

Abstract
Targeted drug delivery remains at the forefront of biomedical research but remains a challenge to date. Herein, we describe the first super-assembly of nanosized metal–organic polyhedra (MOP) and their biomimetic coatings of lipid bilayers to synergistically combine the advantages of micelles and supramolecular coordination cages for targeted drug delivery. The super-assembly technique affords unique hydrophobic features that endow individual MOP to act as nanobuilding blocks and enable their super-assembly into larger and well-defined nanocarriers with homogeneous sizes over a broad range of diameters. Various cargos were controllably loaded into the MOP with high payloads, and the nanocages were then super-assembled to form multi-drug delivery systems. Additionally, functional nanoparticles were introduced into the super-assemblies via a one-pot process for versatile bioapplications. The MOP super-assemblies were surface-engineered with epidermal growth factor receptors and could be targeted to cancer cells. In vivo studies indicated the assemblies to have a substantial circulation half-life of 5.6 h and to undergo renal clearance – characteristics needed for nanomedicines.
Keywords: metal–organic polyhedra, micelles, nanoparticles, super-assembly, targeted drug delivery
Graphical Abstract
A novel and modular super-assembly approach combines the synergistic advantages of MOP super-assembly (e.g., highly controlled architecture, high payloads of multiple cargos) and micelle (e.g., low inherent toxicity) has been reported. The MOP super-assembly-supported micelle can be loaded with various drugs, functional nanoparticles, and can function as multi-drug delivery systems with specific cargo loading amounts. By modification of targeting moieties, targeted drug delivery to cancer cells has been realized.
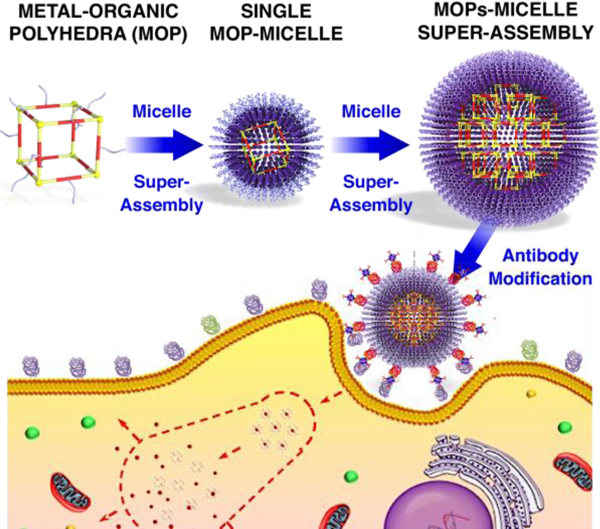
The desire to improve drug efficacy, enhance targeted delivery to specific sites, and reduce side effects by controlling drug pharmacokinetic and biodistribution profiles has remained the main driving force for the development of drug delivery systems over the past two decades.[1] Among all of the developed nanocarriers (e.g., micelles, liposomes, and mesoporous silica nanoparticles (NPs)),[2] nanosized metal–organic polyhedra (MOP) obtained upon metal–ligand coordination have attracted increasing attention for drug delivery owing to the highly designable nanoarchitectures, well-defined pore cavities, as well as diverse chemical properties and functions.[3] In this field, Therrien et al. presented a series of organometallic cages as anticancer drug delivery vehicles for photodynamic therapy.[4a,b] Lippard et al. reported a well-defined metal–organic octahedron that can enhance the delivery of cis-platin prodrugs to cancer cells.[4c] In addition, Isaacs et al. highlighted the potential of host–guest interactions, by combining MOP with cucurbituril, for enhanced delivery of chemotherapeutic drugs.[4d,e] Although many MOP-based supramolecular systems have been developed for drug delivery applications,[5] compared to the well-developed NP-based systems such as mesoporous silica NPs,[5d] the use of MOP for drug delivery is still in its infancy. This is especially the case of research concerning targeted drug delivery for cancer therapy. A major limitation of MOP as nanocarriers is their rapid renal clearance and short circulation time owing to their small size, which is typically below the filtration barrier of the glomerulus (i.e. ~5.5 nm).[4d,e] Furthermore, using coordination complexes for targeted therapy requires their functionalization with various targeting ligands consisting of small molecular moieties or large antibodies. However, the design and synthesis of the required organic linkers are highly challenging.
To overcome these limitations, we describe for the first time the super-assembly of MOP. The proposed fabrication technique affords a simple synthesis process and the organization of individual MOP to form advanced hierarchical structures. Specifically, the super-assembly of MOP supported by micelles (MOPsa@micelle) for targeted drug delivery is described (see Scheme 1a). The key fabrication point is the decoration of the outer surface of the MOP with hydrophobic chains to transform MOP units into nanobuilding blocks that can self-assemble into larger and well-defined super-assemblies within micelles. This approach is different from the reported molecular building block (MBB) approach that is used to construct metal–organic framework (MOF)-based lipid bilayers,[2d] where the open metal sites of MOF units allow further coordination of additional organic ligands to form MOF NPs (Scheme 1b). Nonetheless, the MBB approach typically affords limited size control, whereas the super-assembly approach described herein provides homogeneous size distributions in broad ranges of diameters through the simple control of the MOP concentration to obtain ultrasmall single MOP@micelle and MOPsa@micelle. In addition, owing to the highly porous nature of the MOP, various cancer drugs were controllably loaded into individual MOP units with high payloads and subsequently integrated into MOP super-assemblies to form multiple drug delivery systems. Furthermore, the MOP super-assembly approach enabled the modular assembly of MOPs with additional functional NPs such as fluorescent quantum dots (QDs) for multi-fluorescence imaging or gold NPs for enhanced bio-imaging. To demonstrate the potential of the MOP super-assembly concept for drug delivery, a Fujita-type MOP composed of dozens of dodecyl chains was chosen as a prototype. A dose-dependent assay for MOPsa@micelles of different sizes demonstrated negligible hemolytic activities and long-term colloidal stability in various media, thereby mitigating possible concerns of structure disassembly during circulation. Cell viability tests further demonstrated the good biocompatibility of the MOPsa@micelles that is related to the NP size and the molecular weight of the polyethylene glycol (PEG) that was used for surface modification. Importantly, designing MOPsa@micelle nanocarriers with targeting moieties enabled targeted cancer cell delivery and in vivo experiments using a mouse model also confirmed the good circulation. Taken together, this novel and modular super-assembly approach combines the synergistic advantages of micelles (e.g., low inherent toxicity, and long circulation time) and the MOP super-assembly (e.g., highly controlled architecture, stability and high payloads of multiple cargos), and promotes the design of MOP-inspired nanocarriers for targeted cancer therapies.
Scheme 1.

Schematic illustration of (a) the design and construction of drug delivery nanocarriers (single MOP or MOP super-assembly-supported micelle based on a modular super-assembly approach and (b) the solution synthesis of MOF NP-supported lipid bilayers based on MBB approach.
A giant Pd24L48 Fujita-type spherical framework (~7.2 nm) composed of up to 48 decorations of dodecyl chains (denoted as Pd24L48-C12) was first prepared for proof of concept (Figure 1a).[6] Dipyridyl-3-dodecyl-thiophene was first synthesized as an organic linker (ligand 1) using the Suzuki–Miyaura reaction. To prepare the coordination sphere, ligand 1 (0.1 mmol) and Pd(BF4)2 (50 μmol) were heated in a mixture of acetonitrile and chloroform at 70 °C for 24 h. Then, an excess amount of a mixed solution of ethyl acetate and diethyl ether (1:1 v/v) was added to promote the precipitation. The structure of Pd24L48-C12 MOP was first determined by Fourier-transform infrared spectroscopy. As shown in Figure S1, the characteristic peak at 1589 cm−1 assigned to the vibration of C=N in the pyridine ring was shifted to 1610 cm−1, indicating the coordination of Pd2+ metal ions. Based on chemical analysis, the ratio of organic ligand to Pd2+ was calculated to be ~1.8, which is close to the exact ratio of 2. Moreover, in nuclear magnetic resonance (NMR) Spectra (Figure 1b), the b and c protons of the pyridyl or thiophene groups (Hb and Hc) were shifted downfield by 0.20 and 0.22 ppm, respectively, upon coordination to Pd2+. Compared with the 1H signal of free ligand 1, the 1H signal of Pd24L48-C12 MOP was much broader. Diffusion-ordered NMR measurements also demonstrated a single product given the diffusion coefficient of 5.7 × 10−10 m2 s−1, which was indicative of the formation of larger chemical species. Furthermore, Electrospray ionization and matrix-assisted laser desorption ionization time of flight mass spectrometry techniques were used to confirm the coordination structure. Although the molecular weight of the Pd24L48 cage was determined by a series of prominent peaks in [Pd24L48-(BF4-)m]m+ (m/z = 17, 24, 27, and 33, Figure S2), the molecular weight of the Pd24L48-C12 cage could not be quantified by the same technique, presumably due to presence of the multiple dodecyl chains. Nevertheless, high-resolution transmission electron microscopy (HR-TEM) displayed a dense packing of Pd24L48-C12 MOPs with a distance of 4.8 nm between closest neighbors (Figure 1c). Atomic force microscopy (AFM) imaging also showed single cages with sizes of ~5.0 nm (Figure S3). All this structural information confirmed the successful formation of giant coordination spheres. Based on molecular mechanics (MM) calculations, the optimized structure of Pd24L48-C12 was obtained. As shown in Figure 1d, the rhombicuboctahedral structure of Pd24L48-C12 is highly spherical with an inscribed sphere with a diameter of 3.6 nm and a circumscribed sphere, with alkane chains, of 7.2 nm in diameter. The distance between antipodal palladium atoms was measured to be 4.0 nm.
Figure 1.
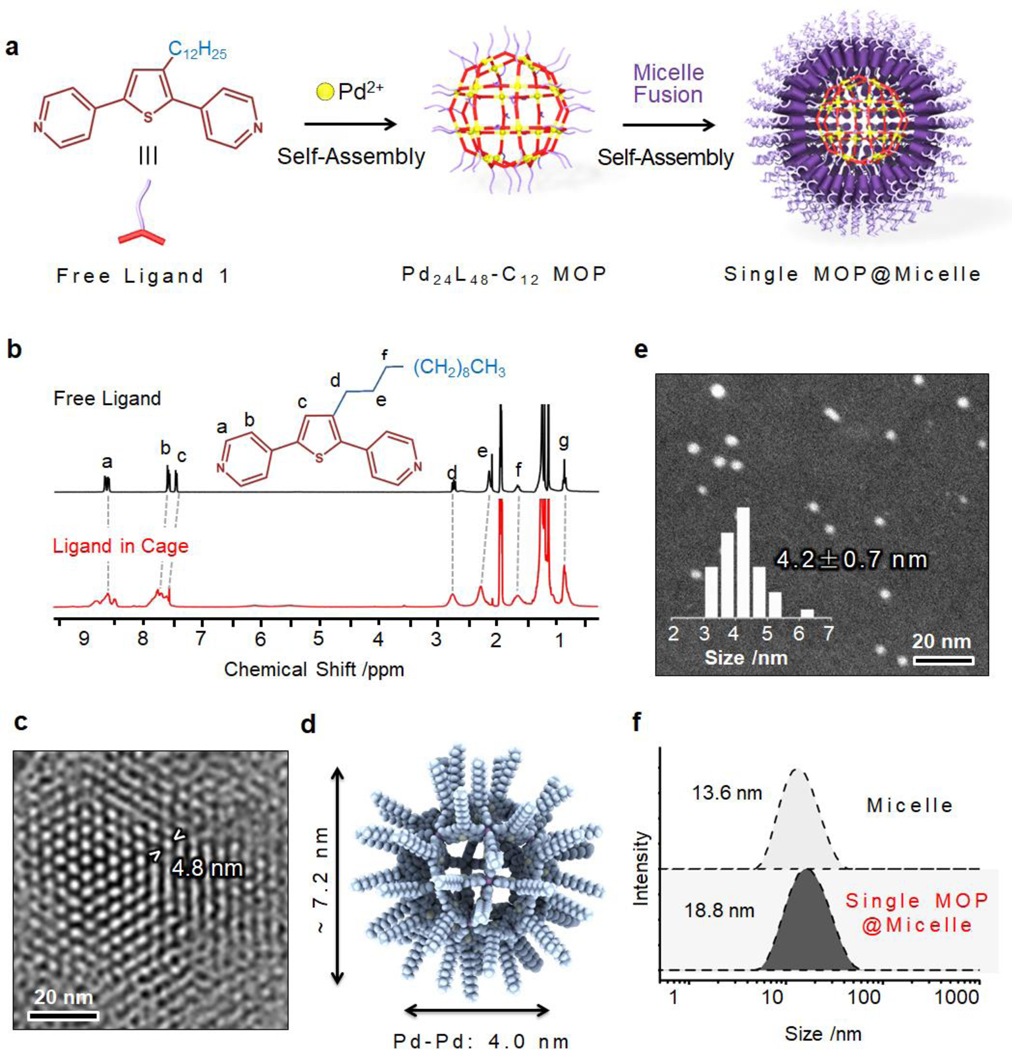
(a) Self-assembly of Pd24L48-C12 MOP with hydrophobic chain decoration and lateral fusion of micelles. (b) 1H NMR spectra of free ligand and Pd24L48-C12 MOP in DMSO-d6 solvent. (c) HR-TEM image of the Pd24L48-C12 MOP assembly; the distance between two closed packed cages is also highlighted. (d) Optimized structures of Pd24L48-C12 MOP based on MM calculations. (e) Dark-field STEM image of single Pd24L48-C12 MOP-supported micelle and the related size distribution. (f) DLS data of P6LEL-based micelle before and after loading with single Pd24L48-C12 MOP NPs.
Subsequently, the preparation of single MOP@micelle NPs followed by MOP super-assembly was performed using the Pd24L48-C12 MOP nanobuilding blocks. First, to promote the super-assembly of the hydrophobic Pd24L48-C12 MOP cages in an aqueous solution, a micellar solution assembled by polyoxyethylene (6) lauryl ether (P6LEL) was added (Figure 1a,e,f). A small drop (1 μL) of a MOP solution (0.17 mM in dimethyl sulfoxide (DMSO)) was added to the micellar solution, and the resulting mixture was sonicated to promote the formation of single MOP supported-micelle NPs (Figure 2a). High-magnification dark-field scanning transmission electron microscopy (STEM) of the super-assembly showed uniform sizes of 4.2 ± 0.7 nm (Figure 1e), well correlated with the molecular shell of Pd24L48-C12 MOP defined by the coordinated palladium ions. The hydrodynamic diameters of the micelle and MOP-micelle conjugate were determined by dynamic light scattering (DLS) to be 13.6 and 18.8 nm (Figure 1f), respectively. These data confirm the successful formation of single MOP supported-micelles. Moreover, with the increase of MOP concentration, larger MOP super-assemblies with nanometer or micrometer sizes were generated (Figure 2a,b). Notably, the size could be easily tuned in a broad range by gradually increasing the MOP concentration (Figure 2c). The monodisperse particle size distributions of the single MOP-supported micelle (6.8 ± 0.6 nm) and MOP super-assemblies (65.8 ± 4.8 nm; 100.4 ± 14.6 nm) were further confirmed by AFM images (Figures 2d and S4). TEM imaging of the air-dried samples revealed a hexagonal packing of MOPs inside the super-assemblies with a long-range order on a scale of 100 nm × 100 nm (Figure 2e). The enlarged image shown in Figure 2f reveals the overlaying hexagons with a 5.4 nm dimension. The hydrophobic interactions from the long alkane chains of closest MOPs are presumably the main driving force for the super-assembly of MOP and may as a result lead to high stability during in vivo circulation. The surface area and pore volume of the MOPsa@micelle NPs were found to be inaccessible using standard argon adsorption–desorption measurements due to the kinetically closed pore of the cages and shell blocking effect as a result of dense packing of the long PEG chains (Figure S5).[7]
Figure 2.
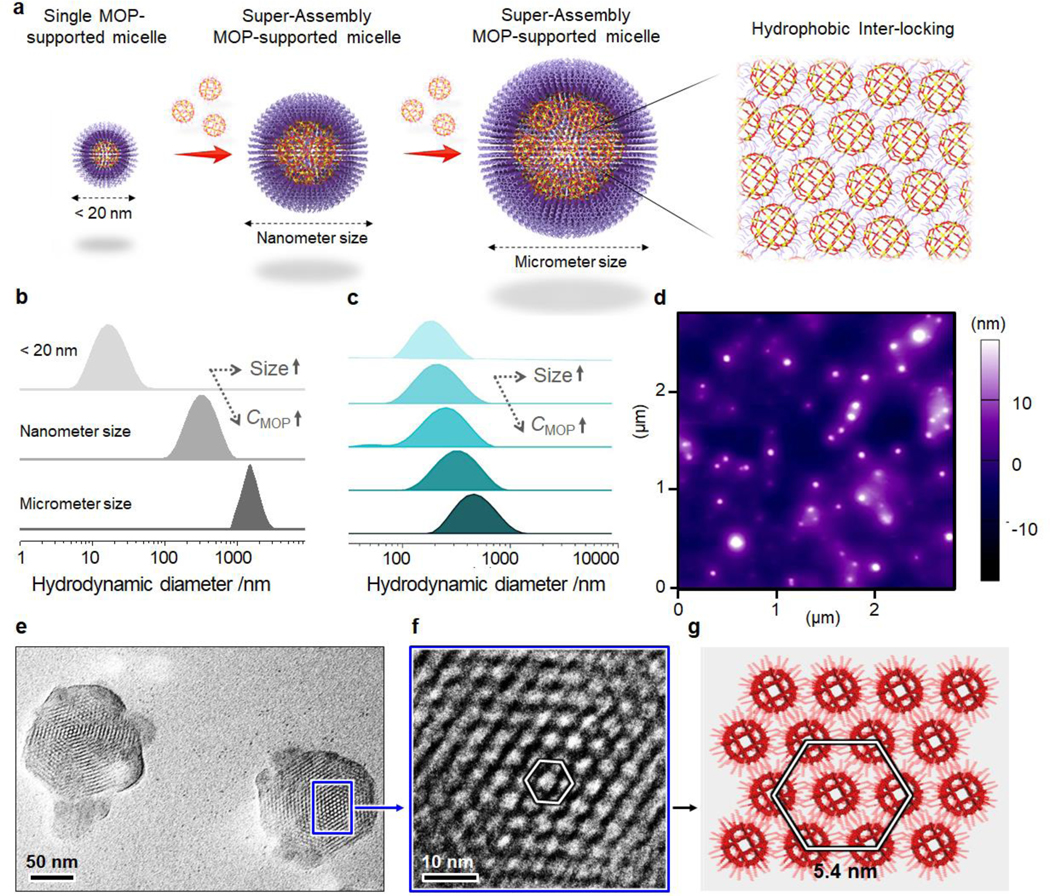
(a) Schematic illustration of the construction of single MOP@micelle and MOPsa@micelles with different size scales. (b) DLS data of Pd24L48-C12 MOP-supported micelles with ultrasmall, nanometer, and micrometer sizes. (c) DLS data showing size control of MOPsa@micelles. (d) AFM images of MOPsa@micelles with nanometer size. (e,f) HR-TEM images and (g) simulated structure of the dense packing of Pd24L48-C12 MOP in the super-assembly.
The cargo loading capabilities of the MOPsa@micelle nanomaterials were then investigated. The high inherent porosity of Pd24L48-C12 as well as the relatively large pore windows (1.4 nm × 1.4 nm) of the cages (Figure S6) suggested that various guest molecules could be loaded into the MOP nanocavity in a controlled manner. To evaluate the cargo loading capacities of our nanocarriers, three types of molecules were separately loaded, including doxorubicin hydrochloride (DOX∙HCl) as a hydrophilic drug for chemotherapy, sulforhodamine B as a fluorescent dye for labeling, and Mn(III)tetra (4-sulfonatophenyl) porphyrin (MnTPPS4) as a contrast agent for magnetic resonance imaging. The successful loading of the guest molecules was confirmed in the particles after cargo loading by the characteristic absorption bands of the guest molecules displayed in the UV-Vis spectra (see 503, 561, and 467 nm in Figure 3a and S7). Based on calculations from the UV-Vis spectra, one Pd24L48-C12 MOP entity was estimated to be loaded with 22 DOX∙HCl, 38 sulforhodamine B, and 13 MnTPPS4, confirming the high loading efficacy. Note that the structure of the cargo-loaded MOPsa@micelle nanocarriers remained unaffected by the cargo loading (see TEM image in Figure S8). In addition, multiple guest molecules were loaded within individual MOP, which were then super-assembled simultaneously with a highly controlled molar ratio. For instance, three fluorescent molecules (6-aminocoumarin, fluorescein isothiocyanate, and sulforhodamine B) were individually loaded into different Pd24L48-C12 MOP samples and then integrated into MOP super-assemblies with tunable sample ratios for potential multi-fluorescence imaging, as confirmed by UV-vis spectroscopy (Figure 3b). This controllable super-assembly scheme provides a powerful approach to prepare multi-drug delivery nanosystems with precise control of the ratio for each drug by simply tuning the MOP sample ratios.
Figure 3.

(a) UV-vis spectra of free DOX, free Pd24L48-C12 MOP, and DOX@Pd24L48-C12 MOP in DMSO solution. (b) UV-vis spectra of the multiple dye-loaded MOPsa@micelles with different dye loading ratios. (c) UV-Vis and emission spectra of CdSe/ZnS QDs@MOPsa@ micelles and UV-vis spectrum of Au NPs@MOPsa@micelles. The insets show the corresponding optical fluorescent and optical images. (d) Schematic illustration of the fabrication of MOP super-assembly-based separation membrane. (e) SEM images of the MOP super-assembly-based coating on a porous polypropylene-based substrate. (f) The use of Pd24L48-C12 MOP super-assembly-based membrane for sulforhodamine B separation.
The super-assembled MOP-micelle particles were then incorporated with functional materials and nanomaterials for a range of proof-of-concept studies. First, several functional nanomaterials, such as fluorescent QDs and gold nanomaterials (Figure S9), were incorporated into the MOPsa@micelle carriers via one-pot during the super-assembly of MOP-micelle nanocarriers. As depicted in Figure 3c, the fluorescent peak at 627 nm for CdSe/ZnS quantum dot@MOPsa@micelle and the typical plasmon resonance peak at 529 nm for Au NPs@ MOPsa@micelle confirmed the successful doping of the MOPsa@micelle particles (Figure S10–11). Such incorporation of functional nanomaterials into the MOPsa@micelle nanocarriers can diversify the utility and potential of our platform. Additionally, we found that the MOP super-assemblies could be deposited onto polypropylene membranes as prototypes for separation applications (Figure 3d). The fabrication of the mixed membrane was realized with a controlled thickness of ~2 μm, as shown by SEM (Figure 3e). After five deposition cycles (Figure 3d), a film consisting of a dense packing of MOP super-assemblies was obtained and was then applied to separate mixtures. Owing to the porosity and the reversible host–guest interactions of MOPs, the resultant membrane showed promising separation properties for sulforhodamine B-containing solutions, even after a few cycles of separation (Figure 3f). All of these results point to multifunctional MOPsa@micelle nanocarriers and novel mixed membranes based on MOP nanocages for biomedical and separation applications.
The biocompatibility and colloidal stability of the MOPsa@micelles were then studied in view of future biomedical applications. To evaluate the concentration-dependent lysis of red blood cells (RBCs), the nanomaterials were incubated with RBCs at concentrations varying from 1 to 200 μg mL−1 in 1x phosphate-buffered saline (PBS) solution for 3 h. As shown in Figure 4a,b, the hemolysis percentage of RBCs for all samples increased in a dose-dependent manner. The smaller single MOP@micelle carriers caused a higher release of hemoglobin than the larger carriers. The same trend was observed with mesoporous silica nanoparticles.[8] Nevertheless, all of the nanocarriers showed negligible hemolytic activities, thus meeting the essential prerequisites of biomedical applications. The colloidal stabilities of the MOPsa@ micelles were then tested in various media (PBS solution, Dulbecco’s modified Eagle medium (DMEM), and F-12K). DLS data of the MOPsa@micelle samples with different sizes showed narrow hydrodynamic size distributions with low PDI values of less than 0.2 (Figure 4c). Notably, long-term exposure in various media (7 days) caused insignificant size changes, indicating long-term colloidal stability. It can be surmised that the polymeric PEG chains act as protective layers to prevent particle aggregation. The cytotoxicity profiles of the single MOP@micelle or MOPsa@micelle-based nanocarriers with different PEG chain lengths and sizes were also evaluated against two human cancer cell lines: A549 cell (lung cancer cell) and HeLa cell (cervical cancer cell). The corresponding IC80 values (concentration required to reduce cell viability to 80%) are summarized in Figure 4e. As shown in Figure 4e and S12, free nanometric MOP exhibited higher cytotoxicity (IC80 = 25 μg mL−1) on A549 cells than MOP super-assemblies with different PEG chain lengths (6, 10, and 23) (IC80 = 62, 103, and 200 μg mL−1, respectively), thus confirming the low inherent toxicity of PEGylation. In addition, higher PEG repetitive unit numbers ranging from 6 to 23 led to considerably reduced cytotoxicity for the MOP super-assemblies, which correlates with the reported molecular weight-dependent cytotoxicity of PEG samples.[9] Additionally, size-dependent cytotoxicity effects were observed (Figure S12). For ultrasmall single MOP@micelle particles, the IC80 value (82 μg mL−1) was much higher than that for MOPsa@ micelles with larger sizes. In summary, increasing the particle size leads to reduced toxicity, and the high cell viabilities of all MOP@micelle-based nanocarriers (even at high concentrations) suggest the good biocompatibility of the hybrid nanocarriers developed herein for biomedical applications.
Figure 4.

(a) Percent hemolysis and (b) photographs of human RBCs incubated with single MOP-supported micelle and MOP super-assembly-supported micelles with different size scales. (c) Long-term colloidal stability of various MOP super-assembly-supported micelles in different media: PBS, DMEM, and F-12K at 25 °C. (d) Time-dependent drug (DOX) release behavior of MOP super-assembly-supported micelles in PBS solution at different pH values. (e) Cytotoxicity profiles of single MOP-supported micelle and MOP super-assembly-supported micelles with different PEG chain lengths (size: ~162 nm) and sizes (PEG23-based) against A549 and HeLa cells.
The potential of the biocompatible MOP-micelle carriers for cargo release was then tested in aqueous solution. The DOX drug was selected as it is a widely used anticancer drug. Drug release experiments were carried at room temperature in fresh PBS buffer solution at pH 7.4 and 5.5. The absorbances of the supernatant collected at fixed time intervals were measured to determine the amount of DOX released under these conditions. As displayed in the release profiles of Figure 4d, nearly 36 % of the total drug release at pH 7.4 and 5.5 occurred within the first 4 h, and 5 % more of the total drug was released after 7 h. In this configuration of MOP and cargo, the pH had negligible influence on the release kinetics. Note that DOX may have quite strong host-guest interactions with Pd24L48-C12 MOP due to collective, weak, non-covalent interactions. When the concentration of DOX inside the MOP cavity was high, the interactions between DOX and MOP may be weakened to a certain extent due to both DOX/DOX and DOX/MOP interactions. Following the release of DOX from the MOP cavity, there may be a balance between releasing and re-binding, which leads to only a partial release of loaded drugs. Nevertheless, given the versatility of MOP and drug structure, controllable release via stimuli-responsive MOPs (e.g. light) could be envisioned to promote the construction of smart drug delivery systems with higher extents of release.[10]
The potential of the MOPsa@micelle carriers for targeted drug delivery was eventually assessed on cancer cells. The A549 cancer cell line was selected owing to the high expression of the epidermal growth factor receptor (EGFR). Targeting was accomplished through the conjugation of biotin-NeutrAvidin-biotin moieties onto MOPsa@micelle with anti-EGFR monoclonal antibodies as depicted in Figure 5a. The preparation involved polyoxyethylene (23) lauryl ether (P23LEL):1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)-2000] (ammonium salt)) (DSPE-PEG-2000-biotin):1,2-distearoyl-sn-glycero-3-phosphoeth-anolamine-N-[methoxy (polyethyleneglycol)-2000] (ammonium salt) (DSPE-PEG2000) with molar ratios of 92:4:4. To examine the targeting specificity, MOPsa@micelles with or without EGFR modification were incubated with A549 cells. The confocal laser scanning microscopy images of A549 cells cultured with both red fluorescent dye sulforhodamine B-labeled MOPsa@micelles (50 μg mL−1) at 37 °C for 45 min are shown in Figure 5b,c. The cellular filamentous actin network and nuclei were stained with fluorescent probes Alexa Fluor 488 phalloidin and Hoechst 33342, respectively. Significant selective binding and internalization of the EGFR-modified MOPsa@micelles to A549 cells were observed. Binding or internalization of particles without EGFR functionalization was not observed. It was thus concluded that the MOPsa@micelles were mostly internalized into the cells via receptor-mediated endocytosis. To elucidate the receptor–ligand binding kinetics, both dye-labeled MOPsa@micelles were incubated with A549 cells for different times. After incubation for 15 min, a small binding shift occurred for the control samples, even after extending the incubation time to 45 min (Figure 5d,e). Regarding the EGFR-modified samples, however, an obvious binding shift was observed within 15 min, and a maximal binding shift was observed after 45 min. The targeted cell killing efficacy of MOPsa@micelles nanocarriers against A549 cells was also measured. The viabilities of the A549 cells after being incubated for 2 h with a series of concentrations of free DOX, and MOPsa@micelles with or without DOX loading were quantified. As shown in Figure S13, free DOX exhibited dose-dependent toxicity to A549 cells. In contrast, MOPsa@micelles with the same DOX loading displayed a lower toxicity, indicating that DOX had been trapped in the MOPsa@micelle nanocarrier and thus reduced the killing efficacy of DOX to the cells. To evaluate the target specific drug delivery, A549 cells were incubated with increasing concentrations of MOPsa@micelles with or without EGFR modification or DOX loading in complete media under standard culturing conditions. In Figure 5f, compared to the MOPsa@micelles with (18.0 %) or without EGFR modification (0.1 %), the DOX-loaded targeted MOPsa@micelle causes much higher cell death of ~42.0 %, indicating an enhanced killing efficacy that was attributed to the successful release of DOX inside A549 cells.
Figure 5.
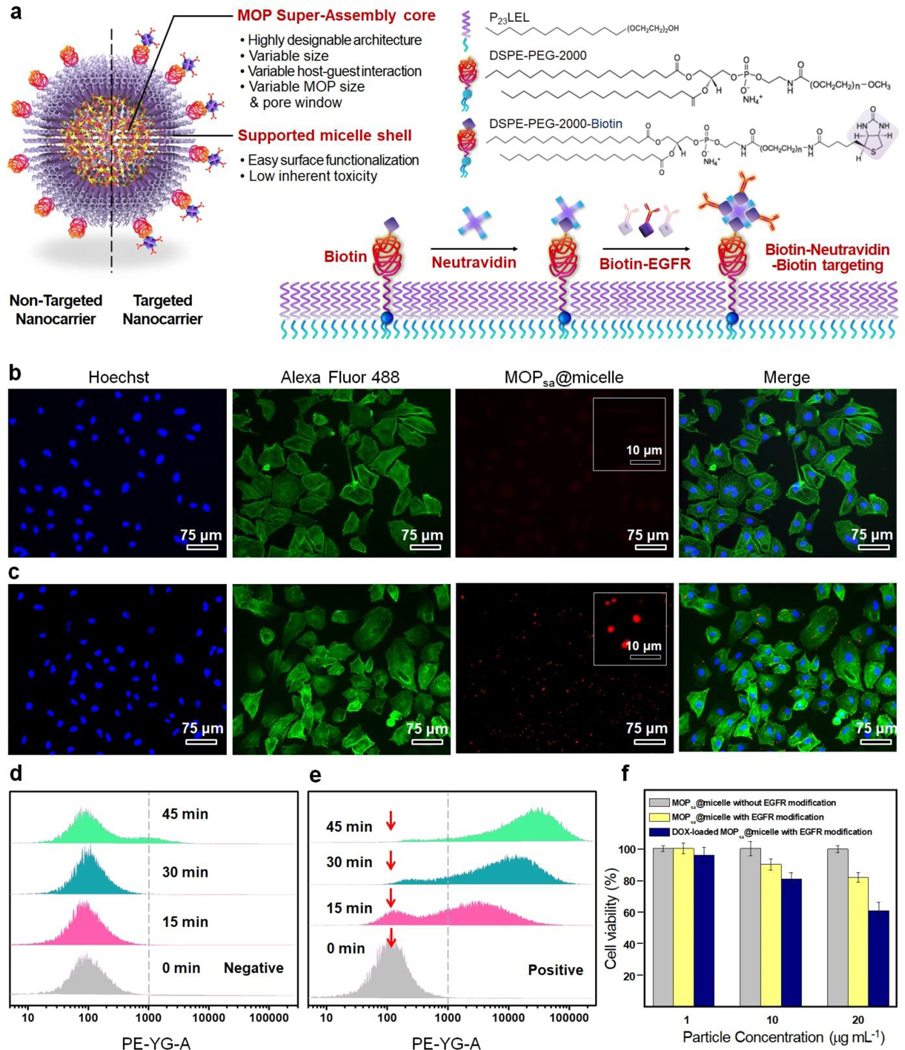
(a) Schematic illustration of the construction of EGFR-modified MOPsa@micelle for targeted drug delivery. Fluorescence microscopy images of A549 cells treated with MOPsa@micelles (b) without and (c) with EGFR modification after incubation for 45 min at 37 °C. Flow cytometry analysis of A549 cancer cells incubated with red fluorescent dye-loaded MOPsa@micelles (d) without or (e) with EGFR modification at multiple time points. (f) Sustained viability of A549 cells after incubation of MOPsa@micelle with or without EGFR modification or DOX loading for 1 h.
Furthermore, to evaluate the potential in vivo application of our newly created MOPsa@micelles, the related circulation and biodistribution were assessed using a mouse model. Mice were injected with CdSe/ZnS QD (627 nm)-labeled MOPsa@ micelles by retro-orbital injection at a dose of 150 μg NPs/mouse. To study the circulation half-life, at various time points following the injection, blood was collected from the eye socket of the mice to evaluate the NPs remaining in circulation. At 24 h and 48 h post injection, the MOPsa@ micelles exhibited 12 % and 3 % overall retention in mice blood (Figure S14), respectively, similar to the 11% and 2% exhibited by PEG-coated NPs.[11] The semilog plot of retention-circulation time (Figure S15) illustrates an exponential decrease in particle concentration over time, indicating that the MOPsa@micelle NP circulation followed a one-way nonlinear clearance model. Based on this pharmacokinetic model, the half-life (i.e., time at which 50% of the particles are cleared) of MOPsa@micelle NP was calculated to be 5.6 h. Moreover, to analyze the related biodistribution, at 6 h, 12 h, 24 h, and 48 h post injection, mice were euthanized and their liver, spleen, kidneys, heart, lungs, and blood were harvested for fluorescence analysis (Figure S16). The majority of fluorescence signal was found in the two primary filtering organs (Figure S17), the liver and spleen at 6 h post injection, supporting removal by the reticuloendothelial system (RES).[12–13] This behavior is consistent with the behavior of nanocarriers employed currently for in vivo delivery.[14] After 24 h a visible signal was found in the feces (Figure S16), indicating the MOPsa@micelle NPs can be cleared from the body. These results demonstrate that our developed MOPsa@micelle NP has good in vivo residence time and clearance behavior needed for targeted cancer therapies.
In summary, we have described the first super-assembly of nanosized metal–organic polyhedra and their biomimetic coatings of lipid bilayers to synergistically combine the advantages of micelles and supramolecular coordination cages for biomedical applications. The hydrophobic self-assembly of the MOP nanocages was facilitated via their surface modification using dodecyl groups, and remarkable long-range hexagonal order was visualized via high-resolution electron micrographs. Homogeneous and tunable size distributions of MOPsa@micelle particles were obtained by simply adjusting the MOP concentration. PEGylated nanomaterials showed good biocompatibility and were stable in bio-relevant media, as demonstrated by systematic in vitro studies. The MOP super-assembly could be loaded with various dyes and drugs, QDs and gold nanoparticles, and could function as multi-drug delivery systems with specific cargo loading amounts through the precise mixing of multiple cargo-loaded MOP samples. Through the surface modification with targeting moieties, the MOPsa@micelles were successfully synthesized and applied to cancer cells where we observed selective cytotoxicity. Finally, in vivo experiments in a mouse model demonstrated good in vivo circulation of MOPsa@micelles. The metal–organic polyhedra supra-assembly within micelles concept has thus been validated and expected to be applicable to a range of biomedical applications as well as for separation, sensing, and catalytic processes.
Supplementary Material
Acknowledgments
C. J. B. and W. Z. acknowledge support from the University of New Mexico Center for Micro-Engineered Materials. C. J. B. and R. S. acknowledge support from NIH RO1 (FP0003261). C. J. B. acknowledges support by the Sandia National Laboratory Laboratory-Directed Research and Development Program and support from the Department of Energy Office of Science, Division of Materials Science and Engineering. This work was supported, in part, by the National Science Foundation (NSF) under Cooperative Agreement No. EEC-1647722. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSF. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honey-well International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DENA-0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. R. S. acknowledge use of the UNM Animal Models and Microscopy facilities, supported by UNM Comprehensive Cancer Center NCI grant 2P30 CA118100-11. Part of this work was conducted and funded by the Australian Research Council (ARC) Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036). F.C. acknowledges the award of a National Health Medical Research Council Senior Principal Research Fellowship (APP1135806).
References
- [1].a) Mitragotri S, Burke PA, Langer R, Nat. Rev. Drug Discovery 2014, 13, 655; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blanco E, Shen H, Ferrari M, Nat. Biotechnol. 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Elsabahy M, Heo GS, Lim S, Sun G, Wooley KL, Chem. Rev. 2015, 115, 10967; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Grim N, Andrade F, Segovia N, Ferrer-Tasies L, Veciana J, Ventosa N, Chem. Soc. Rev. 2016, 45, 6520; [DOI] [PubMed] [Google Scholar]; c) Croissant JG, Fatieiev Y, Khashab NM, Adv. Mater. 2017, 29, 1604634; [DOI] [PubMed] [Google Scholar]; d) Zhu W, Xiang G, Shang J, Guo J, Motevalli B, Durfee P, Agola JO, Coker EN, Brinker CJ, Adv. Funct. Mater. 2018, 28, 1705274; [Google Scholar]; e) Richardson JJ, Cui J, Björnmalm M, Braunger JA, Ejima H, Caruso F, Chem. Rev. 2016, 116, 14828. [DOI] [PubMed] [Google Scholar]
- [3].a) Cook TR, Vajpayee V, Lee MH, Stang PJ, Chi K, Acc. Chem. Res. 2013, 46, 2464; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ahmad N, Younus HA, Chughtai AH, Verpoort F, Chem. Soc. Rev. 2015, 44, 9; [DOI] [PubMed] [Google Scholar]; c) Harris K, Fujita D, Fujita M, Chem. Commun. 2013, 49, 6703; [DOI] [PubMed] [Google Scholar]; d) Grishagin IV, Pollock JB, Kushal S, Cook TR, Stang PJ, Olenyuk BZ, Proc. Natl. Acad. Sci. USA 2014, 111, 18448; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yu G, Cook TR, Li Y, Yan X, Wu D, Shao L, Shen J, Tang G, Huang F, Chen X, Stang PJ, Proc. Natl. Acad. Sci. USA 2016, 113, 13720; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Vardhan H, Yusubov M, Verpoort F, Coord. Chem. Rev. 2016, 306, 171. [Google Scholar]
- [4].a) Therrien B, Süss-Fink G, Govindaswamy P, Renfrew AK, Dyson PJ, Angew. Chem. Int. Ed. Engl. 2008, 47, 3773; Angew. Chem. 2008, 120, 3833; [DOI] [PubMed] [Google Scholar]; b) Schmitt F, Freudenreich J, Barry NPE, Juillerat-Jeanneret L, Süss-Fink G, Therrien B, J. Am. Chem. Soc. 2012, 134, 754; [DOI] [PubMed] [Google Scholar]; c) Zheng Y, Suntharalingam K, Johnstone TC, Lippard SJ, Chem. Sci. 2015, 6, 1189; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Samanta SK, Moncelet D, Briken V, Isaacs L, J. Am. Chem. Soc. 2016, 138, 14488; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Samanta SK, Quigley J, Vinciguerra B, Briken V, Isaacs L, J. Am. Chem. Soc. 2017, 139, 9066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Zhao D, Tan S, Yuan D, Lu W, Rezenom YH, Jiang H, Wang L, Zhou H, Adv. Mater. 2011, 23, 90; [DOI] [PubMed] [Google Scholar]; b) Xu W, Fan Y, Wang H, Teng J, Li Y, Chen C, Fenske D, Jiang J, C. Su. Chem. Eur. J. 2017, 23, 3542; [DOI] [PubMed] [Google Scholar]; c) Rodriguez J, Mosquera J, Couceiro JR, Nitschke JR, Vázquez ME, Mascareñas JL, J. Am. Chem. Soc. 2017, 139, 55; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Croissant JG, Fatieiev Y, Almalik A, Khashab NM, Adv. Healthcare Mater. 2018, 7, 1700831. [DOI] [PubMed] [Google Scholar]
- [6].a) Sun QF, Iwasa J, Ogawa D, Ishido Y, Sato S, Ozeki T, Sei Y, Yamaguchi K, Fujita M, Science 2010, 328, 1144; [DOI] [PubMed] [Google Scholar]; b) Fujita D, Ueda Y, Sato S, Yokoyama H, Mizuno N, Kumasaka T, Fujita M, Chem 2016, 1, 91. [Google Scholar]
- [7].Park J, Sun L, Chen Y, Perry Z, Zhou HC, Angew. Chem. Int. Ed. Engl. 2014, 53, 5842; Angew. Chem. 2014, 126, 5952. [DOI] [PubMed] [Google Scholar]
- [8].Lin Y, Haynes CL, J. Am. Chem. Soc. 2010, 132, 4834. [DOI] [PubMed] [Google Scholar]
- [9].Pozzi D, Colapicchioni V, Caracciolo G, Piovesana S, Capriotti AL, Palchetti S, Grossi SD, Riccioli A, Amenitsch H, Laganà A, Nanoscale 2014, 6, 2782. [DOI] [PubMed] [Google Scholar]
- [10].Han M, Michel R, He B, Chen Y, Stalke D, John M, Clever GH, Angew. Chem. Int. Ed. Engl. 2013, 52, 1319; Angew. Chem. 2013, 125, 1358. [DOI] [PubMed] [Google Scholar]
- [11].Hu CJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Proc. Natl. Acad. Sci. USA 2011, 108, 10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang G, Yang Z, Lu W, Zhang R, Huang Q, Tian M, Li L, Liang D, Li C. Biomaterials 2009, 30, 1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cui J, Rose RD, Alt K, Alcantara S, Paterson BM, Liang K, Hu M, Richardson JJ, Yan Y, Jeffery CM, Price RI, Peter K, Hagemeyer CE, Donnelly PS, Kent SJ, Caruso F. ACS Nano, 2015, 9, 1571. [DOI] [PubMed] [Google Scholar]
- [14].Lipka J, Semmler-Behnke M, Sperling RA, Wenk A, Takenka S, Schleh C, Kissel T, Parak WJ, Kreyling WG, Biomaterials 2010, 31, 6574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.