

Abstract
p53-binding protein 1 (53BP1) exerts distinct impacts in different situations involving DNA double-strand break (DSB) rejoining. Here we focus on how 53BP1 impacts upon the repair of ionising radiation-induced DSBs (IR-DSBs) and how it interfaces with Ku, the DNA end-binding component of canonical non-homologous end-joining (c-NHEJ), the major DSB repair pathway in mammalian cells. We delineate three forms of IR-DSB repair: resection-independent c-NHEJ, which rejoins most IR-DSBs with fast kinetics in G1 and G2, and Artemis and resection-dependent c-NHEJ and homologous recombination (HR), which repair IR-DSBs with slow kinetics in G1 and G2 phase, respectively. The fast component of DSB repair after X-ray exposure occurs via c-NHEJ with normal kinetics in the absence of 53BP1. Ku is highly abundant and has avid DNA end-binding capacity which restricts DNA end-resection and promotes resection-independent c-NHEJ at most IR-DSBs. Thus, 53BP1 is largely dispensable for resection-independent c-NHEJ. In contrast, 53BP1 is essential for the process of rejoining IR-DSBs with slow kinetics. This role requires 53BP1’s breast cancer susceptibility gene I (BRCA1) C-terminal (BRCT) 2 domain, persistent ataxia telangiectasia mutated (ATM) activation and potentially relaxation of compacted chromatin at heterochromatic-DSBs. In distinction, 53BP1 inhibits resection-dependent IR-DSB repair in G1 and G2, and this resection-inhibitory function can be counteracted by BRCA1. We discuss a model whereby most IR-DSBs are rapidly repaired by 53BP1-independent and resection-independent c-NHEJ due to the ability of Ku to inhibit resection, but, if delayed, then resection in the presence of Ku is triggered, the 53BP1 barrier comes into force and BRCA1 counteraction is required for resection.
Keywords: DNA double-strand break repair, ionising radiation, non-homologous end-joining, homologous recombination, 53BP1, DNA-PK
INTRODUCTION
Canonical or classical DNA non-homologous end-joining (c-NHEJ) and homologous recombination (HR) represent the two major DNA double-strand break (DSB) repair pathways. c-NHEJ is a relatively simple process involving factors that co-ordinate DNA end synapsis and protection, DNA end-processing and ligation (see [1, 2] for reviews). HR has also been extensively reviewed and will only be outlined here [3, 4]. HR is more elegant and complex, involving extensive DNA end-resection, binding of replication protein A (RPA) and subsequently RAD51 to the single stranded (ss)DNA tails, invasion of the undamaged sister chromatid forming a hybrid DNA structure, and repair synthesis exploiting the intact homologue. DNA end-resection is an essential early step during HR and importantly represents the step that commits to HR and precludes the ability to use c-NHEJ. Lengthy DNA end-resection necessitates extensive chromatin changes around the DSB, and use of a sister chromatid requires maintaining the sisters in close proximity. In mammalian cells, HR only uses a sister chromatid as a repair template, restricting its function to late S/G2 [5]. Indeed, HR’s essential function lies in promoting replication fork restart or repairing one-ended DSBs that arise at stalled or collapsed replication forks [6]. Nonetheless, HR also contributes to repairing two-ended radiation-induced DSBs in late S/G2 phase [7].
The existence of two efficient DSB repair pathways begs the question of how the choice between them is regulated. Both pathways are exploited in S/G2 cells to repair DSBs induced by ionising radiation (IR-DSBs) [7]. DSBs induced physiologically during V(D)J recombination and class switch recombination (CSR) only use c-NHEJ [8]. In contrast, DSBs that arise following replication fork collapse or stalling are one-ended, since they are in the process of undergoing replication, and repair of such one-ended DSBs and those created during meiosis preferentially undergo repair by HR [9]. An important component in this context is p53-binding protein 1 (53BP1), which is often described as promoting c-NHEJ. The role of 53BP1 in DSB repair during CSR, at one-ended DSBs and at dysfunctional telomere ends, has been well discussed [10–12]. Here, we will consider how 53BP1 impacts upon the repair of IR-DSBs. We will overview the distinct IR-DSB repair pathways, including two forms of c-NHEJ, and consider the key role of DNA-dependent protein kinase (DNA-PK) in promoting this process. We will then discuss two distinct roles by which 53BP1 influences DSB repair. We will present a model for the regulation of pathway usage.
IR-Induced DSB repair pathways: insight from examining the kinetics of DSB repair
Insightful information concerning DSB repair pathways and their usage has emerged from studies examining the kinetics of DSB repair [2]. Such studies revealed that in G2 cells the majority (~70%) of IR-DSBs undergo repair with fast kinetics via c-NHEJ whilst the remaining ~30% are repaired by HR with slower kinetics [7] (Fig. 1A). Thus, after X- or γ-rays, c-NHEJ is the major DSB repair process in G2 phase human cells. It is noteworthy that the contribution of HR to DSB repair in G2 is somewhat greater in rodent cells, most likely due to the lower abundance of Ku. Additionally, HR makes a greater contribution to the repair of the more complex DSBs induced by high linear energy transfer (LET) radiation, demonstrating that DSB end-complexity is a factor regulating pathway choice. Interestingly, DSB repair also occurs with similar biphasic kinetics in G1 [13] (Figure 1B). In G1, as in G2, the fast process represents resection-independent c-NHEJ, requiring solely c-NHEJ proteins, whilst in G1 the slow process is an Artemis and resection-dependent form of c-NHEJ [2, 14]. Artemis is a nuclease that was shown to be required for the cleavage of hairpin-ended DSBs that arise during the process of variable (diversity) joining during immune development [15], It is also required uniquely for this slow process of c-NHEJ. This slow process also requires ataxia telangiectasia mutated (ATM), 53BP1 and the ATM-dependent signalling proteins required for 53BP1 recruitment (all of which are also required for IR-DSB repair by HR repair in G2). Thus, the DSBs that undergo repair by HR in G2 are rejoined by Artemis and resection-dependent c-NHEJ in G1. The length of resection is substantially less in G1 and the promoting mechanisms are similar although distinct (see below) [2, 16].
Fig. 1.
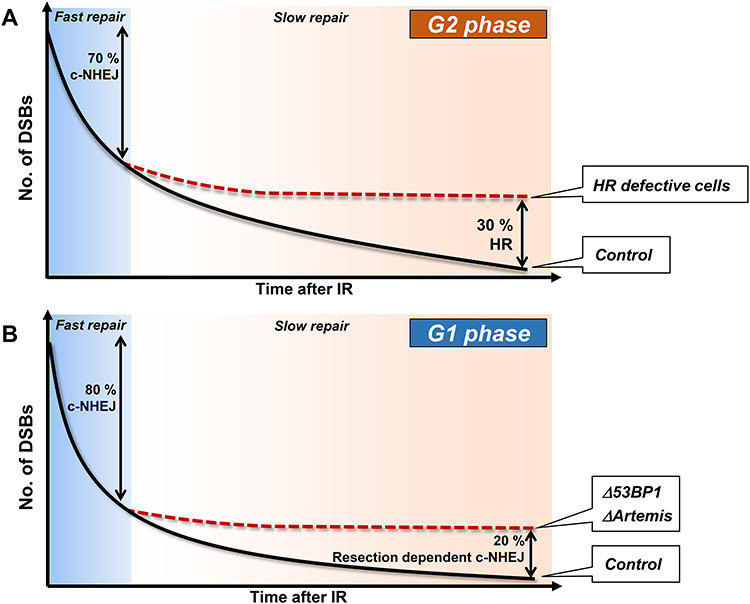
The kinetics and pathway usage for IR-DSB repair in G2 and G1 phase. c-NHEJ repairs the majority of DSBs with fast kinetics in G2 (70%) and G1 (80%) phase. 30% of DSBs are repaired with slow kinetics in G2 phase by HR. The fraction of DSBs repaired with slow kinetics in G1 phase is slightly smaller (20%) than in G2 phase since TA-DSBs are repaired with fast kinetics in G1 phase. The slow process in G1 phase requires Artemis and c-NHEJ proteins. 53BP1 is also required for the slow repair process in G1 phase and also for some (though not necessarily 30%) of the DSBs repaired by HR in G2 phase (not shown in the figure). The time scale on the y-axis represents up to ~ 24 h after 1–5 Gy X-rays.
IR-DSBs within transcriptionally active regions are repaired by HR in G2
The slow repair component in G2 is about one-third greater than in G1 phase (Fig. 1). Important studies have now shown that DSBs repaired by HR in G2 encompass those repaired with slow kinetics in G1 phase plus those within transcriptionally active regions [i.e. transcription-associated DSBs (TA-DSBs)], consistent with earlier findings that TA-DSBs undergo HR [17–19] (Fig. 2). TA-DSB HR requires specific factors distinct from those required for the repair of non-TA DSBs by HR, including xeroderma pigmentosum complementation group G (XPG) and RAD52, which mediate R-loop processing [18]. In the model, R-loops containing DNA–RNA hybrids arise when the transcription machinery encounters DSBs within transcription active loci. DSB-dependent R-loops are recognized by RAD52. Subsequently, RAD52 recruits XPG. XPG endonuclease activity incises DNA within R-loops and initiates 5′ to 3′ resection at TA-regions, most likely representing the transcript arising from ongoing transcription. RAD52 recognises the R-loop and recruits RAD52 to promote R-loop resolution (Fig. 2; see reference [18] for further details). TA-DSBs are reparable even without TA-HR, however, their repair by c-NHEJ results in a pronounced increase in genomic instability. While recognizing its importance, we will not discuss it further.
Fig. 2.
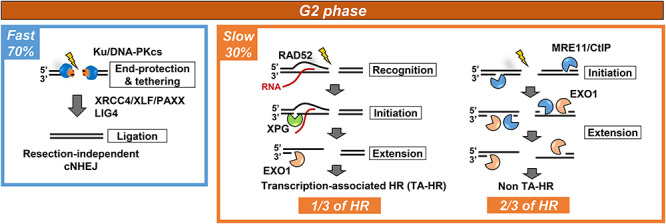
The pathways functioning in G2 phase and their contribution to DSB repair. c-NHEJ repairs 70% of IR-induced DSBs with fast kinetics in G2 phase. 30% of DSBs are repaired with slower kinetics by HR. Of these one-third represent TA-HR requiring the components indicated as well as core HR proteins. The remaining two-thirds are not transcriptionally associated DSBs but undergo resection as indicated.
Ku’s role in regulating pathway choice
Ku, a tightly associated heterodimer of Ku70 and Ku80 subunits, is the DSB recognition factor of c-NHEJ, binding DSBs with avid affinity, aided by its high abundance in human cells. Structural analysis revealed Ku to be cradle-shaped with the base being substantially thicker [1, 20]. The central core can thread onto DSB ends without sequence dependency. Each Ku subunit has an N-terminal von Willebrand type A-like domain (vWA), a central core and a C-terminal region. The extensive dimerization interface involves the central core region. Once bound to DNA ends, the larger C-terminal region of Ku80 interacts with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) creating the DNA-PK holoenzyme [1, 21, 22]. Ku binding protects DSB ends from nucleolytic degradation, contributing to a role in regulating pathway choice, since DNA end-resection is a prerequisite for HR [23]. Although Ku is the major component of c-NHEJ that protects DNA ends from nucleolytic degradation, DNA-PKcs facilitates this impact, in part, by promoting synapsis and the progression of c-NHEJ without end-resection (see [1] for details). Small angle X-ray scattering and cryogenic electron microscopy studies of DNA-PKcs have suggested that the Huntingtin, elongation factor 3, protein phosphatase 2A and yeast kinase TOR1 (HEAT) repeats provide a flexible cradle with the carboxy-terminal kinase domain situated above it [1, 24, 25]. Higher resolution analysis of DNA-PKcs crystals complexed with the Ku80 C-terminus (residues 539–732) have shown that DNA-PKcs encompasses three regions, an N-terminal region, a circular cradle and a head region harbouring a FRAP-ATM-TRRAP (FAT) domain [1, 26, 27]. Hence, DNA-PKcs has been proposed to be a hub on which c-NHEJ components and processing factors assemble, with the kinase activity being regulated via allosteric and conformational changes. Interestingly, the circular cradle forms a ring at the base of the molecule through which Ku may present DNA for repair. Collectively, this suggests that the kinase activity of DNA-PKcs is highly regulated by conformational changes that relay stages of the NHEJ process. These structural studies consolidate cellular and molecular approaches as well as a recent single-molecule approach, showing that DNA-PKcs co-ordinates DNA end-processing, promotes synapsis and acts as a hub allowing the assembly of NHEJ proteins, including XRCC4-like factor (XLF), Paralog of XRCC4 and XLF (PAXX), X-Ray Cross Complementing group 4 (XRCC4) and DNA ligase IV [1, 28].
In summary, Ku’s high abundancy and efficient DNA end-binding capacity empowers it to be the first responder to the majority of DSBs, protecting them from degradation and promoting rejoining by c-NHEJ. Rapid DNA-PK complex assembly contributes to these roles.
Resection in the presence of Ku
If Ku is the first responder at most DSBs and if DNA-PK has a key role in protecting the DNA ends from nucleolytic degradation, then how does resection proceed in its presence? The steps promoting resection in the presence of Ku at two-ended DSBs were initially revealed using G2 cells. Critical to the process of resection at DNA ends is meiotic recombination 11 (MRE11), a nuclease that has both 3′ to 5′ exonuclease as well as endonuclease activity. In G2, resection is initiated by endonucleolytic incision by MRE11 coupled with a requirement for CtBP-interacting protein (CtIP), and then resection is extended in a 5′ to 3′ direction away from the DSB end by exonuclease 1 (EXO1) with overlap from DNA2-like helicase (DNA2) [29–31] (Fig. 2). Recent studies have shown that MRE11 searches for free DNA ends by 1D facilitated diffusion that can take place on nucleosome-coated DNA, and it appears that Ku bound at the DNA end does not block MRE11 endonuclease activity [31]. Following the incision step, MRE11 exonuclease, which has 3′ to 5′ activity, becomes activated and promotes resection towards the DSB end, and potentially displaces Ku from the end. However, there is also evidence that MRE11 endonuclease activity and/or the proteasome contribute to Ku removal [31, 32]. Importantly, inhibition of MRE11 endonuclease activity or CtIP depletion allows DSB repair to progress via c-NHEJ, since resection is not initiated [29]. In contrast, loss of EXO1 or MRE11 exonuclease activity causes a DSB repair defect, since resection is initiated but cannot be completed and hence neither c-NHEJ nor HR can ensue. These studies support a model whereby in G2-phase EXO1 and MRE11 exonucleases function downstream of MRE11 endonuclease activity and that the step committing to HR involves exonucleolytic extension of an upstream initiation step. Importantly, they also show that the DSBs normally repaired by HR can be repaired by c-NHEJ if resection is not initiated (Fig. 2). Thus, at least after X-rays, there is nothing about these DSBs that precludes their repair by c-NHEJ.
As stated above, resection also arises in G1 cells during the slow DSB repair process, albeit less extensively than during HR. How does this resection process occur given that the DSBs are finally repaired by c-NHEJ? Significantly, MRE11 endonuclease activity is dispensable for Artemis-dependent DSB repair in G1 although MRE11 and EXO1 exonuclease activities are required [14]. CtIP is also required but is regulated by Polo-like kinase 3 (PLK3) in G1, distinct from its regulation by CDK1 in G2 [2, 14, 16]. We have proposed that in G1 cells, Ku might inwardly translocate, allowing resection of the free DNA ends without endonuclease activity, and subsequent repair by c-NHEJ (see below). Additionally and importantly, the nuclease Artemis also functions to promote resection during resection-mediated c-NHEJ in G1 cells (Fig. 3). Interestingly, Artemis appears to have a downstream function of removing an intermediate generated by the upstream nucleases, since depletion of CtIP, EXO1, PLK3 or inhibition of MRE11 exonuclease activity can relieve the need for Artemis for DSB repair in G1 phase [2, 14],
Fig. 3.
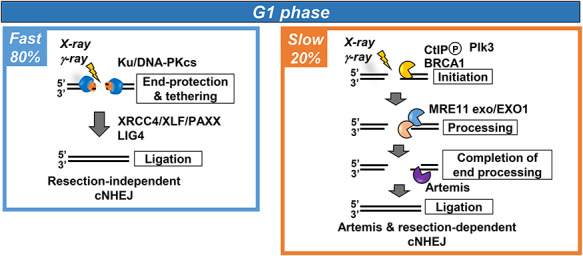
The pathways functioning in G1 phase and their contribution to DSB repair. c-NHEJ repairs 80% of DSBs with fast kinetics in G1 phase. 20% of DSBs are repaired with slow kinetics by Artemis and resection-dependent c-NHEJ. This process requires the genetic components shown. 53BP1 is also required for this process potentially to alter the chromatin structure at DSB. It is not included in the figure since it does not appear to be a core component essential for Artemis and resection-dependent c-NHEJ.
The 53BP1 block to resection
In addition to these nucleases, it has long been known that additional factors, including breast cancer susceptibility gene I (BRCA1), are required for the extensive 3’ ssDNA tails needed to drive HR. It is now appreciated that 53BP1 plays a critical role in inhibiting resection and that BRCA1 has a counteracting function [33].
53BP1 is a large chromatin-associated protein with a tandem BRCA1 C-terminal (BRCT), a glycine/arginine-rich (GAR) domain and a Tudor domain. It binds chromatin minimally without DNA damage but is recruited to DSBs via interactions with nucleosomes containing H4K20me2 and damage-induced ubiquitylated H2K15 [34]. Renowned studies have shown that 53BP1 creates a pro-c-NHEJ environment by restricting resection and that BRCA1 counteracts this resection-inhibitory function [33, 35, 36]. This supports the notion that 53BP1 promotes non-resection-dependent c-NHEJ although it is not a core c-NHEJ protein. Consistent with such a model, BRCA1 promotes the repositioning of 53BP1 in G2 phase at IR-DSBs, enabling resection within a 53BP1-devoid core [37, 38]. This can be visualized using high-resolution microscopy by the substantial enlargement of 53BP1 foci in G2 phase from 30 min to 8 h after IR, creating a ring-shaped structure with a central core lacking 53BP1, where RPA foci form. Such changes do not arise in G1 phase cells and are BRCA1-dependent in G2 phase [37, 38]. The resection-suppressive function of 53BP1 requires interactions with Pax Transactivation-Domain Interacting Protein (PTIP) and replication timing regulatory factor 1 (RIF1) via multiple N-terminal phosphorylation sites (Fig. 4) [39]. RIF1 recruits the Shieldin complex encompassing SHLD1, 2, 3 and REV7 [40–42]. RIF1/Shieldin exerts a major resection-inhibitory impact since RIF1 recruitment and resection are abolished in a 53BP1 28A mutant with mutations in all N-terminal ATM phosphorylation sites [39]. Resection requires BRCA1-dependent 53BP1 dephosphorylation and RIF1 loss [43]. Loss of PTIP binding also impedes resection (due to enhanced RIF1 binding), but some resection, especially at telomeres, ensues [44, 45]. Elucidating the overall function of 53BP1 in regulating the response to DSBs has been complex because it has several distinct functions or impacts. In addition to its impact on resection discussed above, 53BP1 oligomerises via oligomerization (OD) and Dynein light chain 8 (LC8)-binding domains. Recent super-high resolution microscopy [3D structured illumination (3D-SIM) and stimulated emission depletion (STED) microscopy] have highlighted the structure of 53BP1 foci, revealing sub-domains [termed 53BP1-nanodomains (53BP1-NDs)] assembled into higher-order microdomains (53BP1-MDs) in a RIF-dependent manner (Fig. 4) [46]. 53BP1 also enhances the mobility of certain DSBs, potentially contributing with its oligomerisation capacity to the rejoining of distal DSBs [45, 47]. Recent studies have also described the capacity of 53BP1 to undergo phase separation [48] and to have a post-resection function promoting BRCA1-independent RAD51 loading [44]. Finally, 53BP1 also has C-terminal BRCT2 which functions in binding MRE11, RAD50, NBS1 (MRN) and hence ATM, and promotes interaction with γH2AX distinct from its characterised recruitment domains (Fig. 4) [49, 50]. The role of this latter 53BP1 function in IR-DSB repair will be discussed below. Roles for 53BP1 in inhibiting resection and the counteracting function of BRCA1 have been shown from studies of one-ended DSBs induced in BRCA1-deficient cells after poly ADP-ribose polymerase I inhibition (PARP1i) [33]. Studies involving CSR and the fusion of dysfunctional telomeres have also revealed a pro-c-NHEJ role for 53BP1. Loss of 53BP1 increases resection during CSR and promotes telomere fusions due to the lack of end protection [10, 35]. Similar to IR-induced DSB ends, RIF1/Shieldin is involved in the resection-inhibitory process at telomere regions.
Fig. 4.
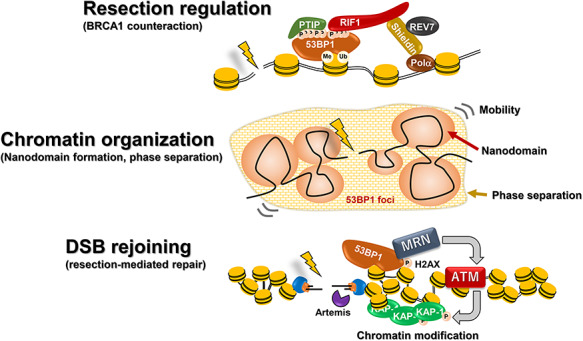
Processes influenced by 53BP1. 53BP1 functions as a barrier to resection via processes involving PTIP, RIF1 and Shieldin. BRCA1 serves to relieve this function of 53BP1. 53BP1 promotes the reorganization of chromatin via nanodomain formation and phase separation. This likely affects its function as a barrier to resection and potentially its ability to enhance the movement of certain IR-induced DSBs. 53BP1 has a distinct function in being essential for the repair of ∼20–30% of DSBs in G1 and G2 phase (those repaired with slow kinetics). For this function, 53BP1’s interaction with MRN, which enhances the tethering of ATM and hence the formation of pKAP1 foci, appears to be important.
Roles for 53BP1 in IR-DSB repair
In this section, we focus on the role of 53BP1 in IR-DSB repair. Importantly, many and indeed most IR-DSBs are repaired with normal kinetics in the absence of 53BP1 via resection-independent c-NHEJ, suggesting that this form of c-NHEJ is 53BP1-independent [13]. Further evidence that 53BP1 is dispensable for c-NHEJ is that mice and indeed, humans, lacking c-NHEJ proteins display severe combined immunodeficiency, due to the role of c-NHEJ during V(D)J recombination. This, however, is not a feature of mice lacking 53BP1. We should stress, however, that 53BP1 could exert a function in promoting the fidelity of resection-independent c-NHEJ.
In contrast, as mentioned above, 53BP1 is essential for DSBs repaired with slow kinetics in G1 and G2, namely Artemis and resection-dependent c-NHEJ in G1 and HR in G2. This role is not counteracted by BRCA1 deficiency and involves the 53BP1 BRCT2 domain rather than interaction with RIF1. Indeed, there is evidence that it requires maintained tethering of ATM at DSBs via an interaction with MRN, which is recruited by 53BP1 BRCT2 [49–51]. This begs asking what is special about these slowly repairing DSBs (Fig. 5). Although further studies are required and direct evidence is lacking, it appears that DSBs repaired with slow kinetics lie within heterochromatic (HC) regions, since in mouse cells where HC regions can be visualised as densely staining 4′.6′-diamidino-2-phenylindole (DAPI) chromocentres, the γ-H2AX foci remaining in the absence of factors required for the slow repair process are predominantly located at the periphery of the chromocentres [52]. Additionally, the requirement for Artemis and, indeed 53BP1, in G1 can be alleviated by depletion of HC-building factors, such as KRAB-associated protein-1 (KAP1) and Heterochromatin Protein 1 (HP1) [52, 53]. ATM phosphorylates KAP1, which has been proposed to promote HC relaxation via an interaction with Chromodomain-helicase-DNA-binding protein 3 [51, 52]. Taken together with the fact that a greater proportion of DSBs induced by high LET radiation are repaired with slow kinetics, this suggests that both chromatin complexity and DSB end-complexity can promote usage of a resection-dependent repair pathway. Importantly, this role of 53BP1 in promoting HC relaxation is genetically distinct to its role in preventing resection.
Fig. 5.
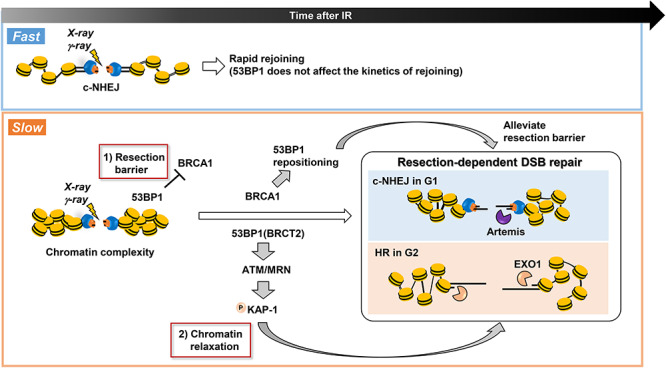
53BP1 has two roles in IR-induced DSB repair: it functions as a resection barrier and is essential for HC-DSB repair. As stated above, most DSBs are repaired with fast kinetics by c-NHEJ in G1 and G2 phase. In G1 phase, 20% of DSBs are repaired by resection-dependent c-NHEJ and a similar subset of DSBs are repaired by HR in G2 phase (note: HR also repairs TA-DSBs in G2 phase). Both HR and resection-dependent c-NHEJ involve some level of resection (greater for HR compared to resection-dependent c-NHEJ) and 53BP1 poses a barrier to both processes, which is relieved by BRCA1 (item 1 and the upper arrow of the figure). Additionally, these DSBs appear to be located in regions of high chromatin complexity (heterochromatin regions) and necessitate chromatin relaxation prior to rejoining. 53BP1 is essential for this process, which involves the tethering of MRN, ATM recruitment and focused phosphorylation of KAP1 (item 2 and the lower arrow of the figure). These two distinct roles of 53BP1 are required for both resection-dependent c-NHEJ in G1 and HR repair of these specific DSBs in G2.
Although this essential role for 53BP1 in the slow component of DSB repair has added confusion, 53BP1 also inhibits resection during the slow repair processes in G1 and G2, with BRCA1 counteracting the inhibitory barrier [13]. In G2, this becomes evident when the essential role of 53BP1 in relaxing HC is relieved via KAP1 depletion [54]. Progression of HR then requires BRCA1 and the requirement for BRCA1 can be alleviated by knocking down 53BP1. Perhaps surprisingly, BRCA1 is also required for slow DSB repair in G1 [14]. As mentioned above, Artemis is an essential downstream factor required for the slow repair process in G1, and depletion of upstream factors, including CtIP, can bypass the need for Artemis, providing genetic evidence for their role in G1 [14]. The lack of a specific requirement for these factors for repair in G1 is because failure to initiate resection still allows resection-independent DSB repair. Similarly, BRCA1 loss, although not conferring a DSB repair defect in G1, relieves the need for Artemis, suggesting an upstream role in promoting resection [13]. This role is relieved by 53BP1 depletion. The repair of closely-spaced site-specific DSBs in constructs where DNA end-joining necessitates the loss of a small intervening fragment (rendering the ends distal) has been exploited to model resection-dependent c-NHEJ. The same factors (Artemis, CtIP, EXO1 and MRE11 exonuclease) needed for the slow DSB repair component were required for such rejoining, with most DNA ends undergoing resection. Significantly, this process also required BRCA1 to counteract 53BP1. Thus, to conclude, BRCA1 also functions during resection-dependent c-NHEJ to counteract the resection-inhibitory function of 53BP1, although the magnitude of resection is substantially less in G1 cells [14].
A model for the regulation of IR-DSB repair
The DNA ends generated by X- or γ-rays predominantly have associated base or sugar damage and require end-processing. Nonetheless, the DNA ends likely remain in close proximity, which is distinct from one-ended DSBs, ends at dysfunctional telomeres and most likely DNA ends generated during CSR. After rapid Ku binding, we suggest that IR-DSBs, if not highly complex and in a non-HC environment, can be rapidly repaired by c-NHEJ even in the absence of 53BP1, since DNA-PK functions to tether the DNA ends, prevent resection and promote c-NHEJ. This likely reflects resection-independent c-NHEJ, the fast repair process in G1 and G2 phase. Unless sequence information is lost from both strands, such DSBs can potentially be repaired with good accuracy.
We propose a time factor whereby if rapid repair by c-NHEJ fails to progress, then steps promoting resection proceed. In G1, repair then ensues via Artemis and resection-dependent c-NHEJ; in G2 HR takes place. Such a pathway switch is likely to take place rapidly because NHEJ factors immediately attempt repair once DSBs occur [55]. DNA end-complexity and highly compacted chromatin are barriers inhibiting rapid rejoining, but end-proximity could be an additional factor. Both the extensive resection in G2 and modest resection in G1 can be inhibited by 53BP1 and necessitate BRCA1 to relieve this inhibitory barrier. We suggest that in G1, Ku might undergo inward translocation to provide resectable DNA ends coupled with Ku’s retention to promote subsequent c-NHEJ. 53BP1-RIF1 could pose a barrier to such inward movement, requiring BRCA1-dependent 53BP1 repositioning, similar to the situation in G2 although smaller in scale. One important question is whether Ku (or DNA-PK) can function alone to prevent resection in the absence of 53BP1. A distinct question is whether 53BP1 enhances DNA end-tethering and hence limits chromosome translocations during resection-independent c-NHEJ. Although this needs further investigation, an important study found reduced translocations arising following IR of 53BP1-deficient G0 cells [52].
However, adding confusion, after X- or γ-rays, the predominant factor delaying resection-independent c-NHEJ is the chromatin environment, and 53BP1 exerts a role requiring its BRCT2 domain to allow the repair of this DSB subset, a role distinct from the inhibition of resection. Thus, we propose a kinetic model in which Ku or DNA-PK is sufficient to promote c-NHEJ in situations where repair can be rapidly effected. However, if delayed due to DNA end complexity or proximity or the chromatin environment, then the 53BP1 barrier is effected and must be relieved for minor end-resection in G1 or more significant resection in G2. Indeed, it is possible that a major role of 53BP1 is to ensure limited resection in G1 phase but allow more substantial resection for HR in G2 phase.
CONCLUSION
We consider here the role of DNA-PK in preventing resection during IR-DSB repair and consider two impacts of 53BP1, one is its role as a resection barrier in G1/G2 and the second is its ability to block HC-DSB repair. Due to the rapid binding and high abundance of DNA-PK proteins, the majority of IR-DSBs are repaired via resection-independent c-NHEJ with no overt requirement for 53BP1 in G1 and G2 phase. In contrast, all DSBs repaired with slow kinetics have an essential requirement for 53BP1 that involves its BRCT2 domain. However, distinctly, 53BP1 can restrict resection-dependent DSB repair in G1 and G2 phase via a RIF1-dependent barrier, with BRCA1 being required to counteract this role. Interestingly, 53BP1 has a range of domains conferring properties such as oligomerisation, phase separation and promoting DSB mobility. Further studies are required to determine if these also contribute to IR-DSB repair. We stress that our conclusions are largely based on the kinetics of DSB repair and do not monitor the fidelity of repair, which may influence only a small subset of DSBs. Thus, an examination of the role of 53BP1 in the fidelity of DSB repair is required. Additionally, 53BP1’s role in the repair of high LET-induced DSBs could also expose additional roles.
FUNDING
This work was supported by Grants-in-Aid from the Japan Society for the Promotion of Science for KAKENHI (JP17H04713 to A.S.) and the Program of the network-type Joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University and Fukushima Medical University.
EDITORIAL NOTE
Prof. Penny A. Jeggo was invited to deliver a lecture in Japan and to write this review article for the international promotion of Journal of Radiation Research. The traveling and publication costs are supported by Grant-in-Aid for Publication of Scientific Research Results - Enhancement of International Dissemination of Information (No. 19HP2003) from Japan Society for the Promotion of Science to Japanese Radiation Research Society.
References
- 1. Wu Q. Structural mechanism of DNA-end synapsis in the non-homologous end joining pathway for repairing double-strand breaks: Bridge over troubled ends. Biochem Soc Trans 2019;47:1609–19. [DOI] [PubMed] [Google Scholar]
- 2. Lobrich M, Jeggo P. A process of resection-dependent nonhomologous end joining involving the goddess Artemis. Trends Biochem Sci 2017;42:690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 2010;11:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet 2010;44:113–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 2000;19:3398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ait Saada A, Lambert SAE, Carr AM. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018;71:135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shibata A, Conrad S, Birraux J et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 2011;30:1079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prochazkova J, Loizou JI. Programmed DNA breaks in lymphoid cells: Repair mechanisms and consequences in human disease. Immunology 2016;147:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shibata A, Jeggo P, Lobrich M. The pendulum of the Ku-Ku clock. DNA Repair 2018;71:164–71. [DOI] [PubMed] [Google Scholar]
- 10. Mirman Z, Lange T. 53BP1: A DSB escort. Genes Dev 2020;34:7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Setiaputra D, Durocher D. Shieldin - the protector of DNA ends. EMBO Rep 2019;20:e47560. doi: 10.15252/embr.201847560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ghodke I, Soutoglou E. 53BP1-RIF1: Sculpting the DNA repair focus in 3D. Nat Struct Mol Biol 2019;26:1087–8. [DOI] [PubMed] [Google Scholar]
- 13. Riballo E, Kuhne M, Rief N et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell 2004;16:715–24. [DOI] [PubMed] [Google Scholar]
- 14. Biehs R, Steinlage M, Barton O et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell 2017;65:671–84 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moshous D, Callebaut I, Chasseval R et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 2001;105:177–86. [DOI] [PubMed] [Google Scholar]
- 16. Barton O, Naumann SC, Diemer-Biehs R et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J Cell Biol 2014;206:877–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aymard F, Bugler B, Schmidt CK et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 2014;21:366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yasuhara T, Kato R, Hagiwara Y et al. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell 2018;175:558–70 e11. [DOI] [PubMed] [Google Scholar]
- 19. Beucher A, Birraux J, Tchouandong L et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 2009;28:3413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001;412:607–14. [DOI] [PubMed] [Google Scholar]
- 21. Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku: Biochemical and atomic-force microscopy studies. EMBO J 1997;16:5098–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005;434:605–11. [DOI] [PubMed] [Google Scholar]
- 23. Pierce AJ, Hu, Han M et al. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 2001;15:3237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 2010;463:118–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dobbs TA, Tainer JA, Lees-Miller SP. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair 2010;9:1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sibanda BL, Chirgadze DY, Ascher DB et al. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 2017;355:520–4. [DOI] [PubMed] [Google Scholar]
- 27. Sharif H, Li Y, Dong Y et al. Cryo-EM structure of the DNA-PK holoenzyme. Proc Natl Acad Sci 2017;114:7367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang JL, Duboc C, Wu Q et al. Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nat Struct Mol Biol 2018;25:482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shibata A, Moiani D, Arvai AS et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 2014;53:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cannavo E, Reginato G, Cejka P. Stepwise 5' DNA end-specific resection of DNA breaks by the Mre11-Rad50-Xrs2 and Sae2 nuclease ensemble. Proc Natl Acad Sci 2019;116:5505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Myler LR, Gallardo IF, Soniat MM et al. Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol Cell 2017;67:891–8 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feng L, Chen J. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat Struct Mol Biol 2012;19:201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bunting SF, Callen E, Wong N et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010;141:243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fradet-Turcotte A, Canny MD, Escribano-Diaz C et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013;499:50–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bothmer A, Robbiani DF, Feldhahn N et al. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 2010;207:855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bouwman P, Aly A, Escandell JM et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 2010;17:688–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chapman JR, Sossick AJ, Boulton SJ et al. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 2012;125:3529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kakarougkas A, Ismail A, Katsuki Y et al. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res 2013;41:10298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Escribano-Diaz C, Orthwein A, Fradet-Turcotte A et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 2013;49:872–83. [DOI] [PubMed] [Google Scholar]
- 40. Dev H, Chiang TW, Lescale C et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 2018;20:954–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mirman Z, Lottersberger F, Takai H et al. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018;560:112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noordermeer SM, Adam S, Setiaputra D et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018;560:117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Isono M, Niimi A, Oike T et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 Dephosphorylation. Cell Rep 2017;18:520–32. [DOI] [PubMed] [Google Scholar]
- 44. Callen E, Zong D, Wu W et al. 53BP1 enforces distinct pre- and post-resection blocks on homologous recombination. Mol Cell 2020;77:26–38 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lottersberger F, Karssemeijer RA, Dimitrova N et al. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell 2015;163:880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ochs F, Karemore G, Miron E et al. Stabilization of chromatin topology safeguards genome integrity. Nature 2019;574:571–4. [DOI] [PubMed] [Google Scholar]
- 47. Dimitrova N, Chen YC, Spector DL et al. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008;456:524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kilic S, Lezaja A, Gatti M et al. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J 2019;38:e101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee JH, Goodarzi AA, Jeggo PA et al. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J 2010;29:574–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baldock RA, Day M, Wilkinson OJ et al. ATM localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with gammaH2AX. Cell Rep 2015;13:2081–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Noon AT, Shibata A, Rief N et al. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol 2010;12:177–84. [DOI] [PubMed] [Google Scholar]
- 52. Goodarzi AA, Noon AT, Deckbar D et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 2008;31:167–77. [DOI] [PubMed] [Google Scholar]
- 53. Goodarzi AA, Kurka T, Jeggo PA. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat Struct Mol Biol 2011;18:831–9. [DOI] [PubMed] [Google Scholar]
- 54. Kakarougkas A, Ismail A, Klement K et al. Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res 2013;41:9719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mari PO, Florea BI, Persengiev SP et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci 2006;103:18597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]