

Abstract
Visualization of in vivo protein levels and localization is essential to analysis and elucidation of Hippo signaling mechanisms and its roles in diverse tissues. This is best done by imaging proteins using fluorescent labels. Fluorescent labeling of a protein can be achieved by direct conjugation to an intrinsically fluorescent protein, like GFP, or by use of antibodies conjugated to fluorescent dyes. Immunofluorescence imaging in Drosophila typically begins with dissection and fixation of a sample tissue, followed by a series of washes and incubations with primary antibodies, directed against proteins of interest, and dye-labeled secondary antibodies, directed against the primary antibodies. This may be followed by fluorescent dyes that label cellular components, such as DNA-labeling dyes to mark nuclei. After staining and washing is completed, samples are placed in a mounting media, transferred to a microscope slide, and imaged on a confocal microscope.
Keywords: Hippo, Yorkie, Antibody, Immunofluorescence, Confocal, GFP
1. Introduction
Detection of protein levels and localization in vivo has had a major impact on our understanding of diverse cellular processes, including Hippo signaling [1, 2]. Fluorescent labeling offers several advantages for detecting proteins, including high sensitivity, large linear range of detection, and the ability to image multiple proteins separately or in combination by using distinct fluorophores. At low resolution, fluorescence imaging enables investigation of tissue expression patterns and relative expression levels, whereas at high resolution, fluorescence imaging enables determination of subcellular localization, which is a crucial parameter of protein function and often dynamically modulated in signal transduction pathways.
The two main approaches to fluorescent labeling are genetic conjugation of a protein of interest to an intrinsically fluorescent protein [3] and indirect attachment of fluorescent labels using antibodies [4]. In practice, many experiments examining protein localization in Drosophila Hippo signaling use both of these approaches in combination within a single experiment. Through spectral separation of different fluorophores, the location of a protein of interest can then be compared to fluorescent markers of cell fate, genotype, gene expression, or subcellular localization. Detection by immunofluorescence bypasses the need to create and introduce unique genetic constructs but requires the creation and validation of antisera or antibodies with high avidity and specificity. Direct conjugation to intrinsically fluorescent proteins, like GFP, bypasses the need to create antibodies and often results in lower background noise but requires the creation and incorporation of appropriate genetic constructs, which must be functionally validated. Tagging with intrinsically fluorescent proteins can also enable live imaging approaches.
A typical Drosophila immunofluorescence staining experiment begins with the creation of flies of the appropriate genotype. The tissues to be analyzed are then dissected out of the animal and subject to histological fixation. Most analysis of Hippo signaling has been performed on imaginal discs [5], which are dissected out of developing larvae, and we have used the basic method described below to study the localization of many Hippo pathway components in discs [6–14]. The fixation and staining methods described below can also be applied to other tissues as well [15–18]. After fixation, samples are washed, incubated in primary antibodies directed against proteins of interest, washed, incubated in secondary antibodies that are directed against the primary antibodies and coupled to fluorescent dyes, washed again, incubated in fluorescent dyes for labeling cellular structures, and mounted on slides for microscopy. A wide-field fluorescence microscope can be used, but most studies employ confocal microscopes due to the improved spatial and spectral resolution they offer.
2. Materials
2.1. Tissue Dissection and Fixation
Flies (see Note 1).
Clear dissection dish (see Note 2).
Fine forceps (see Note 3).
25% sucrose (see Note 4).
Ringers: 111.23 mM NaCl, 1.88 mM KCl, 2.38 mM NaHCO3, 0.09 mM NaH2PO2–2H2O, 0.82 mM CaCl2–H2O (see Note 5).
4% paraformaldehyde (see Note 6).
Dissecting microscope (see Note 7).
2.2. Antibody Incubation
Nutating mixer (see Note 8).
10× PBS (phosphate-buffered saline): 80.6 mM sodium phosphate, 19.4 mM potassium phosphate, 27 mM KCl and 1.37 M NaCl, pH 7.4 (see Note 9).
PBT: PBS + 0.1% (v/v) Triton X-100 + 1% (w/v) BSA + 0.01% (v/v) azide (see Note 10).
Primary antibodies (see Note 11).
Secondary antibodies (see Note 12).
Normal serum from the animal that is the host for the secondary antibodies (typically donkey, sheep, or goat) (see Note 13).
Hoechst 33342 (1 μg/mL in double distilled or deionized water (ddH2O)) (see Note 14).
Phalloidin conjugated to a fluorescent molecule (i.e., Molecular Probes Alexa Fluor dyes) (see Note 15).
2.3. Mounting Tissues
Microscope slides (25 × 75 × 1.0 mm precleaned slides).
No. 1.5 cover glass (22 mm × 22 mm microscope cover glass).
VECTASHIELD Antifade Mounting Media (see Note 16).
Nail polish.
3. Methods
3.1. Tissue Dissection and Fixation
Scoop larvae from food using a metal spatula and gently mix in 25% sucrose (larvae float on sugar water, the food will sink) in a Petri dish. Alternatively, pour 25% sucrose into the fly vial, gently agitate the food while gently stirring with a spatula, and then pour the contents into a Petri dish.
Using dull forceps, pick larvae of the desired age and genotype. This can be done by either picking the desired larvae directly from the Petri dish (i.e., GFP-positive larvae can be directly picked from the sucrose/food) or all of the larvae can be transferred to a spot well dish filled with chilled Ringers and then selected for the correct genotype (this may be an easier method for visualization of some weaker fluorochromes, such as RFP). The larvae to be dissected should all ultimately be placed in a spot well dish filled with chilled Ringers.
Dissect the larvae in Ringers using forceps, while visualizing larvae through the dissecting microscope (Fig. 1). To tear a larva in half, use two forceps and pinch the larva at about one third back from the anterior end. The tips of the forceps should be close together; then move the forceps apart while still pinching the tissue. The larva will now be cleanly torn in half. Discard the posterior half. Remove the protruding guts and fat from the anterior end, although if one has torn the larva in the “perfect” spot, this won’t be necessary. Invert the anterior end of the larva (see Note 17), being careful not to disturb any discs or trachea. Transfer inverted larva (with discs attached) immediately after inversion to approximately 1 mL Ringers on ice, using forceps to gently grab the larval cuticle. Transfer as many larvae as you can invert in 10–15 min, with a maximum of 10–15 larvae per tube.
To fix the larval tissues, remove Ringers from tube containing inverted larvae, and then add 4% paraformaldehyde. Place tube on a Nutator at room temperature for gentle mixing. Immediately start a timer. Typically, fixation time is for 15 min, with some exceptions (see Note 18).
Remove fixative with a Pasteur pipet and discard in hazardous liquid waste container.
-
Rinse larvae twice with Ringers by adding Ringers, closing and inverting the tube, allowing larval cuticles to settle, and removing the liquid.
Note: After this stage, the fixed imaginal discs can be stored in PBT for up to a few days or immediately used for antibody tissue staining (see Note 19).
Fig. 1.
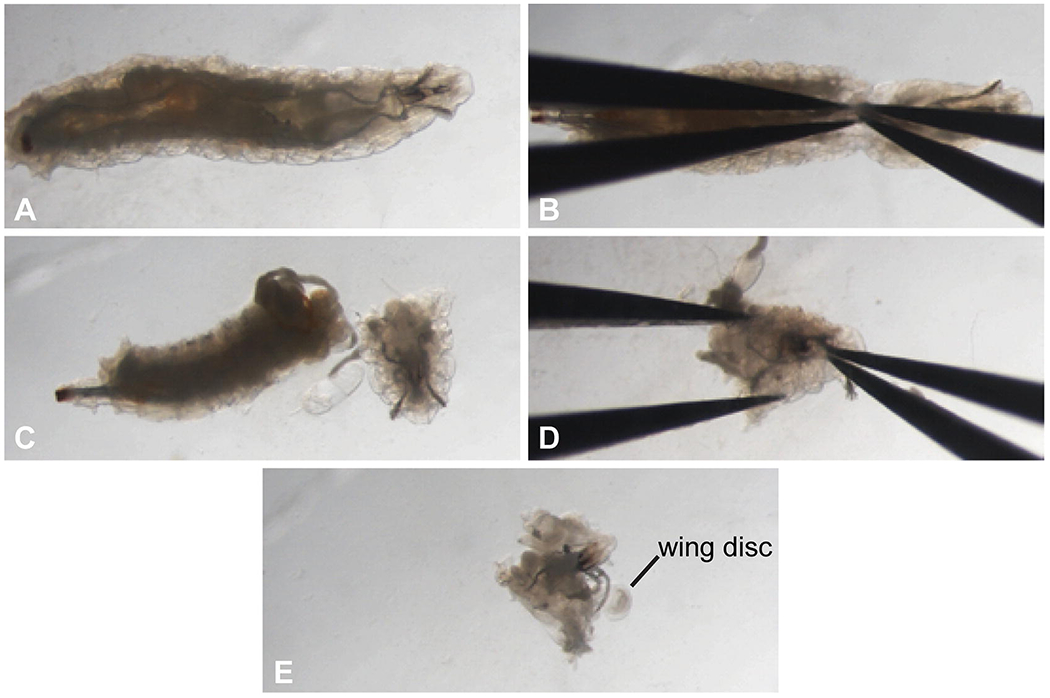
Dissecting and inverting larvae for immunostaining. Images show steps in larval dissection, as described in Subheading 3.1, step 3. (a) Drosophila larva. (b) Positioning of forceps before larva is ripped in half. (c) Larva after the anterior end is separated from the posterior end. (d) Positioning of forceps before the larval head is inverted. (e) Larval head after inversion, with imaginal discs now outside the cuticle. A wing disc is visible
3.2. Staining Tissues
Rinse fixed discs (larval cuticles) twice with PBT, then wash 2 × 20 min each in PBT on Nutator (see Note 20).
Incubate for 30 min in 180 μL PBT + 10 μL Donkey serum (total volume will be ~200 μL, because the larval tissue will be in ~ 10 μL) on Nutator.
-
Incubate with primary antibodies:
180 μL PBT + 10 μL Donkey serum + primary antibody at appropriate dilution, to a total volume of 200 μL (see Note 21).
Incubate overnight at 4 °C on Nutator (see Note 22).
Remove liquid. Rinse in PBT, and then wash four times 10–15 min each in PBT on Nutator.
Incubate for 30 min in 180 μL PBT + 10 μL Donkey serum on Nutator.
-
Incubate with secondary antibodies:
180 μL PBT + 10 μL Donkey serum + secondary antibody at appropriate dilution (see Note 23). Wrap tubes in aluminum foil to keep solutions dark (see Note 24) and place tube on Nutator. Incubate 2 h RT (or overnight 4 °C).
Rinse in PBT, wash 3× 10–15 min each in PBT on Nutator. Keep tubes wrapped in aluminum foil. The immunostained tissue can now be stored in the dark (typically the Eppendorf tube is wrapped in aluminum foil) at 4 °C until ready for removal and mounting of the imaginal discs (see Note 25).
(Optional) Incubate with phalloidin to label F-actin (see Note 26).
(Optional) Incubate with Hoechst to label DNA (see Note 27).
3.3. Dissecting and Mounting Tissues
Place one larval cuticle onto a depression slide in some PBT. Dissection of imaginal discs requires a dissecting microscope. Using transmitted light, locate the larval tissue under the microscope. Using fine forceps, dissect the desired imaginal discs away from the larval cuticle. Imaginal discs are easily damaged and should be separated from the cuticle and other discs without actually grabbing onto the discs. Instead, place the tips of the forceps in between tissues to be separated and gently spread them apart. It may help to pin the larval cuticle with one forceps, and then use the other to gently remove the imaginal disc. Once the discs have been removed, discard the larval cuticle. Add a new larval cuticle into the same well as the previous, and repeat the dissection process. Once all desired discs have been removed from all of the larval tissues, one is ready to mount the imaginal discs.
To mount the imaginal discs onto a microscope slide, place the slide on a black surface. With a Pasteur pipet, suck up the discs in PBT, trying to minimize the amount of liquid. Gently squirt the discs and PBT into the center of the slide. Transfer the slide to the microscope, and while observing, remove some liquid from the slide with the Pasteur pipet. Using one forceps, in the “open” position, push the discs near each other, so that they are within an area that can be covered by the cover glass. For visualization of apical proteins (e.g., Jub, Wts), it is important that the imaginal discs are oriented with the apical side up. The wing disc is slightly convex, and the more curved side will be up. To orient the disc appropriately, use one forceps to try to flip the disc. Do not grasp the tissue, as this will destroy it, but rather capillary action is sufficient to “suck” up the disc, and then flip the disc. Once the discs are all appropriately oriented, remove the last of the liquid, and tip the slide so that the PBT gently runs to the edge of the slide. The discs should stay in place! With a Kimwipe remove any excess PBT. Place a drop (about 8 uL) of mounting media (VECTASHIELD) next to, but not on, the discs. Grab a cover glass with a pair of forceps, and holding it at a 45 angle, place near the VECTASHIELD drop, and then gently lower it, as the VECTASHIELD spreads across the discs. The amount of VECTASHIELD used needs to be such that it spreads under the entire cover glass, but not so much that it, and potentially the dissected imaginal discs, leaks out. Seal the edges of the coverslip with clear nail polish, to prevent mixing of the mounting media and immersion lens oil when imaging. The slides with the mounted tissue can be stored for a few days in the dark at 4 °C prior to imaging.
3.4. Imaging
Image the immunofluorescently stained imaginal discs using a confocal microscope with a 40× or 63× oil-immersion lens (Fig. 2) (see Note 28).
Fig. 2.

Example of confocal microscopy of Hippo pathway components. An example of a confocal micrograph showing multicolor fluorescent labeling of Hippo pathway components in a Drosophila wing imaginal disc. Top left panel shows all three channels together, clockwise from top left show Jub localization, based on a GFP:Jub fusion protein; E-cadherin localization, based on antibody staining; and Wts localization, based on V5 antibody staining of a V5-tagged Wts genomic construct [1]
4. Notes
Drosophila stocks can be obtained from other laboratories or from public stock centers. Large numbers of fly stocks expressing tagged fluorescent proteins are available, and others are being actively generated both by large-scale projects and individual researchers. The main source of information about Drosophila stocks is FlyBase (http://flybase.org).
We use Pyrex 3 depression glass spot plates, which have depressions that are 22 mm wide and 7 mm deep, and can be quickly rinsed and then reused, but other transparent dishes could also be used. Considerations are transparency (larvae are dissected under transmitted light), volume (spot plates allow dissection in less than 1 mL of liquid), and accessibility of larvae to forceps for dissection).
High quality, fine tip forceps, like Dumont #5, are essential for the final stages of dissection, but they are expensive and easily become blunt or bent. Banged-up forceps should be used for earlier stages of dissection, like inverting larvae, and fine, undamaged forceps for later stages, like separating imaginal discs. Forceps can also be resharpened using a sharpening stone, fine sandpaper, or sent to a company specializing in sharpening used forceps.
Dissolve 125 g sucrose in 400 mL water, add water up to a final volume of 500 mL. Filter the solution using a 500 mL filter unit (0.2 μm CN membrane). Store at 4 °C. This 25% sucrose solution does not need to be made from chemical grade reagents; a bag of sugar from the grocery store works fine.
Ringers is an isotonic salt solution used for maintaining tissue integrity during dissections. There are different formulations, the version we use is also known as Becker’s Ringers. Dissolve the following in 800 mL ddH2O. 6.5 g NaCl, 0.14 g KCl, 0.2 g NaHCO3, 0.01 g NaH2PO2–2H2O, and 0.12 g CaCl2–2H2O. Adjust volume to 1 L with ddH2O. Stir until dissolved and then filter the solution using a 1 L Nalgene Rapid-Flow filter unit, 0.2 μm CN membrane. Store stock solution at 4 °C. 50 mL aliquots can be left at room temperature for several days.
For 500 mL, place 450 mL of ddH20 in a glass beaker, insert a thermometer, and heat to 60 °C using a hot plate in the hood, with a stir bar. While stirring, add 20 g of paraformaldehyde powder (weighed in the hood) to the heated water. Cover with aluminum foil, and maintain at 60 °C. Add five drops of 2 N NaOH. The solution should clear within a couple of minutes, albeit there may be a few fine particles that remain. Do not heat above 70 °C as the paraformaldehyde will break down. If necessary, add one 2 N NaOH drop at a time to get most of the powder in solution. Remove the thermometer, and add 50 mL 10× PBS. Stir. Remove from heat, allow solution to cool slightly, and then adjust pH to 7.2 (using a pH meter) by adding HCl, or if too low, NaOH. Adjust the final volume to 500 mL with ddH20. Filter the solution using a 500 mL Nalgene Rapid-Flow filter unit, 0.2 μm CN membrane. Make 20–40 mL aliquots, wrap each tube with aluminum foil, and freeze them at −20 °C. They will last for 4–6 months. Thaw each aliquot as needed, typically overnight at 4 °C. Each thawed tube of paraformaldehyde will be good for 1–2 weeks; any leftover needs to be discarded in liquid hazardous waste. When preparing paraformaldehyde fixative, wear protective clothing including lab coat, eyewear, and gloves, and work in a fume hood. If the pH is not properly adjusted, the wing imaginal discs will become swollen, with the peripodial membrane puffing up from the epidermal cells. Fixative that is no longer “good” will result in tissue disintegration.
Dissecting microscopes have a large working distance and typically have a magnification range from ~8× to 60×. For best visualization of imaginal discs, a dissecting microscope should be equipped with transmitted light. In order to genotype larvae with fluorescent markers (such as balancer chromosomes labeled by GFP), it should also be equipped for epi-fluorescence.
A Clay Adams Nutator (single-speed, 12 rpm) provides a gentle orbital motion that can be used for continuous gentle mixing.
PBS is a blend of phosphate buffers and saline solutions, typically prepared as a 10× concentrated stock. Filter with a Nalgene Rapid-Flow filter unit, 0.2 μm CN membrane, to sterilize. Alternatively, 10× PBS is commercially available.
For 500 mL, mix 50 mL 10× PBS, 5 mL 10% Triton X-100, 5 g BSA (bovine serum albumin Fraction V), and ddH20 to 498 mL. Filter with a Nalgene Rapid-Flow filter unit, 0.2 μm CN membrane, to sterilize. Then add 2.5 mL of 2% sodium azide to the filtered solution. Sodium azide is not essential for immunofluorescent staining but is added as a preservative to inhibit microbiological growth in PBT. Gloves should be used and care should be taken in handling solutions with sodium azide, which is toxic.
Most primary antibodies against Drosophila proteins are made by individual labs. A list of sources for commercially available antibodies that work on Drosophila tissues is maintained at FlyBase (http://flybase.org/wiki/FlyBase:Antibodies). Another useful resource for Drosophila antibodies is the Developmental Studies Hybridoma Bank (http://dshb.biology.uiowa.edu/Antibody-Collections/Drosophila-antigens), a nonprofit NIH-supported resource that distributes over 200 monoclonal antibodies that recognize Drosophila proteins.
Secondary antibodies are antibodies from one animal that recognize antibodies of another animal. Fluorescently labeled secondary antibodies are available from many commercial sources. Our lab mostly uses secondary antisera from Jackson ImmunoResearch. Important considerations in choice of secondary antibodies include host species, fluorescent label, and cross-absorption. Secondary antibodies are made in larger animals (with more sera) like goat, sheep, or donkey. Secondary antisera from the same host should be used in a single experiment (i.e., if staining against mouse and rabbit primary antibodies, use donkey anti-mouse and donkey anti-rabbit, not donkey anti-mouse and goat anti-rabbit, because the secondary antibodies might cross-react with each other). The best fluorescent labels will depend upon your application and available equipment; we would typically use Alexa 405, Alexa 488, Cy3, and Alexa 647 for four-color immunofluorescent labeling. Secondary antibodies will normally be cross-absorbed so that they don’t recognize IgGs of certain species (i.e., an anti-mouse IgG antisera will have been depleted of antibodies recognizing anti-rabbit IgGs). However, there can be substantial differences in the extent of cross-absorption performed for different reagents, so this should be checked carefully. Anti-mouse IgG antisera may be available with or without cross-absorption against rat IgGs, which is essential if an experiment involves both mouse and rat primary antibodies, but will typically result in a weaker signal due to loss of antibodies.
Lyophilized powder can be purchased from Jackson ImmunoResearch. To reconstitute add 10 mL sterile ddH20 to the powder. Aliquot 1 mL into Eppendorf tubes. Store one tube at 4 °C as the working solution; the others should be frozen at —20 °C until ready for use.
Hoechst 33342 is a cell-permeant dye that fluoresces when intercalated into nucleic acids; it is used to label nuclei. DAPI (4–,6-diamidino-2-phenylindole, dihydrochloride) and propidium iodide are alternative dyes for labeling nuclei.
Phalloidin is used to label F-actin and is available conjugated to a variety of fluorochromes, but we typically use the conjugate to Alexa 647.
A variety of mounting media are available, but we use nonhardening VECTASHIELD Antifade Mounting Medium (Vector Labs). Mounting media for immunofluorescence include chemicals that reduce photobleaching of fluorescent protein and fluorescent dyes and can be hardened or nonhardened.
Comments on inverting larvae: if you are inverting larvae for the first time, we recommend that you practice first using wild type. It may take a week to become proficient at inverting larvae, but do not become discouraged. Begin with the wandering third instar larvae that are crawling on the sides of the vial as these are larger and hence easier to invert. For tearing the larva in half: if the larva is ripped too far anterior, the imaginal discs will be lost, while tearing them too far posterior makes the inversion nearly impossible. In older larvae, the imaginal discs are large, such that there is a “clearing” at the anterior end of the larva. You want to tear the larva in half just slightly posterior to this region. A technique for successful inversion is as follows: imagine your lab glove is the anterior end of a larva. With your right-hand pointer and middle finger, pinch the finger end of the glove. With your left hand pointer and middle fingers, insert slightly into the open end of the glove and splay your fingers wide apart. Next step is to invert the glove which is done by moving the right pinched finger end toward the left hand and through the splayed left fingers (the left hand moves slightly right during this process). The glove should now be turned inside out. The procedure is similar for a larva but is done while looking through a dissecting microscope: after the larva is torn in half, orient the larva such that the anterior end is facing right. Use the right-hand forceps to pinch the anterior tip of the larva. Using the left forceps, slightly insert them into the larval opening, and allow them to slightly splay open. With the same motion as with the glove scenario, move the right-hand forceps through the left splayed ones. The larval cuticle should be inside out now.
The typical duration for fixation that we use is 15 min at room temperature, but the optimal fixation time can vary depending upon the protein being examined. Prolonged fixation reduces GFP fluorescence, so for weak GFP signals as short a fixation time as possible should be used. For example, for genomic Wts: GFP we use only 8 min of fixation.
While in most cases fixed tissues can be stored for up to a few days, for sensitive antigens or weak GFP signals it is better to complete the staining and imaging as soon as possible.
To “rinse” means add liquid to the tube containing the fixed larval tissue, allow tissue to settle to bottom of the tube, remove liquid with a Pasteur pipet without disturbing the larval tissue (i.e., don’t create a suction at the bottom of the tube, as then the imaginal discs may be sucked away!), and replace with fresh appropriate liquid. To “wash” means incubate the larval tissue with the appropriate solution for an extended period of time. The period of time for rinsing and washing can be extended, if convenient.
Add in order: PBT first, then serum, then antibodies. The appropriate dilution of the primary antibody will have to be determined by trial-and-error experiments for each antisera. Too high an antisera concentration is wasteful and often gives more non-specific (i.e., background) staining. If background staining persists despite trying a variety of dilutions, non-specific background antibodies (e.g., from other antibodies in the antisera) can be reduced by preabsorption of the antisera against fixed tissues. The procedure for preabsorption is to fix the tissue of interest as per protocol. Rinse the tissue once with PBT, and then wash twice with PBT for 20 min each with the tube placed on the Nutator. Remove the PBT from the tissue, make a 1:10 dilution of your antibody in PBT, and then add it to the fixed tissue. Incubate overnight at 4 °C on a Nutator. After this preabsorption step, the supernatant is ready for tissue staining. Remember that the starting dilution of the antibody is now 1:10.
Typically we incubate overnight at 4 °C, but for most antibodies 2 h at room temperature (25 °C) also works.
The appropriate dilution of 2° antibodies will need to be experimentally determined. For 2° antibodies from Jackson ImmunoResearch, we typically use 1.5 μL of each secondary antibody in a 200 μL staining volume. This sometimes needs to be adjusted for different batches of 2° antibodies, as there can be some lot-to-lot variability. Consideration should also be given to the particular fluorochromes used for each 2° antibody. In our experience, brightest signals are obtained with the Alexa 488, so we would use that for the antibody stain that is expected to be the weakest. Conversely Alexa 405 generally gives a weak signal and so should only be used with primary antibodies that are expected to give a strong signal.
Brief exposure to light will not significantly affect fluorescence (e.g., while dissecting the imaginal discs), but prolonged exposure should be avoided by keeping tubes and slides in the dark as much as is practical.
Confocal imaging of the imaginal discs is best if conducted within 2–3 days of completion of the immunostaining and for weak signals may need to be conducted even sooner.
Phalloidin is used to detect filamentous actin in tissues. Methanol treatment of tissues disrupts filamentous actin, and despite the fact that phalloidin is dissolved in methanol, the following procedure will give excellent visualization of actin in the Drosophila imaginal disc. To stain with phalloidin, premix in a separate Eppendorf tube: 160 μL PBT + 20 μL phalloidin conjugated to a fluorescent dye (i.e., phalloidin-488, phalloidin-555, phalloidin-633, or phalloidin-647). Remove liquid from stained tissue, and replace with premix. Incubate at RT on Nutator for 40 min, tube wrapped in aluminum foil. Rinse in PBT, wash 3× 10–15 min in PBT on Nutator, tube wrapped in aluminum foil.
-
Stain 30 min with a 1:2000 dilution as follows:
1 mE PBT + 0.5 μL Hoechst (kept in small brown bottle at 4 °C).
Incubate for 30 min at RT on Nutator. Tubes wrapped in aluminum foil.
Rinse in PBT, wash 3× 10–15 min in PBT on Nutator. Keep tubes wrapped in aluminum foil.
In theory, a wide-field fluorescence microscope, in combination with deconvolution software, could be used, but we have always used confocal microscopes. For some GFP-tagged proteins, expression levels may be very low, and thus detectors (such as the Leica HyD) with excellent signal-to-noise ratios may be necessary for appropriate imaging.
Acknowledgments
Research in the Irvine laboratory is supported by NIH grants GM78620 and GM121537.
References
- 1.Sun S, Irvine KD (2016) Cellular organization and cytoskeletal regulation of the hippo signaling network. Trends Cell Biol 26:694–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu F-X, Zhao B, Guan K-L (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163:8l1–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chalfie M (1995) Green fluorescent protein. Photochem Photobiol 62:651–656 [DOI] [PubMed] [Google Scholar]
- 4.Fritschy J-M, Härtig W (2001) Immunofluorescence. In: eLS. John Wiley & Sons, Ltd, Hoboken [Google Scholar]
- 5.Held LI (2002) Imaginal discs: the genetic and cellular logic of pattern formation. In: Developmental and cell biology series. Cambridge Press, Cambridge, p 460 [Google Scholar]
- 6.Misra JR, Irvine KD (2016) Vamana couples fat signaling to the Hippo pathway. Dev Cell 39:254–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun S, Reddy BVVG, Irvine KD (2015) Localization of Hippo Signaling complexes and Warts activation in vivo. Nat Commun 6:8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rauskolb C, Sun S, Sun G et al. (2014) Cytoskeletal tension inhibits Hippo signaling through an Ajuba-Warts complex. Cell 158:143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ambegaonkar AA, Pan G, Mani M et al. (2012) Propagation of dachsous-fat planar cell polarity. Curr Biol 22:1302–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauskolb C, Pan G, Reddy BV et al. (2011) Zyxin links fat signaling to the hippo pathway. PLoS Biol 9:e1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao Y, Kucuk B, Irvine KD (2009) Drosophila lowfat, a novel modulator of fat signaling. Development 136:3223–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh H, Irvine KD (2008) In vivo regulation of Yorkie phosphorylation and localization. Development 135:1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Y, Irvine KD (2007) Fat and expanded act in parallel to regulate growth through warts. Proc Natl Acad Sci U S A 104:20362–20367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao Y, Rauskolb C, Cho E et al. (2006) Dachs: an unconventional myosin that functions downstream of Fat to regulate growth, affinity and gene expression in Drosophila. Development 133:2539–2551 [DOI] [PubMed] [Google Scholar]
- 15.Ambegaonkar AA, Irvine KD (2015) Coordination of planar cell polarity pathways through Spiny legs. eLife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy BV, Irvine KD (2011) Regulation of Drosophila glial cell proliferation by Merlin-Hippo signaling. Development 138:5201–5212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staley BK, Irvine KD (2010) Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr Biol 20:1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy BVVG, Rauskolb C, Irvine KD (2010) Influence of Fat-Hippo and Notch signaling on the proliferation and differentiation of Drosophila optic neuroepithelia. Development 137:2397–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]