

Abstract
Background:
Familial Atypical Multiple Mole Melanoma syndrome (FAMMM) is characterized by dysplastic nevi, malignant melanoma, and pancreatic cancer. Given that large deletions involving CDKN2A account for only 2% of cases, we describe a family that highlights the co-occurrence of both melanoma and neural system tumors to aid clinical recognition and propose a management strategy.
Methods:
A patient with multiple neurofibromas was referred with a provisional diagnosis of Neurofibromatosis type 1 (NF1). Prior molecular testing, though, had failed to identify an NF1 mutation by sequencing and MLPA. His family history was significant for multiple in situ/malignant melanomas at young ages and several different cancers reminiscent of an underlying syndrome. A search of the Familial Cancer Database, FaCD Online, highlighted several families with cutaneous melanoma and nervous system tumors who were subsequently identified to have large deletions spanning CDKN2A.
Results:
Although sequencing of CDKN2A and TP53 failed to identify a mutation, a heterozygous CDKN2A deletion was identified by targeted array CGH. Whole-genome oligonucleotide array CGH and SNP analysis identified an interstitial deletion of at least 1.5 Mb within 9p21.3 and spanning approximately 25 genes.
Conclusions:
Identification of the underlying molecular abnormality permits predictive testing for at-risk relatives. Given the young cancer diagnoses, a surveillance regimen was developed and a clinical team organized for ongoing management so that genetic testing could be offered to both adults and minor children. Surveillance recommendations addressed not only cancer risks associated with FAMMM, but also other cancers exhibited by this family with a large contiguous gene deletion.
Keywords: FAMMM, CDKN2A, melanoma, neurofibroma, contiguous gene deletion
INTRODUCTION
Familial Atypical Multiple Mole Melanoma syndrome (FAMMM) is an autosomal dominant cancer predisposition syndrome characterized by multiple dysplastic nevi, malignant melanoma, and, in some families, a potential increased risk for pancreatic cancer. This cancer predisposition syndrome has also been referred to as Dysplastic Nevus syndrome and represents one of several genetic etiologies underlying Familial Cutaneous Malignant Melanoma (FCMM).
The CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, localized to chromosome 9p21, is the major known high-risk susceptibility gene for FAMMM/FCMM. CDKN2A codes for two distinct tumor suppressor proteins, p16INK4A and p14ARF, which are transcribed using alternative first exons, 1α and 1β, and subsequently spliced onto the common exons 2 and 3, but in different reading frames. p16INK4A acts through the pRb pathway and functions normally to inhibit the kinase activity of CDK4. In contrast, p14ARF exerts its biological effects through the p53 pathway. Ultimately, mutations in the CDKN2A gene can cause loss of function of either or both proteins, and so each may contribute to the development of different types of cancer.[1]
Germline CDKN2A mutations have been identified in upwards of 15-40% of patients with FCMM, while an additional 1-2% of familial cases are due to mutations in the CDK4 gene.[2,3]. Recently, three additional high-risk susceptibility genes for familial melanoma were identified, specifically BAP1, TERT, and POT1, all of which are less frequent than CDK4.[4–7] As such, there are likely other genes, not yet identified, that account for the remaining families with a hereditary predisposition to melanoma.
CDKN2A mutations were first reported in kindreds with familial melanoma in 1994 with missense mutations representing the predominant type of mutation identified.[8–9] Nonsense mutations, as well as small deletions and insertions, have also been reported. Large deletions, though, involving 1 or more exons, account for only 2% of all mutations reported in the CDKN2A gene.[3, 10]
Of the small number of families worldwide that have been described with large deletions involving CDKN2A, some have exhibited a predisposition to both melanoma and nervous system tumors (NST), prompting several investigators to propose that this combination of tumors may represent a discrete syndrome.[11–14] Several case reports have specifically described these families as demonstrating tumors characteristic of both FAMMM and Neurofibromatosis type 1 (NF1).[14–17] For example, the family described by Bahuau et al. exhibited many tumors associated with NF1, including both neurofibroma and astrocytoma, as well as features characteristic of FAMMM such as multiple cutaneous malignant melanoma and dysplastic nevi.[14] The family reported by Bahuau et al., though, also exhibited some tumors characteristic of Neurofibromatosis type 2 (NF2) such as schwannomas and meningiomas. We report a family with a large, contiguous gene deletion involving chromosome 9p21.3, and extending beyond CDKN2A to include approximately 25 genes, with tumors characteristic of not only FAMMM and NF1, but several additional tumors including a primitive neuroectodermal tumor (PNET), chondrosarcoma, and leukemia. The constellation of tumors exhibited by this family necessitated the development of a tailored surveillance regimen for ongoing clinical management of both adults, as well as minor children, given the high prevalence of young cancer diagnoses across multiple generations.
METHODS
Patient and Family
A 52 year old Caucasian male with a provisional diagnosis of Neurofibromatosis Type 1 (NF1) was referred to the Penn State Hershey Cancer Genetics Program by his oncologist for cancer genetic counseling and testing. A review of his personal history was significant for a squamous papilloma below the left eye which was diagnosed at 40 years of age, a benign fibrous histiocytoma of the lower back at 43 years of age, and 1 junctional nevus of the right upper arm with moderate to focal severe atypia at 44 years of age. In addition, his personal history was significant for multiple, painful neurofibromas which were diagnosed at 49 years of age, consisting of 2 in the retroperitoneum and 1 excised from the sciatic nerve. Subsequent review of his electronic medical record revealed that he had previously pursued a clinical genetics consult which included testing for NF1. The NF1 gene was analyzed by long-range RT-PCR and sequencing using dye-terminator chemistry on an ABI PRISM capillary sequencer followed by multiplex ligation-dependent probe amplification (MLPA), the results of which were negative. Physical examination by a clinical geneticist documented the presence of multiple hyperpigmented nevi, but there was no evidence, besides the multiple nerve sheath tumors/neurofibromas, of other stigmata characteristic of NF1, such as Lisch nodules or café au lait spots. As such, the proband did not meet clinical diagnostic criteria for NF1.
A review of his family history was significant for multiple relatives, spanning 3 generations, with both in-situ and malignant cutaneous melanomas. His mother was diagnosed with a malignant melanoma at 14 years of age following which she died from metastatic disease at 29 years of age. In addition, his sister was diagnosed with melanoma in situ at 47 years of age and had a history of multiple nevi with atypia. The family history was also significant for a niece with melanoma in situ at 27 years of age. This niece had 4 additional nevi removed, one of which demonstrated slight atypia, and she had 1 lipoma. Of this niece’s 3 daughters, one had a Spitz tumor excised at 5 years of age, possibly representing a precursor lesion to melanoma, and another daughter had 5 lipomas. In addition, the proband’s family history was significant for his son who was diagnosed with a pontomedullary PNET tumor with leptomeningeal spread at 18 years of age, his brother with chondrosarcoma of the inguinal region at 25 years of age followed by the removal of 3 compound nevi at 27 years of age, and his brother’s son with leukemia which was diagnosed at 11 years of age. Other relatives with a history of cancer included his maternal grandmother with a reported diagnosis of cervical cancer at approximately 35 years of age and 4 great aunts and uncles, all with unspecified cancers under 55 years of age. See Figure 1.
Figure 1.
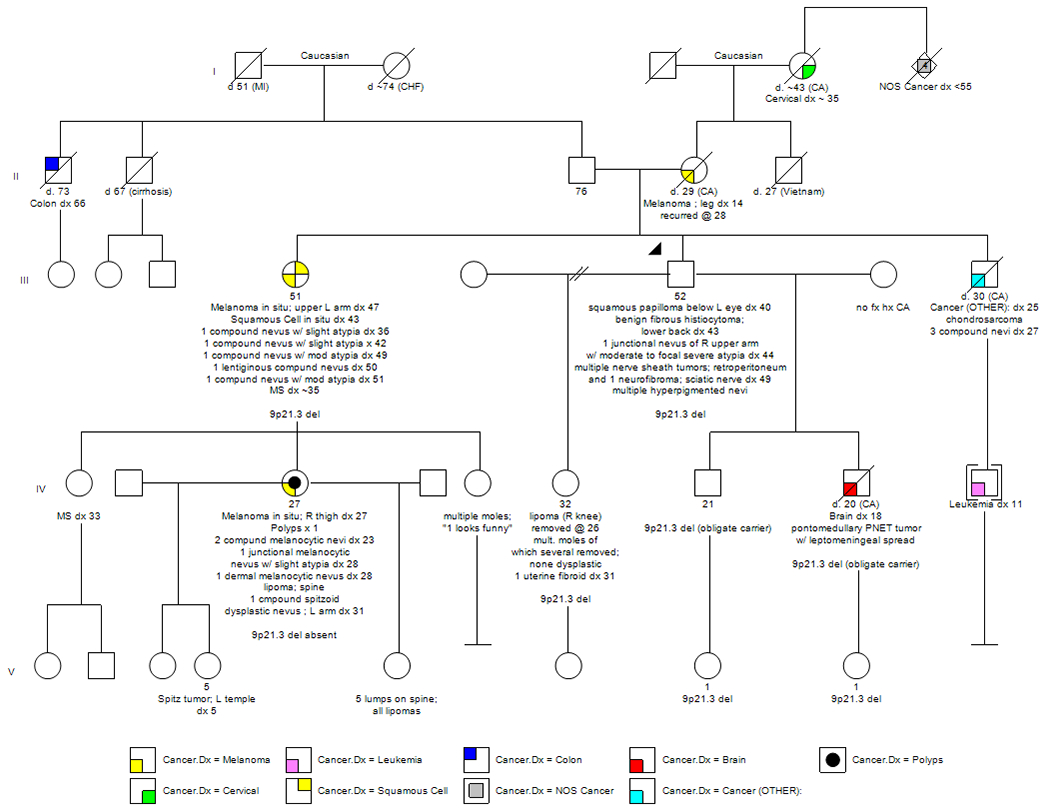
Family Pedigree
Genetic Testing
Given that prior analysis of the NF1 gene was negative by both sequencing and MLPA, the proband was offered testing of the CDKN2A gene, which, at the time, consisted of sequencing only, to address the family history of multiple in situ and malignant melanomas and multiple atypical nevi. Amplified DNA products were sequenced in forward and reverse directions using fluorescent dye-labeled sequencing primers with chromatographic tracings of each amplicon analyzed by a proprietary computer-based review followed by visual inspection and confirmation. In addition, he was offered both sequencing and MLPA analysis of the TP53 gene based on his family history of the PNET tumor, chondrosarcoma, and leukemia, with mutations in this gene responsible for Li-Fraumeni syndrome. Amplified DNA was analyzed by direct DNA sequence analysis on an automated fluorescent sequencer with sequencing of the entire coding region and associated splice junctions performed in both directions. MLPA products were analyzed by DNA fragment analysis on an automated fluorescence sequencer with the absence or presence of deletions/duplications of one or more exons confirmed by MLPA analysis using an independently amplified segment. A search of the Familial Cancer Database, FaCD Online revealed the identification of a small number of reported families worldwide with cutaneous malignant melanoma in the presence of nervous system tumors, features reminiscent of both FAMMM and NF1, in the context of large deletions within and extending beyond CDKN2A that would not be detected by standard sequencing methods.[18–19] As a result, the proband was also offered large rearrangement analysis of the CDKN2A gene via array-based comparative genomic hybridization (aCGH), with the array containing multiple oligonucleotide probes in most exons and/or their flanking intronic regions, through another reference laboratory, if Sanger sequencing proved negative. Hybridization data were analyzed with Genomic Workbench v5 software (Agilent Technologies) to evaluate copy number at the exon level. Targeted exon-level array CGH was followed by whole-genome ologonucleotide array CGH and SNP analysis with the array design based on human genome build GRCh37/hg19.
RESULTS
Genomic DNA was isolated from the proband’s peripheral blood specimen. Subsequent sequencing, though, of the CDKN2A gene, including all exons and adjacent intronic regions, failed to identify a deleterious mutation underlying his personal and family history of cancer. Genetic testing also proved negative for a possible TP53 mutation following both sequencing and MLPA analysis. Targeted array CGH of the CDKN2A gene with exon-level resolution revealed a heterozygous deletion. Although this molecular result was informative for FCMM, the extent of the deletion beyond CDKN2A could not be determined by this assay alone. Subsequent whole-genome oligonucleotide array CGH and SNP analysis identified an interstitial deletion of at least 1.5 Mb within cytogenetic band 9p21.3 with sequence coordinates of chr9:20,951,885-22,447,709 [hg19]. The deleted interval was found to include approximately 25 genes, of which only one, CDKN2A, was known to be associated with a cancer predisposition syndrome, specifically FCMM. See Figures 2A and 2B. Additionally, the proband was identified to have 2 copy number variants of unknown clinical significance: a duplication of at least 229 kb within cytogenetic band 5q35.2 with the duplicated interval including the TSPAN17 and EIF4E1B genes and part of the SNCB and UNC5A genes. The clinical consequence of carrying 3 copies of the SNCB gene, of which missense mutations have been described in 2 unrelated patients with dementia and lewy bodies, or of any of the other genes in this duplicated region has not yet been determined.[11] Further, this region has not been reported to vary in copy number in the normal population.[20] Lastly, the proband was found to have an amplification, consisting of 4 copies of at least 505 kb, in 10q24.32-q24.33. The amplified interval included 10 genes, none of which have been associated with clinical disorders to date.[11] This region in its entirety has not been reported to vary in copy number in the normal population.[20] In summary, the reference laboratory reported the molecular results as arr 5q35.2(176,052,444-176,281,813)x3,9p21.3(20,951,885-22,447,709)x1,10q24.32q24.33(104,853,173-105,357,653)x4 sex: male.
Figure 2.

Array CGH Data. Figure 2A shows the location of the deletion within cytogenetic band 9p21.3. Figure 2B details the approximate 25 genes encompassed within the deleted region as identified in the proband. The green dots represent individual probe locations that are deleted in the proband compared to the reference DNA, based on the relative intensity of the signal. The probe locations are mapped in comparison to the genes in the genomic region. Red dots indicate probe locations whose intensity is increased in the proband relative to the reference DNA. Single probe deviations, whether red or green, represent hybridization noise. Images provided by GeneDx.
Additional relatives tested to date include the proband’s sister with multiple primary melanomas, an unaffected daughter, and 2 unaffected granddaughters. Each of these relatives tested positive for the large interstitial deletion and, in the process, confirmed that both of the proband’s sons were obligate carriers, one of whom was diagnosed with a pontomedullary PNET tumor at 18 years of age. Most recently, the proband’s niece with multiple primary melanomas tested negative for the contiguous gene deletion. This observation adds to the complexity of the family and raises the possibility that the constellation of cancers may result from multiple underlying genetic causes. Alternatively, the niece with melanoma, as well as other relatives, may be at increased risk for melanoma due to the presence of other familial risk factors.
DISCUSSION
We report a family with a large deletion of chromosome 9p21.3, which spans approximately 25 genes and includes CDKN2A. To date, large rearrangement analysis of the CDKN2A gene has not routinely been offered by clinical reference laboratories when CDKN2A is ordered as a standalone test, since large deletions represent only 2% of CDKN2A mutations.[3, 10]. Further, the proband presented for evaluation prior to the clinical availability of multiplex panels in which multiple cancer predisposition genes are analyzed by next generation sequencing, thus routinely permitting the detection of large rearrangements. The molecular etiology underlying this family’s history of tumors was aided by the availability of an online resource called the Familial Cancer Database, which is accessible at www.facd.info, and was primarily developed as a tool to assist healthcare providers in developing a differential diagnosis based on the constellation of tumors and non-tumor features within a family.[18–19]
CDKN2A-Associated Cancer Spectrum
The identification of a large deletion encompassing CDKN2A confirmed a genetic predisposition to melanoma in the proband and a number of his at-risk relatives. In the context of FAMMM/FCMM, lifetime risk estimates for melanoma vary widely, with penetrance estimates ranging from a low of 28% by 80 years of age to a high of 58%-92%, depending on the study design.[21–25] The Melanoma Genetics Consortium, GenoMEL, also found a statistically significant effect when families lived in a geographic area with a high population incidence of melanoma.[26] They concluded that the risk factors which influence the population incidence of melanoma may also mediate the penetrance of CDKN2A mutations.
Pancreatic cancer has also been associated with mutations in the CDKN2A gene with one study estimating a 17% risk to age 75.[27–29] Not all families, though, with a CDKN2A mutation demonstrate an increased risk of pancreatic cancer. Although previous studies have suggested that the development of pancreatic cancer may depend on whether the specific mutation identified impairs the function of the p14ARF protein in addition to p16, definitive evidence for this relationship has not yet been shown.
The melanoma-astrocytoma syndrome, first described in 1993, represents another phenotype postulated to be related to mutations in the CDKN2A gene.[12] Since then, additional studies of families with melanoma have documented the co-occurrence of various neural system tumors, as seen in the proband’s family, including rare solitary internal neurofibromas, as well as cutaneous neurofibromas.[13, 30] Lynch et al also documented the association of sarcoma with malignant melanoma in 2 kindreds with a CDKN2A mutation.[31] Again, however, it has not yet been possible to clearly determine the underlying cause(s) of these rarely co-occurring tumors.
Genotype/Phenotype Correlations with Large CDKN2A Deletions
Although large germline deletions of CDKN2A have only been described in a limited number of families worldwide, the breakpoints of the deletions described thus far and their impact on the function of the gene’s 2 alternative transcripts, p16INK4A and p14ARF, have begun to shed light on the underlying mechanism predisposing to the observed constellation of tumors, including dysplastic nevi, melanoma, and neural system tumors. The first reported individual to carry a large deletion involving part of chromosome 9p, as a result of an unbalanced chromosomal translocation, developed multiple melanomas and a plexiform neurofibroma.[16] It was estimated that the deletion in this patient spanned at least 6 megabases and involved CDKN2A, ARF and CDKN2B. Several years later, Bahuau et al. reported two families with melanoma and various neural system tumors, including astrocytoma, meningioma, schwanomma and neurofibroma, both of which exhibited deletions of a portion of chromosome 9p. [32]
Petronzelli et al proposed that p14ARF was responsible, at least in part, for predisposition to neural system tumors and melanoma based on a family that carried a germline splicing mutation that resulted in a lack of exon 2 sequences, thus rendering both proteins defective. They subsequently concluded that the development of neurofibromas was due to the inactivation of p16INK4A and p14ARF or, alternatively, of p14ARF alone.[17] Pasmant et al detected a large germline deletion, which included the entire p15/CDKN2B-p16/CDKN2A-p14/ARF gene cluster, in a family with cutaneous malignant melanoma and neural system tumors, suggesting a contiguous gene deletion syndrome.[33] However, in their study, they also identified a new long noncoding RNA, within the germline deletion, which they called ANRIL (Antisense Noncoding RNA in the INK4A Locus). More recently, Vanneste et al reported a patient with multiple neurofibromas and a solitary spinal neurofibroma who was found to have a deletion of 14 nucleotides in exon 2 of CDKN2A, providing further evidence that p14ARF, p16INK4A, and/or ANRIL , now designated CDKN2B-AS1, may be specifically involved in the etiology of neurofibromas as a feature of FAMMM.[34] However, numerous FCMM families with large deletions impacting p14ARF do not have neural system tumors. [3, 35, Goldstein et al unpublished data 2015] Thus, further study is required to understand the relationship between CMM, NST, CDKN2A, CDKN2B, CDKN2B-AS1, and other 9p21 genes. Although the contribution of ANRIL expression to neural system tumors remains unknown, single nucleotide polymorphisms (SNPs) which alter its expression, have been associated with numerous diseases including coronary artery disease, stroke, diabetes, as well as melanoma and glioma.[36]. Most recently, Frigerio et al reported a patient with both astrocytoma and multiple melanomas with the largest constitutive deletion described to date involving 9p21.3 and spanning approximately 2,135 Mb.[37] Our proband, reported herein, adds to this growing list of families with large deletions extending beyond CDKN2A, thus raising the question of whether or not this could represent an emerging contiguous gene deletion syndrome.
Clinical Implications
Based on the identification of a molecular deletion encompassing CDKN2A, the proband and his at-risk family members were instructed to follow FAMMM/FCMM surveillance recommendations which typically include total body skin exams every 6 to 12 months by a dermatologist, beginning at 10 years of age and including whole body photography. [24, 38] Given the early-onset skin lesions within the family, though, baseline dermatologic exams were recommended to begin during the first few years of life. Most recently, the proband’s niece with a history of multiple primary melanomas was identified not to carry the large deletion. Given that relatives who test negative for the known CDKN2A mutation remain at increased risk for melanoma due to other familial shared risk factors, though, they should pursue heightened skin surveillance, regardless of their genetic status.[39–40] Lastly, with regards to dermatologic recommendations, family members were educated regarding proper sun protection measures. Current FAMMM/FCMM surveillance recommendations also address the potential increased risk for pancreatic cancer in mutation positive family members. As such, the patient and his at-risk family members were instructed to discuss the role of endoscopic ultrasound (EUS) of the pancreas, as well as possible measurement of the CA-19-9 tumor marker with a gastroenterologist.
Developing a tailored management strategy for this family, which addressed other cancer risks potentially associated with the large deletion, was limited by the fact that several relatives were unavailable for study. As a result, it was not possible to determine whether the brother and the nephew with respective diagnoses of chondrosarcoma and leukemia each carried the large familial deletion. In contrast, the proband’s son with the PNET tumor, although unavailable for study, was determined to be an obligate carrier, given that his daughter tested positive for the familial deletion. As such, the following medical management recommendations were developed based on the family’s specific history of benign tumors/cancers: 1) annual comprehensive physical exam, including a careful neurologic exam, 2) consideration of whole-body MRI, 3) abdominal ultrasound and brain MRI on an annual basis, and 4)bloodwork every 4 months to include complete blood count, erythrocyte sedimentation rate, and lactate dehydrogenase. Given the proband’s son who was diagnosed with a PNET tumor at 18 years of age, baseline brain MRI was recommended beginning at 8 years of age. In addition, annual dilated ophthalmology evaluation by an ophthalmologist was recommended to look for optic glioma or papilledema, a sign of increased intracranial pressure which can occur secondary to the presence of a brain tumor. In summary, the medical team acknowledged that the proposed surveillance regimen was quite intensive. Given the proband’s son, though, who, at the time, was actively dying from leptomeningeal spread of his PNET tumor and the rather young diagnoses of melanoma within the family, this regimen was developed as a starting point for discussion with the clinical geneticist who ultimately would be responsible for overseeing the family’s ongoing medical management. Lastly, given that radiotherapy (RT) may be contraindicated in those with NF1, based on a possible risk of developing malignant peripheral nerve sheath tumors within the field of treatment, it was recommended that any decisions regarding RT be made in the context of a discussion with a radiation oncologist regarding the risks, benefits, and limitations of such treatment. [43–44].
Beyond the challenge of developing a management strategy for the proband and his at-risk relatives, given the paucity of families worldwide with a similar contiguous gene deletion, there was the ethical dilemma of whether pre-symptomatic testing should be offered to minor children. Typically, the appropriateness of offering pre-symptomatic testing to minors depends not only on the specific cancer predisposition syndrome segregating within the family and whether it is known to predispose to childhood cancers, but also the phenotypic variability observed within the family. Given the early-onset cancers exhibited by family members and the development of an intensive management strategy, a number of the proband’s relatives, both affected and unaffected, have since requested testing for both themselves and their minor children. To date, 5 additional relatives, ranging in age from 1 to 52 years of age, have now pursued testing and all but one was confirmed to carry the familial deletion.
Lastly, the proband’s large deletion within 9p21.3 contains a number of candidate genes which make up the interferon gene cluster and have been shown to be important for patient survival and success of interferon therapy, beyond containing genes important in melanoma susceptibility. [45] Linsley et al., for example, linked loss of this locus with reduced immune cell genes within melanoma tumors. [46] They concluded that loss of 9p21.3 may lead or contribute to reduced immune surveillance and/or tumor destruction by the immune system. Thus, the family members described here, who develop melanoma in the context of the large deletion containing the interferon gene cluster, as well as others similarly affected with loss of 9p21.3, may be more likely to suffer metastatic disease and hence a worse prognosis. Chromosomal instability was recently demonstrated to be a mechanism for modulating local cytokine expression in colorectal tumors. [47] Thus, emerging evidence suggests that genomic rearrangements within tumors may represent a broader mechanism for modulating anti-tumor immunity and, as such, could potentially influence the choice of treatment regimen in a family, such as the one presented here with a large deletion within 9p21.3, if and when tumors develop.
Limitations
Our understanding regarding the spectrum of cancers associated with this family’s contiguous gene deletion is limited by the paucity of families described in the literature with similar deletions. Further, within this family, it has yet to be determined whether the brother and nephew’s respective diagnoses of chondrosarcoma and leukemia occurred in conjunction with the familial deletion, whether there is more than one condition segregating within this family, or whether these diagnoses represent sporadic cancers within a family that has a genetic predisposition. For example, some of the features exhibited by the proband and/or his relatives are described in patients with PTEN Hamartoma Tumor syndrome (PHTS) such as the lipomas, the papilloma, and the increased risk for melanoma. A next generation sequencing panel, including PTEN, and potentially other cancer susceptibility genes, could address the possibility of an additional cancer syndrome segregating within the family. In addition, the specific contribution of the other genes within the contiguous deletion, if any, on the phenotype has not been explored. Lastly, although subsequent testing of the proband’s sister identified that she shared both copy number variants (CNVs) of unknown significance in common with her brother, in addition to the large deletion of chromosome 9p21.3, it has yet to be determined from which side(s) of the family the CNVs originated.
CONCLUSION
The family described here has a rare contiguous gene deletion which includes CDKN2A, and predisposes to multiple melanoma/dysplastic nevi characteristic of FAMMM/FCMM. The constellation of additional tumors within this family, some, but not all, of which are reminiscent of Neurofibromatosis Type 1, raises the question as to whether there is another cancer syndrome co-segregating within the family. The identification of the contiguous gene deletion underlying this family’s hereditary predisposition to cancer was aided by the Familial Cancer Database which is a useful online tool to assist clinicians in the development of a differential diagnosis based on a family’s specific history of various benign and malignant tumors. Based on our experience with this family and our review of similar cases within the literature, large rearrangement analysis of the CDKN2A gene should be considered if traditional Sanger sequencing of CDKN2A proves negative, when there is a family history of multiple melanoma concerning for FAMMM/FCMM, in the context of neural system tumors. Likewise, clinicians should consider both sequencing and large rearrangement analysis of the CDKN2A gene in patients suspicious for NF1 whose testing proves negative when there is a personal or family history of melanoma. Improved identification of these families will be further augmented by the increasing utilization of next generation sequencing pan cancer panels, which include CDKN2A, as well as whole genome sequencing, both of which routinely detect large deletions and duplications, thus permitting better characterization of the phenotype associated with this family’s contiguous gene deletion. Lastly, predictive testing of minor children may be warranted in families with large deletions spanning CDKN2A, given the young cancer diagnoses observed in this family and assuming a clinical team can be assembled to develop a surveillance regimen that has the potential to impact prognosis of affected relatives. Further study of additional families with similar deletions spanning CDKN2A and beyond are needed to help guide genetic counseling and anticipatory care for these patients and to better understand this potentially evolving cancer predisposition syndrome.
Acknowlegdements
The authors thank Kathleen Hruska, Ph.D., FACMG with GeneDx of Gaithersburg, MD for providing images illustrating the extent of the deleted region in the proband.
Funding This work was supported in part by the Intramural Research Program of the US National Institutes of Health (NIH), National Cancer Institute (NCI), Division of Cancer Epidemiology and Genetics (DCEG).
Footnotes
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES
- 1.Ghiorzo P, Gargiulo S, Pastorino L, Nasti S, Cusano R, Bruno W, Gliori S, Sertoli MR, Burroni A, Savarino V, Gensini F, Sestini R, Queirolo P, Goldstein AM, Bianchi Scarrà G. Impact of E27X, a novel CDKN2A germ line mutation, on p16 and p14ARF expression in Italian melanoma families displaying pancreatic cancer and neuroblastoma. Hum Mol Genet 2006;15(18):2682–9. [DOI] [PubMed] [Google Scholar]
- 2.Hayward NK. Genetics of melanoma predisposition. Oncogene 2003;22:3053–62. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril M- F, Azizi E, Bianchi-Scarra G, Bishop DT, Bressac-de Paillerets B, Bruno W, Calista D, Cannon Albright LA, Demenais F, Elder DE, Ghiorzo P, Gruis NA, Hansson J, Hogg D, Holland EA, Kanetsky PA, Kefford RF, Landi MT, Lang J, Leachman SA, MacKie RM, Magnusson V, Mann GJ, Niendorf K, Bishop JN, Palmer JM, Puig S, Puig-Butille JA, de Snoo FA, Stark M, Tsao H, Tucker MA, Whitaker L, Yakobson E, The Lund Melanoma Study Group, and the Melanoma Genetics Consortium (GenoMEL). High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res 2006;66(20):9818–28. [DOI] [PubMed] [Google Scholar]
- 4.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, Windpassinger C, Wackernagal W, Wackernagel W, Loy S, Wolf I, Viale A, Lash AE, Pirun M, Socci ND, Rutten A, Palmedo G, Abramson D, Offit K, Ott A, Becker JC, Cerroni L, Kutzner H, Bastian BC, Speicher MR. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 2011;43:1018–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science 2013;339(6122):959–61. [DOI] [PubMed] [Google Scholar]
- 6.Robles-Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, Pooley KA, Pritchard AL, Tiffen JC, Petljak M, Palmer JM, Symmons J, Johansson P, Stark MS, Gartside MG, Snowden H, Montgomery GW, Martin NG, Liu JZ, Choi J, Makowski M, Brown KM, Dunning AM,Keane TM, López-Otín C, Gruis NA, Hayward NK, Bishop DT, Newton-Bishop JA, Adams DJ. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet 2014;46(5):478–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, Ghiorzo P, Bressac-de Paillerets B, Nagore E, Avril MF, Caporaso NE, McMaster ML, Cullen M, Wang Z, Zhang X; NCI DCEG Cancer Sequencing Working Group; NCI DCEG Cancer Genomics Research Laboratory; French Familial Melanoma Study Group, Bruno W, Pastorino L, Queirolo P, Banuls-Roca J, Garcia-Casado Z, Vaysse A, Mohamdi H, Riazalhosseini Y, Foglio M, Jouenne F, Hua X, Hyland PL, Yin J, Vallabhaneni H, Chai W, Minghetti P, Pellegrini C, Ravichandran S, Eggermont A, Lathrop M,Peris K, Scarra GB, Landi G, Savage SA, Sampson JN, He J, Yeager M, Goldin LR, Demenais F, Chanock SJ, Tucker MA, Goldstein AM, Liu Y, Landi MT. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet 2014;46(5):482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W, Hussey C, Tran T, Miki Y, Weaver-Feldhaus J, McClure M, Aitken JF, Anderson DE, Bergman W, Frants R, Goldgar DE, Green A, MacLennan R, Martin NG, Meyer LJ, Youl P, Zone JJ, Skolnick MH, Cannon-Albright LA. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet 1994;8:22–6. [DOI] [PubMed] [Google Scholar]
- 9.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PAT, Ally DS, Sheahan MD, Clark WH Jr, Tucker MA, Dracopoli NC. Germline p16 mutations in familial melanoma. Nat Genet 1994;8:15–21. [DOI] [PubMed] [Google Scholar]
- 10.Mistry SH, Taylor C, Randerson-Moor JA, Harland M, Turner F, Barrett JH, Whitaker L, Jenkins RB, Knowles MA, Newton Bishop JA, Bishop DT. Prevalence of 9p21 deletions in UK melanoma families. Genes Chromosomes Cancer 2005;44(3):292–300. [DOI] [PubMed] [Google Scholar]
- 11.Online Mendelian Inheritance in Man (OMIM) accessed at http://omim.org/entry/155755 on July 29, 2015.
- 12.Kaufman DK, Kimmel DW, Parisi JE, Michels VV. A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology 1993;43:1728–31. [DOI] [PubMed] [Google Scholar]
- 13.Azizi E, Friedman J, Pavlotsky F, Iscovich J, Bornstein A, Shafir R, Trau H, Brenner H and Nass D. Familial cutaneous malignant melanoma and tumors of the nervous system. Cancer 1995;76:1571–8. [DOI] [PubMed] [Google Scholar]
- 14.Bahuau M, Vidaud D, Kujas M, Palangie A, Assouline B, Chaignaud-Lebreton M, Prieur M, Vidaud M, Harpey J-P, Lafourcade J, Caille B. Familial aggregation of malignant melanoma/dysplastic naevi and tumors of the nervous system: an original syndrome of tumor proneness. Ann Genet 1997;40:78–91. [PubMed] [Google Scholar]
- 15.Stokkel MPM, Kroon BBR, van der Sande JJ, Neering H. Malignant cutaneous melanoma associated with neurofibromatosis in two sisters from a family with familial atypical multiple mole melanoma syndrome. Cancer 1993;72(8):2370–5. [DOI] [PubMed] [Google Scholar]
- 16.Petty EM, Gibson LH, Fountain JW, Bolognia JL, Yanng-Feng TL, Housman DE, Bale AE. Molecular definition of a chromosome 9p21 germ-line deletion in a woman with multiple melanomas and a plexiform neurofibroma: implications for 9p tumor suppressor gene(s). Am J Hum Genet 1993;53:96–104. [PMC free article] [PubMed] [Google Scholar]
- 17.Petronzelli F, Sollima D, Coppola G, Martini-Neri ME, Neri G, Genuardi M. CDKN2A germline splicing mutation affecting both p16INK4 and p14ARF RNA processing in a melanoma /neurofibroma kindred. Genes Chromosomes Cancer 2001;31(4):398–401. [DOI] [PubMed] [Google Scholar]
- 18.Sijmons RH, Burger GTN. Familial cancer database: a clinical aide-mémoire. Fam Cancer 2001;1(1):51–5. [DOI] [PubMed] [Google Scholar]
- 19.Sijmons RH, Burger GTN.Rolf H. The use of a diagnostic database in clinical oncogenetics. Hered Cancer Clin Pract, 2003;1(1):31–3. [Google Scholar]
- 20.Database of Genomic Variants; http://projects.tcag.ca/variation.
- 21.Begg CB, Orlow I, Hummer AJ, Armstrong BK, Kricker A, Marrett LD, Millikan RC, Gruber SB, Anton-Culver H, Zanetti R, Gallagher RP, Dwyer T, Rebbeck TR, Mitra N, Busam K, From L, Berwick M for the Genes Environment and Melanoma (GEM) Study Group. Lifetime Risk of Melanoma in CDKN2A Mutation Carriers in a Population-Based Sample. J Natl Cancer Inst 2005;97(20):1507–15. [DOI] [PubMed] [Google Scholar]
- 22.Cannon-Albright LA, Goldgar DE, Meyer LJ, Lewis CM, Anderson DE, Fountain JW, Hegi ME, Wiseman RW, Petty EM, Bale AE, Olopade OI, Diaz MO, Kwiatkowski DJ, Piepkorn MW, Zone JJ, Skolnick MH. Assignment of a locus for familial melanoma, MLM, to chromosome 9p13-p22. Science 1992;258(5085):1148–52. [DOI] [PubMed] [Google Scholar]
- 23.Parker JF, Florell SR, Alexander A, DiSario JA, Shami PJ, Leachman SA. Pancreatic carcinoma surveillance in patients with familial melanoma. Arch Dermatol, 2003;139(8):1019–25. [DOI] [PubMed] [Google Scholar]
- 24.Czajkowski R, Placek W, Drewa G, Czajkowska A, Uchańska G. FAMMM syndrome: pathogenesis and management. See comment in PubMed Commons belowDermatol Surg 2004;30(2 Pt 2): 291–6. [DOI] [PubMed] [Google Scholar]
- 25.Cust AE, Armstrong BK, Goumas C, Jenkins MA, Schmid H, Hopper JL, Kefford RF, Giles GG, Aitken JF, Mann GJ. Sunbed use during adolescence and early adulthood is associated with increased risk of early-onset melanoma. Int J Cancer 2011;128(10):2425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop DT, Demenais F, Goldstein AM, Bergman W, Newton Bishop J, Bressac-de Paillerets B, Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland EA, Mann GJ, Mantelli M, Nancarrow D, Platz A, Tucker MA. Geographical Variation in the Penetrance of CDKN2A Mutations for Melanoma. J Natl Cancer Inst 2002;94(12):894–903. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, Fontaine LS, Organic SM, Dracopoli NC, Clark WH Jr, Tucker MA. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16 (INK4) mutations. New Eng J Med 1995;333: 970–4. [DOI] [PubMed] [Google Scholar]
- 28.Whelan AJ, Bartsch D, Goodfellow PJ. Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2A tumor-suppressor gene. New Eng J Med 1995;333: 975–7 . [DOI] [PubMed] [Google Scholar]
- 29.Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer 2000;87(6):809–11. [PubMed] [Google Scholar]
- 30.Randerson-Moor JA, Harland M, Williams S, Cuthbert-Heavens D, Sheridan E, Aveyard J, Sibley K, Whitaker L, Knowles M, Bishop JN, Bishop DT. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet 2001;10(1):55–62. [DOI] [PubMed] [Google Scholar]
- 31.Lynch HT, Deters CA, Hogg D, Lynch JF, Kinarsky Y, Gatalica Z. Familial Sarcoma: Challenging Pedigrees. Cancer 2003;98(9):1947–57. [DOI] [PubMed] [Google Scholar]
- 32.Bahuau M, Vidaud D, Jenkins RB, Bieche I, Kimmel DW, Assouline B, Smith JS, Alderete B, Cayuela J-M, Harpey J-P, Caille B, Vidaud M. Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res 1998;58(11):2298–303. [PubMed] [Google Scholar]
- 33.Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose experession coclusters with ARF. Cancer Res 2007;67(8):39639. [DOI] [PubMed] [Google Scholar]
- 34.Vanneste R, Smith E, Graham G. Multiple neurofibromas as the presenting feature of Familial Atypical Multiple Malignant Melanoma (FAMMM) syndrome. Am J Med Genet Part A 2013;161A:1425–31. [DOI] [PubMed] [Google Scholar]
- 35.Auroy S, Avril MF, Chompret A, Pham D, Goldstein AM, Bianchi-Scarrà G, Frebourg T, Joly P, Spatz A, Rubino C, Demenais F, Bressac-de Paillerets B; French Hereditary Melanoma Study Group. Sporadic multiple primary melanoma cases: CDKN2A germline mutations with a founder effect. Genes Chromosomes Cancer 2001;32(3):195–202. [DOI] [PubMed] [Google Scholar]
- 36.Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genetics 2010;6(4): e1000899. doi: 10.1371/journal.pgen.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frigerio S, Disciglio V, Manoukian S, Peissel B, Della Torre G, Maurichi A, Collini P, Pasini B, Gotti G, Ferrari A, Rivoltini L, Massimino M, Rodolfo M. A large de novo 9p21.3 deletion in a girl affected by astrocytoma and multiple melanoma. BMC Med Genet. 15:59, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eckerle Mize D, Bishop M, Resse E, et al. Familial Atypical Multiple Mole Melanoma Syndrome In: Riegert-Johnson DL, Boardman LA, Hefferon T, et al. , editors. Cancer Syndromes [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2009-. Available from: http://www.ncbi.nlm.nih.gov/books/NBK7030/ [PubMed] [Google Scholar]
- 39.Kefford RF, Newton Bishop JA, Bergman W, Tucker MA. Counseling and DNA testing for individuals perceived to be genetically predisposed to melanoma: A consensus statement of the Melanoma Genetics Consortium. J Clin Oncol 1999;17(10):3245–51. [DOI] [PubMed] [Google Scholar]
- 40.Hansson J Familial melanoma. Surg Clin North Am. 2008;88(4):897–916, viii. doi: 10.1016/j.suc.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 41.Friedman JM. Neurofibromatosis 1. 1998 Oct 2 [Updated 2014 Sep 4]. In: Pagon RA, Adam MP, Ardinger HH, et al. , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1109/ [Google Scholar]
- 42.Villani A, Tabori U, Schiffman J, Shlien A, Beyene J, Druker H, Novokmet A, Finlay J, Malkin D. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol 2011;12(6):559–67. doi: 10.1016/S1470-2045(11)70119-X. Epub 2011 May 19. [DOI] [PubMed] [Google Scholar]
- 43.Evans D, Baser M, McGaughran J, Sharif S, Howard E, and Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39(5):311–4. doi: 10.1136/jmg.39.5.311. Correction in J Med Genet 2003;40(4):304. doi: 10.1136/jmg.40.4.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME, Evans DG. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol 2006;24(16):2570–5.Format [DOI] [PubMed] [Google Scholar]
- 45.Lenci RE, Bevier M, Brandt A, Bermejo JL, Sucker A, Moll I, Planelles D, Requena C, Nagore E, Hemminki K, Schadendorf D, Kumar R. Influence of Genetic Variants in Type I Interferon Genes on Melanoma Survival and Therapy. PLoS ONE 2012;7(11): e50692. doi: 10.1371/journal.pone.0050692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Linsley PS, Speake C, Whalen E, Chaussabel D. Copy Number Loss of the Interferon Gene Cluster in Melanomas Is Linked to Reduced T Cell Infiltrate and Poor Patient Prognosis. PLoS ONE 2014;9(10): e109760. doi: 10.1371/journal.pone.0109760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, Lafontaine L, Bilocq AM, Kirilovsky A, Tosolini M, Waldner M, Berger A, Fridman WH, Rafii A, Valge-Archer V, Pages F, Speicher MR, Galon J. Functional Network Pipeline Reveals Genetic Determinants Associated with in Situ Lymphocyte Proliferation and Survival of Cancer Patients. Sci Transl Med 2014; 6(228): ra37. doi: 10.1126/scitranslmed.3007240. [DOI] [PubMed] [Google Scholar]