

Abstract
Shwachman–Diamond syndrome (SDS), an autosomal recessive disorder characterized by bone marrow failure, exocrine pancreatic insufficiency, and skeletal abnormalities, is caused by mutations in the Shwachman–Bodian–Diamond syndrome (SBDS) gene, which plays a role in ribosome biogenesis. Although the causative genes of congenital disorders frequently involve regulation of embryogenesis, the role of the SBDS gene in early hematopoiesis remains unclear, primarily due to the lack of a suitable experimental model for this syndrome. In this study, we established induced pluripotent stem cells (iPSCs) from patients with SDS (SDS-iPSCs) and analyzed their in vitro hematopoietic and endothelial differentiation potentials. SDS-iPSCs generated hematopoietic and endothelial cells less efficiently than iPSCs derived from healthy donors, principally due to the apoptotic predisposition of KDR+CD34+ common hemoangiogenic progenitors. By contrast, forced expression of SBDS gene in SDS-iPSCs or treatment with a caspase inhibitor reversed the deficiency in hematopoietic and endothelial development, and decreased apoptosis of their progenitors, mainly via p53-independent mechanisms. Patient-derived iPSCs exhibited the hematological abnormalities associated with SDS even at the earliest hematopoietic stages. These findings will enable us to dissect the pathogenesis of multiple disorders associated with ribosomal dysfunction.
Subject terms: Differentiation, Induced pluripotent stem cells
Introduction
Shwachman–Diamond syndrome (SDS) is a rare autosomal recessive disorder characterized by hematological abnormalities that manifest as cytopenia and progression to myelodysplastic syndrome and acute myeloid leukemia, exocrine pancreatic insufficiency, and skeletal abnormalities1,2. Hematopoietic stem cell transplantation is the sole curative treatment to correct the hematological defect of this syndrome, and mortality remains high due to serious post-transplant complications3.
Mutations in the Shwachman–Bodian–Diamond (SBDS) gene, which is located on chromosome 7q11, are present in approximately 90% of patients with SDS; these mutations commonly arise from gene conversion to the highly similar pseudogene SBDSP4. SBDS mRNA is expressed in a broad range of tissues4. Multiple studies have shown that SBDS protein has a primary function in ribosome assembly5–7. The additional proposed functions for SBDS, such as mitotic spindle stabilization, chemotaxis, cellular stress responses, and apoptosis, reflect indirect downstream effects of perturbing ribosome assembly8–13.
Mutations in genes encoding transcription factors involved in regulating normal development are responsible for a variety of inherited disorders. During embryogenesis, hematopoietic cells (HCs) and endothelial cells (ECs) emerge from common hemoangiogenic progenitors that express vascular endothelial growth factor receptor (VEGFR)-2 (also known as KDR in humans)14–16. Indeed, several HC- and/or EC-related transcriptional factors, such as SCL and RUNX1, are associated with various congenital hematological disorders17. Multiple disorders associated with ribosomal dysfunction (so-called ribosomopathies), including SDS and Diamond–Blackfan anemia (DBA), also present with hematological defects18; however, the pathogenesis of ribosomopathies has not been fully elucidated. Currently, the field lacks an adequate mouse model of the human disease because the most analogous mutant in mouse fails to faithfully recapitulate all disease-associated phenotype19–22: Sbds-/- embryos fail to generate HCs and ECs due to early lethality prior to embryonic day (E) 6.5 before both lineages have developed23.
Induced pluripotent stem cells (iPSCs) are pluripotent stem cell generated by enforced expression of specific transcription factors24. Patient-specific iPSCs, in combination with directed cell differentiation, are a practical source of human embryonic progenitors that can surpass the utility of murine models. Accordingly, these cells have the potential to contribute enormously to patient-oriented research, including disease pathophysiology and drug screening25. In this study, we generated iPSCs from three SDS patients and differentiate them into HCs and ECs using our established differentiation system for human embryonic stem cells (ESCs) and iPSCs26–30.
Results
Generation of iPSCs from SDS patients
Following transduction of peripheral blood cell derived from SDS patients with an episomal plasmid vector encoding Oct3/4, Sox2, Klf4, L-Myc, Lin28, and shRNA against TP53, four clones (SDS1-1 and SDS1-2 from patient 1 and SDS2 from patient 2, and SDS3 from patient 3) were randomly selected for propagation and further analyses, as previously reported29,31. All patient-derived SDS-iPSCs exhibited a characteristic human ESC-like morphology (Fig. 1a, Supplementary Fig. S1a), and were capable of propagating in serial passage. DNA sequencing analysis verified an identical mutation in the SBDS gene in all established SDS-iPSC clones (Fig. 1b, Supplementary Fig. S1b). Chromosomal analysis revealed that all SDS-iPSC clones maintained a normal karyotype (Fig. 1c, Supplementary Fig. S1c). Expression levels of the pluripotency markers Oct3/4, Sox2, Klf4, L-Myc, and Lin28 in all SDS-iPSCs were comparable to those in control iPSCs, although transgene expression was rarely detected (Fig. 1d, Supplementary Fig.S1d). All three primary germ-layer derivatives were detected in cystic teratomas formed after subcutaneous injection of undifferentiated iPSCs into immunocompromised NOD/SCID/γcnull mice (Fig. 1e, Supplementary Fig. S1e).
Figure 1.

Generation of iPSCs from SDS patients. (a) Human ESC-like morphology of SDS-iPSCs. Scale bar: 400 μm. (b, c) DNA sequencing analysis of SBDS RT-PCR product (b) and karyotype analysis (c) of SDS-iPSCs. (d) Expression of KLF4 and SOX2 in SDS and control iPSCs. One primer set detects only the transgene (black bars), whereas the other detects both the transgene and endogenous gene (white bars). (e) Teratoma formation from SDS-iPSCs in NOD/SCID/γcnull mice. Arrows indicate endoderm (respiratory epithelium), mesoderm (cartilage), and ectoderm (pigmented epithelium). Scale bar: 200 μm.
To investigate the pathogenesis of this syndrome, SBDS cDNA and DsRed were transduced into SDS-iPSCs using the PiggyBac transposon system (Fig. 2a). Western blotting revealed a reduction in SBDS protein expression in SDS-iPSCs that was rescued in SBDS-overexpressed iPSCs (Fig. 2b). Polysome profiling demonstrated that ribosomal assembly in SDS-iPSCs was reduced, as evidenced by a decrease in the 80S:40S ratio; this deficiency was reversed by transduction of SBDS cDNA (Fig. 2c,d).
Figure 2.

Transduction of SDS-iPSCs. (a) Construction of lentiviral vectors containing DsRed alone (PBCl-EF1a-DsRed-PuroR-pA) or SBDS cDNA and DsRed (PBCl-EF1aSBDS-IRES2-DsRed-PuroR-pA). (b) Western blotting analysis of SBDS protein in SDS-iPSC clones transduced with SBDS or empty (DsRed alone) vector. TF2B was used as a loading control. “+SBDS” indicates iPSC clones transduced with SBDS vector. (c) Representative polysome profiling of SDS-iPSC clones transduced with SBDS or empty vector. (d) 80S:40S ratio in SDS-iPSC clones transduced with SBDS or empty vector.
Impaired granulopoiesis during in vitro differentiation of SDS-iPSCs
First, using a previously reported in vitro culture system26,29, we investigated whether generated SDS-iPSCs recapitulated the hematological phenotype of the syndrome (Fig. 3a). Floating HCs, which mainly consisted of mature neutrophils, first appeared on day 15 of differentiation of SDS and control iPSCs (Fig. 3b,c, Supplementary Fig. S2a). The remaining HCs consisted of immature myeloid cells and a small number of macrophages. Serial analyses revealed that floating HCs generated from SDS-iPSCs were less abundant than those from control iPSCs (Fig. 3c). Positivity for myeloperoxidase and lactoferrin, the constituent proteins of neutrophil-specific granules, was comparable in neutrophils obtained from SDS-iPSCs and control iPSCs (Supplementary Fig. S2b,c). Similarly, the bactericidal activity of neutrophils from SDS-iPSCs and control iPSCs did not significantly differ (Supplementary Fig. S2d). HC production was comparable between SBDS-overexpressing SDS-iPSCs and control iPSCs (Fig. 3c,d, Supplementary Fig. S1e,f), with no attendant morphological changes (Supplementary Fig. S2g,h). As reported previously9,32, the chemotactic activity of SDS-iPSC–derived neutrophils was severely impaired, and this deficiency was reversed by overexpression of SBDS (Fig. 3e).
Figure 3.

Impaired granulopoiesis during in vitro differentiation from SDS-iPSCs. (a) Outline of the defined, step-wise differentiation protocol for generation of mature neutrophils. (b) May–Giemsa staining of floating HCs obtained from SDS-iPSC clones transduced with SBDS or empty vector, and from control iPSCs on day 30 of differentiation. Scale bar: 100 μm. (c) Sequential analysis of the number of floating HCs generated from control SDS-iPSCs, SBDS-overexpressing SDS-iPSCs, and control iPSCs. (d) Sequential analysis of the number of floating HCs generated from SDS-iPSC clones transduced with SBDS or empty vector. (e) Chemotactic activity of floating HCs generated from SDS-iPSC clones transduced with SBDS or empty vector in response to fMLP. fMLP-treated samples were presented as the fold change to untreated control sample. (f) Number of hematopoietic colonies derived from SDS-iPSC clones transduced with SBDS or empty vector, and from control iPSCs. (g) Light micrographs (upper rows) and May–Giemsa staining of hematopoietic colonies (lower rows) generated from SDS-iPSC clones transduced with SBDS or empty vector, and from control iPSCs. Scale bar: 100 μm. Data represent means ± SEM of triplicate wells; representative results from one of three independent experiments are shown (*P < 0.05).
We then examined the hematological defects of SDS-iPSCs at the clonogenic progenitor level. In methylcellulose colony-forming assays, SDS-iPSCs formed significantly fewer HC colonies, a defect that was rescued by SBDS overexpression (Fig. 3f). The decreased size of HC colonies was also reversed by SBDS overexpression, although the size is not somehow comparable to that of control iPSCs (Fig. 3g). Collectively, these data demonstrated that SDS-iPSCs exhibited reduced HC production, accompanied by impaired neutrophil chemotaxis and limited colony-forming potential, all of which are typical hematological abnormalities of SDS patients.
Reduced HC and EC generation from SDS-iPSC derived hemoangiogenic progenitors
To investigate when the initial pathological events occurred during hematopoietic differentiation of SDS-iPSCs, we focused on the development and differentiation potential of KDR+CD34+ cells (Fig. 4a,b), which represent hemoangiogenic progenitors26,30. On day 6 of initial differentiation, a similar proportion of KDR+CD34+ cells were generated from SDS-iPSCs and control iPSCs (data not shown). After replating onto OP9 cells, however, the KDR+CD34+ cells generated from SDS-iPSCs produced fewer floating HCs than those generated from control iPSCs, a phenotype reversed by SBDS overexpression (Fig. 4c–e, Supplementary Fig. S3a).
Figure 4.

Reduced HC and EC development from SDS-iPSC derived KDR+ CD34+ hemoangiogenic progenitors. (a) Outline of the differentiation protocol for generation of HCs and ECs from iPSC-derived hemoangiogenic progenitors. On day 6 of initial differentiation, sorted KDR+CD34+ cells were transferred onto fresh confluent OP9 cells, and were then cultured in the indicated culture conditions. (b) Isolation of KDR+CD34+ hemoangiogenic progenitors by FACS. Purities of viable KDR+CD34+ cells were 7.1% ± 1.3% in at least three independent experiments. (c) May–Giemsa staining of floating HCs obtained from SDS and control iPSC–derived KDR+CD34+ cells on day 26 of differentiation. Scale bar: 100 mm. (d, e) Sequential analysis of the number of floating HCs generated from SDS and control iPSCs (d), and from SDS-iPSC clones transduced with SBDS or empty vector (e). (f) Immunostaining of CD31 in EC clusters obtained from SDS and control iPSC–derived KDR+CD34+ cells on day 13 of differentiation. Scale bar: 500 μm. (g, h) Numbers (g) and area (h) of EC clusters generated from SDS-iPSC clones transduced with SBDS or empty vector, and from control iPSCs. Data represent means ± SEM of triplicate wells; representative results from one of three independent experiments are shown (*P < 0.05).
We also investigated the capacity of KDR+CD34+ cells to differentiate into ECs. KDR+CD34+ cells generated from SDS-iPSCs produced significantly fewer CD31+ EC clusters than those from control iPSCs (Fig. 4f,g). Furthermore, each EC cluster derived from SDS-iPSCs was smaller (Fig. 4h), suggesting reduced EC differentiation or growth potential. The lower EC production and smaller EC cluster size of SDS-iPSCs were rescued by SBDS overexpression (Fig. 4g,h). EC clusters generated from SDS-iPSCs took up 1,1′-dioctadecyl-1,3,3,3′,3′-tetramethylindocarbocyanine–labeled acetylated low-density lipoprotein (Dil-Ac-LDL); co-expressed various EC-related surface markers, including VE-cadherin, CD31, CD34, CD141, CD146, and KDR; and formed tube networks on Matrigel, similarly to those generated from control iPSCs (Supplementary Fig. S3b–d). Thus, KDR+CD34+ cells generated from SDS-iPSCs had less EC differentiation potential than those generated from control iPSCs, although EC immunophenotype or function did not differ.
Apoptotic predisposition of SDS-iPSC–derived hemoangiogenic progenitors
Given that elevated apoptosis of hematopoietic stem/progenitor cells has been reported in SDS patients10,11,33, we investigated the apoptotic predisposition of hemoangiogenic progenitors or mature cell populations generated during SDS-iPSC differentiation. Detection of caspase 3 and 7 in each population revealed higher proportion of apoptotic cells in SDS-iPSC–derived KDR+CD34+ cell fraction (Fig. 5b). By contrast, we observed no significant changes in the rate of apoptosis in undifferentiated SDS-iPSCs or the neutrophils and ECs derived from them (Fig. 5a,c,d, Supplementary Fig. S4a–c). Overexpression of SBDS of SDS-iPSC–derived KDR+CD34+ cells attenuated the increase in apoptosis (Fig. 5e), suggesting that quantitative differences in SBDS expression were directly linked to the apoptotic predisposition of early hematopoietic progenitors. Bromo-2′-deoxyuridine (BrdU) incorporation assays revealed no difference in proliferation of KDR+CD34+ cells between SDS and control iPSCs (Supplementary Fig. S4d,e). Thus, elevated apoptosis of KDR+CD34+ hemoangiogenic progenitors is primarily responsible for the reduction in HC and EC development from SDS-iPSCs.
Figure 5.

Apoptotic predisposition of SDS-iPSC–derived hemoangiogenic progenitors. (a–d) Proportion of apoptotic cells in undifferentiated SDS and control iPSCs (a) and their derived hemoangiogenic progenitors (b), neutrophils (c), and EC cells (d). (e) Proportion of apoptotic cells among hemoangiogenic progenitors generated from SDS-iPSC clones transduced with SBDS or empty vector. Data represent means ± SEM of triplicate wells (*P < 0.05).
We then investigated the underlying mechanism of the SDS-associated apoptotic predisposition at the hemoangiogenic progenitor stage (Fig. 6a). Flow cytometric analyses revealed that addition of caspase-3 inhibitor (Ac-DEVD-CHO) significantly decreased the proportion of apoptotic KDR+CD34+ cells (Fig. 6b), indicating that apoptosis of hemoangiogenic progenitors was caspase-dependent. Activation of p53 has been proposed as a common mechanism in the pathogenesis of various ribosomopathies, including DBA and SDS21,34. However, the inhibitory effect of p53 inhibitor (pifithrin-α) was smaller than that of Ac-DEVD-CHO (Fig. 6b). Furthermore, phospho-flow cytometry did not detect higher level of p53 phosphorylation in SDS-iPSC–derived KDR+CD34+ cells (Fig. 6c), suggesting that p53-independent apoptosis was at least partially involved in apoptotic predisposition of hemoangiogenic progenitors (Fig. 6d).
Figure 6.
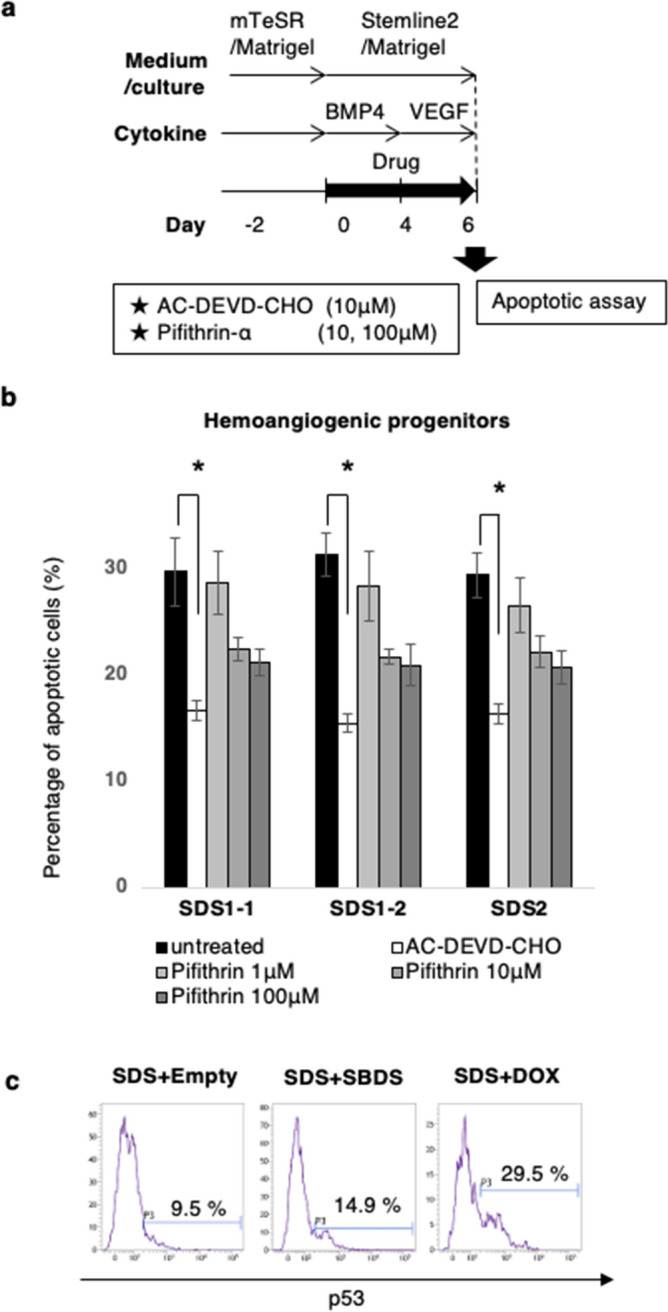
Effect of caspase or p53 inhibitor on the apoptosis of SDS-iPSC–derived hemoangiogenic progenitors. (a) Graphical representation of the experimental procedure. (b) Proportion of apoptotic cells in untreated (black bars), and AC-DEVD-CHO–treated (white bars) or pifithrin-a–treated SDS-iPSC–derived hemoangiogenic progenitors (gray bars). (c) Representative phospho-flow cytometric plot of p53 in SDS-iPSC clones transduced with SBDS or empty vector. Doxorubicin (DOX)-treated SDS-iPSCs was used as a positive control. Data represent means ± SEM of triplicate wells (*P < 0.05).
Discussion
In this study, our in vitro culture system faithfully recapitulated typical hematological abnormalities in SDS patients, including as impaired granulopoiesis, reduced colony-forming potential, and neutrophil chemotaxis dysfunction, in SDS-iPSC clones from patients harboring different SBDS mutations. Furthermore, SBDS overexpression could reverse all disease-related phenotypes, confirming strongly loss of SBDS protein is directly responsible for the hematological defects associated with this syndrome, as reported previously35.
It remains unclear when the initial pathological events leading to hematological defects occur in SDS, primarily because the embryonic lethality of SBDS knock-out mice occurs prior to HC and EC development23, and also due to the ethical restriction for experimental use of human embryonic tissues. This is a first report showing the possibility that the hematopoietic consequences in SDS patients originate from the KDR+CD34+ early hemoangiogenic progenitor stage. Marked reduction of HC and EC development was principally due to the apoptotic predisposition of KDR+CD34+ cells. Previous studies showed that SBDS-deficient cells undergo accelerated apoptosis through the Fas-mediated and generally p53-dependent pathway10,11,33. By contrast, the current study suggested that apoptosis of KDR+CD34+ cells was at least partially p53-independent, which has recently advocated as a different pathway in response to impaired ribosome biogenesis36.
SDS-iPSC–derived KDR+CD34+ cells produced significantly fewer EC clusters of smaller sizes, suggesting that EC differentiation or growth was also impaired. By contrast, the impaired EC development of SDS-iPSCs is inconsistent with clinical observations that SDS patients do not have vascular abnormalities37. One possible explanation for this discrepancy is that SBDS and vasculogenesis-related transcriptional factors are functionally redundant. Alternatively, SDS patients who express detectable levels of SBDS protein expression (see Fig. 2b), as previously reported38, might not display vascular abnormalities postnatally. Nonetheless, patient-specific iPSC technology enables us to obtain transient embryonic progenitors, which allows us to analyze the effect of early hematopoiesis in a variety of congenital hematological disorders.
The effects of proteins related to ribosome biogenesis on early hematopoiesis were recently investigated in zebrafish39,40 and a human iPSC model41. In agreement with the results of this study, Garçon et al. recently reported a significant decrease in the abundance of CD43+ HC progenitors during RPS19-mutated DBA-iPSC differentiation41, largely consistent with the general assumption that loss of ribosome biogenesis principally affects rapidly proliferating progenitors with high translation rates. Notably, SDS-iPSC–derived neutrophils exhibited significantly decreased chemotaxis, whereas no functional differences were observed in ECs generated from SDS-iPSCs and control iPSCs. Thus, although the SBDS gene is ubiquitously expressed in all derivative cells, mutations in this gene can lead to cell- and tissue-specific development or maturation defects, as previously described42.
In conclusion, our results obtained from SDS patient–derived iPSCs revealed that disease onset might occur as early as early hematopoiesis when hemoangiogenic progenitors first emerge. This culture system will serve as a new tool to facilitate disease modeling, drug screening, and cell therapy for ribosomopathies.
Material and methods
Patients
This study was approved by the Ethics Committee of Kyoto University, and informed consent was obtained from a parent and/or legal guardians in accordance with the Declaration of Helsinki. To generate patient-derived iPSCs, peripheral blood cells were obtained from three male SDS patients with the three characteristic clinical signs of the disease (pancytopenia, exocrine pancreatic insufficiency, and short stature). Each patient was a compound heterozygote harboring a splice site mutation of intron 2 (258 + 2 T > C) in conjunction with another previously reported mutation in SBDS (97A > G in patient 1, 184A > T in patient 2, and 183-184TA > CT in patient 3)4,43. Control iPSCs were generated from parents of patient 1 (Ctl1 from the father and Ctl2 from the mother), who carried single mutant alleles and were asymptomatic.
Generation and characterization of SDS-iPSCs
Patient-derived iPSCs were generated as previously reported29,31. In brief, Oct3/4, Sox2, Klf4, L-Myc, Lin28, and shRNA against TP53 were introduced into peripheral blood cells obtained from three SDS patients or their parents using an episomal plasmid vector. The transduced cells were harvested and re-plated onto SNL feeder cells treated with mitomycin C (Kyowa Hakko Kirin). Colonies similar to human ESCs were selected for further cultivation in primate ESC medium (ReproCELL, Yokohama, Japan) supplemented with 5 ng/ml bFGF (R&D Systems). Three weeks later, individual colonies were isolated and expanded. Human iPSCs were maintained on mitotically inactive SNL feeder cells and subcultured onto new SNL feeder cells every 7 days, as described previously22. Culture conditions, characterization of iPSC stemness and pluripotency profile, karyotyping analysis, DNA sequencing analysis, and teratoma formation were performed as previously reported28,29.
Gene transfer in SDS-iPSCs using the PiggyBac transposon system
The CRA236-EF1α-SBDS-IRES2-DsRed-Puro vector was expressed and purified according to a recently published protocol31. SDS-iPSCs were incubated with 10 μM Y-27632 (Wako, Saitama, Japan) for 1 h, and then treated with 0.25% trypsin (Thermo Fisher Scientific, Waltham, MA, USA), 1 mg/ml collagenase IV (Thermo Fisher Scientific), and Accumax (Innovative Cell Technologies, San Diego, CA, USA). The resultant single cells were transduced with the SBDS expression vector and transposase expression vector by electroporation, and then seeded onto fresh mitomycin C–treated SNL76/7 feeder cells in human ESC medium containing 10 μM Y-27632 and 1 μg/ml puromycin (Sigma-Aldrich, St. Louis, MO, USA). One week later, small iPSC colonies were manually picked up and seeded onto fresh mitomycin C–treated SNL76/7 feeder cells. Ectopic expression of SBDS protein in isolates of transduced iPSCs was verified by western blotting analysis.
Initial differentiation of iPSCs using a serum- and feeder-free culture system
Initial differentiation was performed, as previously described29. Briefly, iPSC colonies were cultured on culture dishes coated with growth factor–reduced Matrigel (Becton–Dickinson), in Stemline II hematopoietic stem cell expansion medium (Sigma-Aldrich) containing the insulin–transferrin–selenium (ITS) supplement (Thermo Fisher Scientific) and 20 ng/ml BMP4 (R&D Systems). VEGF (40 ng/ml, R&D Systems) was added on day 4, and the cells were cultivated for 2 more days. To induce neutrophil differentiation, VEGF was replaced with a combination of 50 ng/ml SCF (R&D Systems), 50 ng/ml IL-3 (R&D Systems), 5 ng/ml TPO (R&D Systems), and 50 ng/ml G-CSF (Kyowa Hakko Kirin) on day 6. Thereafter, the medium was replaced every 5 days.
FACS analysis and cell sorting
Staining procedures, FACS analysis, and cell sorting were performed as reported previously27,29. Briefly, cultured cells were harvested with Accumax (Innovative Cell Technologies) and incubated with the indicated primary antibodies for 30 min. Non-viable cells were excluded from analysis by DAPI co-staining. FACS analysis was performed on a Verse flow cytometer with the FACSuite software (Becton–Dickinson). Cell sorting with PE-conjugated CD34 and APC-conjugated VEGFR-2 mAbs was performed on an ARIA II flow cytometer (Becton–Dickinson), as reported previously27,29.
In vitro HC and EC differentiation of hemoangiogenic progenitors
KDR+CD34+ cells were sorted by FACS on day 6 of initial differentiation in a serum-and feeder-free culture system, as previously described26,29. In some experiments, 10 μM Ac-DEVD-CHO (Peptide Institute, Osaka) or 50 μM pifithrin-α (Wako) was added on day 0.
To induce HC and EC differentiation, the sorted KDR+CD34+ cells were then transferred onto fresh confluent OP9 cells26,30. For HC development, the sorted cells were cultured in α-MEM (Thermo Fisher Scientific) supplemented with 10% fetal calf serum (FCS) (Thermo Fisher Scientific), 50 μM 2-mercaptoethanol (ME) (Wako), 10 ng/ml thrombopoietin (TPO) (R&D Systems, Minneapolis MN, USA), 20 ng/ml interleukin-3 (R&D Systems), and 100 ng/ml stem cell factor (SCF) (R&D Systems) until day 20 of differentiation. Subsequently, 10 ng/ml granulocyte colony-stimulating factor (G-CSF, Kyowa Hakko Kirin, Tokyo, Japan) was added instead of SCF and TPO. Methylcellulose colony-forming assays were performed as previously reported29. To analyze EC development, the sorted cells were cultured in α-MEM supplemented with 10% FCS, 50 μM 2-ME, and 20 ng/ml VEGF (R&D Systems).
Characterization of HCs
Serially collected cells were selected using the Dead Cell Removal Kit (Miltenyi Biotec, Bergisch Gladbach, Germany), centrifuged onto glass slides using a Shandon Cytospin® 4 cytocentrifuge (Thermo Fisher Scientific), fixed immediately with PBS containing 4% paraformaldehyde, subjected to May–Giemsa and myeloperoxidase staining and immunostaining for lactoferrin, and analyzed by microscopy, as previously reported27,29.
Chemotaxis was determined using a modified Boyden chamber method, as previously reported20. Briefly, 500 μl of the reaction medium (Hank’s Balanced Salt Solution containing 2.5% FCS) with or without 10 nM formyl-Met-Leu-Phe (fMLP; Sigma-Aldrich) was placed into each well of a 24-well plate, and the cell culture insert (3.0-mm pores; Becton Dickinson) was gently placed into each well to divide the well into upper and lower sections. Floating cells were suspended to the upper well at a concentration of 3.5 × 104 cells per well, allowing the cells to migrate from the upper to the lower side of the membrane for 4 h at 37 °C. After incubation, cells in the lower chamber were collected and counted by flow cytometry. Dihydrorhodamine assay was performed as previously reported27.
Characterization of ECs
Six days after sorting, cells were stained with anti–VE-cadherin and HRP-conjugated anti-mouse IgG, and EC clusters were scored by microscopy. Data analysis was performed using the ImageJ software (https://imagej.nih.gov/ij/) to calculate the size of each EC cluster. For the Dil-Ac-LDL (Thermo Fisher Scientific) incorporation assay, adherent cells were incubated with 10 μg/ml Dil-Ac-LDL, as previously reported44.
The tube formation assay was performed as previously reported45. In brief, cells (1.0 × 105) were seeded on 48-well plates precoated with growth factor–reduced Matrigel, incubated for 24 h at 37℃, and analyzed with the ImageJ software.
RNA isolation and quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis
RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA, USA) and subjected to RT with the Omniscript-RT kit (Qiagen). Quantitative PCR was performed on a 7900HT Fast Real-Time PCR system (Applied Biosystems, Carlsbad, CA, USA) with SYBR Premix Ex Taq II (Takara, Shiga, Japan). Primer sequences are provided in Supplementary Table 1.
Western blot analysis
Cells were lysed, and nuclear fractions of soluble protein were extracted using the NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Fisher Scientific), following the manufacturer’s instructions. Nuclear protein was boiled for 5 min, separated by SDS-PAGE, and transferred to a polyvinylidene fluoride membrane. After blocking with 5% skim milk in TBS-T for 1 h at room temperature, the membrane was incubated overnight at 4℃ with primary antibody diluted in the same solution, washed extensively, and incubated for 1 h at room temperature with secondary antibody diluted in the same solution, followed by chemiluminescence detection.
Polysome profiling
Approximately 80–90% confluent iPSCs were treated with 100 μg/ml cycloheximide (Sigma-Aldrich) for 10 min, harvested, and lysed in 500 μl hypotonic buffer [1.5 mM KCl, 2.5 mM MgCl2, 5.0 mM Tris–Cl (pH 7.5), 100 μg/ml cycloheximide, 1 mM dithiothreitol, 100 units RNase inhibitor, 0.5% Triton X-100, 0.5% sodium deoxycholate, and 1 × protease inhibitor cocktail]. Lysates were centrifuged at 15,000 rpm for 5 min at 4 °C, and the supernatant was loaded onto linear 5–50% sucrose gradients [20 mM HEPES (pH 7.6), 5 mM MgCl2, 100 mM KCl, 10 μg/ml cycloheximide, 10 U/ml RNase inhibitor, and 1 × protease inhibitor cocktail] using a GradientStation (BioComp Frederiction, NB, Canada). Gradients were centrifuged at 35,000 rpm for 2 h at 4 °C in a SW40Ti rotor (Beckman Coulter, Indianapolis, IN, USA), and fractionated on an ISCO fractionator.
Apoptosis assay
To investigate apoptosis, caspases 3 and 7 were detected with the FAM-FLICA in vitro Caspase Detection Kit (ImmunoChemistry TECHNOLOGIES, Bloomington, MN, USA).
Statistical analyses
Differences in mean values between groups were analyzed by Student’s t-test and a Tukey–Kramer multiple comparison test. Statistical significance was defined as P < 0.05.
Supplementary information
Acknowledgments
We thank Ms. Tomomi Sudo, Center for iPS Cell Research and Application, Kyoto University, for her excellent technical assistance, and the Center for Anatomical Studies, Kyoto University Graduate School of Medicine, for immunocytochemical analysis.
Author contributions
T.H. performed experiments; M.O., K.O., K.O., and K.W. established iPS cell lines; T.T., S.N., S.S., S.B., A.N., and M.K.S. assisted with teratoma formation analysis; K.K., T.D., I.K., Y.K., Y.H., A.S., and H.W. assisted with lentiviral transduction of iPS cells; T.H., K.U., and K.W. analyzed and interpreted data; H.H., S.A., T.N., J.T., and T.H. assisted with interpretation of data and provided insightful comments; T.H. prepared figures; T.H., K.U., and K.W. wrote the manuscript; T.H., K.U., K.O., K.W., and T.H. designed the research.
Funding
This study is supported by a Grant-in-Aid for Scientific Research (C) (15K09651).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-71844-8.
References
- 1.Shwachman H, Diamond L, Oski F, Khaw KT. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J. Pediatr. 1964;65:645–663. doi: 10.1016/S0022-3476(64)80150-5. [DOI] [PubMed] [Google Scholar]
- 2.Smith OP, Hann IM, Chessells JM, Reeves BR, Milla P. Haematological abnormalities in Shwachmann-Diamond syndrome. Br. J. Haematol. 1996;94:279–284. doi: 10.1046/j.1365-2141.1996.d01-1788.x. [DOI] [PubMed] [Google Scholar]
- 3.Cesaro R, et al. Haematopoietic stem cell transplantation for Shwachman-Diamond disease: a study from the European Group for blood and marrow transplantation. Br. J. Haematol. 2005;131:231–236. doi: 10.1111/j.1365-2141.2005.05758.x. [DOI] [PubMed] [Google Scholar]
- 4.Boocock GRB, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 5.Menne TF, et al. The Shwachman-Boidan-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 2007;39:486–495. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- 6.Finch AJ, et al. Uncoupling of GTP hydrolysis from elF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25:917–929. doi: 10.1101/gad.623011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weis F, et al. Mechanism of elF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2015;22:914–919. doi: 10.1038/nsmb.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wessels D, et al. The Shwachman-Bodian-Diamond syndrome gene encodes an RNA-binding protein that localizes to the pseudopod of Dictyostelium amoebae during chemotaxis. J. Cell Sci. 2006;119:370–379. doi: 10.1242/jcs.02753. [DOI] [PubMed] [Google Scholar]
- 9.Austin KM, et al. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J. Clin. Invest. 2008;118:1511–1518. doi: 10.1172/JCI33764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rujkijyanont P, et al. SBDS-deficient cells undergo accelerated apoptosis through the Fas-pathway. Haematologica. 2008;93:363–371. doi: 10.3324/haematol.11579. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe K, et al. SBDS-deficiency results in specific hypersensitivity to Fas stimulation and accumulation of Fas at the plasma membrane. Apoptosis. 2009;14:77–89. doi: 10.1007/s10495-008-0275-9. [DOI] [PubMed] [Google Scholar]
- 12.Ball JL, et al. Shwachman-Bodian Diamond syndrome is a multi-functional protein implicated in cellular stress responses. Hum. Mol. Genet. 2009;18:3684–3695. doi: 10.1093/hmg/ddp316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warren AJ. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv. Biol. Regul. 2018;67:109–127. doi: 10.1016/j.jbior.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shalaby F, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 15.Shalaby F, et al. A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis. Cell. 1997;89:981–990. doi: 10.1016/S0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 16.Nishikawa SI, Nishikawa S, Hirashima M, Matsuyoshi N, Kodama H. Progressive lineage analysis by cell sorting and culture identifies FLK1+ VE-cadherin+ cells at diverging point of endothelial and hematopoietic lineages. Development. 1998;125:1747–1757. doi: 10.1242/dev.125.9.1747. [DOI] [PubMed] [Google Scholar]
- 17.Wlison NK, Calero-Nieto FJ, Ferreira R, Göttgens B. Transcriptional regulation of haematopoietic transcription factors. Stem Cell Res. Ther. 2011;2:6. doi: 10.1186/scrt47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tourlakis ME, et al. Deficiency of Sbds in the mouse pancreas leads to features of Shwachman-Diamond syndrome, with loss of zymogen granules. Gastroenterology. 2012;143:481–492. doi: 10.1053/j.gastro.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Tourlakis ME, et al. In vivo senescence in the Sbds-deficient murine pancreas: cell-type specific consequences of translation in sufficiency. PLoS Genet. 2015;11:e1005288. doi: 10.1371/journal.pgen.1005288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zambetti NA, et al. Deficiency of the ribosome biogenesis gene Sbds in hematopoietic stem and progenitor cells causes neutropenia in mice by attenuating lineage progression in myelocytes. Haematologica. 2015;100:1285–1293. doi: 10.3324/haematol.2015.131573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zambetti NA, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19:613–627. doi: 10.1016/j.stem.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 23.Zhang S, Shi M, Hui CC, Rommens JM. Loss of the mouse ortholog of the Shwachman-diamond syndrome gene (Sbds) results in early embryonic lethality. Mol. Cell Biol. 2006;26:6656–6663. doi: 10.1128/MCB.00091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 25.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 26.Niwa A, et al. A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLoS ONE. 2011;6:e22261. doi: 10.1371/journal.pone.0022261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morishima T, et al. Neutrophil differentiation from human-induced pluripotent stem cells. J Cell Physiol. 2011;226:1283–1291. doi: 10.1002/jcp.22456. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka T, et al. Induced pluripotent stem cells from CINCA syndrome patients as a model for dissecting somatic mosaicism and drug discovery. Blood. 2012;120:1299–1308. doi: 10.1182/blood-2012-03-417881. [DOI] [PubMed] [Google Scholar]
- 29.Morishima T, et al. Genetic correction of HAX1 in induced pluripotent stem cells from a patient with severe congenital neutropenia improves defective granulopoiesis. Haematologica. 2014;99:19–27. doi: 10.3324/haematol.2013.083873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki NM, et al. Pluripotent cell models of fanconi anemia identify the early pathological defect in human hemoangiogenic progenitors. Stem Cells Trans. Med. 2015;4:333–338. doi: 10.5966/sctm.2013-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okita K, et al. An efficient nonviral method to generated integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31:458–466. doi: 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- 32.Orelio C, Kuijpers TW. Shwachman-Diamond syndrome neutrophils have altered chemoattractant-induced F-actin polymerization and polarization characteristics. Haematologica. 2009;94:409–413. doi: 10.3324/haematol.13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dror Y, Freedman MH. Shwachman-Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood. 2001;97:3011–3301. doi: 10.1182/blood.V97.10.3011. [DOI] [PubMed] [Google Scholar]
- 34.Sieff CA, et al. Pathogenesis of the erythroid failure in Diamond Blackfan anaemia. Br. J. Haematol. 2010;148:611–622. doi: 10.1111/j.1365-2141.2009.07993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tulpule A, et al. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell. 2013;12:727–736. doi: 10.1016/j.stem.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donati G, Montanaro L, Drerenzini M. Ribosome biogenesis and control of cell proliferation: p53 is not alone. Cancer Res. 2009;72:1602–1607. doi: 10.1158/0008-5472.CAN-11-3992. [DOI] [PubMed] [Google Scholar]
- 37.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24:101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austin KM, Leary RJ, Shimamura A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood. 2005;106:1253–1258. doi: 10.1182/blood-2005-02-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, et al. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev. Cell. 2013;24:411–425. doi: 10.1016/j.devcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan Y, et al. Transcriptome analysis reveal a ribosome constitutes disorder involved in the RPL5 downregulated zebrafish model of Diamond-Blackfan anemia. BMC Med. Genomics. 2016;9:13. doi: 10.1186/s12920-016-0174-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garçon L, et al. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood. 2013;122:912–921. doi: 10.1182/blood-2013-01-478321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mccann KL, Baserga SJ. Genetics. Mysterious ribosomopathies. Science. 2013;341:849–850. doi: 10.1126/science.1244156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shammas C, et al. Structural and mutational analysis of the SBDS protein family. J. Biol. Chem. 2005;280:19221–19229. doi: 10.1074/jbc.M414656200. [DOI] [PubMed] [Google Scholar]
- 44.Umeda K, et al. Identification and characterization of hemoangiogenic progenitors during cynomolgus monkey embryonic stem cell differentiation. Stem Cells. 2006;24:1348–1358. doi: 10.1634/stemcells.2005-0165. [DOI] [PubMed] [Google Scholar]
- 45.Choi KD, et al. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells. 2009;27:559–567. doi: 10.1634/stemcells.2008-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.