

Abstract
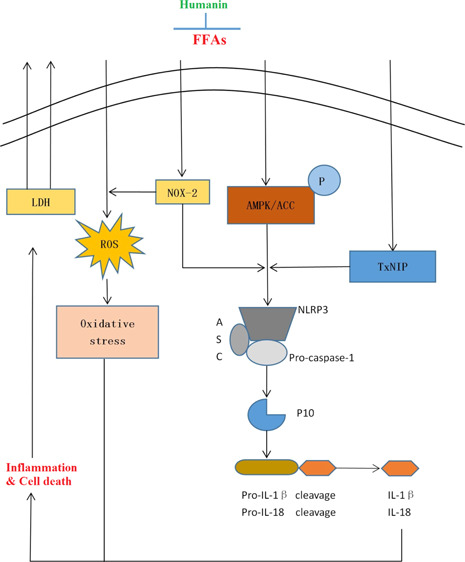
Cardiovascular disease (CVD) has been considered as a major risk factor of death in recent decades. In CVDs, the NLRP3 inflammasome is important for inflammatory response and vascular damage. Therefore, safe and effective treatments to decrease NLRP3 inflammasome activation are required. Increased levels of free fatty acid (FFA) have been associated with the progression of CVD. Humanin, a kind of mitochondrial-derived peptide, has shown its beneficial effects in different types of cells. However, the roles of humanin in the NLRP3 inflammasome induced by FFA are still unknown. Here, we investigated the molecular mechanisms whereby humanin was found to exert protective effects in human aortic endothelial cells (HAECs) against FFA-caused endothelial injury. Here, treatment with humanin inhibited FFA-induced lactate dehydrogenase release, thereby demonstrating a protective capacity against cell death. Humanin also suppressed oxidative stress by downregulating the expression of reactive oxygen species and NOX2. Notably, humanin reduced NLRP3 and p10 and rescued FFA-induced dysfunction of adenosine monophosphate-activated protein kinase. Consequently, humanin inhibited the expression of IL-1β and IL-18. These results conclude that humanin might be a promising therapeutic agent for CVD.
1. Introduction
Cardiovascular disease (CVD) has become a leading cause of death in recent decades and poses an increasing economic burden worldwide. CVD contributes to the obstruction of blood flow and impedes the delivery of nutrients to the heart, which negatively influences the systems of the entire body. Advanced research has indicated that the inflammatory process plays a key role in various aspects of CVD, such as ischemia/reperfusion injury,1 thrombosis,2 and infection.3,4 For example, atherosclerosis (AS) is widespread in the circulatory system with a chronic inflammatory response.5−7 Inflammation is involved in all stages of AS, which eventually results in plaque rupture. These pathological events lead to myocardial infarction and heart failure.8 The NLRP3 inflammasome consists of NLRP3, ASC, and the inactive zymogen pro-caspase-1,9 leading to the maturation of IL-1β and IL-18.10,11 This leads to a robust initiation of the inflammatory process. The NLRP3 inflammasome closely participates in the progression of AS.12,13
Endothelial cells are important for regulating cardiovascular function and homeostasis.14 High free fatty acid (FFA) levels in plasma have been recognized as an essential indicator of CVD, which can induce the activation of the NLRP3 inflammasome.15,16 Endothelial dysfunction induced by FFA is an important event in atherothrombosis. In this study, in order to mimic the inflammatory microenvironment present in CVD, we subjected human aortic endothelial cells (HAECs) to a high concentration of FFA. Oxidative stress has been demonstrated to contribute to the induction of endothelial dysfunction.17 Previous studies have shown that reducing NOX2 suppresses oxidative stress-induced vascular degeneration and decreases the activation of the NLRP3 inflammasome.18 TxNIP can induce activation of the NLRP3 inflammasome, thereby triggering the secretion of IL-1β and IL-18, which are necessary for the progression of inflammation.19 Adenosine monophosphate-activated protein kinase (AMPK) is a key regulator of intracellular energy balance, fat metabolism, and adenosine triphosphate conservation and synthesis. Contemporary studies have provided new evidence that the activation of AMPK signaling leads to inhibition of the NLRP3 inflammasome.20
Humanin is the first discovered mitochondrial-derived peptide. Humanin has displayed its protective effects in various pathologies. For example, studies have shown that humanin protects against stroke in mice by inhibiting ERK activation21 and ameliorates the development of AS.22 Humanin has also been shown to decrease apoptosis in β-cells and to improve glucose tolerance and the onset of diabetes in nonobese diabetic mice.23 However, the function of humanin on the NLRP3 inflammasome remains unknown.
2. Results
2.1. Humanin Prevented High FFA-Induced Cytotoxicity in HAECs
The morphology of HAECs is shown in Figure 1A. As shown in Figure 1B, FFA treatment increased lactate dehydrogenase (LDH) release to 29.6% from 6.5% at the baseline. However, in the presence of 25 μM humanin, FFA treatment only induced 17.8% release of LDH. Moreover, 50 μM humanin further decreased the release of LDH to 13.2%. These data demonstrate that humanin exerts a strong beneficial effect against FFA-induced cytotoxicity in endothelial cells.
Figure 1.

Humanin prevented high FFA-induced cytotoxicity in HAECs. Cells were treated with high FFA (1 mM) with or without humanin (25, 50 μM) for 48 h. (A) Morphology of HAECs; (B) release of LDH (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4).
2.2. Humanin Prevented FFA-Induced Oxidative Stress in HAECs
In order to demonstrate whether humanin can protect HAECs from FFA-caused oxidative stress, we examined the levels of NOX2, reactive oxygen species (ROS), and protein carbonyl. The results in Figure 2A show that the gene level of NOX2 was upregulated significantly from the baseline to 3.7-fold upon exposure to FFA. However, 25 and 50 μM humanin reduced NOX2 expression to 2.6- and 1.9-fold, respectively. The protein of NOX2 was strongly inhibited from an FFA-induced increase of 3.2-fold to 2.4- and 1.5-fold by 25 and 50 μM humanin, respectively. As shown in Figure 3A, humanin had a similar inhibitory effect on FFA-induced increased production of ROS. The results in Figure 3B demonstrate that FFA induced a 2.8-fold increase in the intracellular level of protein carbonyl, which was reduced to 2.0- and 1.6-fold by the two respective doses of humanin. Together, these results demonstrate that humanin exerts a strong inhibitory effect on FFA-induced oxidative stress by suppressing NOX2-mediated ROS production and protein carbonyl in HAECs.
Figure 2.

Humanin reduced high FFA-induced upregulation of NOX2 in HAECs. (A) Gene levels of NOX2; (B) protein of NOX2 (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4–5).
Figure 3.
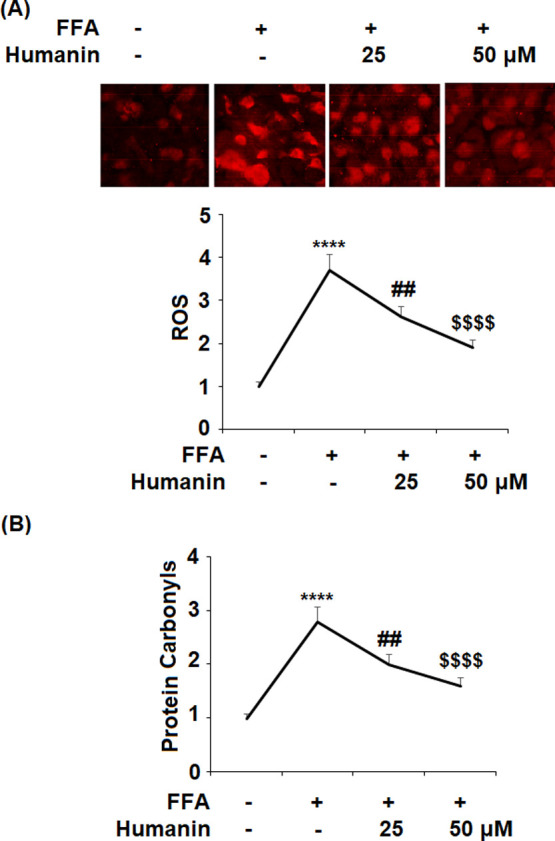
Humanin prevented high FFA-induced oxidative stress in HAECs. (A) Production of ROS; (B) intracellular levels of protein carbonyl (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4–5).
2.3. Humanin Reduced FFA-Induced Upregulation of TxNIP
The effect of humanin on the expression of TxNIP is of considerable importance. The mRNA level of TxNIP was increased significantly to 3.5-fold with FFA treatment, while 25 and 50 μM humanin reduced it to 2.4- and 1.8-fold, respectively (Figure 4A). Moreover, the same doses of humanin inhibited the protein of TxNIP to 2.2- and 1.7-fold compared to 3.2-fold upon exposure to FFA alone (Figure 4B).
Figure 4.

Humanin reduced high FFA-induced upregulation of TxNIP in HAECs. (A) Gene levels of TxNIP; (B) protein levels of TxNIP (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4–5).
2.4. Humanin Inhibited FFA-Induced Activation of the NLRP3 Inflammasome in HAECs
FFA treatment significantly elevated NLRP3 and p10 to 3.2- and 4.6-fold, respectively, which were reduced to 2.3- and 2.7-fold by 25 μM humanin and 1.7- and 1.9-fold by 50 μM humanin, respectively. Thus, humanin exerts an inhibitory action on the NLRP3 inflammasome (Figure 5).
Figure 5.
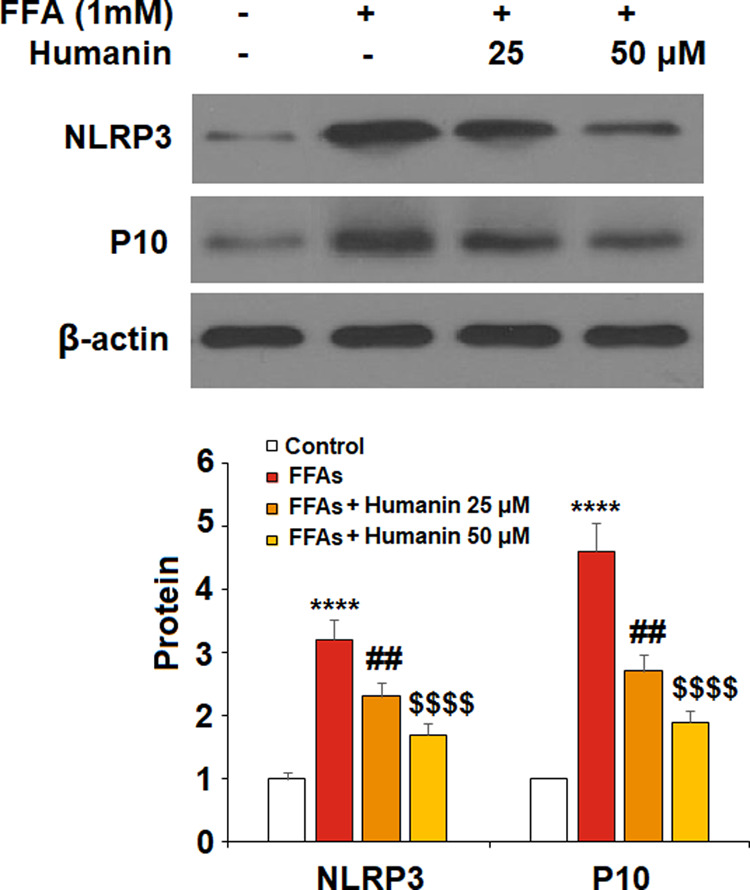
Humanin inhibited high FFA-induced activation of NLRP3 in HAECs. The expression of NLRP3 and p10 were measured (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4–5).
2.5. Humanin Inhibited FFA-Induced Expression of IL-1β and IL-18
The secretion of IL-1β and IL-18 was significantly upregulated from 161.4 and 125.6 to 782.1 and 653.9 pg/mL by FFA, respectively. Meanwhile, 25 μM humanin decreased them to 549.3 and 445.7 pg/mL and 50 μM humanin further reduced the secretion of them to 367.2 and 318.7 pg/mL. Thus, humanin exerts an anti-inflammatory effect by decreasing the expression and secretion of proinflammatory cytokines (Figure 6).
Figure 6.

Humanin inhibited high FFA-induced secretion of IL-1β and IL-18 in HAECs. Cells were treated with high FFA (1 mM) in the presence or absence of humanin (25, 50 μM) for 48 h. (A) Secretion of IL-1β; (B) secretion of IL-18 (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4).
2.6. Humanin Prevented FFA-Induced Inactivation of the AMPK/ACC Signaling Pathway in HAECs
As shown in Figure 7A,B, FFA stimulation reduced the level of phosphorylated AMPKα and ACC to 24 and 39%, respectively, of the baseline. However, 25 μM humanin increased the levels of phosphorylated AMPKα and ACC to 86 and 68%, respectively, while 50 μM humanin further increased them to 1.25- and 1.12-fold, respectively.
Figure 7.

Humanin prevented FFA-induced inactivation of the AMPK/ACC signaling pathway in HAECs. Cells were treated with high FFA (1 mM) with or without humanin (25, 50 μM) for 2 h. (A) Phosphorylated and total levels of AMPKα; (B) phosphorylated and total levels of ACC (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4–5).
2.7. Blockage of AMPK Abolished the Beneficial Effects of Humanin against NLRP3 Inflammasome Activation
To clarify the relationship between AMPK and NLRP3 inflammasome activation, compound C was employed to block the activation of AMPK. NLRP3 was increased significantly upon FFA treatment, while humanin reduced NLRP3, IL-18, and IL-1β. However, the expression of NLRP3 and IL-18 significantly increased in the presence of compound C, which is a specific inhibitor of AMPK (Figure 8). These results show that the effects of humanin against the NLRP3 inflammasome are mediated by AMPK.
Figure 8.

Blockage of AMPK with its specific inhibitor compound C abolished the protective effects of humanin against activation of the NLRP3 inflammasome. Cells were treated with high FFA (1 mM) with or without humanin (50 μM) and compound C for 48 h. (A) NLRP3; (B) secretions of IL-18; (C) secretions of IL-1β (****, P < 0.0001 vs vehicle control; ##, p < 0.01 vs FFA treatment group; $$$$, P < 0.0001 vs FFA + 25 μM humanin group, n = 4).
3. Discussion
CVDs including hyperlipidemia, AS, and hypertension are potentially life-threatening diseases with increased risk in the elderly population. Therapeutic treatments that prevent the progression and development of CVD are in high demand. As a main cause of oxidative stress, increased ROS accumulation induced by high levels of plasma FFA could promote chronic inflammation and endothelial dysfunction in AS.24 NLRP3-dependent maturation of IL-1β and IL-18, induced by oxidative stress, has been demonstrated to relate to the progression of CVD.25 NOX2 regulates the generation of ROS. In addition, NOX2 can promote atherogenesis by causing both epidermic and endothelial inflammation.26,27 Therefore, the inhibition of NOX2 is important for reducing both oxidative stress and the activation of NLRP3. Recent discoveries have shown that humanin protects retinal pigment epithelium cells from oxidative stress.28 In this study, we observed that humanin can suppress oxidative stress by inhibiting the production of ROS induced by FFA in HAECs. Interestingly, the effect of humanin on the production of ROS may be mediated through downregulating the expression of NOX2. Importantly, the cellular release of LDH release by HAECs induced by FFA was significantly decreased with humanin treatment.
Increased TxNIP is recognized as being involved in the modulation of inflammatory response and cellular apoptosis.29 TxNIP also exacerbates the production of ROS by inhibiting thioredoxin (TRX).30,31 Furthermore, TxNIP could modulate the expression of genes that promote atherogenesis.32 In FFA-induced HAECs, excessive expression of TxNIP is a major factor driving the activation of the NLRP3 inflammasome.33 In this study, our findings reveal the inhibitory effect of humanin on TxNIP, thereby downregulating oxidative stress and the NLRP3 inflammasome. Suppressing the NLRP3 inflammasome has been recognized as a therapeutic treatment strategy for various inflammatory diseases, including AS. Once activated, NLRP3 initiates the assembly of NLRP3, ASC, and p10. Ultimately, the activation of the NLRP3 inflammasome complex is mediated by the cleaved form of caspase-1.34
AMPK is a major sensor of metabolism status expressed in different types of tissues. In endothelial cells, AMPK plays a key role in maintaining endothelial function. AMPK regulates the generation of NO in endothelial cells through eNOS activity.35 AMPK has the advantage of monitoring the occurrence of oxidative stress. In addition, research has demonstrated that decreased AMPK activity is associated with the development of AS.36 The AMPK pathway is a major modulator of NLRP3.37,38 Here, we found that compound C abolished the beneficial effects of humanin, indicating that the suppression of NLRP3 inflammasome activation by humanin is mediated by AMPK.
4. Conclusions
In conclusion, our study demonstrates the inhibitory effects of humanin on the activation of the NLRP3 inflammasome, which were found to be mediated by the AMPK pathway. Our data show that humanin treatment is a promising strategy for CVD. However, more investigations are required to further our understanding of the mechanisms behind the beneficial effects of humanin against FFA injury and other types of stimulations.
5. Materials and Methods
5.1. Cell Culture
HAECs were cultured in EGM-2 medium (Lonza, Switzerland) containing 5% fetal bovine serum (FBS). The protocols for all experiments were approved by ethics committee of Ganzhou People’s Hospital. The cells were cultured in the T-75 flasks or 12-well plates, and the medium was changed every 3–4 days. Cells were treated with FFA (1 mM)39 with or without humanin (25, 50 μM)40 for 48 h before experimentation. The humanin used in our study was commercially purchased from GLPBIO (Cat# GC18324).
5.2. LDH Release
The release of LDH from HAECs was assessed using a kit (Cat#C0017, Beyotime). Briefly, 1.5 × 104 HAECs were plated in 96-well plates and subjected to the indicated treatment. From all wells, 50 μL of the culture supernatant was collected and added to a new well, and 50 μL of assay buffer was added to each well. The plate was wrapped with aluminum foil for 1 h. Afterward, 50 μL of stop solution was added to each well. Absorbance at 570 nm was measured to reflect cell death.
5.3. Real-Time PCR Analysis
After treatment, the total RNA was extracted from HAECs using an RNeasy Mini kit (Cat no. 74106, Qiagen, USA). Reverse transcription polymerase chain reaction (RT-PCR) analysis was performed using a GoScript reverse transcription kit following the protocol from the manufacturer. Primers were designed for the target genes, which were combined with 20 μg of cDNA and SYBR Green PCR mix. Real-time PCR was performed on an ABI 7900HT system. The mRNA levels were determined using the 2–ΔΔCt assay. The following primers were used: human NOX2: 5′-CAGCCTGCCTGAATTTCAACT-3′, 5′-GGAGAGGAGATTCCGACACACT-3′ and human GAPDH: 5′-ACCCACTCCTCCACCTTTGA-3′, 5′-CTGTTGCTGTAGCCAAATTCGT-3′.
5.4. Western Blot Analysis
Total protein from HAECs was obtained using radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China). Total proteins (20 μg) were electrically separated on a sodium dodecyl sulfate polyacrylamide gel and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was then blocked with 5% slim milk for 1 h. The PVDF membranes were incubated with a specific primary antibody diluted in block buffer at 4 °C overnight. The following primary antibodies were used: NOX4 (1:2000, Cat#GTX12206, Gene Tax), TxNIP (1:2000, Cat#ab188865, Abcam), NLRP3 (1:2000, Cat#19771-1-AP, Protein Tech), p10 (1:2000, Cat#sc-514, Santa Cruz Biotechnology), phospho-AMPKα (1:2000, Cat #50081, Cell Signaling), AMPKα (1: 2000, Cat#2532, Cell Signaling), phospho-ACC (1:2000 Cat# 3661, Cell Signaling), ACC (1:2000, Cat#3662, Cell Signaling), and β-actin (1:5000, Cat#sc-130656, Santa Cruz Biotechnology). After three washes, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. The protein signals were detected using an enhanced chemiluminescence reagent, and the protein band was analyzed using Quantity One software (Bio-Rad, USA).
5.5. Enzyme-Linked Immunosorbent Assay
L-1β and IL-18 were measured using an IL-1β enzyme-linked immunosorbent assay (ELISA) kit (Cat#SEKH-0002, Solarbio) and a human IL-18 ELISA kit (SEKH-0028, Solarbio) following the protocols from the manufacture. Briefly, 50 μL of the supernatant or prepared standard was added to 96-well plates for 2 h. The antibody (100 μL) was then added for 1 h. After washing four times, 100 μL of diluted HRP-conjugated secondary antibodies was added for 30 min. Then, 100 μL of the chromogenic substrate was added to each well and developed for 30 min. The absorbance at 450 nm was recorded to index the protein concentration.
5.6. Measurement of Intracellular ROS
The ROS level was evaluated using dihydroethidium (DHE) staining (Cat# CAS 104821-25-2, Santa Cruz Biotechnology). After treatment, cells were incubated with 5 μM DHE for 30 min in darkness. After three washes, the fluorescence intensity of the cells was recorded using a fluorescence microscope (excitation/emission wavelengths: 510/610 nm).
5.7. Determination of Protein Carbonyl
Protein carbonyl in HAECs was assessed using ELISA. Briefly, cell homogenates were reacted with 2,4-DNPH. Samples were added into 96-well ELISA plates (Corning Incorporated, USA). Samples were then blocked with 2.5% fish skin gelatin for 1 h and incubated with the primary anti-dinitrophenyl antibody for 1 h. After three washes, the HRP-conjugated secondary antibody and HRP substrate solution (Amresco, USA) were added. The absorbance at 405 nm was recorded to index protein carbonyl.
5.8. Statistical Analysis
The experimental data are presented as mean ± S.E.M. Major statistical analysis was performed using ANOVA using SPSS (Version 19). A P value of < 0.05 was regarded as significant.
Acknowledgments
This study is supported by the “Jiangxi Scientific Support Project (no. 162201340022)”.
Author Present Address
† W.L.: Department of Cardiology, the First Affiliated Hospital of Ji’nan University, Guangzhou Overseas Chinese Hospital.
The authors declare no competing financial interest.
References
- Chaudhari N.; Talwar P.; Parimisetty A.; Lefebvre d’Hellencourt C.; Ravanan P. A.; molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8, 213. 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T.; Endo M.; Oike Y. Endoplasmic reticulum stress-related inflammation and cardiovascular diseases. Int. J. Inflammation 2011, 2011, 259462. 10.4061/2011/259462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolattukudy P. E.; Niu J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ. Res. 2012, 110, 174–189. 10.1161/circresaha.111.243212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker P. M. Inflammation, C-Reactive Protein, and Cardiovascular Disease. Circ. Res. 2014, 114, 594–595. 10.1161/circresaha.114.303215. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Syed T. W.; Liu R.; Yu J. Role of Endoplasmic Reticulum Stress, Autophagy, and Inflammation in Cardiovascular Disease. Front. Cardiovasc. Med. 2017, 4, 29. 10.3389/fcvm.2017.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.-Y.; Li C.-J.; Hou M.-F.; Chu P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. 10.3390/ijms18102034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, 7–12. 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. 10.1186/1741-7015-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam V. A. K.; Vanaja S. K.; Fitzgerald K. A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Zhao Y.; Wang K.; Shi X.; Wang Y.; Huang H.; Zhuang Y.; Cai T.; Wang F.; Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Vanaja S. K.; Rathinam V. A. K.; Fitzgerald K. A. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. 10.1016/j.tcb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek M.; Manduchi E.; Riley R. J.; Stoeckert C. J. Jr.; Davies P. F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. 10.1161/circresaha.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F.; Xing S.; Gong Z.; Mu W.; Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflammation 2014, 2014, 507208. 10.1155/2014/507208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A.; Steven S.; Weber A.; Shuvaev V. V.; Muzykantov V. R.; Laher I.; Li H.; Lamas S.; Münzel T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. 10.1111/bph.13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand-Poels S.; Esser N.; L’homme L.; Scheen A.; Paquot N.; Piette J.; Jacques Free fatty acids as modulators of the NLRP3 Inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 2014, 92, 131–141. 10.1016/j.bcp.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Qi Y.; Du X.; Yao X.; Zhao Y. Vildagliptin inhibits high free fatty acid (FFA)-induced NLRP3 inflammasome activation in endothelial cells. Artif Cells Nanomed. Biotechnol 2019, 47, 1067–1074. 10.1080/21691401.2019.1578783. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Tate M.; Mathew G.; Vince J. E.; Ritchie R. H.; de Haan J. B. Oxidative stress and NLRP3- inflammasome activity as significant drivers of diabetic cardiovascular complications: therapeutic implications. Front. Physiol. 2018, 9, 114. 10.3389/fphys.2018.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M. W.; Wang J.; Dhandapani K. M.; Brann D. W. NADPH Oxidase 2 Regulates NLRP3 Inflammasome Activation in the Brain after Traumatic Brain Injury. Oxid. Med. Cell. Longevity 2017, 2017, 6057609. 10.1155/2017/6057609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Veerdonk F. L.; Netea M. G.; Dinarello C. A.; Joosten L. A. B. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011, 32, 110–116. 10.1016/j.it.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Bullón P.; Alcocer-Gómez E.; Carrión A. M.; Marín-Aguilar F.; Garrido-Maraver J.; Román-Malo L.; Ruiz-Cabello J.; Culic O.; Ryffel B.; Apetoh L.; et al. AMPK Phosphorylation Modulates Pain by Activation of NLRP3 Inflammasome. Antioxid. Redox Signaling 2016, 24, 157–170. 10.1089/ars.2014.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Chua C. C.; Gao J.; Hamdy R. C.; Chua B. H. L. Humanin is a novel neuroprotective agent against stroke. Stroke 2006, 37, 2613–2619. 10.1161/01.str.0000242772.94277.1f. [DOI] [PubMed] [Google Scholar]
- Oh Y. K.; Bachar A. R.; Zacharias D. G.; Kim S. G.; Wan J.; Cobb L. J.; Lerman L. O.; Cohen P.; Lerman A. Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis 2011, 219, 65–73. 10.1016/j.atherosclerosis.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang P. T.; Park P.; Cobb L. J.; Paharkova-Vatchkova V.; Hakimi M.; Cohen P.; Lee K.-W. The neurosurvival factor Humanin inhibits beta-cell apoptosis via signal transducer and activator of transcription 3 activation and delays and ameliorates diabetes in nonobese diabetic mice. Metabolism 2010, 59, 343–349. 10.1016/j.metabol.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaimes E. A.; Hua P.; Tian R. X.; Raij L. Human glomerular endothelium: interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am. J. Physiol.: Renal, Fluid Physiol. 2010, 298, 125–132. 10.1152/ajprenal.00248.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Zeng X.; Li X.; Mehta J. L.; Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2018, 113, 5. 10.1007/s00395-017-0663-9. [DOI] [PubMed] [Google Scholar]
- Lassègue B.; Griendling K. K. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler., Thromb., Vasc. Biol. 2010, 30, 653–661. 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M. W.; Wang J.; Dhandapani K. M.; Brann D. W. NADPH Oxidase 2 regulates NLRP3 inflammasome activation in the brain after traumatic brain injury. Oxid. Med. Cell. Longevity 2017, 2017, 6057609. 10.1155/2017/6057609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Sreekumar P. G.; Peddi S.; Hinton D. R.; Kannan R.; MacKay J. A. The humanin peptide mediates ELP nanoassembly and protects human retinal pigment epithelial cells from oxidative stress. Nanomedicine 2019, 24, 102111. 10.1016/j.nano.2019.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C.; Zhao Y.; Shi X.; Zhang N.; Zu G.; Li Z.; Zhou J.; Gao D.; Lv L.; Tian X.; et al. New insights into salvianolic acid A action: regulation of the TXNIP/NLRP3 and TXNIP/ChREBP pathways ameliorates HFD-induced NAFLD in rats. Sci. Rep. 2016, 6, 28734. 10.1038/srep28734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood D. F. D.; Abderrazak A.; El Hadri K.; Simmet T.; Rouis M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signaling 2013, 19, 1266–1303. 10.1089/ars.2012.4757. [DOI] [PubMed] [Google Scholar]
- Bedarida T.; Baron S.; Vibert F.; Ayer A.; Henrion D.; Thioulouse E.; Marchiol C.; Beaudeux J.-L.; Cottart C.-H.; Nivet-Antoine V. Resveratrol decreases TXNIP mRNA and protein nuclear expressions with an arterial function improvement in old mice. J. Gerontol., Ser. A 2016, 71, 720–729. 10.1093/gerona/glv071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Q.; Nigro P.; World C.; Fujiwara K.; Yan C.; Berk B. C. Thioredoxin interacting protein promotes endothelial cell inflammation in response to disturbed flow by increasing leukocyte adhesion and repressing Kruppel-like factor 2.. Circ. Res. 2012, 110, 560–568. 10.1161/circresaha.111.256362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Joe Y.; Jeong S. O.; Zheng M.; Back S. H.; Park S. W.; Ryter S. W.; Chung H. T. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of NF-κB and inflammasome pathways. Innate Immun. 2014, 20, 799–815. 10.1177/1753425913508593. [DOI] [PubMed] [Google Scholar]
- Jo E.-K.; Kim J. K.; Shin D.-M.; Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou H.; Liu C.; Feng W.; Xiao X.; Tang S.; Mo Z. Role of AMPK in atherosclerosis via autophagy regulation. Sci. China: Life Sci. 2018, 61, 1212–1221. 10.1007/s11427-017-9240-2. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Zhang M.; Wang S.; Liang B.; Zhao Z.; Liu C.; Wu M.; Choi H. C.; Lyons T. J.; Zou M. H. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010, 59, 1386–1396. 10.2337/db09-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H.; Liu Q.; Wen Z.; Feng H.; Deng X.; Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/ GSK3b-Nrf2 signal axis. Redox Biol. 2017, 12, 311–324. 10.1016/j.redox.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero M. D.; Williams M. R.; Ryffel B. AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol. Metab. 2018, 29, 8–17. 10.1016/j.tem.2017.10.009. [DOI] [PubMed] [Google Scholar]
- Qin Q.; Jin J.; He F.; Zheng Y.; Li T.; Zhang Y.; He J. Humanin Promotes Mitochondrial Biogenesis in Pancreatic MIN6 β-Cells. Biochem. Biophys. Res. Commun. 2018, 497, 292–297. 10.1016/j.bbrc.2018.02.071. [DOI] [PubMed] [Google Scholar]
- Jang E.; Shin M.-H.; Kim K.-S.; Kim Y.; Na Y.-C.; Woo H.-J.; Kim Y.; Lee J.-H.; Jang H.-J. Anti-lipoapoptotic effect of Artemisia capillaris extract on free fatty acids-induced HepG2 cells. BMC Complementary Altern. Med. 2014, 14, 253. 10.1186/1472-6882-14-253. [DOI] [PMC free article] [PubMed] [Google Scholar]