

Summary
ER-associated degradation (ERAD) targets misfolded ER proteins for degradation. Retrotranslocation, a key feature of ERAD, entails removal of ubiquitinated substrates into the cytosol for proteasomal destruction. Recently, it has been shown that the Hrd1 E3 ligase forms a retrotranslocation channel for luminal (ERAD-L) substrates. Conversely, our studies found that integral membrane (ERAD-M) substrates exit the ER through a distinct pathway mediated by the Dfm1 rhomboid protein. Those studies also revealed a second, Hrd1-dependent pathway of ERAD-M retrotranslocation can arise in dfm1Δ null. Here we show that, in the dfm1Δ null, the HRD complex undergoes remodeling to a form that mediates ERAD-M retrotranslocation. Specifically, Hrd1's normally present stochiometric partner Hrd3 is efficiently removed during suppressive remodeling, allowing Hrd1 to function in this novel capacity. Neither Hrd1 autoubiquitination nor its cytosolic domain is required for suppressive ERAD-M retrotranslocation. Thus, the HRD complex displays remarkable functional flexibility in response to ER stress.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
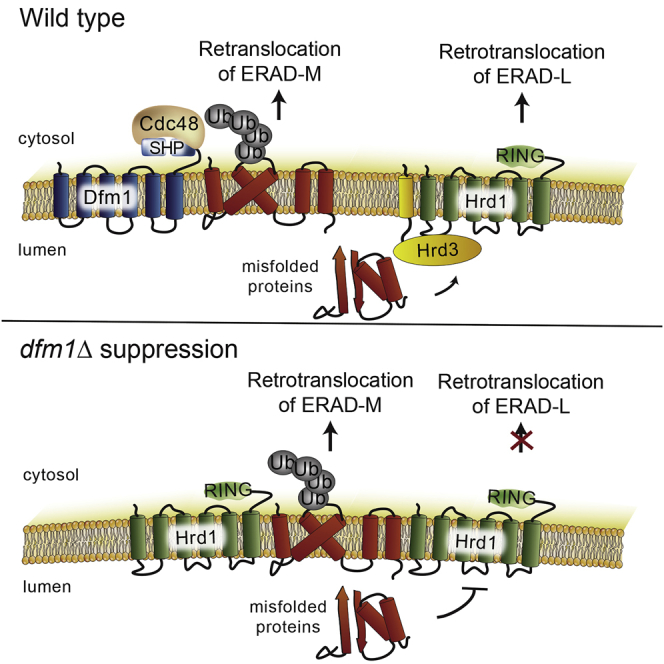
Highlights
-
•
Rhomboid derlin Dfm1 is the major mediator of ERAD-M retrotranslocation
-
•
In dfm1Δ nulls, there is a growth stress imposed by high levels of ERAD-M substrates
-
•
In dfm1Δ, Hrd1 is elevated and Hrd3 is removed to restore ERAD-M retrotranslocation
-
•
Hrd1's autoubiquitination activity is dispensable for restored ERAD-M retrotranslocation
Biological Sciences; Molecular Biology; Cell Biology
Introduction
About one-third of the eukaryotic cellular proteome is folded and matured in the endoplasmic reticulum (ER) (Sicari et al., 2019). Often, proteins fail to fold or assemble properly and are eliminated by the ER-associated degradation (ERAD) pathway (Sun and Brodsky, 2019). ERAD is a highly conserved, quality control pathway that selectively targets misfolded or unassembled proteins for degradation. Failure to remove misfolded proteins from the ER leads to ER stress that is associated with a variety of human maladies including retinal degeneration, neurodegenerative diseases, type II diabetes, and cancer (Bhattacharya and Qi, 2019).
ERAD involves recognition of protein substrates, ubiquitination by ER resident ubiquitin E3 ligases, and transfer of the ubiquitinated clients to the cytosol for degradation by the 26S proteasome (Richly et al., 2005). In S. cerevisiae, ERAD is mediated by the HRD (HMG-CoA Reductase Degradation) and DOA (Degradation of alpha 2) pathways, both of which are conserved in all eukaryotes (Carvalho et al., 2006; Chen et al., 2006; Foresti et al., 2013; Hampton and Garza, 2009). In the HRD pathway, the E3 ubiquitin ligase Hrd1 recognizes and ubiquitinates a variety of substrates, including misfolded membrane substrates (ERAD-M), lumenal substrates (ERAD-L), and some normal proteins such as the Hmg2 isozyme of HMG-CoA reductase (Plemper et al., 1998; Vashist and Ng, 2004; Wangeline et al., 2017). In the DOA pathway, the E3 ubiquitin ligase Doa10 recognizes and ubiquitinates misfolded soluble and membrane proteins, often with lesions in the cytosolic domain (ERAD-C) (Huyer et al., 2004; Nakatsukasa et al., 2008).
A unifying feature of all ERAD pathways is the requirement for moving substrates from the ER to the cytosol for proteasomal degradation. This transport process is broadly referred to as retrotranslocation or dislocation (Baldridge and Rapoport, 2016; Garza et al., 2009a; Hiller et al., 1996; Nakatsukasa et al., 2008; Neal et al., 2018). Retrotranslocation requires the Cdc48 ATPase (p97 in mammals) as an energy source for substrate removal, entailing extraction of integral membrane substrates from ER membrane or movement of luminal ERAD-L substrates across the ER membrane (Braun et al., 2002; Meyer et al., 2000; Neal et al., 2017; Ye et al., 2001). The generality and high conservation of retrotranslocation has resulted in many studies on the mechanism(s) at play in this process.
Recent work has cast needed light on the machinery involved in this critical transfer process, although many questions remain. Because retrotranslocation of either lumenal (ERAD-L) or integral membrane (ERAD-M) substrates were expected to require a channel or other catalytic enhancement, much work has been focused on identifying candidate molecules involved (Carvalho et al., 2010; Garza et al., 2009a; Hampton and Sommer, 2012; Nakatsukasa et al., 2016; Plemper et al., 1997; Scott and Schekman, 2008; Wahlman et al., 2007). From in vitro, in vivo, and structural studies, it now appears that the multi-spanning E3 ligase Hrd1 serves as a channel for lumenal ERAD-L substrates (Baldridge and Rapoport, 2016; Peterson et al., 2019; Schoebel et al., 2017; Vasic et al., 2020). Until recently, the identity of an analogous channel for ERAD-M substrates had remained unclear. Our earlier work demonstrated that Hrd1 is dispensable for the full retrotranslocation of a self-ubiquitinating substrate (SUS), which allowed for the possibility of a Hrd1-independent route out of the ER membrane for actual ERAD-M substrates (Garza et al., 2009a). Going forward with this idea, we employed SUS-GFP to screen the complete collection of yeast mutants with the Single Plate Orf Compendium Kit (SPOCK) array, consisting of a 5,808 yeast strain array of non-essential gene deletion mutants and essential DAmP gene mutants (Jaeger et al., 2018; Neal et al., 2018). This work led to our identification of Dfm1 as an independent, dedicated, and specific mediator for retrotranslocating ERAD-M substrates including those from both the HRD and DOA pathways, and Hrd1 itself in circumstances when it is rapidly degraded by self-ubiquitination (Neal et al., 2018).
Dfm1's role in ERAD-M was initially perplexing because we had much earlier (2006) reported that Dfm1 was not involved in either HRD or DOA-dependent ERAD (Goder et al., 2008; Sato and Hampton, 2006), whereas others had published a role for Dfm1 in only DOA-dependent branches of ERAD (Avci et al., 2014; Stolz et al., 2010). We resolved this discrepancy by showing loss of Dfm1 function along with the burden of strong ERAD-M substrate expression typical of the way we curated strains leads to rapid suppression, masking the phenotype within nine passages of a test culture (Neal et al., 2018). Importantly, retrotranslocation and degradation of ERAD-M substrates are completely restored in the suppressees. Accordingly, we characterized the nature of suppression to discover the apparent alternate route of ER membrane exit offered to ERAD-M substrates in the suppressed state (Neal et al., 2018). We found that the HRD1 gene was required for suppression of dfm1Δ nulls; the absence of Hrd1 resulted in no suppression of dfm1Δ strains, even after many passages. Furthermore, suppressees always had a full duplication of yeast chromosome XV, on which the HRD1 locus resides. However, providing Hrd1 on an autonomously maintained ARS/CEN plasmid resulted in suppression without duplication of chromosome XV, implying that the sole reason for the duplication was to amplify the HRD1 locus (Neal et al., 2018). Finally, all suppressed dfm1Δ null strains showed significant elevation of the Hrd1 protein, to approximately 5-fold the levels above normal expression from the single locus (Neal et al., 2018). Taken together, these results indicate that Hrd1 could adopt a new action as a retrotranslocation factor for ERAD-M, despite the absence of this ability in normal strains. A number of questions arose from these intriguing observations, including the nature of the stress that favors the chromosome XV aneuploids, the sufficiency of Hrd1 to allow ERAD-M dislocation, what prohibits Hrd1 from mediating ERAD-M retrotranslocation in normal circumstances, and the extent to which this new action of Hrd1 is related to its established role in the biophysically distinct process of ERAD-L retrotranslocation recently discovered.
In the studies below, we have thoroughly explored these and related questions. We have discovered that a novel and profound stress is imposed by high levels of ERAD-M substrates in the absence of Dfm1 and that elevated levels of Hrd1 can effectively alleviate this stress by providing a separate route of retrotranslocation. Hrd1 itself appears sufficient to perform this function, but the HRD complex underwent drastic remodeling for ERAD-M retrotranslocation to occur. This new action of Hrd1 could occur in the absence of its ubiquitination activity. Thus, the autoubiquitination mechanism that is critical for ERAD-L was not required for Hrd1-mediated ERAD-M retrotranslocation. These studies demonstrate the critical importance of managing membranes substrates in the ER and imply that the HRD machinery has unanticipated plasticity and breadth of function to meet the changing stresses of this key organelle.
Results
Integral Membrane Substrates Cause a Growth Defect in dfm1Δ
dfm1Δ null strains can undergo rapid phenotypic suppression, causing our early misimpression of Dfm1's non-involvement in ERAD (Neal et al., 2018). Rapid suppression of dfm1Δ strains requires high expression levels of ERAD-M substrates like Hmg2 or SUS-GFP, whereas long-term passage of dfm1Δ strains (over 30 passages tested, Neal et al., 2018) in the absence of strongly expressed substrates maintain a ERAD-M-deficient phenotype. We noted that suppressees acquire a duplicated chromosome XV as a path to suppression, which is rarely observed in normal strains (Neal et al., 2018). These observations imply that strong expression of ERAD-M substrates in dfm1Δ cells induces a growth stress that brings about selection for rare chromosome XV duplicant, suppressed strains. To test this idea, we developed a dilution plate assay to directly examine the effect of strongly expressing ERAD-M substrates on dfm1Δ null growth. The expression of each ERAD-M substrate was controlled by the strong, inducible GAL1-10 promoter (GAL-10pr) that is turned off when cells are grown on glucose medium and strongly induced when cells are grown on galactose. Liquid cultures of cells grown in glucose (no substrate expression) were plated at decreasing dilution onto solid medium with galactose or glucose as the carbon source to evaluate the effect of strong expression on cell growth. Plates were incubated for 3–7 days as indicated (Figures 1A–1F) to evaluate capacity for growth. We first tested the effect of Hmg2, a classic HRD pathway substrate (Hampton et al., 1996). As expected from the suppression experiments, galactose-mediated induction of Hmg2 expression resulted in strikingly slow growth of dfm1Δ null strains (Figure 1A). The slow growth phenotype did not occur in dfm1Δ cells with an empty vector or wild-type (WT) cells with or without galactose-driven Hmg2 (Figures 1A and S1A). Slow growth was also observed with high levels of a variety of other ERAD-M substrates including the constitutively degraded 6myc-Hmg2, Sec61-2, Pdr5∗, or SUS (Figures 1B–1E and S1B–S1E). Interestingly, there was no growth defect observed in dfm1Δ nulls containing overexpressed ERAD-L substrate, CPY∗, or wild-type Sec61 (Figures 1F, 1G, and S1F) suggesting the growth defect in dfm1Δ is specific to integral membrane ERAD substrates targeted for ubiquitination and degradation. Importantly, the growth defect observed is highly specific to dfm1Δ nulls: no other ERAD stabilizing mutants tested, including the HRD ligase mutants (hrd1Δ), exhibited slow growth with strongly expressed integral membrane substrates (Figures 1A–1E and S1A–S1E).
Figure 1.

Strongly Expressed Integral Membrane Substrates Cause a Growth Defect in dfm1Δ Cells
(A) Galactose-induced Hmg2-GFP overexpression causes a growth defect in dfm1Δ cells. WT, dfm1Δ, and hrd1Δ cells either containing empty vector or GAL-driven Hmg2-GFP were compared for growth by dilution assay. Each strain was spotted 5-fold dilutions on glucose or galactose-containing plates to drive Hmg2-GFP overexpression, and plates were incubated at 30°C.
(B–F) WT, dfm1Δ, and hrd1Δ cells were spotted in 5-fold dilutions on glucose or galactose-containing plates to drive overexpression of 6xmyc-Hmg2-GFP, Sec61-2-GFP, Pdr5∗-HA, SUS-GFP, and CPY∗-HA.
(G and H) Same as (A) except non-passaged dfm1Δ and dfm1Δ hrd1Δ non-passaged cells (P0) or cells passaged to suppression (P11) were assessed for growth defect in the dilution assay.
(I) Same as (A) except dilution assay was performed using galactose-induced overexpression of WT Sec61-GFP or mutant Sec61-2-GFP.
(J) Same as (A) except dilution assay was performed on WT, dfm1Δ, ire1Δ, dfm1Δ ire1Δ cells using GAL-driven Hmg2-GFP.
dfm1Δ null strains strongly expressing ERAD-M substrates exhibited a strong growth defect owing to retrotranslocation being compromised. We surmised that suppressed strains with restored retrotranslocation function would no longer exhibit growth stress and have normal growth restored. To test this idea, dfm1Δ null strains were passaged in liquid culture containing galactose as the carbon source for turning on strong expression of ERAD-M substrate, Hmg2. These cells were passaged to suppression with retrotranslocation function completely restored and then subjected to the plate dilution assay to assess for growth fitness. As expected, no growth defect was observed in passaged suppressed dfm1Δ cells (P11) in comparison with non-passaged non-suppressed dfm1Δ cells (P0) (Figure 1H) demonstrating that suppressed dfm1Δ null strains with alleviated retrotranslocation function has normal growth fitness.
It has been established that strongly expressed ERAD-L substrates induce ER stress (Kimata and Kohno, 2011; Travers et al., 2000). However, the extreme stress observed by overexpressed ERAD-M substrates in dfm1Δ nulls is entirely new. We have not observed this effect in any ERAD mutants previously studied (Federovitch et al., 2008). This begs the question as to what pathway(s) are involved in this novel substrate-induced growth stress. One reasonable candidate is the Unfolded Protein Response (UPR), a well-known ER stress response pathway activated by accumulation of misfolded lumenal proteins within the ER lumen (Hetz et al., 2015; Huyer et al., 2004; Ron and Walter, 2007; Schröder and Kaufman, 2005; Walter and Ron, 2011). In yeast, Ire1 senses misfolded lumenal proteins and responds by upregulating ER factors to alleviate ER stress. Furthermore, expression of lumenal ERAD-L substrates in an ire1Δ null causes extreme growth stress (Liu and Chang, 2008). We wondered if Ire1 was similarly involved in sensing ERAD-M substrates in the suppression of dfm1Δ nulls. We first examined whether ire1Δ exhibited a similar slow growth phenotype as a dfm1Δ null with similarly high expression of ERAD-M substrates. When the ERAD-M substrate Hmg2 was strongly expressed, there was no growth stress observed in ire1Δ alone. However, a strong genetic interaction was observed for Dfm1 and Ire1: dfm1Δ ire1Δ cells were killed with high levels of integral membrane substrates (Figure 1I), rather than the slow growth caused in the single dfm1Δ null. This dfm1Δire1Δ synthetic lethality caused by strongly expressed Hmg2 suggests that Ire1 plays a role in the induced dfm1Δ suppression pathway, possibly by regulating the expression of a protein required for the restoration of ERAD-M in the dfm1Δ null suppressees.
Elevated Hrd1 Levels Were Sufficient for dfm1Δ Suppression
We previously reported that Hrd1 is absolutely required for dfm1Δ suppression and restoration of ERAD-M retrotranslocation function. Indeed, a growth defect was still observed in passaged dfm1Δ hrd1Δ cells (P11) in comparison with dfm1Δ cells (P11) (Figure 1J) demonstrating that Hrd1 is required to alleviate retrotranslocation function and normal growth fitness in dfm1Δ null strains. Furthermore, Hrd1 is found on chromosome XV, which was duplicated in dfm1Δ suppressees. Hrd1 appears to be the only chromosome XV gene that requires increased gene dosage to suppress dfm1Δ since providing Hrd1 on an ARS/CEN plasmid, which can stably exist in varying copy numbers within the cell (Flagg et al., 2019), allows suppression that no longer results in duplication of chromosome XV (Neal et al., 2018). This implies that elevated Hrd1 is sufficient for suppressing dfm1Δ and that duplicating chromosome XV is the quickest way for dfm1Δ cells to increase Hrd1 in a typical suppression experiment. Accordingly, we surmised that, if simply attaining high enough levels of Hrd1 was sufficient for restoring retrotranslocation in a dfm1Δ null, then supplying multiple copies of ARS/CEN HRD1 plasmid to dfm1Δ nulls would result in immediate suppression and degradation of SUS-GFP, without the need for outgrowth of rare cells with duplicated chromosome XV. To test this idea, we developed an “instant suppression assay” in which the ARS/CEN HRD1 plasmid is transformed into dfm1Δhrd1Δ cells strongly expressing SUS-GFP (Figure 2A). We reasoned that, if simply having high levels of HRD1 was sufficient to suppress the dfm1Δ deficiency, then those colonies with enough copies of the HRD1-expressing plasmid would immediately show the suppressed phenotype of restored SUS-GFP degradation. Indeed, approximately 10% of total transformants had low colony florescence, as would be the case for isolates that had restored SUS-GFP retrotranslocation and degradation. According to our model, we would expect a subset of colonies that were scored as suppressees to have a higher copy number of the HRD1 plasmid, which would meet the threshold requirement for suppression. Consistent with this idea, western blotting of Hrd1-myc levels in transformants scored as dark suppressees were ∼3- to 6-fold above the isolates that remained bright and were not suppressed (Figure 2B). Importantly, several colonies from the same transformation plate with high SUS-GFP signal indeed had low levels of Hrd1 (Figure 2B). Furthermore, biochemical analysis confirmed that retrotranslocation of SUS-GFP was restored in the instant suppressees (Figure 2D, lanes 5–8). These results indicate that increased HRD1 gene dosage was adequate for suppressing the dfm1Δ phenotype.
Figure 2.

Amplified Hrd1 Levels Are Sufficient for Suppressing dfm1Δ Cells
(A) Depiction of HRD1 gene dosage reporter assay. To assess HRD1 dosage required for instant suppression of dfm1Δ cells, we used an ARS/CEN plasmid containing myc-tagged HRD1 driven by its native promoter. The HRD1 plasmid was transformed into dfm1Δhrd1Δ cells overexpressing SUS-GFP. HRD1 plasmid copy number was quantified by western blotting with α-myc.
(B) Increased HRD1 copy number instantly suppressed dfm1Δhrd1Δ cells. dfm1Δhrd1Δ cells overexpressing SUS-GFP were transformed with an ARS/CEN HRD1 plasmid. Subsequently, transformants were screened for instantly suppressed non-fluorescent dark colonies. Both non-suppressed (bright colonies; n = 9) and suppressed cells (dark colonies; n = 9) were grown to log phase, lysed, and analyzed by SDS-PAGE and immunoblotted for steady-state levels of Hrd1 with α-myc and SUS with α-GFP. Mean ± SEM, ∗∗∗p < 0.001, nested t test.
(C and D) (C) Same as (B), except HRD1 plasmid was transformed into dfm1Δhrd1Δire1Δ cells overexpressing SUS-GFP. Mean ± SEM, ∗∗∗p < 0.001, Nested t test (D) in vivo SUS-GFP retrotranslocation is restored in instantly suppressed dfm1Δhrd1Δ cells. Crude lysate was prepared from the indicated strains treated with vehicle or MG132 (25 μg/mL). Lysates were ultracentrifuged to discern ubiquitinated SUS-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, SUS-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE and immunoblotted with α-GFP and α-Ubi.
(E) Same as (D), except non-suppressed and instantly suppressed dfm1Δhrd1Δire1Δ transformants were used in the in vivo retrotranslocation assay.
(F and G) (F) Strongly expressed Hrd1 instantly suppresses dfm1Δhrd1Δ + SUS-GFP cells. dfm1Δhrd1Δ +SUS-GFP cells were transformed with empty vector or TDH3pr-Hrd1. SUS-GFP levels were analyzed by flow cytometry. Histograms of 10,000 cells are shown, with the number of cells versus GFP fluorescence. Note: panels are aligned so all fluorescent histograms are comparable between panels. (G) Same as (B), except Hrd1 was strongly expressed in dfm1Δhrd1Δ cells containing SUS-GFP.
(H) Same as (C), except dfm1Δhrd1Δire1Δ cells with empty vector or strongly expressed Hrd1 was used in the in vivo retrotranslocation assay. Band intensities of Hrd1-Myc levels were quantified by ImageJ. Data are represented as pixel intensities of Hrd1-Myc levels (n = 9) from at least three experiments.
We previously reported that Hrd1-myc levels became elevated ∼5- to 10-fold in the course of passage-dependent suppression of dfm1Δ (Neal et al., 2018). Thus, a salient question is: how does duplication of chromosome XV during this suppression generate an ∼5- to 10-fold increase in Hrd1 levels when the HRD1 locus has only been doubled? One possibility is that ER stress regulator Ire1, which induces the expression of many genes including Hrd1 (Travers et al., 2000; Walter and Ron, 2011), is involved in the passage-dependent suppression of dfm1Δ. This suggests a model in which suppressive levels of Hrd1 are attained though a combination of duplicating the HRD1 locus combined with Ire1-dependent upregulation of the duplicated HRD1 gene. We next determined whether Ire1 had other roles in dfm1Δ suppression distinct from upregulated HRD1 expression. Accordingly, we asked if Ire1 was dispensable for dfm1Δ suppression when sufficient levels of Hrd1 were provided by alternate means. We used the above instant suppression assay to specifically ask if instant suppressees caused by transformation with the ARS/CEN HRD1 plasmid could still arise in a dfm1Δire1Δ cells. This was indeed the case: 10% of these transformants showed the tell-tale low colony fluorescence and complete restoration of SUS-GFP ERAD (Figures 2C and 2E). Accordingly, our results suggest Ire1 is only needed in passage-dependent suppression to allow the needed expression of Hrd1 from its duplicated locus.
The above experiments suggest that suppression of dfm1Δ requires only elevated Hrd1. In the initial passage-dependent suppression protocols described in recent work and above (Neal et al., 2018), the mechanism of Hrd1 elevation entails duplication of the HRD1 genomic locus combined with increased expression of HRD1 through Ire1 signaling. In the above “instant suppression” assay (Figures 2B and 2C), strains with sufficient expression of HRD1 from a variable copy plasmid were immediately suppressed without need for either multiple passages or the IRE1 gene. As a final test of the sufficiency of elevating Hrd1 as a route to dfm1Δ suppression, we reasoned that Hrd1 expression alone would similarly generate stably suppressed dfm1Δ cells if an appropriately strong promoter was used. To address this, we used a myc-tagged HRD coding region driven by a strong heterologous promoter, TDH3pr. We quantified the steady-state levels of SUS-GFP using flow cytometry and compared SUS-GFP levels in dfm1Δ cells containing empty vector control versus strongly expressed Hrd1. Indeed, dfm1Δ cells with Hrd1 expressed from the strong TDH3 promoter displayed complete suppression: rapid degradation of SUS-GFP (Figure 2F) was observed in dark transformation colonies and by measurement of fluorescence by flow cytometry. Complete suppression was confirmed by biochemical analysis showing complete restoration of SUS-GFP retrotranslocation (Figure 2G, lanes 5–8). Thus, sufficient expression of Hrd1 in dfm1Δ nulls generated stably suppressed strains immediately upon transformation without the need for passaging. As would be expected from the above experiments with the ARS/CEN plasmid-based “instant suppression,” the TDH3pr-driven Hrd1 expression plasmid also suppressed an ire1Δdfm1Δ double null, consistent with the only role of Ire1 in the original passage-based suppression experiments to allow heightened expression of the duplicated HRD1 genomic locus (Figure 2H).
Hrd3 and USA1 Were Not Required for Suppression
Hrd1's ability to substitute for Dfm1 raises the question of how this action relates to the known functions of the HRD complex. Normally, the core HRD complex is composed of the Hrd1 E3 ligase, with a cytoplasmic RING motif required for ubiquitination, the mostly lumenal Hrd3, and Usa1, with a cytoplasmic Ubiquitin-like (Ubl) domain (Carroll and Hampton, 2010; Hampton et al., 1996; Mehnert et al., 2015; Vashistha et al., 2016) (Figure 3A). Both Hrd3 and Usa1 are required for optimal Hrd1 function and stability when expressed at normal genomic levels (Vashistha et al., 2016). We next tested if these key components were required for Hrd1-mediated restoration of ERAD-M in the dfm1Δ nulls. We used the passaging-based suppression assay to test the requirement for Hrd3 and Usa1 in Hrd1-mediated suppression by asking if nulls of either gene still allowed suppression. Bright P0 dfm1Δusa1Δ or dfm1Δhrd3Δ cells expressing SUS-GFP were repeatedly passaged in fresh minimal media. As seen in previous runs of this experiment, dark suppressees emerged in each strain as indicated by the flow cytometry histograms, and at a rate identical to the controls with HRD3 and USA1 intact. Thus, neither Usa1 nor Hrd3 was required in the dfm1Δ suppression pathway (Figure 3B).
Figure 3.

Hrd3 Is Rapidly Degraded in dfm1Δ Suppressees
(A) Depiction of the HRD complex that comprises the E3 ligase Hrd1, its partner, Hrd3, and Usa1, which recognizes and ubiquitinates ERAD-M and ERAD-L substrates.
(B) Hrd3 and Usa1 is dispensable for suppression. The indicated strains overexpressing SUS-GFP were passaged to suppression. Cells were passaged and SUS-GFP levels were analyzed by flow cytometry. Histograms of 10,000 cells are shown, with the number of cells versus GFP fluorescence.
(C) Hrd3 is rapidly degraded in dfm1Δ suppressees. Steady-state levels of the indicated proteins were analyzed in dfm1Δ P0 and dfm1Δ P11 cells strongly expressing Hmg2-GFP. Cells were lysed and analyzed by SDS-PAGE and immunoblotted for Hrd1 with α-myc, Hmg2 with α-GFP, α-Cdc48, and Hrd3 with α-HA.
(D) Hrd3 is degraded by the proteasome in dfm1Δ suppressees. Steady-state levels of the indicated proteins were analyzed in dfm1Δ P0 and dfm1Δ P11 strains treated with vehicle or MG132 (25 μg/mL). Cells were lysed and analyzed by SDS-PAGE and immunoblotted for Hrd1 with α-myc and Hrd3 with α-HA.
(E) Degradation of Hrd3 was measured by CHX-chase assay in passaged suppressed dfm1Δ P11 strains treated with vehicle or MG132 (25 μg/mL). After CHX addition, cells were lysed at the indicated times, analyzed by SDS-PAGE, and immunoblotted for Hrd3 with α-HA. Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t = 0 was taken as 100%, and data are represented as mean ± SEM from at least three experiments, ∗∗∗p < 0.001, repeated measures ANOVA.
Hrd3 Is Rapidly Degraded upon dfm1Δ Null Suppression
Because Hrd1 is significantly elevated in suppressed dfm1Δ nulls, we examined the levels of the other core components of the HRD complex. Since Cdc48 recruitment by Dfm1 is essential for retrotranslocation (Neal et al., 2018), we also examined Cdc48 levels in suppressees. Immunoblotting of Cdc48 showed that steady-state levels of these components were unchanged between non-suppressed P0 and suppressed dfm1Δ P11 cells (Figure 3C). In striking contrast, Hrd3 was completely absent in the suppressed cells (Figure 3C). Moreover, the addition of proteasome inhibitor MG132 to suppressed cells restored Hrd3 levels indicating the normally highly stable Hrd3 was undergoing highly efficient proteasomal degradation in the suppressees, resulting in undetectable steady-state levels (Figures 3D and 3E), presumably due to Hrd1-mediated degradation, as discussed below. Because Hrd3 was not required for Hrd1-mediated suppression, the observed loss of Hrd3 by degradation might not be expected to have consequences in the acquisition of the suppressed state. However, this drastic change in Hrd3 levels upon suppression suggested two possibilities. It could be that Hrd3's loss was simply a side result of the suppressed state and not mechanistically important for Hrd1's acquired ability to mediate ERAD-M retrotranslocation. Alternatively, it could be that profound loss of Hrd3 was required for Hrd1-mediated restoration of ERAD-M in dfm1Δ suppressees. We next tested the mechanistic importance of Hrd3's loss in the suppressed state.
Forced Hrd3 Expression Reverses Hrd1-Mediated Suppression of dfm1Δ Cells
The above experiments indicate that suppression of dfm1Δ nulls occurs in response to a strong growth stress imposed on the null cells when high levels of ERAD-M substrate are expressed (Figures 1A–1E). The slow growth allows for selection of relatively rare chromosome XV duplicants that, in conjunction with Ire1 activation, allows sufficient expression of Hrd1 to alleviate the loss of retrotranslocation and thus restore normal growth. During this transition, we noted above that normally highly stable Hrd3 was efficiently removed by degradation to the point of undetectability (Figures 3C–3E). We next tested the importance of Hrd3 removal in dfm1Δ suppression; we tested the effect of reintroducing Hrd3 in already-suppressed dfm1Δ nulls. This was accomplished by using a GAL1-10 promoter (GAL-10pr)-inducible HRD3 expression plasmid, which was introduced to suppressed cells in promoter-suppressing glucose medium. Plasmid-bearing suppressees were then switched to galactose medium to observe the effects of strong Hrd3 expression. We hypothesized that, if Hrd3's presence blocks Hrd1-medated retrotranslocation, then galactose-induced elevation of Hrd3 in dfm1Δ suppressees would cause the strong growth defect seen when ERAD-M retrotranslocation is blocked. We used the dilution plate assay to determine whether overexpressing Hrd3 caused resumption of growth stress in suppressed dfm1Δ nulls expressing either ERAD-M substrates SUS-GFP (Figure 4A) or Hmg2-GFP (Figure 4B). In both cases, the presence of the galactose-inducible HRD3 coding region caused a drastic growth defect that was observed neither with empty vector nor when HRD3 was similarly elevated in WT cells or an ERAD-deficient hrd1Δ (Figures 4 and 4B). This Hrd3 toxicity implied an antagonistic role for Hrd3 for Hrd1-mediated retrotranslocation in the suppression pathway.
Figure 4.

Hrd3 Antagonizes Hrd1's Suppressive Retrotranslocation Function
(A and B) Galactose-induced Hrd3 overexpression causes cell lethality in dfm1Δ suppressed cells. WT, hrd1Δ, and dfm1Δ suppressed cells overexpressing SUS-GFP (A) or Hmg2-GFP (B) and harboring either empty vector or GAL-driven Hrd3 were compared for growth by dilution assay. Each strain was spotted 5-fold dilutions on glucose-containing or galactose-containing plates to drive Hrd3 overexpression, and plates were incubated at 30°C.
(C) Hrd3 overexpression blocks restoration of ERAD-M retrotranslocation. WT and dfm1Δ cells overexpressing SUS-GFP were grown in the presence of glucose to turn off Hrd3 expression and passaged to suppression at the indicated number of times into fresh minimal media (P11). P11WT and dfm1Δ cells were then passaged into minimal media containing galactose as a sole carbon source to trigger Hrd3 overexpression (P12). Flow cytometry was used to assess SUS-GFP steady-state levels. Histograms of 10,000 cells are shown, with the number cells versus GFP fluorescence.
(D) Degradation of SUS was measured by CHX-chase assay in passaged suppressed dfm1Δ strains containing either empty vector or GAL-driven Hrd3. After CHX addition, cells were lysed at the indicated times, analyzed by SDS-PAGE, and immunoblotted for SUS with α-GFP. Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t = 0 was taken as 100%, and data are represented as mean ± SEM from at least three experiments, ∗∗∗p < 0.001, Repeated measures ANOVA.
If the loss of Hrd3 was important for Hrd1 to perform suppressive retrotranslocation in the dfm1Δ null, then addition of sufficient levels of Hrd3 would be expected to block the restored degradation of ERAD-M substrates. We next employed the suppressed strains that could express Hrd3 under the control of the GAL1pr promoter described above to examine this question. When suppressed dfm1Δ cells harboring a GAL1-driven HRD3 coding region were grown in glucose so that no Hrd3 was expressed, suppressive retrotranslocation proceeded as expected, as indicated by low mean fluorescence and degradation of SUS-GFP and a normal growth rate (Figures 4B–4D). When these cells were placed in galactose medium to cause strong expression of Hrd3, the majority of the cell population displayed sudden lethality, which would be expected if the stress-alleviating suppression was suddenly compromised. As a control, strong expression of Hrd3 in WT cells caused no growth defect and did not lead to lethality. The fraction of cells that survived with severe growth stress showed strongly stabilized SUS-GFP indicating there was a block in retrotranslocation upon forced expression of Hrd3 (Figures 4C and 4D).
Hrd1's Ubiquitination Activity Was Dispensable for Suppressive ERAD-M Retrotranslocation
We previously reported that Hrd1's ubiquitination activity was required for dfm1Δ-associated suppression. Specifically, an inactive Hrd1 mutant (C399S) with ablated ubiquitination function is unable to support dfm1Δ’s route to suppression (Neal et al., 2018 and Figure 5A). One possibility is the requirement for Hrd1's ubiquitination function in catalyzing the retrotranslocation process. An alternative possibility is the requirement for Hrd1's ubiquitination activity in the ubiquitination and removal of Hrd3. Since the above studies indicate that Hrd3 removal was critical to permit Hrd1-mediated suppressive ERAD-M retrotranslocation, perhaps the reason inactive Hrd1 (C399S) could not support suppression, as we reported in our original studies of dfm1Δ suppression, was due to its inability to allow degradative removal of Hrd3. If that was true, then the prediction would be that in a hrd3Δ null background, C399S-Hrd1 would in fact be able to support dfm1Δ suppression. Accordingly, we repeated the dfm1Δ suppression experiment in a hrd3Δ null strain: dfm1Δhrd3Δ C399S-Hrd1 cells expressing SUS-GFP were passaged repeatedly in fresh minimal media. Remarkably, dark suppressees emerged at a rate identical to dfm1Δ control strains, whereas identical C399S-Hrd1 strains with the HRD3 gene intact resulted in no suppression (Figures 5B, S2A and S2B). Hence, inactive C399S-Hrd1 is able to support substrate-induced dfm1Δ suppression, as long as Hrd3 is not present. To test this idea further, we utilized a well-characterized truncated version of Hrd1, hemi-Hrd1, with the catalytic RING domain completely removed, leaving Hrd1's transmembrane domain intact (Gardner et al., 2000 and Figure 5A). Again, HRD3 was removed beforehand to test the requirement of hemi-Hrd1 for substrate-induced dfm1Δ suppression, and again, dfm1Δhrd3Δ hemi-Hrd1 cells showed fully restored degradation of SUS-GFP by nine passages (P9) and thus, the transmembrane domain of Hrd1 alone could support restoration of degradation in the dfm1Δ strains, so long as HRD3 was absent (Figure 5B). Importantly, SUS degradation is unaffected in the control single mutant backgrounds hrd3Δ, C399S-HRD1, and hemi-HRD1, indicating that the HRD complex mutants tested herein do not affect SUS in normal circumstances but only participate in the suppression pathway when Dfm1 is absent (Figure S2C).
Figure 5.

Hrd1's Ubiquitination Activity Is Dispensable for Suppressive ERAD-M Retrotranslocation
(A) Depiction of E3 ligase Hrd1, C399S-Hrd1, and hemi-Hrd1. C399S-Hrd1 is an inactive version of Hrd1 in which the essential cysteine is mutated in the RING finger. Hemi-Hrd1 is a truncated version Hrd1 only containing the transmembrane domain.
(B) Removal of Hrd3 allows RING-dead C399S and hemi-Hrd1 to support suppression and degradation of SUS-GFP when Dfm1 is absent. The indicated strains overexpressing SUS-GFP were passaged to suppression. Cells were passaged and SUS-GFP levels were analyzed by flow cytometry. Histograms of 10,000 cells are shown, with the number of cells versus GFP fluorescence.
(C) Overexpression of C399S-Hrd1 is sufficient for restoring retrotranslocation of SUS-GFP in dfm1Δhrd3Δ cells. Crude lysate was prepared from the indicated strains treated with vehicle or MG132 (25 μg/mL). Lysates were ultracentrifuged to discern ubiquitinated SUS-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, SUS-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE, and immunoblotted with α-GFP and α-Ubi.
(D) Strongly expressed hemi-Hrd1 is sufficient for restoring retrotranslocation of SUS-GFP in dfm1Δhrd3Δ cells. Same as (C) except dfm1Δhrd3Δ cells with empty vector or strongly expressed hemi-Hrd1 was used in the in vivo retrotranslocation assay.
(E) hemi-Hrd1 restores Cdc48 recruitment to ER membrane. Total cell lysate (T) from the indicated strains were separated into soluble cytosolic fraction (S) and pellet microsomal fraction (P) upon centrifugation at 14,000 × g. Each fraction was analyzed by SDS-PAGE and immunoblotted for Cdc48 with α-Cdc48 and PGK1 with α-PGK1. The graph shows the quantification of Cdc48 in the pellet fractions of the respective cells as measured from ImageJ. Data are represented as percentage of Cdc48 that is bound to pellet fraction and is shown as mean ± SEM from three independent experiments, ∗∗∗∗p < 0.0001, unpaired t test.
The above studies indicate that Hrd1's ubiquitination function is not required for restoring SUS-GFP retrotranslocation and degradation but rather serves to remove Hrd3, which impedes this action of Hrd1. We then performed a series of biochemical experiments to directly test if inactive C399S-Hrd1 and truncated hemi-Hrd1 indeed support the restoration of membrane substrate retrotranslocation. As demonstrated above, direct strong expression (from TDH3 promoter) of Hrd1 allowed rapid suppression of dfm1Δ strains without passaging of cells to attain suppression. This simpler approach allows for more direct analysis of the suppressed state and more rapid study of various questions. Hence, both inactive C399S-Hrd1 and hemi-Hrd1 expressed under control of the TDH3 promoter were transformed into dfm1Δ strains and directly tested for restored retrotranslocation of SUS-GFP. As expected, overexpression of C399S-Hrd1 or hemi-Hrd1 dfm1Δ cells with HRD3 intact failed to restore retrotranslocation, indicated by a buildup of ubiquitinated SUS-GFP in the pellet fraction in both untreated and MG132 treated cells (Figures 5C and 5D, lanes 1–4). The same experiment done with inclusion of the hrd3Δ null resulted in normal retrotranslocation of SUS-GFP, which was enhanced by MG132 (Figures 5C and 5D, lanes 5–8). This experiment indicates restoration of ERAD-M retrotranslocation does not require Hrd1's ubiquitination activity.
Previously, we developed an assay for Cdc48 binding to microsomal membranes and have shown that Cdc48 recruitment to the ER surface relied mainly on Dfm1 (Neal et al., 2018). In the case of suppressed cells, we observed restored Cdc48 binding even with no Dfm1 present. Accordingly, we employed the Cdc48 microsomal association assay to see if hemi-Hrd1 similarly restores Cdc48 recruitment in the suppressed state. Briefly, lysates from WT, dfm1Δhrd1Δ, and dfm1Δhrd1Δhrd3Δ cells with overexpressed hemi-Hrd1 were centrifuged at 20,000 × g to separate membrane pellet (P) from supernatant (S) (Figure 5E). Fractions were analyzed on SDS-PAGE and immunoblotted for Cdc48 and the cytoplasmic enzyme PGK1. In all strains, cytosolic PGK1 remained in the supernatant and no PGK1 associated with the microsomal pellet. Hrd1 overexpression in dfm1Δhrd1Δ cells showed the expected Cdc48 binding, with ∼20% of total Cdc48 associating with the microsome pellet. In contrast, hemi-Hrd1 overexpression alone did not support Cdc48 binding to microsomes in dfm1Δ hrd1Δ cells (Figure 5E). However, once Hrd3 was removed, hemi-Hrd1 showed full restoration of Cdc48 recruitment to the microsome in comparison with non-suppressed cells (Figure 5E). Overall, by all criteria examined, the rapid suppression by overexpressed catalytically dead Hrd1 was complete, in terms of substrate degradation, Cdc48 recruitment, and restored retrotranslocation. So long as the substrate is polyubiquitinated by its fused ubiquitinating domain (SUS-GFP) or by the Doa10 E3 ligase (Ste6∗-GFP), suppressive retrotranslocation does not require Hrd1's ligase activity.
Hrd1's Ubiquitination Activity Is Dispensable for Suppressive Retrotranslocation of DOA-Dependent Membrane Substrates
So far, by using SUS-GFP, we demonstrated that Hrd1-dependent retrotranslocation can occur in the absence of its ubiquitination activity. SUS-GFP contains Hrd1's catalytic RING domain, allowing it to undergo rapid self-ubiquitination independent of Hrd1. This valuable tool allows separate analysis of Hrd1's function distinct from its well-characterized self- or substrate ubiquitination. However, because the Hrd1 RING domain is present on the SUS substrate, it is formally possible this trans-expressed RING domain could interact with mutant Hrd1 derivatives (C399S and hemi-Hrd1) and thus create a “de facto” trans-associated Hrd1, as we have shown can occur in vivo (Gardner et al., 2000). To address this issue, we tested the ability of Hrd1 to restore retrotranslocation of ERAD-M substrates ubiquitinated by the Doa10 E3 ligase, thus obviating concerns surrounding inadvertent trans-reconstitution of full-length Hrd1. We first examined Ste6∗ in our suppression experiments, a classic multispanning DOA substrate, which mainly relies on E3 ligase Doa10 for ubiquitination and degradation (Figure S2D) (Huyer et al., 2004; Ravid et al., 2006). Because this substrate is retrotranslocated by Dfm1, but ubiquitinated primarily by Doa10, it allows examination of Hrd1-mediated suppressive retrotranslocation in the absence of the Hrd1 RING domain present on SUS-GFP. Briefly, CHX-chase was performed on dfm1Δ nulls strongly expressing inactive C399S or hemi-Hrd1 and Ste6∗-GFP degradation was monitored over time. By simply overexpressing wild-type Hrd1 in dfm1Δhrd1Δ double null cells, we observed instant suppression and degradation of Ste6∗-GFP (Figure 6A, left panel). When Hrd3 was normally expressed, strong expression of neither C399S nor hemi-Hrd1 could support suppression, as expected from the need for Hrd3 removal to allow Hrd1-mediated retrotranslocation (Figure 6A, left panel). As predicted from this model, strongly expressed C399S and hemi-Hrd1 were each able to support Ste6∗-GFP in the dfm1Δ, when HRD3 was removed beforehand indicating Hrd1's ubiquitination function is also dispensable for removing a DOA substrate, Ste6∗-GFP, from the ER membrane (Figure 6A, right panel). Importantly, Ste6 degradation was unaffected in DFM1 cells containing hrd3Δ or cells strongly expressing C399S or hemi-Hrd1 alone (Figure S2D).
Figure 6.

Hrd1's Ubiquitination Activity Is Not Required for Suppressive Retrotranslocation of DOA-Dependent Substrates, Ste6∗ and Vma12
(A) Inactive C399S-Hrd1 and hemi-Hrd1 restores degradation of Ste6∗-GFP in dfm1Δhrd3Δ cells. Degradation of Ste6∗-GFP was measured by CHX-chase assay in the indicated strains that either contained empty vector or C399S-Hrd1 and hemi-Hrd1 driven by a strong TDH3 promoter. After CHX addition, cells were lysed at the indicated times, analyzed by SDS-PAGE, and immunoblotted for Ste6∗ with α-GFP. Band intensities were normalized to PGK1 loading control and quantified by ImageJ. t = 0 was taken as 100%, and data are represented as mean ± SEM from at least three experiments, ∗∗∗p < 0.001, repeated measures ANOVA.
(B) VMA12-GFP fluorescence of indicated strains was examined by fluorescence microscopy. Equivalent number of cells were captured in each panel using identical settings. Scale bar, 5 μm.
(C) Microsomes were isolated from the indicated strains, and the steady-state levels of VMA12 were analyzed by SDS-PAGE and immunoblotted for VMA12 with α-GFP.
(D) dfm1Δ suppression results in restoration of VMA12 retrotranslocation and degradation. Steady-state levels of VMA12 was measured by fluorescence microscopy in the indicated strains that is either not passaged (P0) or passaged to suppression (P15).
(E) Steady-state levels of VMA12 was measured by western blotting in the indicated strains that is either not passaged (P0) or passaged to suppression (P15). Scale bar, 5 μm.
(F) C399S-Hrd1 and hemi-Hrd1 restores degradation of VMA12-GFP in dfm1Δhrd3Δ cells. VMA12-GFP steady-state levels were analyzed by western blotting with α-GFP in the indicated strains.
We next tested another well-characterized DOA-dependent membrane substrate, Deg1-Vma12-GFP, in our suppression experiments. Deg1-Vma12-GFP is an ER membrane protein with Deg1-a degron recognized by E3 ligase Doa10-fused to its N terminus, which renders it a DOA10 substrate (Ravid et al., 2006). Importantly, Deg1-Vma12-GFP degradation is entirely Doa10 mediated, with no dependence on Hrd1, allowing totally unambiguous separation of Hrd1's retrotranslocation function from its classic ubiquitination activity. Deg1 imparts remarkably rapid degradation to the fusion, such that the GFP signal is almost entirely absent in a wild-type strain where Doa10-mediated degradation occurs (Figure 6B). However, the Deg1-Vma12-GFP signal is easily visible and equally strong in either the doa10Δ or the dfm1Δ null but undetectable in a hrd1Δ null (Figure 6B). We also evaluated degradation of Deg1-Vma12-GFP by immunoblotting. In our wild-type strains, steady-state levels of Deg1-Vma12-GFP were undetectable from whole-cell yeast lysates but were detectable in isolated microsomes. Using microsomes, we observed a dramatic elevation of Deg1-Vma12-GFP steady-state levels in doa10Δ cells, whereas WT or hrd1Δ cells had undetectable levels of Deg1-Vma12-GFP (Figure 6C, left panel). Hence, in agreement to previous studies, this substrate is targeted by E3 ligase Doa10 for ubiquitination and does not require Hrd1 for ubiquitination or degradation (Ravid et al., 2006). Furthermore, in pdr5Δ cells treated with proteasome-inhibitor, MG132, Deg1-Vma12-GFP levels were stabilized indicating this substrate is ultimately degraded by the proteasome (Figure 6C, right panel). The extent of Deg1-Vma12-GFP stabilization caused by doa10Δ was as strong as that caused by dfm1Δ (Figures 6B and 6C). This is consistent with our hypothesis that Dfm1's involvement in ERAD-M retrotranslocation extends to both HRD and DOA branches (Neal et al., 2018). We used both microscopy and microsome immunoblotting to study the role of Hrd1 in dfm1Δ suppression of this Hrd1-independent ERAD-M substrate. As indicated in both assays, by fifteen passages (P15), retrotranslocation and degradation of Deg1-Vma12-GFP were restored, as indicated by undetectable steady-state levels (Figures 6D and 6E). Despite the complete non-involvement of Hrd1 ubiquitination in Deg1-Vma12 degradation, the suppression was entirely dependent on Hrd1: the dfm1Δhrd1Δ double null failed to undergo suppression: even after 15 passages, the levels of Deg1-Vma12-GFP remained stabilized in the double null (Figures 6D and 6E).
The independence of Deg1-Vma12-GFP degradation from Hrd1 ubiquitination activity allowed us to test if Hrd1 supports retrotranslocation of Deg1-Vma12-GFP when its ligase activity is ablated in the absence of the Hrd1-RING domain usually found in SUS-GFP. We used “instant” suppression brought about by direct strong expression of the various Hrd1 test constructs from the TDH3 promoter, as described above. As expected from the above studies with SUS or Ste6∗-GFP, TDH3-driven native Hrd1 could support Deg1-Vma12-GFP degradation in the hrd1Δdfm1Δ strain, whereas neither C399S- nor hemi-Hrd1 could support restored degradation when HRD3 was present (Figure 6F “hrd1Δ dfm1Δ” group). In striking contrast, when HRD3 was absent from these same strains (right panel “hrd3Δ”), degradation of Deg1-Vma12-GFP was fully restored by strong expression of either inactive Hrd1 variant, resulting in low steady-state levels due to rapid degradation (Figure 6F). Altogether, all HRD and DOA pathways membrane substrates so far tested undergo suppressive Hrd1-dependent retrotranslocation in the absence of its catalytic ubiquitination function, so long as HRD3 is absent.
Remodeled HRD Complex no Longer Functions in ERAD-L
Our findings reveal Hrd1-mediated translocation of membrane substrates is dispensable of its partner component, Hrd3, as well as its ubiquitination activity. Recent studies suggest Hrd1-mediated translocation of ERAD-L occurs through self-ubiquitination activity of Hrd1, which has also been shown to be regulated by Hrd3 (Baldridge and Rapoport, 2016; Peterson et al., 2019; Vasic et al., 2020). This led us to posit whether HRD complex remodeling in the suppression pathway negatively affects ERAD-L. CHX-chase assay was used to monitor the degradation rate of ERAD-L substrates, CPY∗ and KHN, in dfm1Δ suppressed cells overexpressing SUS-GFP as well (Figures S3A and S3B). Interestingly, both CPY∗ and KHN degradation was stabilized in suppressed cells. Hence, ERAD-M retrotranslocation is restored in the suppression pathway, whereas ERAD-L is compromised.
Discussion
This study adds a new dimension to our understanding of the Hrd1 E3 ligase and the functional plasticity of the HRD complex. In normal circumstances, Dfm1 appears to be the only mediator of ERAD-M retrotranslocation, with a broad range of integral membrane substrates from both the HRD or DOA pathways (Neal et al., 2018). As shown in our first studies of Dfm1, its dedicated route of ERAD-M even applies to Hrd1 itself: in conditions where Hrd1 self-ubiquitination leads to its very rapid degradation, Dfm1 is absolutely required for Hrd1 retrotranslocation, despite Hrd1's clearly demonstrated role as a ERAD-L retrotranslocon (Neal et al., 2018). This strict boundary makes sense, considering that Hrd1 undergoes an elaborate cycle of self-ubiquitination and de-ubiquitination in the course of its dual roles as a ligase and a lumenal retrotranslocon (Peterson et al., 2019; Vasic et al., 2020). If Hrd1 could mediate its own retrotranslocation in normal circumstances, that might lead to catastrophic loss of this key quality control factor in the course of its normal molecular duty cycle. There may be other evolutionary reasons for separate routes of membrane and lumenal dislocation as well; the approaches and information from our and other's recent studies set the stage for exploring these intriguing questions.
Our discovery of Dfm1's broad and critical role in ERAD-M retrotranslocation was delayed for over 10 years by what we now know to be a rapid suppression mechanism that in the correct circumstances masks the critical role of Dfm1 in ERAD (Neal et al., 2018). In dfm1Δ null cells that have undergone suppression, retrotranslocation of ERAD-M substrates is fully restored, implying a separate and very efficient route of retrotranslocation could occur. This suppression mechanism was first described and characterized in our original description of Dfm1 as an ERAD-M retrotranslocon. In that work, we showed that elevation of Hrd1 was a central feature of suppression, indicating that Hrd1 might be able to participate in ERAD-M retrotranslocation in some conditions despite its usual non-participation in this branch of ERAD (Neal et al., 2018). This surprising finding demanded investigation both to understand the details of Dfm1 action and to more broadly explore the functional plasticity of the HRD machinery.
The suppression of dfm1Δ strains was originally characterized in a simple passaging assay (Neal et al., 2018). In null strains strongly expressing an ERAD-M substrate, suppression occurs rapidly, typically in less than 10 passages. The studies above show that in passage-derived suppressees, a fairly byzantine mechanism allows amplification of Hrd1 to levels sufficient for restoration of ERAD-M: the entire Hrd1 chromosome is amplified, combined with Ire1-stimulated HRD1 expression to bring about the >5-fold increase on Hrd1 needed for suppression. Thus, a key feature of the original passage-based suppression is the uniform appearance of the duplicated chromosome XV. This is normally a very rare event that we have been unable to observe in simple plating assays for a GFP-labeled chromosome XV to track for frequency of duplication (S. Neal, D. Lam, unpublished observation). In exploring how such a rare duplication can occur with total uniformity, we discovered that loss of Dfm1 function caused a drastic cellular stress when present in conjunction with elevated levels of an ERAD-M substrate. The growth stress is profound (over 600-fold by plating) and thus allows for a strong selection for the rare duplicant suppressees, which did indeed have a restored normal growth rate along with restored ERAD-M retrotranslocation. Remarkably, this strong growth phenotype is unique to dfm1Δ strains: other equally strong ERAD-deficient mutants both upstream or downstream of Dfm1 (hrd1Δ or cdc48-2) show no growth stress upon similar elevation of ERAD-M substrates. Thus, the growth effects above suggest the intriguing possibility that Dfm1 has a unique role in this novel ER stress. This possible function for Dfm1 in managing membrane-protein stress may be part of a larger role for rhomboids in stress physiology. For example, mitochondrial rhomboid protease PARL is a key participant in mitochondrial stress response (Hill and Pellegrini, 2010). Whether the above growth effects represent a stress unique to Dfm1 function, or are a more general phenomenon of rhomboid biology, the assays above provide a facile and genetically tractable way to explore this aspect of Dfm1 action. It is critical to highlight that, although strong expression of ERAD-L substrates has been known to trigger the UPR (Kimata and Kohno, 2011; Ron and Walter, 2007; Schröder and Kaufman, 2005; Walter and Ron, 2011), the extreme stress caused by strongly expressed ERAD-M substrates in dfm1Δ seems to be something distinct. There appears to be no activation of the classical UPR upon initial strong expression of ERAD-M substrates (our observation) in the dfm1Δ nulls. Several questions are raised from this dfm1Δ-associated stress state: (1) How do membrane substrates cause the growth defect phenotype? (2) How are cells negatively affected from strong expression of membrane substrates when Dfm1 is absent? Intriguingly, SUS-GFP, which contains a stably folded domain, Hmg1, also elicits growth stress in the absence of Dfm1. This raises the possibility that the source of stress is not from substrate buildup but from excessive ubiquitination of substrates at the ER membrane. Understanding the extent that protein misfolding and/or ubiquitination is the source of Dfm1-mitigated stress will reveal new features of dfm1Δ-associated stress physiology.
A variety of experiments indicated that the only outcome of these complex events needed for re-establishment of ERAD-M retrotranslocation is elevation of Hrd1 levels; neither UPR nor any other portion of chromosome XV was needed for Hrd1-dependent suppression. Although IRE1 was indeed needed for the original growth-based assays when DFM1 was absent, this was due to the contribution that the UPR made to establishing elevated Hrd1 levels expressed from the duplicated chromosome with its native UPR-responsive promoter. If Hrd1 was instead expressed from a strong promoter to sufficient levels, then the requirement for IRE1 was removed. It is worth noting, however, that the requirement for IRE1 is consistent with the inability of the remodeled, dfm1Δ suppressing HRD complex to support ERAD-L as demonstrated above, since loss of ERAD-L can lead to UPR activation (Kimata and Kohno, 2011; Ron and Walter, 2007; Schröder and Kaufman, 2005; Walter and Ron, 2011).
The above studies of dfm1Δ suppressees revealed that the HRD complex undergoes surprising remodeling concomitant with the restoration of ERAD-M retrotranslocation. The highly stable Hrd3 protein that is normally present at identical levels to Hrd1 undergoes drastic loss owing to proteasome-dependent degradation, which is dependent on Hrd1 ubiquitination activity. Based on our current model of the suppression pathway, Hrd3 appears to be the “first victim” of elevated Hrd1. Specifically, as Hrd1 elevates above the 1:1 Hrd1:Hrd3 stoichiometry, Hrd3 becomes an ERAD-M substrate and constitutively degraded. So far, very little is known about the feature requirement for Hrd3 degradation. Moreover, HRD complex remodeling leads to complete stabilization of ERAD-L, CPY∗ and KHN, degradation. This is distinct from earlier results showing CPY∗ degradation in cells that are overexpressing Hrd1 even when Hrd3 is absent. This discrepancy supports the idea that there are functional and, in all likelihood, compositional changes in the HRD complex that allow ERAD-M supported by elevated Hrd1 with absence of ERAD-L. In fact, our previous studies have shown Der1, the HRD complex component involved in ERAD-L retrotranslocation, is dispensable in the suppression pathway. Clearly these differences demand a detailed proteomic analysis of the ERAD-M permissive HRD complex, and integrating the resulting information into the natural biology of ERAD in normal and stressed cells.
Moreover, we have observed loss of Hrd3 was absolutely required for Hrd1-dependent suppression and retrotranslocation. Consistent with this model, simple removal of Hrd3 by use of a hrd3Δ strain allowed catalytically inactive C399S-Hrd1 or even Hrd1 missing its entire cytoplasmic region to function in suppressive ERAD-M retrotranslocation. Forced reintroduction of Hrd3 to suppressees by activation of a strong inducible promoter caused immediate cessation of restored ERAD-M retrotranslocation and recapitulation of the drastic growth stress as seen in unsuppressed dfm1Δ strains with high levels of ERAD-M substrates. This result could mean that under normal circumstances, Hrd3 prohibits Hrd1 from mediating ERAD-M retrotranslocation. It is unclear what features of Hrd3 are blocking suppressive ERAD-M retrotranslocation. Because our earlier studies demonstrated that the luminal domain of Hrd3 is sufficient for functioning with Hrd1, along with the fact that functional Hrd3 homologs in other organisms are purely only luminal, we believe the luminal region of Hrd3 is important for controlling Hrd1's suppressive function. Furthermore, a cryo-EM structure of Hrd1-Hrd3 complex shows a portion of Hrd3 binds to TMD 1&2 of Hrd1-regions suggested to serve as a lateral gate for incoming integral membrane substrates (Schoebel et al., 2017). It is possible that the presence of Hrd3 blocks lateral gate function and prevents dislocation of ERAD-M substrates. In addition, recent studies have shown that a subpopulation of mammalian HRD complex can arise without SEL1 (SEL1 is the mammalian homolog of Hrd3) and this complex was more efficient in retrotranslocating ER membrane clients, further implying an antagonistic or regulatory role of Hrd3 in ERAD-M retrotranslocation (Hwang et al., 2017). Future studies will include analysis and understanding of Hrd3's action in its natural versus suppressed state using the tools and approaches gleaned from its specific role in dfm1Δ suppression.
We previously reported Cdc48 recruitment to the ER surface relies mainly on Dfm1 (Neal et al., 2018). Surprisingly, the dfm1Δ suppressees show restored Cdc48 recruitment without Dfm1. This implies that Cdc48 recruitment can be mediated by a number of mechanisms. For example, polyubiquitinated Hrd1 has been shown to mediate the binding of Cdc48 (Baldridge and Rapoport, 2016; Stein et al., 2014). However, in the suppressed dfm1Δ cells, a Hrd1 devoid of its RING domain was still able to support Cdc48 recruitment, indicating that other effective modes of ER localization are yet to be discovered.
It would appear from these studies that elevation of Hrd1 can cause a remarkable remodeling of the HRD complex leading to different actions and specificity. These insights were made possible by use of the unique tool SUS-GFP, which provides powerful self-ubiquitinating activity (Garza et al., 2009b; Neal et al., 2018). With this special substrate, it is possible to separate and examine other functions of Hrd1, in particular its Hrd3-gated retrotranslocation of ERAD-M substrates. However, most studies use more traditional substrates that are ubiquitinated by Hrd1, and thus separable observation of Hrd1's other actions are not facile. Many earlier studies from our and other groups have shown that in normal strains, Hrd1 overexpression will bypass the requirement of other upstream components (Hrd3, Usa1, and Der1), suggesting Hrd1 might have a primordial function in both branches of ERAD (Carvalho et al., 2010; Vashistha et al., 2016). However, these observations need to be integrated with the above and other studies showing that that HRD complex function and composition is variable. Because many studies including our own have capitalized on Hrd1 overexpression for studying aspects of ERAD (Garza et al., 2009a; Vashistha et al., 2016), caution must be used in the interpretation of results so obtained with the growing understanding of HRD complex's complex behavior.
Several interesting questions arising from this study demand further investigation. First, how is it that elevation of Hrd1 leads to conversion of Hrd3 from a stable ERAD co-conspirator to a hapless, rapidly degraded substrate? Also, how does Hrd1 remain stable in the absence of Hrd3, which is normally absolutely required for Hrd1 stability (Vashistha et al., 2016)? How does the suppressed state restore the significant ER membrane association of Cdc48 in the absence of its normal Dfm1-mediated association with the ER surface, and more generally, what is the composition of the remodeled HRD complex that arises in suppression? What features of Hrd1 are important for replacing Dfm1, and do any structural features of rhomboids, which are not known to form channels, also exist in the Hrd1 transmembrane domain to allow suppression? Finally, and most generally, are there normal physiological circumstances in which elevation of Hrd1 occurs to bring about the remodeling observed in these studies? Are there stress response elements in the HRD1 promoters of yeast or mammals that are regulated by still-unknown signals of ER membrane stress? Whatever the answers to these questions, it is now clear from this work that the ERAD machinery is capable of surprising plasticity and functional flexibility to accommodate the large number of known and novel stresses that arise during cellular life and may lead to new ways to modify ER-based quality control in fundamental and translational approaches to cellular quality control.
Limitations of the Study
We have shown that overexpression of membrane substrates in dfm1Δ cells elicits a profound growth stress. For example, misfolded Sec61-2 is toxic to dfm1Δ cells, whereas its folded counterpart, Sec61, has no impact on the growth of dfm1Δ null cells implying misfolding of membrane substrates is the source of stress. In the contrary, SUS, the substrate initially used to characterize this pathway, has a stably folded domain, Hmg1, suggesting accumulation of its ubiquitinated forms and not protein misfolding is the source of growth toxicity. In fact, an earlier work by Metzger and Michaelis supports this idea of a similar stress caused by ubiquitination of ERAD-M substrates (Metzger and Michaelis, 2009). Although the degree to which ubiquitination and/or misfolding contributes to dfm1Δ null-associated stress has not been addressed in this study, studying the nature and features of this stress pathway opens up an entire line inquiry for future studies.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sonya Neal (seneal@ucsd.edu).
Materials Availability
Plasmids and yeast strains generated in this study is available from our laboratory.
Data and Code Availability
This study did not generate/analyze dataset/code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Tom Rapoport (Harvard Medical School), Davis Ng (National University of Singapore), Randy Schekman (University of California, Berkeley), Susan Michaelis (Johns Hopkins University), and Mark Hochstrasser (Yale School of Medicine) for providing plasmids and antibodies. We thank Sascha Duttke for illustrations and Darren Lam for initial aneuploid studies. We also thank the Neal and Hampton lab members for in depth discussions and technical assistance, and R.Y.H. specifically acknowledges Carbon Hampton for assistance in a variety of interpretive endeavors. These studies were supported by NIH grant 1R35GM133565-01 and Burroughs Wellcome Fund 1013987 (to S.E.N.) and 5R37DK051996-18 (to R.Y.H.).
Author Contributions
S.E.N. and R.Y.H. designed research; S.Neal, D.S., A.N. and S.Nadeau. performed research; S.E.Neal, R.Y.H., A.N., and D.S., analyzed data; and S.Neal. and R.Y.H. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Declaration of Interests
The authors declare they have no competing interests within the contents of this article.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101493.
Supplemental Information
References
- Avci D., Fuchs S., Schrul B., Fukumori A., Breker M., Frumkin I., Chen C., Biniossek M., Kremmer E., Schilling O. The yeast ER-intramembrane protease Ypf1 refines nutrient sensing by regulating transporter abundance. Mol. Cell. 2014;56:630–640. doi: 10.1016/j.molcel.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Baldridge R.D., Rapoport T.A. Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell. 2016;166:394–407. doi: 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A., Qi L. ER-associated degradation in health and disease – from substrate to organism. J. Cell Sci. 2019;132:jcs232850. doi: 10.1242/jcs.232850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S., Matuschewski K., Rape M., Thoms S., Jentsch S., Bays N., Gardner R., Seelig L., Joazeiro C., Hampton R. Role of the ubiquitin-selective CDC48(UFD1/NPL4 )chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 2002;21:615–621. doi: 10.1093/emboj/21.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll S.M., Hampton R.Y. USA1p is required for optimal function and regulation of the Hrd1p endoplasmic reticulum-associated degradation ubiquitin ligase. J. Biol. Chem. 2010;285:5146–5156. doi: 10.1074/jbc.M109.067876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P., Goder V., Rapoport T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Carvalho P., Stanley A.M., Rapoport T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Mariano J., Tsai Y.C., Chan A.H., Cohen M., Weissman A.M. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl. Acad. Sci. U S A. 2006;103:341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federovitch C.M., Jones Y.Z., Tong A.H., Boone C., Prinz W.A., Hampton R.Y. Genetic and structural analysis of Hmg2p-induced endoplasmic reticulum remodeling in Saccharomyces cerevisiae. Mol. Biol. Cell. 2008;19:4506–4520. doi: 10.1091/mbc.E07-11-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagg M.P., Kao A., Hampton R.Y. Integrating after CEN Excision (ICE) Plasmids: combining the ease of yeast recombination cloning with the stability of genomic integration. Yeast. 2019;36:593–605. doi: 10.1002/yea.3400. [DOI] [PubMed] [Google Scholar]
- Foresti O., Ruggiano A., Hannibal-Bach H.K., Ejsing C.S., Carvalho P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife. 2013;2:e00953. doi: 10.7554/eLife.00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R.G., Swarbrick G.M., Bays N.W., Cronin S.R., Wilhovsky S., Seelig L., Kim C., Hampton R.Y. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza R.M., Sato B.K., Hampton R.Y. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J. Biol. Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza R.M., Sato B.K., Hampton R.Y. In vitro analysis of Hrd1p-mediated retrotranslocation of its multSUSispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J. Biol. Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V., Carvalho P., Rapoport T.A. The ER-associated degradation component Der1p and its homolog Dfm1p are contained in complexes with distinct cofactors of the ATPase Cdc48p. FEBS Lett. 2008;582:1575–1580. doi: 10.1016/j.febslet.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton R.Y., Garza R.M. Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem. Rev. 2009;109:1561–1574. doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton R.Y., Sommer T. Finding the will and the way of ERAD substrate retrotranslocation. Curr. Opin. Cell Biol. 2012;24:460–466. doi: 10.1016/j.ceb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Hampton R.Y., Gardner R.G., Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C., Chevet E., Oakes S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015;17:829–838. doi: 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill R.B., Pellegrini L. The PARL family of mitochondrial rhomboid proteases. Semin. Cell Dev. Biol. 2010;21:582–592. doi: 10.1016/j.semcdb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M.M., Finger A., Schweiger M., Wolf D.H. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- Huyer G., Piluek W.F., Fansler Z., Kreft S.G., Hochstrasser M., Brodsky J.L., Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- Hwang J., Walczak C.P., Shaler T.A., Olzmann J.A., Zhang L., Elias J.E., Kopito R.R. Characterization of protein complexes of the endoplasmicreticulum-associated degradation E3 ubiquitin ligase Hrd1. J. Biol. Chem. 2017;292:9104–9116. doi: 10.1074/jbc.M117.785055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger P.A., Ornelas L., McElfresh C., Wong L.R., Hampton R.Y., Ideker T. Systematic gene-to-phenotype Arrays: a high-throughput technique for molecular phenotyping. Mol. Cell. 2018;69:321–333.e3. doi: 10.1016/j.molcel.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y., Kohno K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 2011;23:135–142. doi: 10.1016/j.ceb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Liu Y., Chang A. Heat shock response relieves ER stress. EMBO J. 2008;27:1049–1059. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert M., Sommermeyer F., Berger M., Kumar Lakshmipathy S., Gauss R., Aebi M., Jarosch E., Sommer T. The interplay of Hrd3 and the molecular chaperone system ensures efficient degradation of malfolded secretory proteins. Mol. Biol. Cell. 2015;26:185–194. doi: 10.1091/mbc.E14-07-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger M.B., Michaelis S. Analysis of quality control substrates in distinct cellular compartments reveals a unique role for Rpn4p in tolerating misfolded membrane proteins. Mol. Biol. Cell. 2009;20:1006–1019. doi: 10.1091/mbc.E08-02-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H.H., Shorter J.G., Seemann J., Pappin D., Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K., Huyer G., Michaelis S., Brodsky J.L. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K., Kamura T., Bagola K., Mehnert M., Jarosch E., Sommer T., Xie W., Ng D., Hampton R., Sommer T. Subcellular fractionation analysis of the extraction of ubiquitinated polytopic membrane substrate during ER-associated degradation. PLoS One. 2016;11:e0148327. doi: 10.1371/journal.pone.0148327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S., Mak R., Bennett E.J., Hampton R. A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated Degradation (ERAD-M) substrates. J. Biol. Chem. 2017;292:3112–3128. doi: 10.1074/jbc.M116.770610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S., Jaeger P.A., Duttke S.H., Benner C.K., Glass C., Ideker T., Hampton R. The Dfm1 derlin is required for ERAD retrotranslocation of integral membrane proteins. Mol. Cell. 2018;69:306–320.e4. doi: 10.1016/j.molcel.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson B.G., Glaser M.L., Rapoport T.A., Baldridge R.D. Cycles of autoubiquitination and deubiquitination regulate the erad ubiquitin ligase hrd1. Elife. 2019;8:e50903. doi: 10.7554/eLife.50903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper R.K., Böhmler S., Bordallo J., Sommer T., Wolf D.H. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- Plemper R.K., Egner R., Kuchler K., Wolf D.H. Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J. Biol. Chem. 1998;273:32848–32856. doi: 10.1074/jbc.273.49.32848. [DOI] [PubMed] [Google Scholar]
- Ravid T., Kreft S.G., Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly H., Rape M., Braun S., Rumpf S., Hoege C., Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Sato B.K., Hampton R.Y. Yeast derlin Dfm1 interacts with Cdc48 and functions in ER homeostasis. Yeast. 2006:1053–1064. doi: 10.1002/yea.1407. [DOI] [PubMed] [Google Scholar]
- Schoebel S., Mi W., Stein A., Ovchinnikov S., Pavlovicz R., DiMaio F., Baker D., Chambers M.G., Su H., Li D. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature. 2017;548:352–355. doi: 10.1038/nature23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder M., Kaufman R.J. The mammalian UNFOLDED protein response. Annu. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Scott D.C., Schekman R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J. Cell Biol. 2008;181:1095–1105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicari D., Igbaria A., Chevet E. Control of protein homeostasis in the early secretory pathway: current status and challenges. Cells. 2019;8:1347. doi: 10.3390/cells8111347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A., Ruggiano A., Carvalho P., Rapoport T.A. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A., Schweizer R.S., Schäfer A., Wolf D.H. Dfm1 forms distinct complexes with Cdc48 and the ER ubiquitin ligases and is required for ERAD. Traffic. 2010;11:1363–1369. doi: 10.1111/j.1600-0854.2010.01093.x. [DOI] [PubMed] [Google Scholar]
- Sun Z., Brodsky J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019;218:3171–3187. doi: 10.1083/jcb.201906047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K.J., Patil C.K., Wodicka L., Lockhart D.J., Weissman J.S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Vashist S., Ng D.T.W. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashistha N., Neal S.E., Singh A., Carroll S.M., Hampton R.Y. Direct and essential function for Hrd3 in ER-associated degradation. Proc. Natl. Acad. Sci. U S A. 2016;113:5934–5939. doi: 10.1073/pnas.1603079113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasic V., Denkert N., Schmidt C.C., Riedel D., Stein A., Meinecke M. Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. 2020:1–8. doi: 10.1038/s41556-020-0473-4. [DOI] [PubMed] [Google Scholar]
- Wahlman J., DeMartino G.N., Skach W.R., Bulleid N.J., Brodsky J.L., Johnson A.E. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell. 2007;129:943–955. doi: 10.1016/j.cell.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wangeline M.A., Vashistha N., Hampton R.Y. Proteostatic tactics in the strategy of sterol regulation. Annu. Rev. Cell Dev. Biol. 2017;33:467–489. doi: 10.1146/annurev-cellbio-111315-125036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Meyer H.H., Rapoport T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze dataset/code.