

Abstract
Covalent co-assembly holds great promise for the fabrication of hydrogels with controllable nanostructure, versatile chemical composition, and enhanced mechanical properties given its relative simplicity, high efficiency, and bond stability. This report describes our approach to designing functional multicomponent hydrogels based on photo-induced chemical interactions between an acrylamide-functionalized resilin-like polypeptide (RLP) and a peptide amphiphile (PA). Circular dichroism (CD) spectroscopy, electron microscopy, and amplitude sweep rheology were used to demonstrate that the co-assembled hydrogel systems acquired distinct structural conformations, tunable nanostructures, and enhanced elasticity in a PA concentration-dependent manner. We envisage the use of these materials in numerous biomedical applications such as controlled drug release systems, microfluidic devices, and scaffolds for tissue engineering.
Keywords: co-assembly, peptide amphiphiles, resilin, thiol-ene click chemistry, elastic biomaterials
Introduction
Over the past decades, fabrication of nanostructured materials from molecular building blocks by self-assembly has been an exciting development in chemistry.1 Inspired by nature’s machinery such as viral capsids and the DNA double helix, it has become possible to generate a wide variety of ordered nanostructures.2 Key to this structural formation is the use of multiple non-covalent interactions such as hydrogen bond, ionic, van der Waals, and π-π interactions to facilitate self-assembly of a single molecule or co-assembly of multiple molecular building blocks in a thermodynamically controlled manner. Unfortunately, supramolecular nanostructures tend to exhibit insufficient robustness and have limited practical utility as a result of the weak and reversible non-covalent interactions that underpin their formation. Consequently, there has been an increasing focus on the use of irreversible covalent interactions to drive self-assembly of molecular building blocks into more functional materials.3 For examples, the use of covalent interactions to facilitate co-assembly of self-assembling molecules with polymers towards the creation of robust and functional hybrid hydrogels is currently a topic of intense focus.4
Peptides and proteins represent a rich source of inspiring building blocks for fabricating diverse self-assembled nanostructures due to their exquisite structures and functions.5 These building blocks have been used for the design of flexible,6 tough,7 dynamic,8 auxetic,9 and bioactive1b, 10 materials. Resilin is a structural intrinsically disordered protein present in insects that exhibits outstanding mechanical properties including rubber-like elasticity,11 low stiffness, exceptional resilience,12 efficient energy storage,13 and fatigue lifetime.14 Consequently, resilin-like polypeptides (RLPs) have attracted increasing attention in the past decades.15 As the natural protein, RLPs are also disordered proteins with the capability to respond to multiple stimuli including temperature, light, moisture, pH, and ions.14, 16 These molecules have been exploited for diverse applications as biomaterials15, 17 and templating agents for nanoparticle synthesis.18
In order to impart RLPs with improved properties that can be used to design hydrogel materials, their structure and conformation have been manipulated by covalent co-assembly with other proteins and polymers. For example, Kiick and co-workers have fabricated RLP-based hydrogels with tunable mechanical properties,19 precise microstructures,20 and excellent biocompatibility21 by covalent conjugation of RLP and modified polyethylene glycols (PEG) through a Michael-type addition reaction as well as through thiol-ene click chemistry. In other reported studies, Pepe and co-workers described a chimeric RLP-silk-like polypeptide-collagen system that is able to co-assemble into a matrix of nanofibers.22 Similarly, Xia et al reported on a genetically engineered conjugate of RLP and silk-like polypeptide (SLP) and their hierarchical co-assembly into self-supporting hydrogels in a temperature- and time-dependent manner.23 However, the use of modular, biocompatible, and easy-to-synthesize building blocks such as peptides can serve as simpler and more predictable components to co-assemble with RLPs.
Self-assembling peptides are an attractive alternative to be used as precise co-assembling building blocks of RLPs to control their hierarchical organization into practical materials. Peptide amphiphiles (PAs) are a class of self-assembling peptides with high propensity to self-assemble into extracellular matrix-like nanofibers in aqueous environments.24 Stupp and co-workers first demonstrated the possibility to co-assemble PAs with large polysaccharide chains to generate ordered hierarchical structures.25 Inspired by this work, and taking advantage of the modular nature of PAs, we have recently reported on the possibility to use PAs as molecular manipulators to co-assemble with26 and modulate27 the conformation of proteins to generate materials with emergent properties including morphogenesis, elasticity, and structural hierarchy. However, for many applications, these materials remain fragile or require post-assembly modifications,28 and consequently have found limited applicability. Strategies involving hybridization of PA with polymers have been developed to overcome this limitation.29 In this study, we report a covalent co-assembly strategy based on PAs and RLPs to enable both control over the hierarchical assembly of RLPs and enhancement of mechanical properties of the resulting hybrid material. We describe the assembling mechanism and demonstrate the capacity to generate different hydrogels having different nanostructures and exhibiting tuneable mechanical properties.
Materials and Methods
Synthesis and Characterization of RLP-4Ac
Resilin-like polypeptides (RLPs) were expressed and purified as previously described.1–4 The RLPs were functionalized with acrylamide groups via modification of regularly positioned lysine residues on the polypeptide chain as previously reported.5 In brief, the RLP proteins were dissolved in PBS with concentration of 10 mg/mL. Solution (50 mg/mL) of acrylic acid N-hydroxysuccinimide ester (NHS-Ac) was prepared in dimethyl sulfoxide (DMSO) and added drop-wise into the RLP solution. The molar ratio of NHS-Ac to lysine was 0.5 was required to yield the desired functionality of the RLP-4Ac conjugation. The reaction was stirred at room temperature for approximately 4 h. This reaction solution was diluted 8 times with DI water to prevent precipitation and dialyzed with membrane (Snakeskin, 3.5kDa, Thermo Scientific) against DI water at 4 °C (in a cold room) to remove by-products and DMSO. The purified RLP-Ac was filtered and lyophilized and stored at −20 °C for further characterization. Yield: 95%.
The functionality of the RLP-Ac was characterized with 1H NMR spectroscopy. The purified RLP-4Ac (~2 mg) was dissolved in D2O (600 μL) (Cambridge Isotope Laboratories, Tewksbury, MA) and analyzed on an AVIII 600MHz NMR spectrometer (Bruker Daltonics, Billerica, MA). The protons from the eight phenylalanine residues per RLP molecule were used as an internal reference for the quantification of acrylamide group functionality. The integration of the aromatic protons of phenylalanine (1H NMR (600 MHz, D2O, δ): 7.15–7.40 (m, 5H)) was compared to the integration of the vinylic protons of the acrylamide that resulted from the reaction of the acrylamide and lysine amine groups (1H NMR (600 MHz, D2O, δ): 5.65–6.30 (d, 3H)).
Synthesis and purification of C15H31CO-VVVAAAEEECY (E3CY)
The peptide amphiphile (PA), E3CY, was synthesised using solid phase peptide synthesis (SPPS) on Liberty Blue automated microwave peptide synthesizer (CEM, UK). The standard 9-fluorenylmethoxycarbonyl (Fmoc) protection chemistry on a 4-methylbenzhydrylamine (MBHA). Rink Amide resin (Novabiochem Corporation, UK) was employed. Amino acid couplings were performed using 4 mmol equivalent of Fmoc-protected amino acids (Novabiochem Corporation, UK), 4 mmol equivalents of 1-hydroxybenzotriazole (HOBt, Carbosynth Limited, UK) and 6 mmol equivalents of N,N’-diisopropylcarbodiimide (DIC, Sigma-Aldrich, UK) for 1 h. Fmoc deprotections were performed with 20% piperidine (Sigma-Aldrich, UK) in N,N-dimethylformamide (DMF, Alfa Aesar, UK). Following Fmoc removal from the final amino acid residue, the alkyl tail moiety (from palmitic acid, C16H32O2, Calbiochem, UK) was conjugated to the free N-terminus. The alkylation reaction was accomplished by using palmitic acid (4 mmol), HOBt (4 mmol), and DIC (6 mmol) in DMF/dichloromethane. The reaction was allowed to proceed at room temperature for 4 h or until obtaining a negative Kaiser test. Cleavage of the PA cleavage from the resin and deprotection of the side chains was done with a mixture of trifluoroacetic acid (TFA, Sigma-Aldrich, UK), triisopropylsilane (TIS, Alfa Aesar, UK), ethanedithiol (Sigma-Aldrich, UK) and water in a ratio of 91:3:3:3 for 3h at room temperature. After filtration of the cleavage mixture and two subsequent TFA washings, TFA was removed by rota-evaporation and the resulting solution was triturated with cold diethylether. The precipitate was collected by centrifugation, washed twice with cold diethylether, air-dried, dissolved in deionised water and lyophilized. The crude PA powder (100 mg) was dissolved in water (10 mL) with the addition of NH4OH and DTT (100 mg) to reduce all cysteine amino acids to free thiol which was then purified using a preparative High-Performance Liquid Chromatography (Waters, USA) with reverse-phase Xbridge C18 column (Waters, USA) and water/acetonitrile (0.1% NH4OH) binary mobile phase. The HPLC fractions were evaporated to dryness to remove the remove acetonitrile. Finally, the PA was lyophilized to obtain a white fluffy pure powder. Yield: 85%. PA was characterized by electrospray ionization mass spectrometry (ESI-MS). ESI-MS (m/z) calc. for C67H110N12O19S 1419.7729; found 1420.7729 (100%[M+H]+). Analytical HPLC was used to assess the purity (95%) of the peptide.
RLP-4AC and E3CY co-assemblies
Our initial studies involved co-assembling RLP-4AC with E3CY by incubation at basic pH condition (thiol-Michael addition reaction). In this case, solutions of RLP-4AC (20 wt %) in PBS were mixed aqueous solutions of E3CY (2 wt %, pH 8) in PBS. The resulting mixtures were incubated at 37 °C for 6 h. On the other hand, co-assembling of RLP-4AC and E3CY by thiol-ene photoclick reaction was carried out by photo-irradiation. To this end, solutions of RLP-4AC (20 wt %) and E3CY (2 wt %) were mixed with the photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, 4.6 mM). The resulting mixtures were photo-irradiated using long wavelength UV light (5 mW/cm2 at 365 nm). Omnicure S1500 mercury lamp (208–600 nm) was used as the source of UV irradiation. Hydrogels of pure RLP-4AC (10 wt %) were similarly prepared by photo-irradiation as described above. For E3CY, the hydrogels were prepared by adding CaCl2 (0.5 mM, 20 mL) to aqueous solutions of E3CY (1 wt %) at room temperature.
Electron microscopy characterization
Samples were prepared by mixing 0.05 wt % of E3CY (50 μL, pH 8) with 0.5 wt % of RLP-4AC (50 μL) and incubating at 37 °C for 6 h. The thiol-ene photoclicked co-assembled RLP-4AC_E3CY were also prepared with the same concentration of RLP-4AC and E3CY but were photo-irradiated using Omnicure S1500 mercury lamp with long wavelength UV light (5 mW/cm2 at 365 nm) for 5 min in the presence of LAP photoinitiator (46 μM). Samples of individual components E3CY and RLP-4AC (crosslinked and uncrosslinked) were also prepared as described above. Samples prepared under various conditions were mounted on copper TEM plasma etched holey carbon-coated copper grid (Agar Scientific, UK). The grids were immerse in the sample solutions for 30 s. Excess samples were removed on filter paper before incubation and grids were washed with ultrapure water for 30 s. Grids were incubated with 2% uranyl acetate solution for 30 s and air dried for 24 h at room temperature. Bright-field transmission electron microscopy (TEM) was performed on a JEOL 1230 Transmission Electron Microscope operated at an acceleration voltage of 80 kV. All the images were recorded by a Morada CCD camera (Germany).
Microstructures of the macroscopic hydrogels were observed using scanning electron microscopy (SEM). Hydrogels were prepared as described above. The hydrogels were prepared for SEM firstly by gradual dehydration with increasing concentrations of ethanol (20, 50, 70, 80, 90, 96, and 100 v/v %). Dehydrated samples were then subjected to critical point drying (K850, Quorum Technologies, UK). The SEM micrographs of the xerogels were acquired on Inspect F50 (FEI Comp, the Netherlands) after sputter-coating with gold (10 nm thick).
Circular dichroism measurements
Circular dichroism (CD) was measured with Chirascan™ circular dichroism spectrometer (Applied Photophysics Limited, UK) using quartz cell with 1 mm path length and the following parameters: data pitch − 0.5 nm, scanning mode- continuous, scanning speed − 100 nm/min, bandwith − 2 nm and accumulation − 5. All CD data are presented as ellipticity and recorded in millidegree (mdeg). CD spectra were obtained by signal integrating 3 scans, from 190 to 260 nm at speed of 50 nm/min. Data were processed by a simple moving average and smoothing method. Background scans of PBS 1x were recorded and automatically subtracted from the sample scans. Samples were prepared by mixing 0.02, 0.05 and 0.1 wt % of E3CY (200 μL, pH 8) with 0.5 wt % of RLP-4AC (200 μL) and incubate at 37 °C for 6 h. Samples were transferred into a 1 mm length path length quartz cuvette. Spectra were acquired using the above parameters. Similarly, the cross-linked RLP-4AC_E3CY samples were prepared by adding LAP (46 μM) to the mixtures of RLP-4AC (0.5 wt %) and various concentrations of E3CY (0.05, 0.1 and 0.2 wt %, pH 7.6). Samples were photo-irradiated using Omnicure S1500 mercury lamp (Poly Dispensing Systems, France) with long wavelength UV light (5 mW/cm2 at 365 nm) for 5 min prior CD measurements. CD spectra of the individual components E3CY (0.025, 0.05 and 0.1 wt %) and RLP-4AC (cross-linked and uncross-linked) were also acquired as detailed above.
Dynamic rheological measurements
Rheological measurements were performed using a Discovery Hybrid Rheometer, Rheo-DHR3 (TA Instruments) with a UV accessory. The upper parallel plate and the bottom quartz window (which allows transmittance of UV light) are 20 mm in diameter. Omnicure S1500 mercury lamp (208–600 nm) (Poly Dispensing Systems, France) was used as the source of UV irradiation. UV light intensity was calibrated with radiometer and controlled by the advanced TRIOS software during measurements. All data were collected at 25 °C. For the co-assembled RLP-4AC_E3CY hydrogels, first, various concentrations of E3CY (1 and 2 wt %) were prepared in PBS and pH of the E3CY solutions was adjusted with NaOH (0.5 M) to aid solubility. RLP-4AC solutions (20 wt %) was also prepared in PBS (pH 7.3). Then aliquots of E3CY (pH 7.6, 20 μL) and RLP-4AC (20 wt %, 20 μL) were mixed to attain a final concentrations of 0.5 and 1 wt % for E3CY and 10 wt % for RLP-4AC. After the addition of the photoinitiator LAP (4.6 mM) to the RLP-4AC_E3CY mixtures and vortexing to ensure complete solubility of LAP, the mixtures were carefully pipetted onto the bottom quartz window for in situ rheological characterization. Time sweep measurements were performed at constant frequency (1 Hz) while strain sweeps were performed from 0.1 to 1000% strain at constant frequency (1 Hz) to determine the strain-to-break value. Similarly, RLP-4AC hydrogels were characterized with the same protocol and concentration of 10 wt %. Self-healing was assessed initially at 10% strain for 100 s, then at 1000% strain for 200 s, 10% strain for 200 s, 1000% strain for 200 s and 10% strain for 400 s.
Raman spectroscopy measurements
Raman spectra were acquired using a confocal WITEC Alpha300 system utilising 785 nm excitation with an incident laser power of 63 mW. The microscope objective used was a 20x (S Plan Fluor, NA 0.45, ELWD) lens. Samples were prepared by placing a small amount of the solid sample on a microscope slide which had been previously cleaned with a methanol-soaked tissue, with a new slide used for each sample. The integration time was varied depending on the sample and details are provided in the associated figure caption. All spectra obtained were baseline corrected and any erroneous cosmic ray peaks were removed. Data processing was carried out using spectrographic software SpectraGryph 1.2.
Results and Discussions
Rationale of design
Our design aims to integrate the emergent complexity provided by multicomponent self-assembly with the functionality of covalent interactions. In this context, we take advantage of the inherent properties of both PAs and RLPs to develop a co-assembling system that uses a thiol-ene photoclick reaction between the two components to manipulate their molecular conformation, control their nano and microstructure, and enhance the resulting hydrogel properties (Fig. 1). The co-assembled hydrogels (RLP-4AC_E3CY) were designed to form by covalent interactions between an RLP functionalized with acrylamide moiety and a PA with sulfhydryl group. The RLP is a polypeptide containing 12 repeats of the putative consensus sequence (GGRPSDSYGAPGGGN) derived from Drosophilia melanogaster (fruit fly) and 5 repeats of lysine-rich segments (GGKGGKGGKGG) (Fig. 1b). The RLP was expressed according to procedures extensively used by Kiick and co-workers17c and was further functionalized with four acrylamide groups to facilitate photopolymerization with and without PA (Fig. 1e-g). On the other hand, the PA (E3CY) was designed with the inclusion of one cysteine residue on the C-terminus. On the basis of previous studies using other molecules with sulfhydryl moieties,21, 30 we expected the E3CY to form a covalent co-assembly with RLP-4AC via a thiol-ene click reaction.
Figure 1.
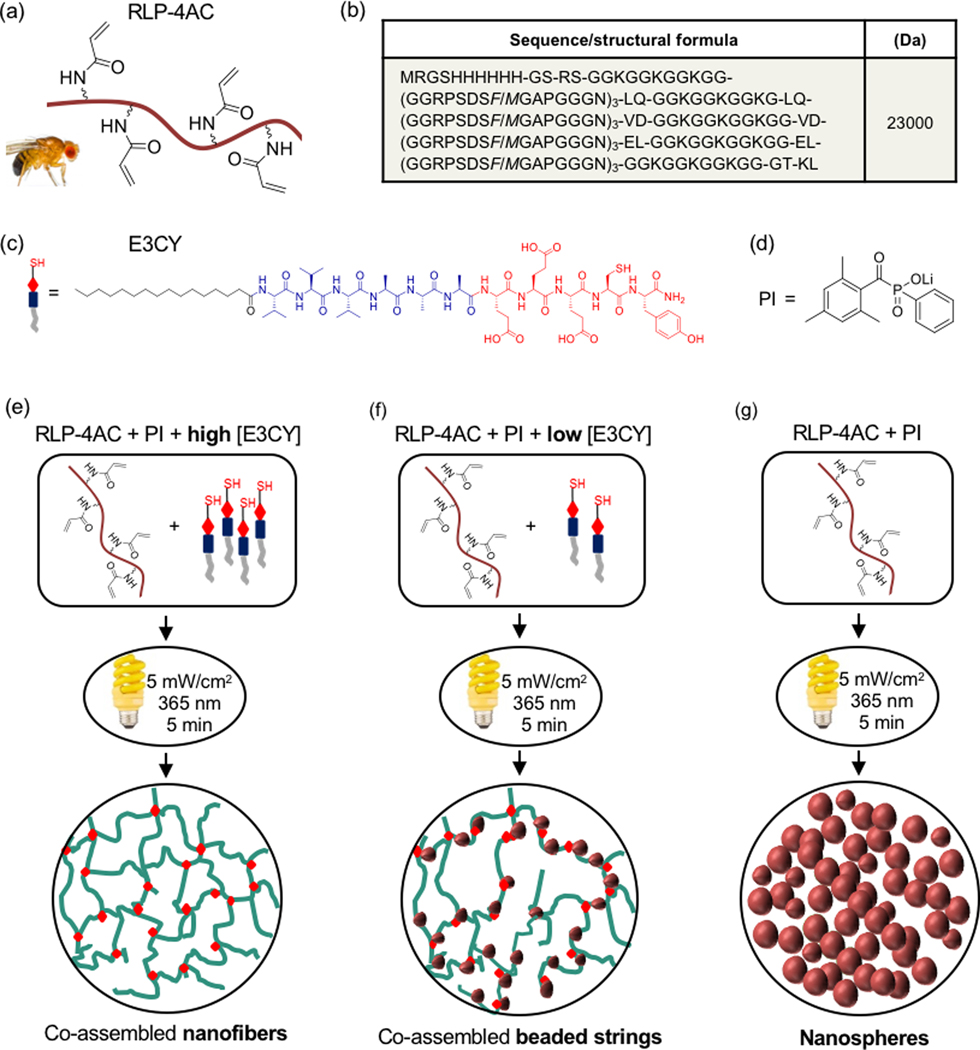
(a) Schematic representation of RLP structure functionalized with four acrylamide moieties, (b) amino acid sequence of RLP, (c) structure of E3CY peptide amphiphile and (d) structure of the photoinitiator (PI) used. Schematic representation of photopolymerization of (e) RLP-4AC and high concentration of E3CY (f) RLP-4AC and low concentration of E3CY and (g) RLP-4AC to generate nanostructures co-assembled hydrogels with nanofibers and beaded strings and, single-component hydrogels with nanospheres nanostructures.
Formation of hydrogels by covalent self-assembly
In order to synthesize co-assembled hydrogels of RLP-4AC (20 wt %, pH 8) and E3CY (1 and 2 wt %, pH 8), we first attempted simple mixing of both components such that the effective concentrations of RLP-4AC and E3CY were 10 and 0.5 or 1 wt %, respectively. After incubating the mixture of both components in phosphate saline buffer (PBS) at 37 °C for 6 h, formation of cloudy colloids or partial hydrogels was observed, suggesting an interaction (thiol-Michael addition reaction) between RLP-4AC and E3CY. Given that the zeta potential (ζ) values of RLP-4AC and E3CY are +0.57 and −30 mV, respectively, we do not expect a significant electrostatic interaction to occur between the two components. To verify this assumption, we mixed RLP-4AC with an E3Y analogue without the cysteine amino acid residue. However, the solution remains transparent after 6 h incubation. Raman spectroscopy was used to confirm the interaction between RLP-4AC and E3CY. The Raman spectrum of E3CY exhibited a peak at 2570 cm−1 due to the S-H vibrational stretching frequency (υSH) of the cysteine residue (Supplemental information, Fig. S1). In addition to this peak, bands were observed at 2930 cm−1 due to C-H stretching (υCH), 1737 cm−1 due to C=O, 1495 cm−1 due to C=C (aromatic), 1422 cm−1 and 1217 cm−1 due to CH2, and 1050 cm−1 due to C-O. In the Raman spectrum of RLP-4AC, a peak corresponding to C=C stretching (υC=C) of an acryloyl group was expected to be apparent at ~1632 cm−1 but was not visible. We assume that this is due to overlapping within the broad peak between 1570 and 1736 cm−1, which corresponds to multiple functional groups (C=C, C=O and CH2=COOR). Raman spectra of the incubated RLP-4AC (20 wt %) and E3CY (2 wt %) mixtures (see RLP_E3CY@ 37 °C) did not show a band typical of S-H, indicating that the thiol-Michael addition reaction mediated the partial hydrogel formation. Traditionally, thiol-Michael addition reactions are base catalysed and reaction kinetics and yield of the thioether product depend on a number of factors such as the strength of a base catalyst, steric accessibility of the thiol group, and nature of the electron withdrawing moiety conjugated to the ene group.30 Therefore, we reason that one or more of these factors might be responsible for the inability to form robust hydrogels between RLP-4AC and E3CY with this approach.
To further promote the formation of new covalent bonds in our system, we employed a photoinitiated polymerization approach to support the co-assembly of RLP-4AC and E3CY into self-supporting hydrogels. The thiol-ene photoclick reaction (for ~5 min) between the acrylamide moiety of RLP-4AC (20 wt %) and the thiol group of E3CY (2 wt %) using long wavelength UV light (5 mW/cm2 at 365 nm) and the photoinitiator (PI, lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP)) produced flexible, yet self-supporting hydrogels (Fig. 2). The co-assembled hydrogels prepared with 1 and 2 wt % E3CY (which corresponds to 1:1 and 1:2 ene to thiol ratios) were opaque (Fig. 2a,b). Similarly, free radical polymerization of the acrylamide moiety of pure RLP-4AC (10 wt %) using similar initiation conditions also yielded flexible hydrogels (Fig. 2c). Unlike the co-assembled hydrogels, the RLP-4AC hydrogels were less opaque, suggesting that the RLP-4AC and the co-assembled RLP-4AC_E3CY hydrogels might have distinct internal nanostructures. In addition, we used dynamic time-sweep rheology to monitor the evolution of storage (G’) and loss (G”) moduli as evidence of in situ gelation for both RLP-4AC and the RLP-4AC_E3CY co-assemblies. The results confirmed that gelation reached a plateau within 45 s in both cases (Fig. 2e), indicating that the cross-linking of the two-component system is as rapid as that of the pure RLP-4AC. The observed increased G’ and G” values of RLP-4AC_E3CY in the time sweep rheographs than in the time sweep rheograph of RLP-4AC alone shows that there was an initial formation of a viscous material upon mixing the RLP-4AC and E3CY and prior the photoinitiation. As a control, hydrogels of pure E3CY were synthesized by using a metal coordination strategy with CaCl2 (0.5 mM). Unlike the RLP-4AC and RLP-4AC_E3CY hydrogels, the E3CY (1 wt %) formed transparent but weak hydrogels (Fig. 2d) within a few seconds.
Figure 2.
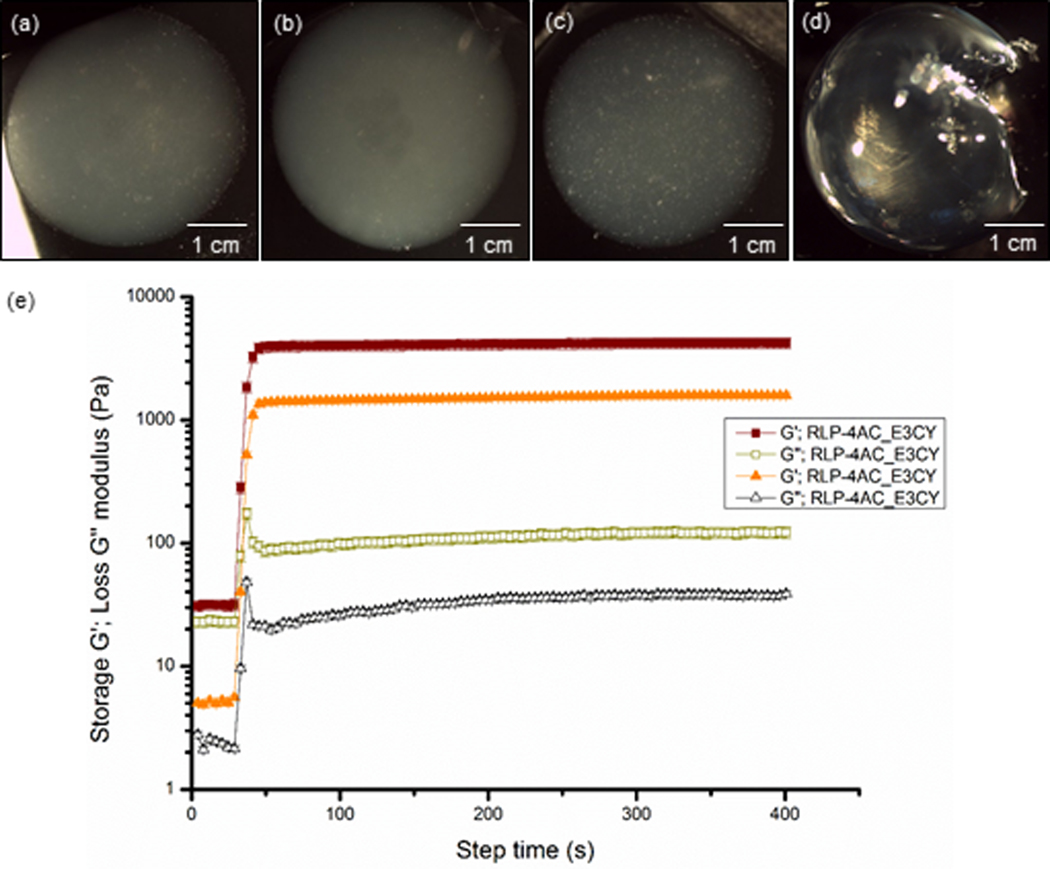
Optical image of hydrogels prepared with (a) thiol-ene photoclicked RLP-4AC (20 wt %)_E3CY (1 wt % ), (b) RLP-4AC (20 wt %)_E3CY (2 wt %), (c) photopolymerized RLP-4AC (10 wt %) and (d) E3CY- Ca2+ (1 wt %). [Scale bar: 1 cm]. (e) Time sweep rheographs showing evolution of G’ and G”, confirming in situ hydrogel formation with 2 wt % E3CY.
Again, we used Raman spectroscopy to confirm that the thiol-ene photoclick reaction between RLP-4AC and E3CY underpins co-assembly of both components (Supplementary Information, Fig, S1). The co-assembled systems exhibit distinct Raman spectra from those of the individual components. Similar to the Raman spectra of the incubated RLP-4AC_E3CY@37oC mixture, Raman spectra of the thiol-ene photoclicked hydrogels (see RLP-4AC_E3CY photoclicked) did not exhibit any band typical of S-H, suggesting the observed hydrogel formation was not mainly due to homopolymerization of RLP-4AC.
Hydrogel co-assembly, structure, and properties
The nanostructures of RLP-4AC_E3CY hydrogels prepared with and without photoinitiation were investigated using transmission electron microscopy in order to elucidate possible differences in underlying structures. For all samples, 0.5 wt % of RLP-4AC and 0.05 wt % of E3CY were used so as to reproduce the same acrylamide:thiol ratio used in the preparation of the macroscopic hydrogels (Fig. 2a). First, we incubated a mixture of RLP-4AC and E3CY at 37 °C for 6 h prior to TEM imaging. As shown in Fig. 3a and 3ai, the co-assembled RLP-4AC_E3CY samples exhibited bundles of aligned nanofibers of several microns in length.
Figure 3.

TEM image of (a) mixture of E3CY (0.05 wt %) and RLP-4AC (0.5 wt %) reacted for 6 h, (ai) higher magnification of (a), (b) photo-crosslinked RLP-4AC (0.5 wt %)_E3CY (0.05 wt %), (c) aqueous solution of RLP-4AC (0.25 wt %), (d) aqueous suspension of photo-crosslinked RLP-4AC (0.25 wt %), and (e) aqueous solution of E3CY (0.025 wt %).
In contrast, the RLP-4AC_E3CY hydrogels prepared by thiol-ene photoclick reaction formed nanobeads with diameters ranging between 10 and 50 nm and some short nanofibers (≤ 100 nm in length) (Fig. 3b). To provide insight into the mechanism of hydrogel formation, we also acquired TEM images of both components on their own. First, RLP-4AC solutions alone (0.25 wt %) comprised disordered aggregates (Fig. 3c), which transformed into nanospheres with diameters ranging between 10 and 30 nm upon photopolymerization (Fig. 3d). This nanosphere formation is reminiscent of the microspheres reported by Kiick and co-workers visualizing temperature-triggered phase transitions of a RLP.31 We also confirmed in our systems that the disordered aggregates observed in RLP-4AC solutions were not present in the TEM images of the co-assembled systems as well as the photopolymerized pure RLP-4AC. These results suggest a conformational transformation of RLP-4AC to ordered nanostructures as a result of both their co-assembly and photopolymerization. Furthermore, E3CY (0.025 wt %) molecules assembled into the expected PA cylindrical micelles that were ~500 nm in length and ~10 nm in diameter (Fig. 3e). The difference in morphology between these nanofibers and those from the co-assembled system suggests that the incubation-induced (thiol-Michael addition reaction) co-assembly of E3CY with RLP-4AC facilitates the formation of elongated, aligned, and bundled nanofibers. It is worth mentioning that E3CY retains its nanofiber morphology after irradiation under the same experimental conditions, suggesting that the observed short nanofibers are not due to UV-induced fragmentation.
We used CD spectroscopy to explore the effect of co-assembly on the secondary structures of RLP-4AC and E3CY in the resulting hybrid structures prepared with and without the photoinitiated reaction. First, as expected, the CD spectrum of an aqueous solution of E3CY (0.05 wt %) alone depicts a negative band at 218 nm, which indicates a β-sheet conformation (Fig. 4a, blue diamond trace).24a On the other hand, the CD spectrum of an aqueous solution of RLP-4AC (0.25 wt %) alone resembles a random coil signature with negative peak at ~ 196 nm (Fig. 4a), suggesting that the modification with acrylamide moiety did not affect the intrinsic conformation of the protein. Upon mixing various concentrations of E3CY (0.025, 0.05, and 0.1 wt %) with a fixed concentration of RLP-4AC (0.5 wt %) and incubating for 6 h at 37 ˚C, the peak corresponding to the RLP-4AC random coil signature was not apparent in the spectra of the RLP-4AC_E3CY co-assembly (Fig. 4a). Moreover, a red shift (~4 nm) was observed in the absorption wavelength from 218 nm to 221 nm, especially, with 0.01 and 0.025 wt % of E3CY, indicating that there is an interaction between RLP-4AC and E3CY. However, with 0.05 wt % E3CY, spectrum of the co-assembled RLP-4AC_E3CY is dominated by the spectra features of E3CY. Put together, this result is also consistent with the morphological transformation revealed by TEM (Fig. 3). We also acquired the CD spectra of the photo-crosslinked systems. In this case, both the solutions of pure RLP-4AC and its mixtures with various concentrations of E3CY were not incubated prior photoinitiated polymerization. As shown in Fig. 4b-d (green triangular traces), the photopolymerized RLP-4AC (0.25 wt %) did not display the characteristic random coil signature of RLP-4AC at 196 nm but exhibited a new CD spectrum with the characteristic β-sheet signature. We also confirmed in the co-assembled RLP-4AC_E3CY systems synthesized via thiol-ene photoclick reaction that neither the random coil signature of RLP-4AC at 196 nm nor the β-sheet peak of E3CY at 218 nm were present, but the β-sheet-like spectra red-shifted to 227 nm while the intensity of the band increased with increased concentration (0.025, 0.05 and 0.1 wt %) of E3CY (Fig. 4a-c, brown circular traces). Previous studies have suggested that a red-shift in β-sheet spectra is indicative of a twisted and distorted arrangement of the β-sheets, which would result in weaker bonds between the PA molecules on the nanofiber surfaces.32 More so, an increase in the intensity of a β-sheet-like spectra is representative of longer nanofibers or high-order assemblies.32b, 33 Therefore, we reasoned that the covalent interactions between RLP-4AC and E3CY may impact the secondary structure of both components, which are critical in dictating the resulting nanostructures and corresponding bulk properties of the co-assembled RLP-4AC_E3CY hydrogels.
Figure 4.

Circular dichroism spectra for (a) E3CY (blue diamond trace), RLP-4AC (green open circular trace), incubated mixture of RLP-4AC with 0.01 (brown triangular trace), 0.025 (brown diamond trace) and 0.05 wt % (brown open square trace) of E3CY. (b)-(d) CD spectra for E3CY (blue square-line trace), photocrosslinked RLP-4AC (green triangular trace) and co-assembled RLP-4AC_E3CY by thiol-ene photoclick reaction with 0.025, 0.05 and 0.1 wt % of E3CY (brown circular traces). Total concentration of RLP-4AC in all cases was 0.25 wt %.
Nano and microstructure of the co-assembled hydrogel
Unlike the TEM images, which were obtained from diluted systems, we used scanning electron microscopy (SEM) to characterize the nanostructures of the macroscopic co-assembled RLP-4AC_E3CY hydrogels synthesized by thiol-ene photoclick reaction between RLP-4AC (20 wt %) and E3CY (1 and 2 wt %). The results revealed that the co-assembled nanostructures transitioned from “beaded strings” when constructed with 1 wt % E3CY (Fig. 5a) to nanofibers when prepared with 2 wt % E3CY (Fig. 5b). In contrast, photopolymerized hydrogels of pure RLP-4AC exhibited nanospheres having diameters between 50 and 90 nm (Fig. 5c) while hydrogels of metal coordinated E3CY presented nanofibrous networks similar to classical PA hydrogels (Fig. 5d). Put together, these findings show how a PA molecule can be used to engineer the nanostructure of a protein-based hydrogel in a facile, one-pot, and concentration-dependent manner. This type of hydrogel microstructure tunability from spheres to “beaded-string” has recently been reported by Heilshorn and co-workers using controlled chemical crosslinking and physical coacervation of an elastin-like polypeptide (ELP).34 The authors adduced the underlying mechanism for their tunable hydrogel microstructures to the chain mobility and thermo-responsiveness of the ELP. We have previously demonstrated how PA molecules can be used to facilitate spatio-temporal control of ELP molecules towards the supramolecular fabrication of dynamic tubular macrostructures.27 Taking inspiration from this, we hypothesized that the structural tunability observed in the current study is due to the ability of the rigid PA nanofibers to facilitate spatio (and perhaps temporal) control of the chain mobility and disordered conformation of RLP-4AC in the RLP-4AC_E3CY hydrogels.
Figure 5.

SEM image of xerogels of (a) thiol-ene photoclicked RLP-4AC (20 wt %)_E3CY (1 wt %), (b) thiol-ene photoclicked RLP-4AC (20 wt %)_E3CY (2 wt %), (c) photo-crosslinked RLP-4AC (10 wt %) and (d) E3CY- Ca2+ (1 wt %).
Mechanical properties of the co-assembled hydrogel
To confirm the impact of the proposed structural modification on the mechanical properties of the co-assembled RLP-4AC_E3CY hydrogels, dynamic amplitude sweep rheology at 1 Hz frequency and 25 °C was performed on the preformed hydrogels (Fig. 6). The rheological data demonstrated that G’/G” > 10 in all the rheographs, suggesting that solid-like hydrogels were formed by the co-assembled system as well as control hydrogels, regardless of their nanostructures and conformations. The G’ values for the control E3CY (1 wt %) (prepared with CaCl2) and RLP-4AC (10 wt %) (photo-crosslinked) hydrogels were 0.8 kPa and 1.6 kPa, respectively (Fig 6a). In contrast, the co-assembled RLP-4AC_E3CY hydrogels (thiol-ene photoclicked) exhibited a G’ value of ~ 4.5 kPa, which suggests the predominance of covalent interaction in the co-assembling system. In addition, RLP-4AC hydrogels exhibited linear stress-strain behaviours up to ~250% strain whereas E3CY could only maintain linear stress-strain behaviours until 2.5% strain. In comparison, the co-assembled RLP-4AC_E3CY hydrogels behaved similarly to the RLP-4AC hydrogels, maintaining linear stress-strain characteristics until ~250% critical strain and withstanding strains up to 850%. Additionally, the loss angle, δ, which gives a relative measure of viscous to elastic properties in a material (δ = 0˚ indicates an elastic solid and δ = 90˚ indicates a Newtonian viscous fluid) < 1˚ for both RLP-4AC and RLP-4AC_E3CY hydrogels until 1000% strain whereas, E3CY hydrogels only exhibit δ < 1˚ until 2.5% (Fig. 6b). This data demonstrates that the stiffness of the co-assembled RLP-4AC_E3CY hydrogels is more than the sum of the stiffness of the individual components, suggesting that the co-assembled RLP-4AC_E3CY hydrogels have a more rigid internal nanostructures than the hydrogels of the individual components. The observed critical strain values show that E3CY hydrogels are extremely brittle while both RLP-4AC and the co-assembled RLP-4AC_E3CY hydrogels are highly elastic and energy-storing solids. Such magnitude of extensibility has previously been reported by Kiick and co-workers with another variant of RLP.17a It is worth noting that a slight strain stiffening effect was observed in the strain-sweep rheographs of RLP-4AC and RLP-4AC_E3CY hydrogels before failure. However, this effect was more prolonged in the rheographs of RLP-4AC hydrogels, perhaps due to the complex internal nanostructuring of RLP-4AC_E3CY compared to RLP-4AC hydrogels. Also, an electrostatic interaction-driven co-assembly between an unmodified RLP (20 wt %) analogue and E3CY (2 wt %) at pH 7.5 produced weak hydrogels (G’ ~ 800 Pa) with lower critical strain value (10%) (Fig. 6a) than the photopolymerized RLP-4AC and photoclicked RLP-4AC_E3CY, but higher than the critical strain value (2.5%) for E3CY-Ca2+ hydrogels. Thus, lending support to the importance of covalent interactions in designing practical multicomponent hydrogels with improved mechanical properties.
Figure 6.

(a) Amplitude sweep rheographs for the photopolymerized RLP-4AC (10 wt %), E3CY- Ca2+ (1 wt %), thiol-ene photoclicked RLP-4AC (20 wt %)_E3CY (2 wt %), and RLP (20 wt %)_E3CY (2 wt %). (b) Tan (δ) versus strain rheographs for the photopolymerized RLP-4AC (10 wt %), E3CY-Ca2+ (1 wt %) and thiol-ene photoclicked RLP-4AC (20 wt %)_E3CY (2 wt %), and RLP (20 wt %)_E3CY (2 wt %). Step-strain measurements with applied oscillatory strain alternated between 10 and 1000% for 100, 200, 200, 200 and 400 s (f = 1 Hz, 20 °C) on (c) photopolymerized RLP-4AC and (d) photoclicked RLP-4AC_E3CY hydrogels.
In an effort to assess reversibility and self-healing behaviour of the covalent hydrogels, we carried out a step strain measurement where alternating strain of 10 (within the linear visco-elastic regions of the hydrogels) and 1000% (outside the linear visco-elastic regions of the hydrogels) were applied at various time intervals and 3 cycles. Our results revealed that photopolymerized RLP-4AC hydrogels displayed a ~ 60 and 50% recovery after the first and second strain cycles, respectively (Fig. 6c). On the other hand, photoclicked RLP-4AC_E3CY exhibited 75% recovery after the first and second strain cycles (Fig. 6d). It is important to mention that although our co-assembled RLP-4AC_E3CY hydrogels were unable to exhibit 100% recovery after the first cycle under such extremely high strain, 100% recovery to the preceding G’ values was attained after the second cycle. Under high strain (1000%), both photopolymerized RLP-4AC and photoclicked RLP-4AC_E3CY hydrogels underwent internal breakage as indicated by the significant decrease of G′ and G″ values. While there was an inversion of G’ and G” graphs in the case of RLP-4AC hydrogels, which means that the liquid-like behaviour of the hydrogels dominates under high strain, no inversion was observed with RLP-4AC_E3CY hydrogels. We reasoned that the different performance of these two types of photosynthetic hydrogels might be due to the difference in their internal nanostructures. We envisioned that the photoclicked RLP-4AC_E3CY hydrogels resistance to G’-G” inversion arises from energy dissipative mechanisms provided by the complex and nanofibrous morphology, as against the RLP-4AC single-component nanospheres. It is noteworthy that hydrogels (both covalent and non-covalently synthesized) that can exhibit a significant degree of recovery under this magnitude of high strain amplitude (1000 %) are rare.35 Both polymerization and thiol-ene photoclick reaction of RLP-4AC with E3CY can alter the intrinsic random coil conformation of RLP-4AC. However, such alteration did not hinder its chain mobility, which is the underlying factor for the exceptional elasticity and self-healing property exhibited by our RLP-based hydrogels.
Conclusion
We have demonstrated the potential to combine self-assembly with covalent-crosslinking as a strategy to develop hydrogel materials with enhanced complexity and functionality in a PA concentration-dependent manner. This approach opens the possibility to combine the inherent properties of PAs with those of RLPs more broadly. The hydrogels exhibit properties such as tunable conformation, nano/microstructures, self-healing and high extensibility, which resulted from covalent interactions between the two molecular building blocks. We also established that the high elasticity of the co-assembled hydrogels can be attributed to the retention of the RLP’s chain mobility in the hydrogels. These properties could open opportunities in applications such as the fabrication of tissue engineering scaffolds or sustained drug release systems.
Supplementary Material
Acknowledgments
The work was supported by the ERC Starting Grant (STROFUNSCAFF) and the UK Regenerative Medicine Platform (UKRMP2) Acellular/Smart Materials. The authors gratefully acknowledge funding support from the National Institute on Deafness and Other Communication Disorders (RO1 DC011377A) and from the National Science Foundation (DMR-1609544). Instrument resources from the Delaware COBRE program were supported by grants from the National Institute of General Medical Sciences (1-P30-GM110758-01 and 1-P20-RR017716). The views represented do not necessarily reflect those of the National Science Foundation nor the National Institutes of Health. We thank Dr Giulia Mastroianni in the School of Biological and Chemical Sciences, Queen Mary University of London, for technical supports.
Footnotes
Conflicts of interest
We wish to confirm that there are no known conflicts of interest associated with this publication.
References
- 1.(a) Sang Y. and Liu M, Mol. Syst. Des. Eng, 2019, 4, 11–28; [Google Scholar]; (b) Okesola BO, Wu Y, Derkus B, Gani S, Wu D, Knani D, Smith DK, Adams DJ and Mata A, Chem. Mater, 2019, 31, 7883–7897; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hirst AR, Escuder B, Miravet JF and Smith DK, Angew. Chem. Int. Ed, 2008, 47, 8002–8018. [DOI] [PubMed] [Google Scholar]
- 2.(a) Luo Q, Hou C, Bai Y, Wang R. and Liu J, Chem. Rev, 2016, 116, 13571–13632; [DOI] [PubMed] [Google Scholar]; (b) Tominaga M, Suzuki K, Kawano M, Kusukawa T, Ozeki T, Sakamoto S, Yamaguchi K. and Fujita M, Angew. Chem. Int. Ed, 2004, 43, 5621–5625. [DOI] [PubMed] [Google Scholar]
- 3.Baek K, Hwang I, Roy I, Shetty D. and Kim K, Acc. Chem. Res, 2015, 48, 2221–2229. [DOI] [PubMed] [Google Scholar]
- 4.(a) Cornwell DJ and Smith DK, Materials Horizons, 2015, 2, 279–293; [Google Scholar]; (b) Abul-Haija YM and Ulijn RV, Biomacromolecules, 2015, 16, 3473–3479; [DOI] [PubMed] [Google Scholar]; (c) Noteborn WEM, Zwagerman DNH, Talens VS, Maity C, van der Mee L, Poolman JM, Mytnyk S, van Esch JH, Kros A, Eelkema R. and Kieltyka RE, Adv. Mater, 2017, 29, 1603769; [DOI] [PubMed] [Google Scholar]; (d) Cornwell DJ, Okesola BO and Smith DK, Angew. Chem. Int. Ed, 2014, 53, 12461–12465. [DOI] [PubMed] [Google Scholar]
- 5.Okesola BO and Mata A, Chem. Soc. Rev, 2018, 47, 3721–3736. [DOI] [PubMed] [Google Scholar]
- 6.Annabi N, Shin SR, Tamayol A, Miscuglio M, Bakooshli MA, Assmann A, Mostafalu P, Sun J-Y, Mithieux S, Cheung L, Tang X, Weiss AS and Khademhosseini A, Adv. Mater, 2016, 28, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandal BB, Grinberg A, Seok Gil E, Panilaitis B. and Kaplan DL, Proc. Natl. Acad. Sci. U S A, 2012, 109, 7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galland R, Leduc P, Guérin C, Peyrade D, Blanchoin L. and Théry M, Nat. Mater, 2013, 12, 416. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki Y, Cardone G, Restrepo D, Zavattieri PD, Baker TS and Tezcan FA, Nature, 2016, 533, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Micol LA, Ananta M, Engelhardt E-M, Mudera VC, Brown RA, Hubbell JA and Frey P, Biomaterials, 2011, 32, 1543–1548; [DOI] [PubMed] [Google Scholar]; (b) Hamley IW, Chem. Rev, 2017, 117, 14015–14041. [DOI] [PubMed] [Google Scholar]
- 11.(a) Burrows M, BMC Biology, 2009, 7, 27; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qin G, Hu X, Cebe P. and Kaplan DL, Nat. Commun, 2012, 3, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanami M, Shtein Z, Sweeney I, Sorushanova A, Rivkin A, Miraftab M, Shoseyov O, O’Dowd C, Mullen AM, Pandit A. and Zeugolis DI, Biomedical Materials, 2015, 10, 065005. [DOI] [PubMed] [Google Scholar]
- 13.(a) Burrows M, Shaw SR and Sutton GP, BMC Biology, 2008, 6, 41; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burrows M. and Sutton GP, The Journal of Experimental Biology, 2012, 215, 3501. [DOI] [PubMed] [Google Scholar]
- 14.Balu R, Whittaker J, Dutta NK, Elvin CM and Choudhury NR, J. Mater. Chem. B, 2014, 2, 5936–5947. [DOI] [PubMed] [Google Scholar]
- 15.Li L. and Kiick KL, ACS Macro Letters, 2013, 2, 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balu R, Bourgeois L, Elvin CM, Hill AJ, Choudhury NR and Dutta NK, J. Mater. Chem. B, 2015, 3, 6580–6586. [DOI] [PubMed] [Google Scholar]
- 17.(a) Charati MB, Ifkovits JL, Burdick JA, Linhardt JG and Kiick KL, Soft Matter, 2009, 5, 3412–3416; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McGann CL, Levenson EA and Kiick KL, Macromolecular Chemistry and Physics, 2013, 214, 203–213; [Google Scholar]; (c) Li L, Tong Z, Jia X. and Kiick KL, Soft Matter, 2013, 9, 665–673; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Su RSC, Kim Y. and Liu JC, Acta Biomaterialia, 2014, 10, 1601–1611. [DOI] [PubMed] [Google Scholar]
- 18.Mayavan S, Dutta NK, Choudhury NR, Kim M, Elvin CM and Hill AJ, Biomaterials, 2011, 32, 2786–2796. [DOI] [PubMed] [Google Scholar]
- 19.McGann CL, Akins RE and Kiick KL, Biomacromolecules, 2016, 17, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Lau HK, Li L, Jurusik AK, Sabanayagam CR and Kiick KL, ACS Biomaterials Science & Engineering, 2017, 3, 757–766; [DOI] [PubMed] [Google Scholar]; (b) Lau HK, Paul A, Sidhu I, Li L, Sabanayagam CR, Parekh SH and Kiick KL, Advanced Science, 2018, 5, 1701010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGann CL, Dumm RE, Jurusik AK, Sidhu I. and Kiick KL, Macromolecular Bioscience, 2016, 16, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bracalello A, Santopietro V, Vassalli M, Marletta G, Del Gaudio R, Bochicchio B. and Pepe A, Biomacromolecules, 2011, 12, 2957–2965. [DOI] [PubMed] [Google Scholar]
- 23.Huang S-C, Qian Z-G, Dan A-H, Hu X, Zhou M-L and Xia X-X, ACS Biomaterials Science & Engineering, 2017, 3, 1576–1585. [DOI] [PubMed] [Google Scholar]
- 24.(a) Hendricks MP, Sato K, Palmer LC and Stupp SI, Acc. Chem. Res, 2017, 50, 2440–2448; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mata A, Palmer L, Tejeda-Montes E. and Stupp SI, in Nanotechnology in Regenerative Medicine: Methods and Protocols, eds. Navarro M. and Planell JA, Humana Press, Totowa, NJ, 2012, DOI: 10.1007/978-1-61779-388-2_3, pp. 39–49. [DOI] [Google Scholar]
- 25.Capito RM, Azevedo HS, Velichko YS, Mata A. and Stupp SI, Science, 2008, 319, 1812. [DOI] [PubMed] [Google Scholar]
- 26.Hedegaard CL, Collin EC, Redondo-Gómez C, Nguyen LTH, Ng KW, Castrejón-Pita AA, Castrejón-Pita JR and Mata A, Adv. Funct. Mater, DOI: 10.1002/adfm.201703716, 1703716-n/a. [DOI] [Google Scholar]
- 27.Inostroza-Brito KE, Collin E, Siton-Mendelson O, Smith KH, Monge-Marcet A, Ferreira DS, Rodríguez RP, Alonso M, Rodríguez-Cabello JC, Reis RL, Sagués F, Botto L, Bitton R, Azevedo HS and Mata A, Nat. Chem, 2015, 7, 897. [DOI] [PubMed] [Google Scholar]
- 28.Inostroza-Brito KE, Collin EC, Poliniewicz A, Elsharkawy S, Rice A, del Río Hernández AE, Xiao X, Rodríguez-Cabello J. and Mata A, Acta Biomaterialia, DOI: 10.1016/j.actbio.2017.05.043. [DOI] [PubMed] [Google Scholar]
- 29.(a) Goktas M, Cinar G, Orujalipoor I, Ide S, Tekinay AB and Guler MO, Biomacromolecules, 2015, 16, 1247–1258; [DOI] [PubMed] [Google Scholar]; (b) Boekhoven J, Zha RH, Tantakitti F, Zhuang E, Zandi R, Newcomb CJ and Stupp SI, RSC Adv., 2015, 5, 8753–8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR and Bowman CN, Chem. Mater, 2014, 26, 724–744. [Google Scholar]
- 31.Li L, Luo T. and Kiick KL, Macromolecular Rapid Communications, 2015, 36, 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Paramonov SE, Jun H-W and Hartgerink JD, J. Am. Chem. Soc, 2006, 128, 7291–7298; [DOI] [PubMed] [Google Scholar]; (b) Pashuck ET, Cui H. and Stupp SI, J. Am. Chem. Soc, 2010, 132, 6041–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke DE, Parmenter CDJ and Scherman OA, Angew. Chem. Int. Ed, 2018, 57, 7709–7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Paul A, Nguyen D, Enejder A. and Heilshorn SC, ACS Appl. Mater. Interfaces, 2018, 10, 21808–21815. [DOI] [PubMed] [Google Scholar]
- 35.Wu Y, Shah DU, Liu C, Yu Z, Liu J, Ren X, Rowland MJ, Abell C, Ramage MH and Scherman OA, Proc. Natl. Acad. Sci. U S A, 2017, DOI: 10.1073/pnas.1705380114, 201705380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.