. Cell Stress Chaperones. 2010;15:487–495. doi: 10.1007/s12192-009-0162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palubinsky AM, et al. CHIP is an essential determinant of neuronal mitochondrial stress signaling. Antioxid. Redox Signal. 2015;23:535–549. doi: 10.1089/ars.2014.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stankowski JN, et al. C-terminus of heat shock cognate 70 interacting protein increases following stroke and impairs survival against acute oxidative stress. Antioxid. Redox Signal. 2011;14:1787–1801. doi: 10.1089/ars.2010.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cabral-Miranda F, et al. rAAV8-733-mediated gene transfer of CHIP/Stub-1 prevents hippocampal neuronal death in experimental brain ischemia. Mol. Ther. 2017;25:392–400. doi: 10.1016/j.ymthe.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geisler S, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 36.Geisler S, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6:871–878. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- 37.Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 2011;14:1929–1938. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh D, Mehra S, Sahay S, Singh PK, Maji S. K. alpha-synuclein aggregation and its modulation. Int. J. Biol. Macromol. 2017;100:37–54. doi: 10.1016/j.ijbiomac.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 39.Mehra S, Sahay S, Maji SK. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019;1867:890–908. doi: 10.1016/j.bbapap.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005;280:23727–23734. doi: 10.1074/jbc.M503326200. [DOI] [PubMed] [Google Scholar]
- 41.Tetzlaff JE, et al. CHIP targets toxic alpha-Synuclein oligomers for degradation. J. Biol. Chem. 2008;283:17962–17968. doi: 10.1074/jbc.M802283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalia LV, et al. Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5) PloS ONE. 2011;6:e14695. doi: 10.1371/journal.pone.0014695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimprich A, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 44.Valente EM, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 45.Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 46.Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nature reviews. Nat. Rev. Neurol. 2020;16:97–107. doi: 10.1038/s41582-019-0301-2. [DOI] [PubMed] [Google Scholar]
- 47.Ko HS, et al. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc. Natl Acad. Sci. USA. 2009;106:2897–2902. doi: 10.1073/pnas.0810123106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding X, Goldberg MS. Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PloS ONE. 2009;4:e5949. doi: 10.1371/journal.pone.0005949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Funayama M, et al. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport. 2007;18:273–275. doi: 10.1097/WNR.0b013e32801254b6. [DOI] [PubMed] [Google Scholar]
- 50.Rudenko IN, et al. The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem. J. 2012;446:99–111. doi: 10.1042/BJ20120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rudenko IN, et al. The G2385R risk factor for Parkinson’s disease enhances CHIP-dependent intracellular degradation of LRRK2. Biochem. J. 2017;474:1547–1558. doi: 10.1042/BCJ20160909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morett E, Bork P. A novel transactivation domain in parkin. Trends Biochem. Sci. 1999;24:229–231. doi: 10.1016/s0968-0004(99)01381-x. [DOI] [PubMed] [Google Scholar]
- 53.Imai Y, et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 54.Imai Y, et al. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol. cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- 55.Yoo L, Chung KC. The ubiquitin E3 ligase CHIP promotes proteasomal degradation of the serine/threonine protein kinase PINK1 during staurosporine-induced cell death. J. Biol. Chem. 2018;293:1286–1297. doi: 10.1074/jbc.M117.803890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, et al. Drosophila CHIP protects against mitochondrial dysfunction by acting downstream of Pink1 in parallel with Parkin. FASEB J. 2017;31:5234–5245. doi: 10.1096/fj.201700011R. [DOI] [PubMed] [Google Scholar]
- 57.Oboudiyat C, Glazer H, Seifan A, Greer C, Isaacson RS. Alzheimer’s disease. Semin. Neurol. 2013;33:313–329. doi: 10.1055/s-0033-1359319. [DOI] [PubMed] [Google Scholar]
- 58.Harris LD, Jasem S, Licchesi JDF. The Ubiquitin system in Alzheimer’s disease. Adv. Exp. Med. Biol. 2020;1233:195–221. doi: 10.1007/978-3-030-38266-7_8. [DOI] [PubMed] [Google Scholar]
- 59.Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32:150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 60.Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J. Biol. Chem. 2010;285:21978–21987. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dickey CA, et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl Acad. Sci. USA. 2008;105:3622–3627. doi: 10.1073/pnas.0709180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saidi LJ, et al. Carboxy terminus heat shock protein 70 interacting protein reduces tau-associated degenerative changes. J. Alzheimer’s Dis. 2015;44:937–947. doi: 10.3233/JAD-142094. [DOI] [PubMed] [Google Scholar]
- 64.Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- 65.Goryunov D, Liem RK. CHIP-ping away at tau. J. Clin. Investig. 2007;117:590–592. doi: 10.1172/JCI31505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dickey CA, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sahara N, et al. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005;94:1254–1263. doi: 10.1111/j.1471-4159.2005.03272.x. [DOI] [PubMed] [Google Scholar]
- 68.Lee S, et al. Sulforaphane upregulates the heat shock protein co-chaperone CHIP and clears amyloid-beta and Tau in a mouse model of Alzheimer’s Disease. Mol. Nutr. food Res. 2018;62:e1800240. doi: 10.1002/mnfr.201800240. [DOI] [PubMed] [Google Scholar]
- 69.Murphy MP, LeVine H., 3rd Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Zhou W, Tong Y, He G, Song W. Control of APP processing and Abeta generation level by BACE1 enzymatic activity and transcription. FASEB J. 2006;20:285–292. doi: 10.1096/fj.05-4986com. [DOI] [PubMed] [Google Scholar]
- 71.Cai H, et al. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 72.Del Prete D, Rice RC, Rajadhyaksha AM, D’Adamio L. Amyloid Precursor Protein (APP) may act as a substrate and a recognition unit for CRL4CRBN and Stub1 E3 ligases facilitating ubiquitination of proteins involved in presynaptic functions and neurodegeneration. J. Biol. Chem. 2016;291:17209–17227. doi: 10.1074/jbc.M116.733626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar P, et al. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum. Mol. Genet. 2007;16:848–864. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- 74.Singh AK, Pati U. CHIP stabilizes amyloid precursor protein via proteasomal degradation and p53-mediated trans-repression of beta-secretase. Aging Cell. 2015;14:595–604. doi: 10.1111/acel.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 76.Bertram L, et al. Family-based association between Alzheimer’s disease and variants in UBQLN1. N. Engl. J. Med. 2005;352:884–894. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- 77.Oddo S, et al. Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J. Neurosci. 2008;28:12163–12175. doi: 10.1523/JNEUROSCI.2464-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meng Y, et al. Toll-like receptor-2 ligand peptidoglycan upregulates expression and ubiquitin ligase activity of CHIP through JNK pathway. Cell. Physiol. Biochem. 2013;32:1097–1105. doi: 10.1159/000354509. [DOI] [PubMed] [Google Scholar]
- 79.Hiyoshi H, et al. 2-(4-Hydroxy-3-methoxyphenyl)-benzothiazole suppresses tumor progression and metastatic potential of breast cancer cells by inducing ubiquitin ligase CHIP. Sci. Rep. 2014;4:7095. doi: 10.1038/srep07095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheng XR, et al. A co-module regulated by therapeutic drugs in a molecular subnetwork of Alzheimer’s disease identified on the basis of traditional chinese medicine and SAMP8 mice. Curr. Alzheimer Res. 2015;12:870–885. doi: 10.2174/1567205012666150710111858. [DOI] [PubMed] [Google Scholar]
- 81.Zhang GR, Cheng XR, Zhou WX, Zhang YX. Age-related expression of STUB1 in senescence-accelerated mice and its response to anti-Alzheimer’s disease traditional Chinese medicine. Neurosci. Lett. 2008;438:371–375. doi: 10.1016/j.neulet.2008.04.075. [DOI] [PubMed] [Google Scholar]
- 82.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 83.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Urushitani M, et al. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 2004;90:231–244. doi: 10.1111/j.1471-4159.2004.02486.x. [DOI] [PubMed] [Google Scholar]
- 86.Niwa J, et al. Dorfin ubiquitylates mutant SOD1 and prevents mutant SOD1-mediated neurotoxicity. J. Biol. Chem. 2002;277:36793–36798. doi: 10.1074/jbc.M206559200. [DOI] [PubMed] [Google Scholar]
- 87.Ishigaki S, et al. Dorfin-CHIP chimeric proteins potently ubiquitylate and degrade familial ALS-related mutant SOD1 proteins and reduce their cellular toxicity. Neurobiol. Dis. 2007;25:331–341. doi: 10.1016/j.nbd.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 88.Niwa J, et al. A novel centrosomal ring-finger protein, dorfin, mediates ubiquitin ligase activity. Biochem. Biophys. Res. Commun. 2001;281:706–713. doi: 10.1006/bbrc.2001.4414. [DOI] [PubMed] [Google Scholar]
- 89.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sakamoto KM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteom. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 91.Oyake D, Nishikawa H, Koizuka I, Fukuda M, Ohta T. Targeted substrate degradation by an engineered double RING ubiquitin ligase. Biochem. Biophys. Res. Commun. 2002;295:370–375. doi: 10.1016/s0006-291x(02)00673-3. [DOI] [PubMed] [Google Scholar]
- 92.Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 93.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 94.Nagai Y, Popiel HA. Conformational changes and aggregation of expanded polyglutamine proteins as therapeutic targets of the polyglutamine diseases: exposed beta-sheet hypothesis. Curr. Pharm. Des. 2008;14:3267–3279. doi: 10.2174/138161208786404164. [DOI] [PubMed] [Google Scholar]
- 95.Takeuchi T, Nagai Y. Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Res. 2017;7:128. doi: 10.3390/brainsci7100128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morishima Y, et al. CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum. Mol. Genet. 2008;17:3942–3952. doi: 10.1093/hmg/ddn296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Adachi H, et al. CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J. Neurosci. 2007;27:5115–5126. doi: 10.1523/JNEUROSCI.1242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Al-Ramahi I, et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J. Biol. Chem. 2006;281:26714–26724. doi: 10.1074/jbc.M601603200. [DOI] [PubMed] [Google Scholar]
- 99.Miller VM, et al. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J. Neurosci. 2005;25:9152–9161. doi: 10.1523/JNEUROSCI.3001-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jana NR, et al. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005;280:11635–11640. doi: 10.1074/jbc.M412042200. [DOI] [PubMed] [Google Scholar]
- 101.Choi JY, et al. Co-chaperone CHIP promotes aggregation of ataxin-1. Mol. Cell. Neurosci. 2007;34:69–79. doi: 10.1016/j.mcn.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 102.Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL. In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol. Dis. 2009;33:342–353. doi: 10.1016/j.nbd.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Holmes G. A form of familial degeneration of the cerebellum. Brain. 1908;30:466–489. [Google Scholar]
- 104.Shi CH, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet. 2014;23:1013–1024. doi: 10.1093/hmg/ddt497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Margolin DH, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cordoba M, Rodriguez-Quiroga S, Gatto EM, Alurralde A, Kauffman MA. Ataxia plus myoclonus in a 23-year-old patient due to STUB1 mutations. Neurology. 2014;83:287–288. doi: 10.1212/WNL.0000000000000600. [DOI] [PubMed] [Google Scholar]
- 107.Depondt C, et al. Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology. 2014;82:1749–1750. doi: 10.1212/WNL.0000000000000416. [DOI] [PubMed] [Google Scholar]
- 108.Chiu HH, et al. Clinical and genetic characterization of autosomal recessive spinocerebellar Ataxia Type 16 (SCAR16) in Taiwan. Cerebellum. 2020;19:544–549. doi: 10.1007/s12311-020-01136-4. [DOI] [PubMed] [Google Scholar]
- 109.Olszewska DA, Kinsella JA. Extending the phenotypic spectrum associated with STUB1 mutations: a case of dystonia. Mov. Disord. Clin. Pract. 2020;7:318–324. doi: 10.1002/mdc3.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shi Y, et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PloS ONE. 2013;8:e81884. doi: 10.1371/journal.pone.0081884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gazulla J, Izquierdo-Alvarez S, Sierra-Martinez E, Marta-Moreno ME, Alvarez S. Inaugural cognitive decline, late disease onset and novel STUB1 variants in SCAR16. Neurological Sci. 2018;39:2231–2233. doi: 10.1007/s10072-018-3545-5. [DOI] [PubMed] [Google Scholar]
- 112.Synofzik M, et al. Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J. Rare Dis. 2014;9:57. doi: 10.1186/1750-1172-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Turkgenc B, et al. STUB1 polyadenylation signal variant AACAAA does not affect polyadenylation but decreases STUB1 translation causing SCAR16. Hum. Mutat. 2018;39:1344–1348. doi: 10.1002/humu.23601. [DOI] [PubMed] [Google Scholar]
- 114.Hayer SN, et al. STUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations. Orphanet J. Rare Dis. 2017;12:31. doi: 10.1186/s13023-017-0580-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bettencourt C, et al. Clinical and neuropathological features of spastic Ataxia in a Spanish family with novel compound heterozygous mutations in STUB1. Cerebellum. 2015;14:378–381. doi: 10.1007/s12311-014-0643-7. [DOI] [PubMed] [Google Scholar]
- 116.Heimdal K, et al. STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity. Orphanet J. Rare Dis. 2014;9:146. doi: 10.1186/s13023-014-0146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garcia AM, et al. The cerebellum and embodied semantics: evidence from a case of genetic ataxia due to STUB1 mutations. J. Med. Genet. 2017;54:114–124. doi: 10.1136/jmedgenet-2016-104148. [DOI] [PubMed] [Google Scholar]
- 118.Madrigal SC, et al. Changes in protein function underlie the disease spectrum in patients with CHIP mutations. J. Biol. Chem. 2019;294:19236–19245. doi: 10.1074/jbc.RA119.011173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Genis D, et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48) Neurology. 2018;91:e1988–e1998. doi: 10.1212/WNL.0000000000006550. [DOI] [PubMed] [Google Scholar]
- 120.Clarkson YL, Gillespie T, Perkins EM, Lyndon AR, Jackson M. Beta-III spectrin mutation L253P associated with spinocerebellar ataxia type 5 interferes with binding to Arp1 and protein trafficking from the Golgi. Hum. Mol. Genet. 2010;19:3634–3641. doi: 10.1093/hmg/ddq279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lise S, et al. Recessive mutations in SPTBN2 implicate beta-III spectrin in both cognitive and motor development. PloS Genet. 2012;8:e1003074. doi: 10.1371/journal.pgen.1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Palvadeau R, et al. Cerebellar cognitive-affective syndrome preceding ataxia associated with complex extrapyramidal features in a Turkish SCA48 family. Neurogenetics. 2020;21:51–58. doi: 10.1007/s10048-019-00595-0. [DOI] [PubMed] [Google Scholar]
- 123.Mol MO, et al. Clinical and pathologic phenotype of a large family with heterozygous STUB1 mutation. Neurol. Genet. 2020;6:e417. doi: 10.1212/NXG.0000000000000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen DH, et al. Heterozygous STUB1 missense variants cause ataxia, cognitive decline, and STUB1 mislocalization. Neurol. Genet. 2020;6:1–13. doi: 10.1212/NXG.0000000000000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.De Michele G, et al. Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: a report of two Italian families. Parkinsonism Relat. Disord. 2019;65:91–96. doi: 10.1016/j.parkreldis.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 126.Lieto M, et al. The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur. J. Neurol. 2020;27:498–505. doi: 10.1111/ene.14094. [DOI] [PubMed] [Google Scholar]