

Abstract
We report a case of a 6-year old girl with known type 3 Gaucher's Disease on enzyme replacement therapy who developed bilateral, symmetric osteolytic lesions in her humeri and femurs. While this manifestation of Gaucher's disease has been previously documented, it is an exceedingly rare variation. We observe that this patient shares 2 commonalities with 3 other patients reported in the literature to present with this phenotype. First, the patient's L444P/L444P genotype, present in approximately 11% of all Gaucher's patients, was also seen in these other patients. Second, like the other patients, this patient was treated with enzyme replacement therapy. It is unknown whether there is a correlation between these 2 independent variables and this rare phenotype, and further investigation may be warranted.
Keywords: Gaucher's disease, Lucent Bone Lesions, Type 3 Gaucher's Disease, Chronic Neuronopathic Gaucher's Disease, Enzyme Replacement Therapy
Introduction
Gaucher disease (GD), the most prevalent lysosomal storage disorder, is an autosomal recessive, multiorgan disease with a variety of genotypic underpinnings and phenotypic expressions. It results from the accumulation of undegraded glucosylceramide in the lysosomes of monocytes and macrophages in the reticuloendothelial system. The affected leukocytes, known as Gaucher cells, preferentially accumulate in the bone marrow, liver, spleen, and lungs [1], [2], [3], with corresponding clinical manifestations including a variety of osseous abnormalities, pancytopenia, hepatosplenomegaly, and neurological symptoms [4]. The menagerie of common osseous manifestations in GD includes osteopenia, insufficiency fractures, acute bone crises, and so-called “Erlenmeyer flask deformities.”
Case report
A 6-year-old female with Type 3 Gaucher's disease (L444P/L444P mutation) complicated by tracheostomy and gastrostomy tube dependence, seizures, progressive early onset neuromuscular scoliosis, rickets, and 8 lower extremity insufficiency fractures with associated bowing of the femoral shaft presented to the emergency department with oxygen desaturations. The course of her Gaucher's Disease had been managed with the gold-standard enzyme replacement therapy, a twice monthly IV infusion of imiglucerase (60 units/kg). She had begun enzyme replacement therapy at 10 months of age.
Physical exam was notable for lethargic appearance, tachycardia, abdominal distension with intermittent tensing during exam, decreased bowel sounds, abnormal reflexes, abnormal muscle tone with clonus in the upper extremities, contractures, and twitching. Notably, the patient had undergone T3-T5 and L4-S2 spinal fusion surgery approximately 1 month earlier, and there was “bogginess” at the surgical site on presentation. The chest radiograph was positive for new pulmonary opacities, concerning for multifocal pneumonia and/ or aspiration. Her blood culture was positive for methicillin resistant staphylococcus aureus, and the region of concern at the surgical site was revealed to contain an abscess on ultrasound, which was aspirated and consistent with MRSA. She was placed on IV antibiotics and improved, with discharge after a few days’ hospitalization.
A chest radiograph incidentally revealed bubbly lucencies in the bilateral humeral diaphyses with cortical thinning involving the mid shaft on the right (Figs. 1, 5, and 6). An abdominal radiograph during the same admission also incidentally noted similar bubbly lucencies in the bilateral proximal femoral shafts (Figs. 2-4), with further evaluation of the right femur lesion on a separate radiograph (Fig. 3).
Fig. 1.

Bubbly lucencies are noted in the bilateral humeral diaphysis with cortical thinning involving the mid shaft on the right.
Fig. 5.

Zoomed-in image of the chest radiograph showing the right humeral lucencies.
Fig. 6.
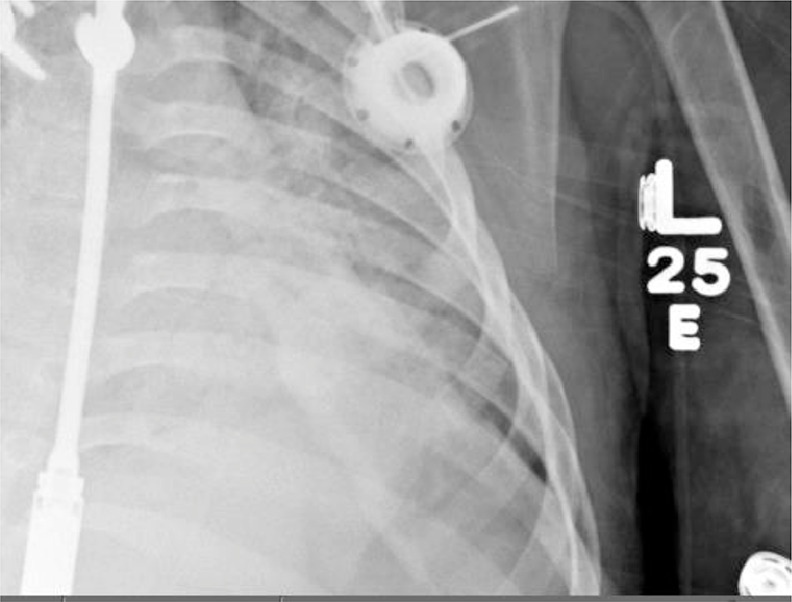
Zoomed-in image of the chest radiograph showing the left humeral lucency.
Fig. 2.

Similar bubbly lucencies are present in the bilateral proximal femoral shafts on the abdominal radiograph.
Fig. 4.
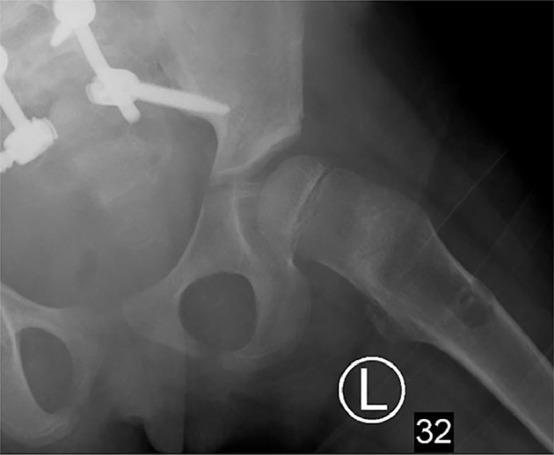
Zoomed-up view of the left femoral lucency, taken from the abdominal radiograph.
Fig. 3.

Right femoral diaphyseal lucency is characterized to better advantage on this dedicated right femur radiograph a few days later.
Discussion
Overall, Gaucher's disease is clinically heterogenous with 3 subtypes. Type 1, known as non-neuronopathic GD, is the mildest and most common form. Type 2, known as acute neuronopathic GD, demonstrates the most severe clinical course, featuring rapid progression of neurologic symptoms with high fatality rates in early childhood. Type 3, or chronic neuronopathic, is characterized by late onset of neurological symptoms and a protracted clinical course [7,8]. Despite the apparent rigidity of this schema, in clinical practice the phenotypic expression within each subtype is highly variable [9].
Common osseous manifestations of Gaucher's Disease include avascular necrosis, bone infarcts, osteosclerosis, and insufficiency fractures. Patient morbidity, with impaired mobility and performance status, occurs due to fractures, joint collapse, and pain, often leading to impaired mobility and performances status. The pathophysiology of the osseous manifestations is not entirely understood. One hypothesis is that Gaucher cells accumulating in the bone marrow disrupt the vascularity at the cortical surface. Another is that the presence of glucocerebroside leads to macrophage and cytokine activation, resulting in osseous insult.
Enzyme replacement therapy (ERT) is the standard of care for patients with types 1 and 3 GD and frequently ameliorates the extent of visceral, hematologic and bony manifestations [10], [11], [12]. It consists of long-term biweekly infusions of a recombinant analogue of the missing human enzyme glubocerebrosidase that catalyzes the hydrolysis of glucocerebroside to glucose and ceramide [7]. ERT has been demonstrated to improve hematological parameters such as hemoglobin concentration and platelet counts, organomegaly and bone involvement [13,14].
However, there are limitations to the efficacy of ERT in the bone. Gaucher cells in bone marrow exhibit less uptake of the replaced enzyme compared to cells in other organ systems, perhaps due to factors within their vascular milieu or possibly due to adjacent fibrosis [7]. Therefore, while ERT is generally effective in treating bone pain, bone crises, and osteoporosis, and while AVN in GD patients post-ERT initiation typically occurs in patients with late treatment initiation, AVN has been known to occur despite high-dose ERT [7].
As would be expected, the specific genetic mutation underpinning a patient's illness impacts on the patient's phenotypic expression. The most common genotypic mutation (N370S/N370S), seen in 38% of patients, is known to cause the classic bone symptoms (avascular necrosis, osteopenia, etc) and does so in 92% of patients [15]. The clinical significance of the lesions seen in this patient is unknown.
The patient in this case report with Type 3 Gaucher's Disease possessed the L444P/L444P mutation, overall seen in 11% of Gaucher's patients. Of considerable interest is that in 2 separate case reports involving 3 other Gaucher's patients with bilateral symmetric lucencies, all 3 also had the L444P/L444P genotype [5,6]. All 3 had also been diagnosed with Type 3 Gaucher's Disease [5,6].
Also of interest is that all of these patients had been on enzyme replacement therapy at an early age [5,6]. This commonality has also been previously observed in one of the prior case reports on this subject [6]. This raises questions about whether enzyme replacement therapy, along with the L444P/L444P genotype, is associated with this rare phenotypic expression. Further investigation may be warranted.
Conclusion
There are a variety of characteristic and common osseous manifestations of Gaucher's Disease, including “Erlenmeyer flask deformities,” osteoporosis and resulting insufficiency fractures, and avascular necrosis. This case presents a very rare osseous manifestation of Gaucher's disease, with bilateral symmetric lucencies in the humeri and femurs, in a patient with L444P/L444P mutation and on enzyme replacement therapy. It is uncertain what, if any, clinical significance these lesions have. Given the commonality of genetics and history of enzyme replacement in 3 other, separately reported patients in the literature, continued inquiry into the existence of a potential link between these 2 independent factors with this rare osseous phenotype may be warranted.
Patient consent statement
The study was approved by the Yale University Institutional Review Board (IRB). It was determined that consent was not required from either the patient or the patient's family (the patient is a minor).
References
- 1.Wenstrup RJ, Roca-Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol. 2002;75(Suppl 1):A2–12. doi: 10.1259/bjr.75.suppl_1.750002. [DOI] [PubMed] [Google Scholar]
- 2.Marcucci G, Zimran A, Bembi B, Kanis J, Reginster J, Rizzoli R. Gaucher disease and bone manifestations. Calcif Tissue Int. 2014;95(6):477–494. doi: 10.1007/s00223-014-9923-y. [DOI] [PubMed] [Google Scholar]
- 3.Andersson H, Kaplan P, Kacena K, Yee J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008;122(6):1182–1190. doi: 10.1542/peds.2007-2144. [DOI] [PubMed] [Google Scholar]
- 4.Ceravolo F, Grisolia M, Sestito S, Falvo F, Moricca MT, Concolino D. Combination therapy in a patient with chronic neuronopathic Gaucher disease: a case report. J Med Case Rep. 2017;11(1):19. doi: 10.1186/s13256-016-1147-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oppenheim IM, Canon AM, Barcenas W, Groden C, Goker-Alpan O, Resnik C. Bilateral symmetrical cortical osteolytic lesions in two patients with Gaucher disease. Skeletal Radiol. 2011;40(12):1611–1615. doi: 10.1007/s00256-011-1260-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teefe E, Kim J, Lopez G, Sidransky E. Bilateral femoral osteolytic lesions in a patient with type 3 Gaucher Disease." 2015. Molecular Genetics and Metabolism Reports. [DOI] [PMC free article] [PubMed]
- 7.Potnis KC, Flueckinger LB, Ha CI, Upadia J, Frush DP, Kishnani PS. Bone manifestations in neuronopathic Gaucher disease while receiving high-dose enzyme replacement therapy. Mol Genet Metab. 2019;126(2):157–161. doi: 10.1016/j.ymgme.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Grabowski GA. Recent clinical progress in Gaucher disease. Curr Opin Pediatr. 2005;17(4):519–524. doi: 10.1097/01.mop.0000172702.33128.19. [DOI] [PubMed] [Google Scholar]
- 9.Goker-Alpan O, Schiffmann R, Park JK, Stubblefield BK, Tayebi N, Sidransky E. Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr. 2003;143(2):273–276. doi: 10.1067/S0022-3476(03)00302-0. [DOI] [PubMed] [Google Scholar]
- 10.Razek A, Abdalla A, Gaber N, Fathy A, Megahed A, Barakat T. Proton MR Spectroscopy of the brain in children with neuronopathic Gaucher’s disease. Eur Radiol. 2013;23(11):3005–3011. doi: 10.1007/s00330-013-2924-9. [DOI] [PubMed] [Google Scholar]
- 11.Erikson A, Bembi B, Schiffmann R. Neuronopathic forms of Gaucher's disease. Baillieres Clin Haematol. 1997;10(4):711–723. doi: 10.1016/s0950-3536(97)80035-2. [DOI] [PubMed] [Google Scholar]
- 12.Mota RM, Mankin H. Use of plain radiography to optimize skeletal outcomes in children with type 1 Gaucher disease in Brazil. J Pediatr Orthop. 2007;27(3):347–350. doi: 10.1097/BPO.0b013e3180340d9f. [DOI] [PubMed] [Google Scholar]
- 13.Drelichman G, Ponce E, Basack N, Freigeiro D, Aversa L, Graciela E. Clinical consequences of interrupting enzyme replacement therapy in children with type 1 Gaucher disease. J Pediatr. 2007;151(2):197–201. doi: 10.1016/j.jpeds.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 14.Gervas-Arruga J, Cebolla JJ, de Blas I, Roca M, Pocovi M, Giraldo P. The influence of genetic variability and proinflammatory status on the development of bone disease in patients with Gaucher disease. PLoS One. 2015;10(5) doi: 10.1371/journal.pone.0126153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams DR. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J. 2006;36(10):652–660. doi: 10.1111/j.1445-5994.2006.01153.x. [DOI] [PubMed] [Google Scholar]