

Abstract
Race and ancestry have long been associated with differential risk and outcomes to disease as well as responses to medications. These differences in drug response are multifactorial with some portion associated with genomic variation. The field of pharmacogenomics aims to predict drug response in patients prior to medication administration and to uncover the biological underpinnings of drug response. The field of human genetics has long recognized that genetic variation differs in frequency between ancestral populations, with some single nucleotide polymorphisms found solely in one population. Thus far, most pharmacogenomic studies have focused on individuals of European and East Asian ancestry, resulting in a substantial disparity in the clinical utility of genetic prediction for drug response in US minority populations. In this review, we discuss the genetic factors that underlie variability to drug response and known pharmacogenomic associations and how these differ between populations, with an emphasis on the current knowledge in cardiovascular pharmacogenomics.
Keywords: pharmacogenomics, cardiovascular, polymorphism, genetic variation, minority populations
1. INTRODUCTION
Adverse drug reactions (ADRs), which account for approximately 100,000 deaths per year in the United States (1), are thought to be multifactorial, with the patient’s genome accounting for varying proportions of the risk. Pharmacogenomics is aimed at identifying genetic variation [e.g., single nucleotide polymorphisms (SNPs)] that influences interindividual differences in drug response and adverse events, which has widespread clinical relevance. Its application promises to enable targeted drug administration, improved therapeutic outcome, and informed drug development. Pharmacogenomic insights have also improved our understanding of the underlying pathways and mechanisms behind ADRs. Genome-wide association studies (GWAS) for pharmacogenomic traits are increasingly being conducted to identify loci that affect either drug response or susceptibility to ADRs. Figure 1 shows all the current GWAS associations related to drug responses. The findings from pharmacogenomic GWAS have uncovered genetic associations to rare ADRs (2). A prime example of the ability of pharmacogenomics to uncover important aspects of our biology is the discovery of the SNP rs4363657, which is in nearly complete linkage disequilibrium with the nonsynonymous SNP rs4149056, located in SLCO1B1 (encoding the organic anion transporter OAT1B1). This gene is involved in the hepatic transport of statins. The presence of the SNP rs4149056 T>C has been associated with statin-induced myopathy with an odds ratio (OR) of 4.5 per copy of the risk allele (C) and an OR of 16.9 for the homozygous CC genotype as compared with TT homozygotes (3). Unlike disease traits where genetic variants may have small effects across several loci, pharmacogenetic traits can be influenced by relatively common alleles with large effect sizes (4). The overall contribution of SNPs to drug response is highly variable and dependent on the drug and phenotype being measured. Importantly, the allele frequency of associated variants contributes to the ability of a study to identify associations and to correctly ascertain the proportion of variability explained in the pharmacogenomic phenotype. These findings continue to grow with the addition of new drug response phenotypes and larger cohorts; however, the clinical application of these findings has been slow. The reasons for this are multifactorial, and questions about the generalizability of findings across racial and ethnic groups persist.
Figure 1.
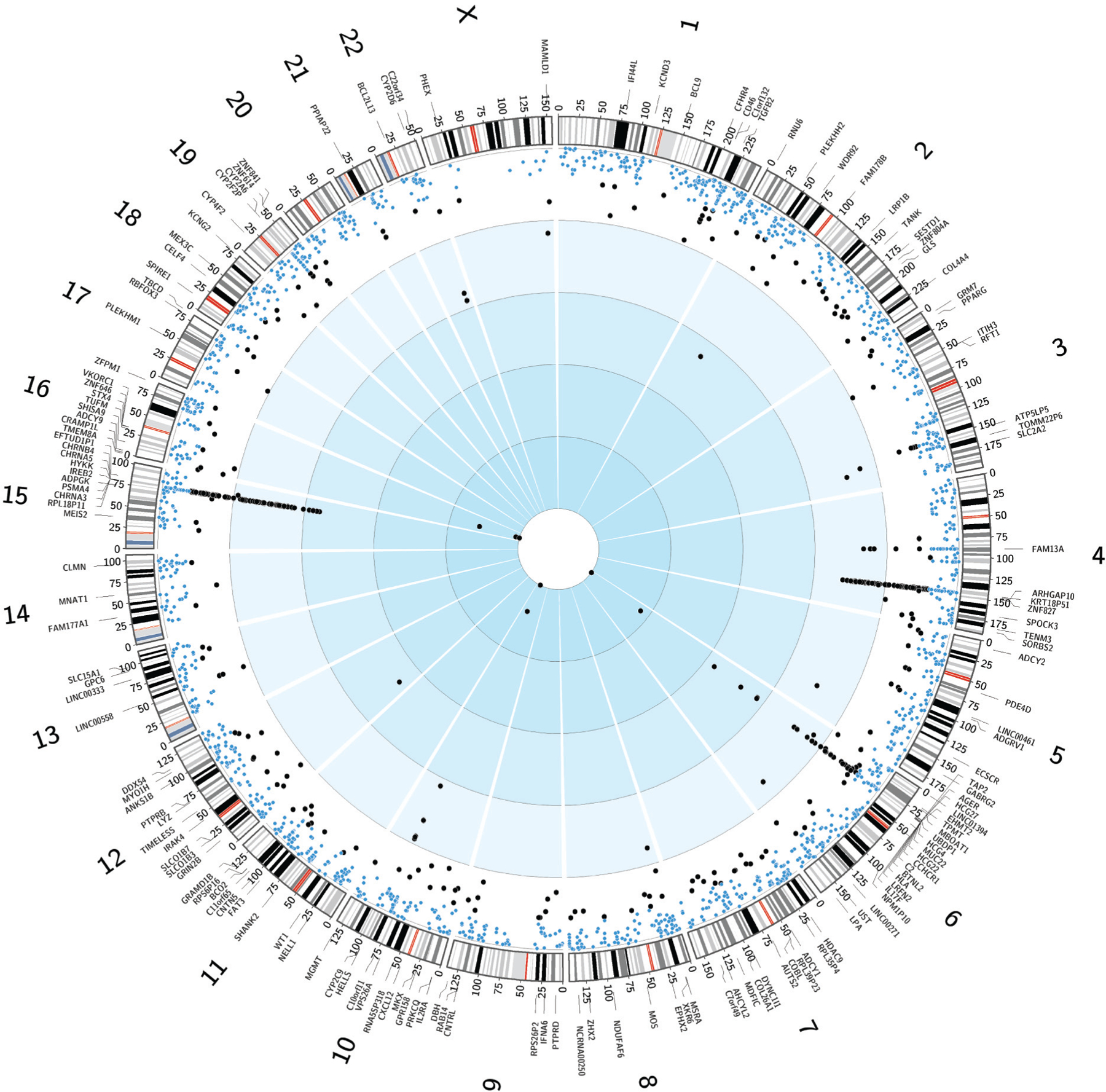
Circos plot of GWAS-identified SNPs related to drug response. The inner circle depicting 2,610 nominally associated SNPs (blue circles: 5.0 × 10−8 < p < 9 × 10−6) and genome-wide significant SNPs (black circles: p < 5.0 × 10−8) scaled from 0 to 1 based on p-value. Gene names are shown in the outermost circle corresponding to genome-wide significant SNP trait–associated loci followed by karyotype. Abbreviations: GWAS, genome-wide association studies; SNP, single nucleotide polymorphism. Data for figure provided by NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas) (166).
Although pharmacogenomics has made strides in our understanding of serious ADRs such as Stevens-Johnson syndrome with carbamazepine (5) and statin-induced myopathy (6), US minority populations have largely been excluded from these studies (7). The vast majority of the genetic association studies used in developing clinical pharmacogenomic guidelines, such as the Clinical Pharmacogenetics Implementation Consortium (CPIC) (8), were conducted in populations of European or East Asian descent. Some of the SNPs characterized as clinically important in these studies are rare or absent in minority populations. This means that the SNPs recommended for clinical implementation may not adequately explain the interindividual variation in drug response or adverse events in non-European populations as well as they can in populations of European descent.
Many pharmacogenetic polymorphisms differ in frequency to some degree among populations (9). Additionally, medication dosing recommendations have historically been based on clinical drug trials conducted in mainly European populations (10). As different ethnic groups have been incorporated into clinical research studies, it has become clear that they may differ in their response to drugs. If, for example, drug metabolism differs between populations, then the data generated in one population may not be directly extrapolated to another. Understanding the role of genomics in these differences is the goal of pharmacogenomics. In this review we focus on the types of genetic variation that affect drug response and the population differences that may account for differential response in selective cardiovascular therapies.
2. HEALTH DISPARITIES IN CARDIOVASCULAR OUTCOMES
Despite the reduction in cardiovascular mortality over the past 50 years, cardiovascular disease (CVD) continues to be the leading cause of death across all populations in the United States (11). Racial disparity in CVD burden has been observed for decades (12–14). Asian Americans, the fastest-growing minority in the United States (15), have a greater incidence of hemorrhagic strokes such as the subarachnoid and intracerebral hemorrhage subtypes than whites (16). CVD is also the leading cause of death among Hispanic adults. However, the overall rates of coronary heart disease and overall cardiac mortality are lower in Hispanics compared to European Americans (17). People of African ancestry have a higher incidence of coronary heart disease, heart failure, stroke, and overall CVD mortality as compared to other populations (14). The high prevalence of cardiovascular risk factors such as hypertension, diabetes mellitus, and obesity underlies the increased incidence of CVDs among African Americans (18, 19). African Americans are one of the oldest racial groups in the United States, constituting 13.3% of the US population, and they are the second largest minority after Hispanics/Latinos (20). Yet marked disparities exist in CVD treatments and outcomes, signifying a need for health care to alleviate disparities with comprehensive screening, enhanced specificity of diagnoses, and tailored disease and therapeutic management.
Genetic variability between populations can result in significant differences in disease characteristics and treatment outcomes. A study investigating the power of available SNPs to detect disease associations compared SNPs in 50 genes in African Americans versus European Americans and observed that only 52% of the SNPs were common in both populations (21). Of these SNPs, 22% were found only in African Americans and 5% only in European Americans. Furthermore, 36% of the SNPs had significant differences in frequencies between the two populations. CVD, being a multifactorial disease, has both genetic and environmental determinants (22), and genetic differences in disease pathogenesis can contribute to variability in therapeutic response. Studies have shown an increased genetic susceptibility in African Americans to CVDs (19, 23) and substantial differences in their response to cardiovascular drugs, compared to other ethnicities (24, 25). However, guidelines for the therapeutic management of CVD in African Americans do not differ from those in other ethnic groups.
3. GENETIC BASIS OF POPULATION DIFFERENCES IN DRUG RESPONSE
Variability in drug response is well known, with epidemiological and clinical studies identifying clinical and demographic factors, including age, comorbidities, comedications, and disease subtypes, that all contribute to differences in response. Genetic variation may serve as one of the major determinants of population differences in drug response, and most pharmacogenomic studies strive to include known clinical and demographic factors that affect drug response in their association analysis. The inclusion of these nongenomic factors is key to uncovering true genetic effects, as confounding leads to false discovery as well as a loss of power to detect true genetic associations.
The key components of genetically driven nonresponse or ADRs are polymorphisms in the genes responsible for metabolizing drugs [pharmacokinetic (PK) variation], variation in drug targets [pharmacodynamic (PD) variation], and SNPs that underlie disease pathogenesis (disease subtype response variability) (26). The frequency of such polymorphisms has been found to vary between populations. Table 1 shows variants within the pharmacogenomics knowledgebase of very important pharmacogenes (27) related to cardiovascular medications and the average population allele frequency of these variants. Many SNPs show marked differences between global populations. An example of this can be seen in Figure 2, which shows the global allele frequency of rs2242480, a SNP associated with increased warfarin clearance and reduced glycoprotein IIb/IIIa activation (important in platelet activation during clopidogrel treatment). This SNP varies widely between populations; hence, it can only be identified as associated with drug response in populations in which the allele frequency is high enough to achieve adequate statistical power.
Table 1.
Allele frequency distribution of variants of the very important pharmacogenes associated with cardiovascular medications across different populations
| Gene | Varianta | Allele frequencyb | Consequence (Reference) | PharmGKB level of evidence | CPIC level | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EUR | AFR | ASN | AMR | |||||||||
| CEU | GBR | ASW | YRI | EAS | SAS | MXL | PEL | |||||
| ACE | rs4359 C>T | 0.54 | 0.49 | 0.37 | 0.43 | 0.39 | 0.36 | 0.39 | 0.30 | Increased response to ramipril in people with hypertension (111) | 3 | NA |
| rs4291 T>C | 0.37 | 0.34 | 0.08 | 0.08 | 0.35 | 0.28 | 0.23 | 0.17 | Increased risk of aspirin intolerance when exposed to aspirin in people with asthma (112) | 3 | ||
| ADRB1 | rs1801252 A>G | 0.12 | 0.12 | 0.17 | 0.21 | 0.14 | 0.13 | 0.26 | 0.38 | Decreased blood pressure reduction with metoprolol (98, 113) | 3 | NA |
| rs1801253 G>C | 0.67 | 0.70 | 0.63 | 0.52 | 0.79 | 0.73 | 0.83 | 0.89 | Reduced response to metoprolol and atenolol therapy (113–115) | 3 | ||
| ADRB2 | rs1042713 G>A | 0.35 | 0.42 | 0.55 | 0.53 | 0.55 | 0.45 | 0.48 | 0.44 | Increased mortality due to β-blocker adverse effect (102) | 3 | NA |
| rs1042714 G>C | 0.54 | 0.60 | 0.88 | 0.88 | 0.93 | 0.81 | 0.85 | 0.87 | Increased mortality due to β-blocker adverse effect (102) | 3 | ||
| CYP2A6 | rs1801272 A>T | 0.04 | 0.03 | 0.01 | 0 | 0 | 0.01 | 0.02 | 0.01 | Decreased metabolism of coumarin (116) | 4 | NA |
| rs28399433 A>C | 0.05 | 0.04 | 0.10 | 0.10 | 0.23 | 0.15 | 0.10 | 0.09 | Decreased metabolism of coumarin (117) | 4 | ||
| CYP3A4 | rs2242480; part of the CYP3A4*4 haplotype C>T | 0.06 | 0.07 | 0.75 | 0.84 | 0.27 | 0.37 | 0.39 | 0.58 | Increased clearance of warfarin (118) Reduced GP IIb/IIIa activation, lower platelet activation in clopidogrel-treated coronary artery disease patients (119) | 3 | NA |
| CYP2B6 | rs3211371 C>T | 0.09 | 0.11 | 0.03 | 0.009 | 0.003 | 0.09 | 0.08 | 0.03 | High on-treatment platelet reactivity (120) | 3 | NA |
| CYP2C19 | CYP2C19*2 (rs4244285 G>A) | 0.13 | 0.14 | 0.14 | 0.17 | 0.31 | 0.36 | 0.13 | 0.06 | Diminished platelet response to clopidogrel treatment and poorer cardiovascular outcomes (121) | 1A | CYP2C19-clopidogrel: CPIC level A |
| Lower warfarin maintenance dose requirement in patients with nonvalvular atrial fibrillation (122) | 3 | |||||||||||
| CYP2C19*17 (rs12248560 C>T) | 0.22 | 0.24 | 0.20 | 0.25 | 0.02 | 0.14 | 0.12 | 0.04 | Enhanced clopidogrel response and an increased bleeding risk in ACS/PCI patients (123) | 1A | ||
| CYP2C19*4 (rs28399504) | 0 | 0 | 0 | 0 | 0.001 | 0 | 0.008 | 0 | Poor metabolism of clopidogrel and increased risk for secondary cardiovascular events (124) | 1A | ||
| CYP2C19*3 (rs4986893 G>A) | 0 | 0 | 0 | 0 | 0.056 | 0.012 | 0 | 0 | Poor metabolism of clopidogrel (125) | 1A | ||
| Lower warfarin maintenance dose requirement in patients with nonvalvular atrial fibrillation (122) | 3 | |||||||||||
| rs3814637 C>T | 0.08 | 0.06 | 0.09 | 0.11 | 0.09 | 0.12 | 0.02 | 0.01 | Decreased dose of warfarin requirement (118) | 3 | ||
| rs11568732 T>G | 0.06 | 0.06 | 0.05 | 0.06 | 0.09 | 0.12 | 0.02 | 0.012 | Increased risk of hemorrhage in patients on clopidogrel (126) | 3 | ||
| CYP2C9 | rs28371685 C>T | 0 | 0 | 0.008 | 0.05 | 0 | 0.001 | 0 | 0 | Decreased warfarin dose requirement (127) | 3 | |
| CYP2C9*3 (rs1057910 A>C) | 0.07 | 0.07 | 0.02 | 0 | 0.03 | 0.11 | 0.02 | 0.01 | Sensitivity to the anticoagulant effect of warfarin, with increased risk of bleeding (128, 129) | 1A | ||
| Decreased dose of acenocoumarol or closer INR monitoring (130) | 2A | |||||||||||
| CYP2C9*2 (rs1799853 C>T) | 0.15 | 0.09 | 0.04 | 0 | 0.001 | 0.04 | 0.10 | 0.02 | Decreased metabolism of warfarin (131) | 1A | ||
| Increased risk of a gastrointestinal hemorrhage from acenocoumarol (132) | 2A | |||||||||||
| CYP2C9*8 (rs7900194 G>A) | 0.005 | 0 | 0.03 | 0.05 | 0 | 0.001 | 0 | 0 | Lowest warfarin maintenance dose requirement (133) | 2A | ||
| rs7089580 A>T | 0.22 | 0.23 | 0.17 | 0.22 | 0.01 | 0.13 | 0.11 | 0.04 | Lower dose of warfarin requirement (48) | 2A | ||
| rs28371686 C>G | 0 | 0 | 0.02 | 0.02 | 0 | 0 | 0 | 0 | Increased dose of warfarin requirement (127) | 2A | ||
| rs4917639 A>C | 0.22 | 0.16 | 0.22 | C: 0.19 | 0.08 | 0.15 | 0.13 | 0.03 | Increased dose of warfarin requirement (134) | 2A | ||
| CYP2C locus | rs12777823 G>A | 0.15 | 0.15 | 0.21 | 0.28 | 0.31 | 0.36 | 0.13 | 0.06 | Decreased warfarin dose requirement (73) | 2B | NA |
| CYP2D6*4 (rs3892097 C>T) | 0.23 | 0.24 | 0.12 | 0.06 | 0.00 | 0.11 | 0.13 | 0.07 | Increased plasma concentrations of metoprolol (135) | 2A | CYP2D6-metoprolol: CPIC level C | |
| Decreased clearance of carvedilol in people with heart diseases (136) | 3 | |||||||||||
| CYP2D6*6 (rs5030655 A>deletion) | 0.02 | 0.01 | 0.008 | 0 | 0 | 0.001 | 0 | 0 | Increased plasma concentrations of metoprolol (137) | 2A | ||
| CYP2D6*9 (rs5030656 CTT>deletion) | 0.02 | 0.04 | 0.008 | 0 | 0 | 0 | 0.008 | 0 | Decreased metabolism/clearance of metoprolol (138) | 2A | ||
| CYP2D6*35 (rs769258 C>T, haplotype: rs1058164, rs1135840, rs16947) | 0.05 | 0.05 | 0 | 0 | 0.003 | 0.006 | 0.02 | 0.006 | Increased metabolism/clearance of metoprolol (138) | 2A | ||
| CYP2D6*35 (rs769258 C>T, haplotype: rs1058164, rs1135840, rs16947) | CYP2D6*3 [rs35742686 T>deletion] | 0.020 | 0.03 | 0.016 | 0 | 0 | 0.002 | 0 | 0 | Decreased metabolism/clearance of metoprolol (128) | 2A | |
| CYP2D6*41 (rs28735595 C>T, haplotype: rs16947, rs1058164, rs1080996, rs1080997, rs1080998, rs1081000, rs1135840, rs1080995, rs2837172, rs2862481, rs28633410, rs28695233, rs28735595, rs29001518) | 0.43 | 0.44 | 0.43 | 0.32 | 0.27 | 0.47 | 0.59 | 0.6 | Decreased metabolism/clearance of metoprolol (138) | 2A | ||
| CYP2D6*17 (rs16947 A>G, haplotype: rs1058164, rs1135840, rs28371706) | 0.67 | 0.69 | 0.58 | 0.44 | 0.86 | 0.64 | 0.74 | 0.67 | Decreased metabolism/clearance of metoprolol (138) | 2A | ||
| CYP2D6*10 (rs1065852 G>A, haplotype: rs1058164, rs1135840) | 0.24 | 0.24 | 0.15 | 0.10 | 0.57 | 0.16 | 0.15 | 0.07 | Decreased metabolism/clearance of metoprolol (138) | 2A | ||
| Decreased clearance of carvedilol (139) | 3 | |||||||||||
| CYP1A2 | rs762551 A>C | 0.27 | 0.27 | 0.36 | 0.46 | 0.33 | 0.47 | 0.27 | 0.14 | Decreased on-treatment platelet reactivity (140) | 3 | NA |
| CYP4F2 | rs2108622 C>T | 0.25 | 0.29 | 0.09 | 0.06 | 0.21 | 0.41 | 0.25 | 0.12 | Increased warfarin dose requirement (141, 142) | 1B |
CYP4F2- warfarin: CPIC level B CYP4F2- acenocoumarol CPIC level B |
| Increased acenocoumarol dose requirement (141, 142) | 2A | |||||||||||
| F5 | rs6025 C>T | 0.03 | 0.01 | 0 | 0 | 0 | 0.01 | 0.008 | 0.01 | Increased risk of stroke when treated with hormonal contraceptives for systemic use in women (143) | 2A | F5-hormonal contraceptives: CPIC level C |
| KCNH2 | rs3807375 C>T | 0.35 | 0.30 | 0.73 | 0.77 | 0.76 | 0.39 | 0.57 | 0.72 | Increased QT interval (144) | 4 | NA |
| rs12720441 G>C | 0 | 0 | 0 | 0 | 0.001 | 0 | 0 | 0 | Increased risk of torsades de pointes when treated with amiodarone (145) | 4 | ||
| rs1805123 T>G | 0.25 | 0.27 | 0.04 | 0.00 | 0.05 | 0.25 | 0.16 | 0.08 | Increased QT interval (144) | 4 | ||
| rs36210421 C>A | 0.05 | 0.02 | 0 | 0 | 0.001 | 0.003 | 0.01 | 0 | Decreased catalytic activity when exposed to dofetilide (146) | 4 | ||
| MTHFR | rs1801133 | 0.29 | 0.32 | 0.14 | 0.10 | 0.29 | 0.12 | 0.47 | 0.44 | Increased response to benazepril in people with hypertension (147) | 3 | NA |
| NQO1 | rs1800566 G>A | 0.36 | 0.20 | 0.17 | 0.19 | 0.42 | 0.36 | 0.38 | 0.42 | Increased warfarin dose requirement (66) | 3 | NA |
| rs10517 G>A | 0.07 | 0.13 | 0.11 | 0.10 | 0.35 | 0.14 | 0.078 | 0.02 | Decreased dose of warfarin requirement (148) | 3 | ||
| P2RY1 | rs1065776 C>T | 0.06 | 0.02 | 0.21 | 0.22 | 0.04 | 0.13 | 0.05 | 0.07 | Increased risk of aspirin resistance in aspirin-treated patients with myocardial infarction (149) | 3 | NA |
| rs701265 A>G | 0.17 | 0.13 | 0.67 | 0.81 | 0.25 | 0.20 | 0.20 | 0.31 | Increased risk of aspirin resistance in aspirin-treated patients with myocardial infarction (149) | 3 | ||
| P2RY12 | rs2046934 G>A | 0.80 | 0.78 | 0.86 | 0.84 | 0.85 | 0.90 | 0.89 | 0.93 | Increased platelet reactivity when treated with aspirin and clopidogrel (150) | 3 | NA |
| PTGS2 | rs20417 C>G | 0.17 | 0.14 | 0.31 | 0.38 | 0.04 | 0.19 | 0.21 | 0.21 | Aspirin resistance (151) | 3 | NA |
| SLCO1B1 | rs4149056 T>C | 0.14 | 0.14 | 0.06 | 0.009 | 0.12 | 0.04 | 0.07 | 0.14 | Increased likelihood of cough when treated with enalapril (152) | 3 | NA |
| Increased concentrations of ticagrelor in acute coronary syndrome patients (153) | 3 | |||||||||||
| VDR | rs4760658 A>G | 0.29 | 0.28 | 0.18 | 0.10 | 0.02 | 0.16 | 0.23 | 0.16 | Increased warfarin doses phenotype (154) | 3 | NA |
| rs11168293 G>T | 0.28 | 0.27 | 0.18 | 0.10 | 0.02 | 0.16 | 0.23 | 0.16 | Increased warfarin doses phenotype (154) | 3 | NA | |
| VKORC1 | rs7294 C>T | 0.31 | 0.42 | 0.47 | 0.49 | 0.11 | 0.24 | 0.35 | 0.44 | High warfarin dose requirement (155, 156) | 1A | VKORC1-warfarin: CPIC level A |
| rs9923231 C>T | 0.43 | 0.36 | 0.15 | 0.28 | 0.12 | 0.15 | 0.47 | 0.38 | Low warfarin dose phenotype (61) | 1A | ||
| Low acenocoumarol dose phenotype (157) | 1B | |||||||||||
| rs17708472 G>A | 0.25 | 0.21 | 0.07 | 0.02 | 0.003 | 0.09 | 0.15 | 0.04 | Higher warfarin dose phenotype (158) | 2A | NA | |
| rs2359612 G>A | 0.43 | 0.35 | 0.22 | 0.17 | 0.88 | 0.14 | 0.47 | 0.39 | Decreased warfarin dose requirement (65) | 2A | NA | |
| rs8050894 G>C | 0.56 | 0.63 | 0.29 | 0.23 | 0.11 | 0.85 | 0.49 | 0.61 | Decreased warfarin dose requirement (65) | 2A | NA | |
| rs9934438 G>A | 0.43 | 0.36 | 0.15 | 0.03 | 0.88 | 0.15 | 0.46 | 0.38 | Increased warfarin dose requirement (159) | 2A | NA | |
| rs2884737 A>C | 0.27 | 0.21 | 0.06 | 0 | 0.001 | 0.067 | 0.14 | 0.03 | Decreased warfarin dose requirement (160) | 2A | NA | |
| rs7196161 G>A | 0.56 | 0.65 | 0.54 | 0.55 | 0.11 | 0.85 | 0.51 | 0.61 | Increased warfarin dose requirement (161) | 2B | NA | |
| rs61162043 G>A | 0.56 | 0.65 | 0.5 | 0.5 | 0.11 | 0.85 | 0.51 | 0.61 | Increased warfarin dose requirement (48) | 3 | NA | |
| rs7200749 G>A | 0 | 0 | 0.20 | 0.24 | 0 | 0 | 0.008 | 0.012 | Increased warfarin dose requirement (162) | 3 | NA | |
| rs17880887 G>T | 0.34 | 0.32 | 0.09 | 0.02 | 0.003 | 0.12 | 0.17 | 0.07 | Lower levels of transthyretin precursor when exposed to warfarin (163) | 3 | NA | |
| rs17886199 G>T | 0 | 0 | 0.04 | 0.05 | 0 | 0 | 0 | 0 | Decreased warfarin dose requirement (164) | 3 | NA | |
| rs11150606 T>C | 0.03 | 0.03 | 0.04 | 0 | 0.7 | 0.01 | 0.31 | 0.34 | Decreased warfarin dose requirement (165) | 3 | NA | |
The PharmGKB levels of evidence are as follows: high literature evidence, CPIC guideline and known clinical implementation (1A, 1B); moderate literature evidence, variant in PharmGKB VIP (2A, 2B); low literature evidence (3); and preliminary literature evidence (4). The CPIC levels include the following: prescribing action recommended, alternate therapies or dosing are highly likely to be effective and safe (A or B); no prescribing change based on genetics, alternatives are unclear or evidence is weak, but testing is common or gene is CPIC level A or B for other drugs (C). Abbreviations: ACS, acute coronary syndrome; AFR, African populations; AMR, admixed American populations; ASN, Asian populations; ASW, African ancestry in southwest United States; CEU, Utah residents with northern and western European ancestry; CPIC, Clinical Pharmacogenetics Implementation Consortium; EAS, East Asian; EUR, European populations; GBR, British in England and Scotland; INR, international normalized ratio; MXL, Mexican ancestry in Los Angeles, California; NA, not available; PCI, percutaneous coronary intervention; PEL, Peruvians in Lima, Peru; PharmGKB, pharmacogenomics knowledgebase; SAS, South Asian; YRI, Yoruba in Ibadan, Nigeria.
Rs numbers and allele change from dbSNP.
Allele frequencies as reported by 1000 Genomes.
Figure 2.

The global allele frequency difference for rs2242480, a single nucleotide polymorphism associated with increased warfarin clearance and lower platelet activation in clopidogrel-treated coronary artery disease patients. As shown, the frequency of the T allele (risk allele) varies widely between populations. Although the frequency is very high in African ancestry populations, followed by South American and Asian populations, it is comparatively very low in European populations.
In addition to allele frequency differences, population-specific SNPs may also help to explain a portion of drug response phenotypes. Only one or a few populations carry these variants, making their discovery dependent on the population studied. As an example, individuals of African ancestry carry more genetic variation than other populations; hence, it is plausible that they also carry more variation that may affect drug response. Hispanic/Latino individuals in the United States are also genetically diverse as their genome is trichotomous, with ancestral contributions of indigenous American, European, and African ancestry (28). The proportion of genetic ancestry derived from each of these lineages varies substantially among and within ethnic groups from different countries in Latin America and in US Hispanic/Latino populations (29, 30). Therefore, these admixed minorities may differ in allele frequency, distribution, and the combination of allelic variants when compared to other populations. By studying the unique genomic architecture of admixed populations, we can discover novel population-specific genetic variations affecting drug response. In a recent transethnic study of asthma response to bronchodilators, the investigators identified population-specific and transethnic genetic variants associated with drug response (31). This finding suggests that for each ethnic group different combinations of SNPs contribute to the drug response phenotype. Because of the rarity of minority-focused studies such as this, it remains difficult to truly ascertain the proportion of variability that non-European genomes contribute to drug response phenotypes.
4. VARIABILITY IN DRUG PHARMACOKINETICS AND PHARMACODYNAMICS
Considerable variability in drug response can exist between individuals receiving similar doses of a drug. Such variability may result in a lack of efficacy in some patients while leading to overdose and/or toxicity in others. The efficacy of a drug is determined by its PK and PD properties. PK variability arises from variability in the delivery of drugs or their active metabolites to the target tissues or in the clearance of drugs (via elimination and metabolism to inactive metabolites) from the body. It is important to remember that the effect of SNPs associated with PK genes may be broader than the specific drugs studied thus far. PD variability is mainly due to variability in the relationship between drug concentration and effect (e.g., the binding of a drug to its drug targets). Among the several factors that can influence population differences in drug PK and PD, the prevalence of genetic polymorphisms in drug targets and drug metabolism pathways or transport pathways can affect parameters such as area under the curve (representing the total body exposure to drug), steepness of the dose-response relationship (e.g., nonresponse at therapeutic doses), and altered drug disposition (e.g., toxicity) (Figure 3).
Figure 3.

The potential effects of genetic variation on variability in drug (a) pharmacokinetics and (b) pharmacodynamics. (a) Although patients with homozygous wild-type alleles (WT/WT) can have normal therapeutic concentrations of a drug (hence normal metabolizers), the presence of one (WT/V) or two copies (V/V) of polymorphic allele(s) can lead to subtherapeutic concentrations due to increased metabolism (hence ultrarapid metabolizers) or supratherapeutic concentrations due to decreased metabolism (poor/intermediate metabolizers), as illustrated by the change in area under the curve for each genotype. (b) A drug within the normal therapeutic dosing range in patients with wild-type alleles (WT/WT) will have an efficacious response with low to no toxicity and hence no adverse drug event (ADE), while the presence of one copy of a variant allele (WT/V) can have decreased drug efficacy, which may lead to increased dosing and toxicity. The presence of two copies of variant allele (V/V) can lead to either the least or no efficacy with the risk of drug toxicity (usually related to adverse drug reactions from off-target drug effects) or no effect and no toxicity (usually seen in drugs in which the therapeutic effect and toxicity are related to the drug target).
A prime example of PK variability can be seen in the case of CYP2D6, which is one of the most extensively characterized polymorphic drug-metabolizing enzymes, with at least 100 allelic variants reported to date (32). These variants are classified as functional, nonfunctional, and reduced-function groups based on their effect on CYP2D6 activity, from poor to ultrarapid metabolism of substrates (33). There are vast ethnic differences in the frequency of these variants, with CYP2D6*3, CYP2D6*4, CYP2D6*5, CYP2D6*6 null-function alleles, and CYP2D6*41 reduced-function alleles more common in Caucasians, CYP2D6*10 (decreased activity) more common in Asians, and CYP2D6*17 (decreased activity) common in people of African ancestry but with considerably lower prevalence or absence in other ethnic groups (34, 35). CYP2D6 gene duplications (at least three copies) with the ultrarapid metabolizer phenotype are present in approximately 28% of North Africans, Ethiopians, and Arabs; 10% of Caucasians; 3% of African Americans; and 1% of Hispanics, Chinese, and Japanese (35). Therefore, drugs like tricyclic antidepressants or β-blockers that undergo CYP2D6-mediated biotransformation to inactive metabolites can cause adverse effects in poor metabolizers and a lack of efficacy in ultrarapid metabolizers (36). In contrast, drugs that are biotransformed by CYP2D6 to active metabolites (e.g., codeine into morphine) can have a lack of efficacy in poor metabolizers and exaggerated effects in ultrarapid metabolizers (37, 38). Differences in the presence and frequency of these SNPs and copy number variants may contribute to the complex genotype-phenotype relationships that have been observed and may well explain ethnicity-specific benefits and toxicity.
Studies of African Americans have shown differences in the expression and function of key drug-metabolizing enzymes that have been linked to population-specific genetic variation; a prime example is the CYP3A5*3 polymorphism, which truncates the CYP3A5 protein. The allele leading to protein truncation is found at a high frequency in individuals of European ancestry but at a much lower allele frequency in African Americans. This difference in allele frequency results in a functional CYP3A5 enzyme in African Americans (reviewed in 39), while Caucasians and Asian populations do not have a functional enzyme. A recent study of tacrolimus dosing in 50 African Americans showed that to achieve a therapeutic trough concentration, significantly higher plasma concentrations of drug were required in African Americans carrying a functional CYP3A5 enzyme (40), with higher variability in tacrolimus dose associated with greater acute organ rejection in African Americans (41). A study exploring population differences in acetaminophen PK parameters showed that African Americans oxidize acetaminophen more slowly than European Americans, which may be partially explained by the CYP2E1*1D polymorphism that is more prevalent in people of African ancestry (42). Similar population-specific findings can also be seen in the sulfotransferase enzymes (reviewed in 43) and CYP2A6 (44). Well-documented differences between African Americans and Europeans in drug dosing have been observed in drugs such as angiotensin-converting enzyme (ACE) inhibitors (reviewed in 45) and warfarin (46–48), as well as in the expression of coregulators of CYP activity (one subtype of drug-metabolizing enzymes) (49). These differences in dose and expression may be the result of altered drug PK related to genetic variants that are population specific.
5. CARDIOVASCULAR PHARMACOGENOMICS
5.1. Oral Anticoagulant
Warfarin, the most widely prescribed oral anticoagulant, contributes to 15–20% of cases of bleeding per year, with life-threatening bleeding accounting for as much as 1–3% (50). However, these values are based on trials conducted predominantly in patients of European ancestry and do not account for differences in responsiveness to warfarin in other populations (51). Patients of African descent are at a 58% higher risk of major bleeding from warfarin compared to patients of European descent (52, 53). They also require higher warfarin doses compared to patients of European or Asian origin to achieve and maintain the same international normalized ratio (INR) (51, 54). Most dosing algorithms recommend a standard 5 mg loading dose regimen based on an estimation of the initiation dose in whites (55–57), which could result in an extreme dose in patients of Asian ancestry and an insufficient dose in patients of African ancestry. Since the incidence of hemorrhage is highest during the initial stage of warfarin therapy, a more accurate prediction of initial dosing may potentially decrease the rates of hemorrhagic and thrombotic complications of this narrow therapeutic index drug.
Besides clinical factors such as age, weight, height, smoking status, and comedications, warfarin dosing can be influenced by SNPs in genes encoding cytochrome P450 2C9 (CYP2C9), which metabolizes the drug and vitamin K epoxide reductase complex 1 (VKORC1) that serves as the drug target (58). Both candidate gene studies and GWAS have consistently shown that VKORC1 and CYP2C9 genotypes explain up to 30% of the total variability in warfarin dose requirements in people of European or South and East Asian origins (59–63). The International Warfarin Pharmacogenetics Consortium has reported that the VKORC1-1639 G→A (rs9923231), CYP2C9*2 (rs1799853), and CYP2C9*3 (rs1057910) SNPs can be used with clinical variables to predict warfarin dose, with 47% of the variability explained in European ancestry patients (64, 65). However, given the differences in CYP2C9 and VKORC1 allele frequencies between ethnic groups (see Table 1), differences in the amount of dosing variability explained by these SNPs are observed between populations. In a Hispanic population, these SNPs accounted for 58% of the dose variability (66), similar to what was seen in Caucasians (57%). However, only 31% of the variability in dosing was accounted for in African Americans (61). Several clinical trials were performed to determine the benefit of using pharmacogenetic-guided warfarin dosing in clinical practice. The multicenter European Pharmacogenetics of Anticoagulant Therapy trial (NCT01119300) randomized patients to pharmacogenetic-guided dosing or a fixed dosing regimen with a primary outcome of percentage of time in therapeutic range (PTTR) during the first 12 weeks of therapy (67). Patients in the genotype-guided dosing group received warfarin loading doses for the first 5 days based on algorithms that included pharmacogenetic variants, while patients in the control group received a fixed loading dose regimen for the first 3 days, with subsequent dosing based on routine practice for both groups. Pharmacogenetic-guided dosing resulted in significantly greater PTTR, fewer patients with INR ≥ 4, and a reduction in the time required to achieve therapeutic INR levels, demonstrating that pharmacogenetic-guided dosing performed better than fixed-dosing regimens in achieving therapeutic INRs in patients of European ancestry on warfarin. Another multicenter, multiethnic trial, Clarification of Optimal Anticoagulation Through Genetics (NCT00839657), randomized patients to warfarin dosing with either pharmacogenetic-based algorithms or clinical algorithm dosing for the first 5 days of therapy, with a primary outcome of PTTR within the first month (68). No significant differences were observed in PTTR, time to first therapeutic INR, or in adverse drug event rates. However, one of the most informative findings was the significant interaction detected between dosing strategy and ethnicity. African American patients, who constituted 27% of study subjects, had lower PTTR, more INRs above therapeutic range, and increased time to therapeutic INR in the genotype-guided group than in the clinical algorithm group. This may be due to the exclusion of population-specific SNPs from the dosing algorithm used in this trial (69). The most recent trial of warfarin pharmacogenomics, the Genetics Informatics Trial (GIFT) (NCT01006733), assessed the safety and effectiveness of pharmacogenetic-guided warfarin dosing for the reduction of deep venous thrombosis compared with clinical algorithm dosing following total hip or knee repair (70). In contrast to previous trials, the GIFT was stratified by race, and pharmacogenetic dosing algorithms guided therapy for the first 11 days of treatment. The primary end point was a composite of venous thromboembolism, major hemorrhage, INR ≥ 4, or death. Genotype-guided warfarin dosing, compared to dosing based on clinical algorithms, performed better in reducing the risk of major bleeding, INR ≥ 4, venous thromboembolism, and death.
CYP2C9*2 and *3 are common in Caucasians, but less frequent in African Americans and East Asians (71), whereas CYP2C9*8 (rs7900194), which is associated with a low warfarin dose requirement, is found exclusively in people of African ancestry (72). Additionally, an African American GWAS found that rs12777823, present in the CYP2C gene cluster, was associated with low warfarin dose requirements in African Americans but had no influence on warfarin dosing in individuals of European, Japanese, or Egyptian descent (73). Likewise, different VKORC1 haplotypes differ by population as well (60). Asian populations have a higher proportion of patients with the VKORC1 haplotypes that require low warfarin doses, while patients from African population more frequently carry the VKORC1 haplotypes that require high-dose warfarin (60). In addition to CYP2C9 and VKORC1, polymorphisms in genes encoding γ-glutamyl carboxylase (GGCX), vitamin K–dependent coagulation factors and inhibitors, and microsomal epoxide hydrolase also contribute to the variability in the warfarin anticoagulation effect (74). Hispanic-specific genetic variation has also been identified. In a study of both African American and inner-city Hispanics, SNPs in CYP4F2 and NQO1 were associated with increased warfarin dose, and the addition of these SNPs to known algorithms increased the proportion of dose variability that is explained with both clinical and genetic factors from 58% to 63% (66). Thus, genetic variation with differing allele frequencies or population-specific effects on PK and PD may contribute to the interpopulation variability in warfarin clearance and dosing and hence patient response.
5.2. Antiplatelet Therapy
Antiplatelet therapy with clopidogrel has been the mainstay for the thromboprophylaxis of CVDs (75). Unfortunately, significant interindividual variability in drug response affects both the efficacy and safety profile (76). The clinical consequence of inadequate response is recurrent cardiovascular events (CVEs) that can lead to myocardial infarction (MI), stroke, and death. The rates of such adverse events vary among different geographical regions and populations. A study conducted across 24 US hospitals showed a 1-year mortality rate of 7.2% in clopidogrel-treated African Americans versus 3.6% in Caucasians on clopidogrel (77). The global Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance trial (NCT00050817) (78) explored ethnic differences in adverse cardiovascular outcomes and bleeding complications in clopidogrel-treated patients across 32 countries from six continents and reported a higher risk of cardiovascular and all-cause mortality among African American and Hispanic patients compared to Asians and whites. Although there was no significant difference in severe bleeding from clopidogrel between the populations, moderate bleeding was observed more frequently among African American and Asian patients than whites, and intracranial hemorrhage occurred more frequently among Hispanic patients. The underlying mechanism for clopidogrel-associated adverse events is multifactorial (76) with genetic polymorphisms as one of the causes of variable drug response within an individual or across populations (77, 79).
Previous studies showed that loss-of-function (LOF) CYP2C19*2 (rs4244285) and CYP2C19*3 (rs4986893) allele carriers display significantly reduced responses to clopidogrel and a higher rate of recurrent CVEs compared to noncarriers (80–82). However, the overall contribution of these variants to all CVEs across populations is unknown. A multicenter study assessed outcomes with the clinical implementation of CYP2C19 genotype–guided antiplatelet therapy after percutaneous coronary intervention (PCI) and reported increased risk of major CVEs in patients with an LOF allele who were prescribed clopidogrel versus an alternative therapy (83). In contrast, CYP2C19*17 (rs12248560) gain-of-function allele carriers have lower CVE risk, but they are at a higher risk of bleeding due to increased metabolism of clopidogrel and enhanced platelet inhibition (84). This is due to the bioconversion of clopidogrel to its active metabolite via the liver enzyme CYP2C19. Individuals who carry one or two reduced-activity or nonfunctioning CYP2C19*2 or CYP2C19*3 alleles are classified as intermediate or poor metabolizers, while individuals with one or two copies of the *17 allele are typically classified as ultrarapid metabolizers (85). Approximately 2% of Caucasians, 4% of African Americans, and 14% of Chinese are CYP2C19 poor metabolizers (85). The prevalence of these risk alleles varies by ancestry (Table 1). Compared to other populations, the frequencies of the CYP2C19*2 and CYP2C19*3 LOF alleles are higher in Asian populations, suggesting that individuals of Asian descent are more likely to be resistant to clopidogrel therapy (82). A study by Cresci et al. (77) compared the effect of CYP2C19 polymorphism on CVEs among clopidogrel-treated Caucasian and African American patients with acute MI; they observed that the CYP2C19*2 allele was significantly associated with an increased 1-year mortality rate and a trend towards an increased rate of recurrent MI in Caucasians, while the CYP2C19*17 allele was associated with an increased 1-year mortality rate and an increased risk of bleeding in African Americans. Because of the well-established association and clear outcome benefit of genotype-guided therapy in patients receiving clopidogrel, several institutions have begun offering genotype-guided therapy for clopidogrel based on CPIC guidelines (86), with alternative antiplatelet agents like P2Y12 inhibitors (e.g., prasugrel, ticagrelor) offered to those that carry the LOF alleles. However, a comparison trial of ticagrelor and clopidogrel in patients with acute coronary syndrome (ACS) (NCT00391872) and a comparison trial of prasugrel and clopidogrel in ACS subjects who were to undergo PCI (NCT00097591) showed that ticagrelor and prasugrel are associated with an increased risk of fatal and life-threatening bleeding compared to clopidogrel (87, 88). On the other hand, Steg et al. (89) demonstrated that the bleeding risk is lower in the first 30 days after stent thrombosis when treated with ticagrelor versus clopidogrel. Nevertheless, both ticagrelor and prasugrel P2Y12 inhibitors are substantially more expensive than clopidogrel, and ticagrelor is also associated with more frequent discontinuation because of side effects compared to clopidogrel (87). Several ongoing randomized clinical trials now aim to determine the clinical utility of genotype-guided therapy, including the potential economic benefits. The multisite Tailored Antiplatelet Therapy Following PCI trial (NCT01742117) will determine the best antiplatelet therapy (ticagrelor versus clopidogrel) for patients with CYP2C19 LOF alleles in preventing major adverse CVEs at 1 year (90). A second trial, named the Cost-effectiveness of Genotype Guided Treatment With Antiplatelet Drugs in STEMI Patients: Optimization of Treatment trial (NCT01761786), will assess the efficacy, safety, and cost-effectiveness of the CYP2C19 genotype–guided antiplatelet treatment strategy using clopidogrel in noncarriers of a CYP2C19 LOF allele and ticagrelor or prasugrel in carriers of a CYP2C19 LOF allele in ST-elevation myocardial infarction (STEMI) patients. The primary net clinical benefit end point is the composite of death, recurrent MI, definite stent thrombosis, stroke, and major bleeding complications (91). The cost-effectiveness of genotype-guided therapy compared to a nontailored strategy will also be determined. A third trial, the Assessment of Prospective CYP2C19 Genotype Guided Dosing of Anti-Platelet Therapy in Percutaneous Coronary Intervention (NCT02508116), will evaluate the cost-effectiveness of genotype-guided antiplatelet therapy by randomizing patients undergoing PCI to CYP2C19 genotyping or usual care groups. The findings from all these trials will facilitate the widespread adoption of personalized antiplatelet therapies. Much like the case of warfarin before it, most genomic association and outcome studies of clopidogrel were conducted in populations of European descent, and none of the discovery efforts have been conducted in minority populations; therefore, the scientific community does not know if other populations carry unique variation that may predispose them to nonresponse or adverse outcomes from this medication.
5.3. Antihypertensive Agents
The frequency of hypertension, mortality from hypertensive heart disease, stroke, and hypertensive renal disease is higher in people of African ancestry than in other ethnicities (92, 93). Rostand and colleagues (94) examined the clinical course of 94 patients with treated primary hypertension and initial normal serum creatinine concentrations for 12–174 months of follow-up. Despite adequate diastolic blood pressure control, African Americans were twice as likely as whites to have elevated serum creatinine. In addition, several observations showed that African Americans respond less favorably to monotherapy with β-blockers compared to whites (24, 93). β-blocker efficacy is attenuated in African Americans, likely due to lower plasma renin activity and higher proportions of low renin hypertension (95). However, a greater reduction in blood pressure and plasma renin release in response to propranolol was observed in Chinese men compared with US white men (96). The noted population differences in response to antihypertensive drugs could reflect genetic alterations in sensitivity to β1-adrenergic receptor blockade. Two common nonsynonymous polymorphisms, rs1801252 (Ser49Gly) and rs1801253 (Arg389Gly), in the β1 Adrenergic Receptor gene ADRB1 have been reported and are thought to be functionally important based on in vitro studies (97). In hypertensive patients treated with metoprolol, a threefold greater reduction in daytime diastolic blood pressure in Arg389 homozygous wild-type patients was observed compared to those who carried the Gly389 variant allele (98). Inclusion of the Ser49Gly polymorphism showed a gradation to the hypotensive response, which was highest for double homozygous wild-type (Arg389/Ser49) carriers and negligible in double carriers of the risk alleles (389G/49G). The prevalence of the SNPs varies significantly among ethnic groups (Table 1). The higher frequency of the Gly389 allele in African Americans compared with whites could be a possible explanation for their reduced response to β-blockers. Although ethnicity and ADRB1 polymorphisms have been reported to be independent predictors of response to β-blockers (99), further prospective studies elucidating the roles of these genetic variants on ethnic-specific responses to β-blockers are warranted.
Polymorphisms in the β2 Adrenergic Receptor gene ADRB2 have also been reported to be associated with the response to β-blockers. Three nonsynonymous polymorphisms have been identified in the human ADRB2 gene: rs1042713 (Gly16Arg), rs1042714 (Gln27Glu), and rs1800888 (Thr164Ile). In vitro and in vivo studies confirmed the functional importance of these polymorphisms, which affect vasodilation, cardiac responses to the agonist, and the desensitization of these responses (97, 100, 101). A study assessing the effect of ADRB2 Gly16Arg and Gln27Glu polymorphisms on survival among patients discharged on β-blockers after ACS reported increased mortality associated with Gln27 and with Arg16 homozygosity (102), both polymorphisms being more common in African Americans. Mortality was not observed in patients not treated with β-blockers, suggesting that these polymorphisms modulate the response to β-blockers rather than the progression of disease.
Population differences in response to ACE inhibitors and ACE receptor blockers (ARBs) have also been observed. Like β-blockers, ACE inhibitors and ARBs are less efficacious in African Americans than in whites (103–105). Additionally, people of African ancestry are at a greater risk of angioedema from ACE inhibitors (106). As ACE is a major therapeutic target of antihypertensives, a better understanding of genetic differences is essential to assess the risk of hypertension and response to therapy. Functional polymorphisms in the ACE gene may account for differences in disease susceptibility or response to ACE inhibitor therapy. As allele frequencies at the ACE locus vary greatly between African Americans and European Americans (107), a study investigated the effect of polymorphisms in the ACE gene and adverse cardiovascular outcomes in African American hypertensive patients with coronary disease (108). A marked allelic expression imbalance associated with SNPs (rs7213516, rs7214530, and rs4290) residing in conserved regions 2–3 kb upstream of ACE was detected, suggesting that these regulatory SNPs affect the expression of ACE. The SNPs rs4290 and rs7213516 were associated with adverse cardiovascular outcomes, which were largely attributable to nonfatal MI in African Americans. The high allele frequency of these variants in African Americans compared to other ethnic groups suggests that these alleles could contribute to variation between populations in cardiovascular risk and treatment outcomes.
In contrast to their responses to other antihypertensives, patients of African ancestry respond better to diuretics than whites. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial, which compared the effectiveness of a calcium channel blocker and ACE inhibitor against diuretics in lowering the incidence of chronic heart disease and other CVEs, was the first study to have data on African American hypertensive patients (106). These patients were shown to have similar results in diastolic blood pressure decline with the amlodipine (calcium channel blocker) versus chlorthalidone (thiazide-type diuretic) treatment. However, blood pressure decline while receiving lisinopril (ACE inhibitor) was significantly less pronounced in African American patients compared to patients of other ethnicities, collectively referred as nonblacks. Furthermore, blood pressure reduction was better achieved with chlorthalidone than lisinopril in African Americans, as there was 4 mm Hg higher systolic blood pressure, a 19% higher risk of all CVEs, including a 40% higher risk of stroke, and a 30% greater risk of heart failure in lisinopril-versus chlorthalidone-treated groups. A recent GWAS investigating the impact of genetic variation on blood pressure response to chlorthalidone identified an intronic SNP (rs79237970) in the WDR92 gene associated with better diastolic blood pressure response to chlorthalidone. The SNP rs79237970 is an expression quantitative trait locus for PPP3R1, the regulatory subunit of calcineurin, whose inhibition results in hypertension (4).
Hydrochlorothiazide (HCTZ), which is very similar to chlorthalidone, is the most commonly prescribed thiazide diuretic and antihypertensive agent in the United States and is currently recommended as a first-line treatment, or as an add-on, for most individuals with essential hypertension (109). However, HCTZ use is associated with a series of adverse effects, including an increased risk of developing diabetes and hyperuricemia (110). A GWAS investigating HCTZ-induced changes in uric acid identified five unique gene regions associated with HCTZ-induced uric acid elevations in African Americans (LUC7L2, COX18/ANKRD17, FTO, PADI4, and PARD3B), and one region associated with these elevations in Caucasians (GRIN3A) (109). Therefore, although diuretics or calcium channel blockers are considered to be a better choice than ACE inhibitors or β-blockers as a first-line monotherapy for African Americans, genetic testing prior to prescribing diuretics can help to advance personalized antihypertensive therapy approaches and prevent drug-associated adverse events in these patients.
6. CONCLUSIONS
We are now in an age in which genomics findings may help to guide therapy decisions and lessen the frequency of drug-related adverse events. Although controversy persists on how to properly implement and use genomics information and who will ultimately bear the cost of genetic testing, optimism remains as to the utility of precision medicine. National efforts such as All of Us (https://allofus.nih.gov/) and U-PGx (http://upgx.eu/) have brought attention to the need for large diverse cohorts with the implementation of precision medicine. Additionally, efforts by the National Institute for Minority Health and Health Disparities have worked to fund the acceleration of findings in underrepresented minorities. With increased access to high-dimensional data in minorities, we will begin to realize the promise of precision medicine for all.
ACKNOWLEDGMENTS
We would like to gratefully thank Mr. Edmund Perera for assistance with the figures used in this manuscript.
Glossary
- SNP
single nucleotide polymorphism
- GWAS
genome-wide association study
- PK
pharmacokinetics
- PD
pharmacodynamics
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Lazarou J, Pomeranz BH, Corey PN. 1998. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 279:1200–5 [DOI] [PubMed] [Google Scholar]
- 2.Crowley JJ, Sullivan PF, McLeod HL. 2009. Pharmacogenomic genome-wide association studies: lessons learned thus far. Pharmacogenomics 10:161–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Group SC, Link E, Parish S, Armitage J, Bowman L, et al. 2008. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N. Engl. J. Med 359:789–99 [DOI] [PubMed] [Google Scholar]
- 4.Wadelius M, Chen LY, Lindh JD, Eriksson N, Ghori MJ, et al. 2009. The largest prospective warfarin-treated cohort supports genetic forecasting. Blood 113:784–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu K, Reynolds NJ. 2012. Pharmacogenetic screening to prevent carbamazepine-induced toxic epidermal necrolysis and Stevens-Johnson syndrome: a critical appraisal. Br. J. Dermatol 166:7–11 [DOI] [PubMed] [Google Scholar]
- 6.Talameh JA, Kitzmiller JP. 2014. Pharmacogenetics of statin-induced myopathy: a focused review of the clinical translation of pharmacokinetic genetic variants. J. Pharmacogenom. Pharmacoproteom 5:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popejoy AB, Fullerton SM. 2016. Genomics is failing on diversity. Nature 538:161–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Relling MV, Klein TE. 2011. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther 89:464–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suarez-Kurtz G, ed. 2007. Pharmacogenomics in Admixed Populations. Austin, TX: Landes Biosci. [Google Scholar]
- 10.Dickmann LJ, Schutzman JL. 2018. Racial and ethnic composition of cancer clinical drug trials: How diverse are we? Oncologist 23:243–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. 2016. Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation 133(4):e38–360 [DOI] [PubMed] [Google Scholar]
- 12.Chaturvedi N 2003. Ethnic differences in cardiovascular disease. Heart 89:681–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. 2015. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation 131:e29–322 [DOI] [PubMed] [Google Scholar]
- 14.Pool LR, Ning H, Lloyd-Jones DM, Allen NB. 2017. Trends in racial/ethnic disparities in cardiovascular health among US adults from 1999–2012. J. Am. Heart Assoc 6(9):e006027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palaniappan LP, Araneta MR, Assimes TL, Barrett-Connor EL, Carnethon MR, et al. 2010. Call to action: cardiovascular disease in Asian Americans: a science advisory from the American Heart Association. Circulation 122:1242–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klatsky AL, Friedman GD, Sidney S, Kipp H, Kubo A, Armstrong MA. 2005. Risk of hemorrhagic stroke in Asian American ethnic groups. Neuroepidemiology 25:26–31 [DOI] [PubMed] [Google Scholar]
- 17.Balfour PC Jr., Ruiz JM, Talavera GA, Allison MA, Rodriguez CJ. 2016. Cardiovascular disease in Hispanics/Latinos in the United States. J. Lat. Psychol 4:98–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark LT, Ferdinand KC, Flack JM, Gavin JR 3rd, Hall WD, et al. 2001. Coronary heart disease in African Americans. Heart Dis. 3:97–108 [DOI] [PubMed] [Google Scholar]
- 19.Carnethon MR, Pu J, Howard G, Albert MA, Anderson CAM, et al. 2017. Cardiovascular health in African Americans: a scientific statement from the American Heart Association. Circulation 136:e393–423 [DOI] [PubMed] [Google Scholar]
- 20.US Census Bur. 2012. National population projections tables. Data Set, US Census Bur., Washington, DC [Google Scholar]
- 21.Carlson CS, Eberle MA, Rieder MJ, Smith JD, Kruglyak L, Nickerson DA. 2003. Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans. Nat. Genet 33:518–21 [DOI] [PubMed] [Google Scholar]
- 22.O’Donnell CJ, Nabel EG. 2011. Genomics of cardiovascular disease. N. Engl. J. Med 365:2098–109 [DOI] [PubMed] [Google Scholar]
- 23.Gibbons GH. 2004. Physiology, genetics, and cardiovascular disease: focus on African Americans. J. Clin. Hypertens 6:11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson JA. 2008. Ethnic differences in cardiovascular drug response: potential contribution of pharmacogenetics. Circulation 118:1383–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muszkat M 2007. Interethnic differences in drug response: the contribution of genetic variability in β adrenergic receptor and cytochrome P4502C9. Clin. Pharmacol. Ther 82:215–18 [DOI] [PubMed] [Google Scholar]
- 26.Burroughs VJ, Maxey RW, Levy RA. 2002. Racial and ethnic differences in response to medicines: towards individualized pharmaceutical treatment. J. Natl. Med. Assoc 94:1–26 [PMC free article] [PubMed] [Google Scholar]
- 27.Hernandez-Boussard T, Whirl-Carrillo M, Hebert JM, Gong L, Owen R, et al. 2008. The pharmacogenetics and pharmacogenomics knowledge base: accentuating the knowledge. Nucleic Acids Res. 36:D913–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conomos MP, Laurie CA, Stilp AM, Gogarten SM, McHugh CP, et al. 2016. Genetic diversity and association studies in US Hispanic/Latino populations: applications in the Hispanic community health study/study of Latinos. Am. J. Hum. Genet 98:165–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manichaikul A, Palmas W, Rodriguez CJ, Peralta CA, Divers J, et al. 2012. Population structure of Hispanics in the United States: the multi-ethnic study of atherosclerosis. PLOS Genet. 8:e1002640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gravel S, Zakharia F, Moreno-Estrada A, Byrnes JK, Muzzio M, et al. 2013. Reconstructing Native American migrations from whole-genome and whole-exome data. PLOS Genet. 9:e1004023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mak AC, White MJ, Eckalbar WL, Szpiech ZA, Oh SS, et al. 2018. Whole genome sequencing of pharmacogenetic drug response in racially diverse children with asthma. Am. J. Respir. Crit. Care Med 197:1552–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sim SC, Ingelman-Sundberg M. 2013. Update on allele nomenclature for human cytochromes P450 and the Human Cytochrome P450 Allele (CYP-allele) Nomenclature Database. Methods Mol. Bio 987:251–59 [DOI] [PubMed] [Google Scholar]
- 33.Zhou SF. 2009. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin. Pharmacokinet 48:689–723 [DOI] [PubMed] [Google Scholar]
- 34.Crews KR, Gaedigk A, Dunnenberger HM, Leeder JS, Klein TE, et al. 2014. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin. Pharmacol. Ther 95:376–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dean L 2012. Codeine therapy and CYP2D6 genotype In Medical Genetics Summaries, ed. Pratt V, McLeod H, Dean L, Malheiro A, Rubinstein W. Bethesda, MD: Natl. Cent. Biotechnol. Inf. [Google Scholar]
- 36.Ahmed S, Zhou Z, Zhou J, Chen SQ. 2016. Pharmacogenomics of drug metabolizing enzymes and transporters: relevance to precision medicine. Genom. Proteom. Bioinformat 14:298–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasche Y, Daali Y, Fathi M, Chiappe A, Cottini S, et al. 2004. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N. Engl. J. Med 351:2827–31 [DOI] [PubMed] [Google Scholar]
- 38.Dalen P, Frengell C, Dahl ML, Sjoqvist F. 1997. Quick onset of severe abdominal pain after codeine in an ultrarapid metabolizer of debrisoquine. Ther. Drug Monit 19:543–44 [DOI] [PubMed] [Google Scholar]
- 39.Lamba JK, Lin YS, Schuetz EG, Thummel KE. 2002. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev 54:1271–94 [DOI] [PubMed] [Google Scholar]
- 40.Trofe-Clark J, Brennan DC, West-Thielke P, Milone MC, Lim MA, et al. 2018. Results of ASERTAA, a randomized prospective crossover pharmacogenetic study of immediate-release versus extended-release tacrolimus in African American kidney transplant recipients. Am. J. Kidney Dis 71:315–26 [DOI] [PubMed] [Google Scholar]
- 41.Taber DJ, Su Z, Fleming JN, McGillicuddy JW, Posadas-Salas MA, et al. 2017. Tacrolimus trough concentration variability and disparities in African American kidney transplantation. Transplantation 101:2931–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Court MH, Zhu Z, Masse G, Duan SX, James LP, et al. 2017. Race, gender, and genetic polymorphism contribute to variability in acetaminophen pharmacokinetics, metabolism, and protein-adduct concentrations in healthy African-American and European-American volunteers. J. Pharmacol. Exp. Ther 362:431–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowell S, Falany CN. 2006. Pharmacogenetics of human cytosolic sulfotransferases. Oncogene 25:1673–78 [DOI] [PubMed] [Google Scholar]
- 44.Fukami T, Nakajima M, Higashi E, Yamanaka H, McLeod HL, Yokoi T. 2005. A novel CYP2A6*20 allele found in African-American population produces a truncated protein lacking enzymatic activity. Biochem. Pharmacol 70:801–8 [DOI] [PubMed] [Google Scholar]
- 45.Brown NJ, Vaughan DE. 1998. Angiotensin-converting enzyme inhibitors. Circulation 97:1411–20 [DOI] [PubMed] [Google Scholar]
- 46.Cavallari LH, Langaee TY, Momary KM, Shapiro NL, Nutescu EA, et al. 2010. Genetic and clinical predictors of warfarin dose requirements in African Americans. Clin. Pharmacol. Ther 87:459–64 [DOI] [PubMed] [Google Scholar]
- 47.Limdi NA, McGwin G, Goldstein JA, Beasley TM, Arnett DK, et al. 2008. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin. Pharmacol. Ther 83:312–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perera MA, Gamazon E, Cavallari LH, Patel SR, Poindexter S, et al. 2011. The missing association: sequencing-based discovery of novel SNPs in VKORC1 and CYP2C9 that affect warfarin dose in African Americans. Clin. Pharmacol. Ther 89:408–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang N, Agrawal V, Giacomini KM, Miller WL. 2008. Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. PNAS 105:1733–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zareh M, Davis A, Henderson S. 2011. Reversal of warfarin-induced hemorrhage in the emergency department. West. J. Emerg. Med 12:386–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dang MT, Hambleton J, Kayser SR. 2005. The influence of ethnicity on warfarin dosage requirement. Ann. Pharmacother 39:1008–12 [DOI] [PubMed] [Google Scholar]
- 52.Limdi NA, Brown TM, Shendre A, Liu N, Hill CE, Beasley TM. 2017. Quality of anticoagulation control and hemorrhage risk among African American and European American warfarin users. Pharmacogenet. Genom 27:347–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen AY, Chen W, Yao JF, Brar SS, Wang X, Go AS. 2008. Effect of race/ethnicity on the efficacy of warfarin: potential implications for prevention of stroke in patients with atrial fibrillation. CNS Drugs 22:815–25 [DOI] [PubMed] [Google Scholar]
- 54.Blann A, Hewitt J, Siddiqui F, Bareford D. 1999. Racial background is a determinant of average warfarin dose required to maintain the INR between 2.0 and 3.0. Br. J. Haematol 107:207–9 [DOI] [PubMed] [Google Scholar]
- 55.Harper P, Monahan K, Baker B. 2005. Warfarin induction at 5 mg daily is safe with a low risk of anticoagulant overdose: results of an audit of patients with deep vein thrombosis commencing warfarin. Intern. Med. J 35:717–20 [DOI] [PubMed] [Google Scholar]
- 56.Crowther MA, Ginsberg JB, Kearon C, Harrison L, Johnson J, et al. 1999. A randomized trial comparing 5-mg and 10-mg warfarin loading doses. Arch. Intern. Med 159:46–48 [DOI] [PubMed] [Google Scholar]
- 57.Harrison L, Johnston M, Massicotte MP, Crowther M, Moffat K, Hirsh J. 1997. Comparison of 5-mg and 10-mg loading doses in initiation of warfarin therapy. Ann. Intern. Med 126:133–36 [DOI] [PubMed] [Google Scholar]
- 58.Jonas DE, McLeod HL. 2009. Genetic and clinical factors relating to warfarin dosing. Trends Pharmacol. Sci 30:375–86 [DOI] [PubMed] [Google Scholar]
- 59.Cha PC, Mushiroda T, Takahashi A, Kubo M, Minami S, et al. 2010. Genome-wide association study identifies genetic determinants of warfarin responsiveness for Japanese. Hum. Mol. Genet 19:4735–44 [DOI] [PubMed] [Google Scholar]
- 60.Rieder MJ, Reiner AP, Gage BF, Nickerson DA, Eby CS, et al. 2005. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med 352:2285–93 [DOI] [PubMed] [Google Scholar]
- 61.Gage BF, Eby C, Johnson JA, Deych E, Rieder MJ, et al. 2008. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin. Pharmacol. Ther 84:326–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, et al. 2009. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLOS Genet. 5:e1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Limdi NA, Beasley TM, Crowley MR, Goldstein JA, Rieder MJ, et al. 2008. VKORC1 polymorphisms, haplotypes and haplotype groups on warfarin dose among African-Americans and European-Americans. Pharmacogenomics 9:1445–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Int. Warfarin Pharmacogenet. Consort., Klein TE, Altman RB, Eriksson N, Gage BF, et al. 2009. Estimation of the warfarin dose with clinical and pharmacogenetic data. N. Engl. J. Med 360:753–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Limdi NA, Wadelius M, Cavallari L, Eriksson N, Crawford DC, et al. 2010. Warfarin pharmacogenetics: A single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood 115:3827–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bress A, Patel SR, Perera MA, Campbell RT, Kittles RA, Cavallari LH. 2012. Effect of NQO1 and CYP4F2 genotypes on warfarin dose requirements in Hispanic-Americans and African-Americans. Pharmacogenomics 13:1925–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pirmohamed M, Burnside G, Eriksson N, Jorgensen AL, Toh CH, et al. 2013. A randomized trial of genotype-guided dosing of warfarin. N. Engl. J. Med 369:2294–303 [DOI] [PubMed] [Google Scholar]
- 68.Kimmel SE, French B, Kasner SE, Johnson JA, Anderson JL, et al. 2013. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med 369:2283–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cavallari LH, Kittles RA, Perera MA. 2014. Genotype-guided dosing of vitamin K antagonists. N. Engl. J. Med 370:1761–66 [DOI] [PubMed] [Google Scholar]
- 70.Gage BF, Bass AR, Lin H, Woller SC, Stevens SM, et al. 2017. Effect of genotype-guided warfarin dosing on clinical events and anticoagulation control among patients undergoing hip or knee arthroplasty: the GIFT randomized clinical trial. JAMA 318:1115–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie HG, Prasad HC, Kim RB, Stein CM. 2002. CYP2C9 allelic variants: ethnic distribution and functional significance. Adv. Drug Deliv. Rev 54:1257–70 [DOI] [PubMed] [Google Scholar]
- 72.Scott SA, Jaremko M, Lubitz SA, Kornreich R, Halperin JL, Desnick RJ. 2009. CYP2C9*8 is prevalent among African-Americans: implications for pharmacogenetic dosing. Pharmacogenomics 10:1243–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perera MA, Cavallari LH, Limdi NA, Gamazon ER, Konkashbaev A, et al. 2013. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. Lancet 382:790–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wadelius M, Chen LY, Eriksson N, Bumpstead S, Ghori J, et al. 2007. Association of warfarin dose with genes involved in its action and metabolism. Hum. Genet 121:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ray S 2014. Clopidogrel resistance: the way forward. Indian Heart J. 66:530–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nguyen TA, Diodati JG, Pharand C. 2005. Resistance to clopidogrel: a review of the evidence. J. Am. Coll. Cardiol 45:1157–64 [DOI] [PubMed] [Google Scholar]
- 77.Cresci S, Depta JP, Lenzini PA, Li AY, Lanfear DE, et al. 2014. Cytochrome P450 gene variants, race, and mortality among clopidogrel-treated patients after acute myocardial infarction. Circ. Cardiovasc. Genet 7:277–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mak KH, Bhatt DL, Shao M, Hankey GJ, Easton JD, et al. 2009. Ethnic variation in adverse cardiovascular outcomes and bleeding complications in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study. Am. Heart J 157:658–65 [DOI] [PubMed] [Google Scholar]
- 79.Pendyala LK, Torguson R, Loh JP, Devaney JM, Chen F, et al. 2013. Racial disparity with on-treatment platelet reactivity in patients undergoing percutaneous coronary intervention. Am. Heart J 166:266–72 [DOI] [PubMed] [Google Scholar]
- 80.Hulot JS, Bura A, Villard E, Azizi M, Remones V, et al. 2006. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood 108:2244–47 [DOI] [PubMed] [Google Scholar]
- 81.Guo B, Tan Q, Guo D, Shi Z, Zhang C, Guo W. 2014. Patients carrying CYP2C19 loss of function alleles have a reduced response to clopidogrel therapy and a greater risk of in-stent restenosis after endovascular treatment of lower extremity peripheral arterial disease. J. Vasc. Surg 60:993–1001 [DOI] [PubMed] [Google Scholar]
- 82.Zhu WY, Zhao T, Xiong XY, Li J, Wang L, et al. 2016. Association of CYP2C19 polymorphisms with the clinical efficacy of clopidogrel therapy in patients undergoing carotid artery stenting in Asia. Sci. Rep 6:25478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cavallari LH, Lee CR, Beitelshees AL, Cooper-DeHoff RM, Duarte JD, et al. 2018. Multisite investigation of outcomes with implementation of CYP2C19 genotype-guided antiplatelet therapy after percutaneous coronary intervention. JACC Cardiovasc. Interv 11:181–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harmsze AM, van Werkum JW, Hackeng CM, Ruven HJ, Kelder JC, et al. 2012. The influence of CYP2C19*2 and *17 on on-treatment platelet reactivity and bleeding events in patients undergoing elective coronary stenting. Pharmacogenet. Genom 22:169–75 [DOI] [PubMed] [Google Scholar]
- 85.Dean L 2012. Clopidogrel therapy and CYP2C19 genotype In Medical Genetics Summaries, ed. Pratt V, McLeod H, Dean L, Malheiro A, Rubinstein W. Bethesda, MD: Natl. Cent. Biotechnol. Inf. [Google Scholar]
- 86.Scott SA, Sangkuhl K, Stein CM, Hulot JS, Mega JL, et al. 2013. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin. Pharmacol. Ther 94:317–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, et al. 2009. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med 361:1045–57 [DOI] [PubMed] [Google Scholar]
- 88.Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, et al. 2007. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med 357:2001–15 [DOI] [PubMed] [Google Scholar]
- 89.Steg PG, Harrington RA, Emanuelsson H, Katus HA, Mahaffey KW, et al. 2013. Stent thrombosis with ticagrelor versus clopidogrel in patients with acute coronary syndromes: an analysis from the prospective, randomized PLATO trial. Circulation 128:1055–65 [DOI] [PubMed] [Google Scholar]
- 90.Pereira NL, Sargent DJ, Farkouh ME, Rihal CS. 2015. Genotype-based clinical trials in cardiovascular disease. Nat. Rev. Cardiol 12:475–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bergmeijer TO, Janssen PW, Schipper JC, Qaderdan K, Ishak M, et al. 2014. CYP2C19 genotype-guided antiplatelet therapy in ST-segment elevation myocardial infarction patients—rationale and design of the Patient Outcome after primary PCI (POPular) genetics study. Am. Heart J 168:16–22.e1 [DOI] [PubMed] [Google Scholar]
- 92.Gravlee CC, Dressler WW, Bernard HR. 2005. Skin color, social classification, and blood pressure in southeastern Puerto Rico. Am. J. Public Health 95:2191–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jamerson K, DeQuattro V. 1996. The impact of ethnicity on response to antihypertensive therapy. Am. J. Med 101:22S–32S [DOI] [PubMed] [Google Scholar]
- 94.Rostand SG, Brown G, Kirk KA, Rutsky EA, Dustan HP. 1989. Renal insufficiency in treated essential hypertension. N. Engl. J. Med 320:684–88 [DOI] [PubMed] [Google Scholar]
- 95.Williams SF, Nicholas SB, Vaziri ND, Norris KC. 2014. African Americans, hypertension and the renin angiotensin system. World J. Cardiol 6:878–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou HH, Koshakji RP, Silberstein DJ, Wilkinson GR, Wood AJ. 1989. Racial differences in drug response. Altered sensitivity to and clearance of propranolol in men of Chinese descent as compared with American whites. N. Engl. J. Med 320:565–70 [DOI] [PubMed] [Google Scholar]
- 97.Small KM, McGraw DW, Liggett SB. 2003. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu. Rev. Pharmacol. Toxicol 43:381–411 [DOI] [PubMed] [Google Scholar]
- 98.Johnson JA, Zineh I, Puckett BJ, McGorray SP, Yarandi HN, Pauly DF. 2003. β1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin. Pharmacol. Ther 74:44–52 [DOI] [PubMed] [Google Scholar]
- 99.Kurnik D, Li C, Sofowora GG, Friedman EA, Muszkat M, et al. 2008. Beta-1-adrenoceptor genetic variants and ethnicity independently affect response to beta-blockade. Pharmacogenet. Genom 18:895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bruck H, Leineweber K, Buscher R, Ulrich A, Radke J, et al. 2003. The Gln27Glu beta2-adrenoceptor polymorphism slows the onset of desensitization of cardiac functional responses in vivo. Pharmacogenetics 13:59–66 [DOI] [PubMed] [Google Scholar]
- 101.Dishy V, Sofowora GG, Xie HG, Kim RB, Byrne DW, et al. 2001. The effect of common polymorphisms of the β2-adrenergic receptor on agonist-mediated vascular desensitization. N. Engl. J. Med 345:1030–35 [DOI] [PubMed] [Google Scholar]
- 102.Lanfear DE, Jones PG, Marsh S, Cresci S, McLeod HL, Spertus JA. 2005. β2-adrenergic receptor genotype and survival among patients receiving β-blocker therapy after an acute coronary syndrome. JAMA 294:1526–33 [DOI] [PubMed] [Google Scholar]
- 103.van Rijn-Bikker PC, Ackaert O, Snelder N, van Hest RM, Ploeger BA, et al. 2013. Pharmacokinetic-pharmacodynamic modeling of the antihypertensive effect of eprosartan in Black and White hypertensive patients. Clin. Pharmacokinet 52:793–803 [DOI] [PubMed] [Google Scholar]
- 104.Ferdinand KC, Armani AM. 2007. The management of hypertension in African Americans. Crit. Pathw. Cardiol 6:67–71 [DOI] [PubMed] [Google Scholar]
- 105.Cohn JN, Julius S, Neutel J, Weber M, Turlapaty P, et al. 2004. Clinical experience with perindopril in African-American hypertensive patients: a large United States community trial. Am. J. Hypertens 17:134–38 [DOI] [PubMed] [Google Scholar]
- 106.Wright JT Jr., Dunn JK, Cutler JA, Davis BR, Cushman WC, et al. 2005. Outcomes in hypertensive black and nonblack patients treated with chlorthalidone, amlodipine, and lisinopril. JAMA 293:1595–608 [DOI] [PubMed] [Google Scholar]
- 107.Rieder MJ, Taylor SL, Clark AG, Nickerson DA. 1999. Sequence variation in the human angiotensin converting enzyme. Nat. Genet 22:59–62 [DOI] [PubMed] [Google Scholar]
- 108.Johnson AD, Gong Y, Wang D, Langaee TY, Shin J, et al. 2009. Promoter polymorphisms in ACE (angiotensin I-converting enzyme) associated with clinical outcomes in hypertension. Clin. Pharmacol. Ther 85:36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vandell AG, McDonough CW, Gong Y, Langaee TY, Lucas AM, et al. 2014. Hydrochlorothiazide-induced hyperuricaemia in the pharmacogenomic evaluation of antihypertensive responses study. J. Intern. Med 276:486–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakanishi N, Okamoto M, Yoshida H, Matsuo Y, Suzuki K, Tatara K. 2003. Serum uric acid and risk for development of hypertension and impaired fasting glucose or Type II diabetes in Japanese male office workers. Eur. J. Epidemiol 18:523–30 [DOI] [PubMed] [Google Scholar]
- 111.Bhatnagar V, O’Connor DT, Schork NJ, Salem RM, Nievergelt CM, et al. 2007. Angiotensin-converting enzyme gene polymorphism predicts the time-course of blood pressure response to angiotensin converting enzyme inhibition in the AASK trial. J. Hypertens 25:2082–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim TH, Chang HS, Park SM, Nam BY, Park JS, et al. 2008. Association of angiotensin I-converting enzyme gene polymorphisms with aspirin intolerance in asthmatics. Clin. Exp. Allergy 38:1727–37 [DOI] [PubMed] [Google Scholar]
- 113.Liu J, Liu ZQ, Yu BN, Xu FH, Mo W, et al. 2006. β1-Adrenergic receptor polymorphisms influence the response to metoprolol monotherapy in patients with essential hypertension. Clin. Pharmacol. Ther 80:23–32 [DOI] [PubMed] [Google Scholar]
- 114.Liu J, Liu ZQ, Tan ZR, Chen XP, Wang LS, et al. 2003. Gly389Arg polymorphism of β1-adrenergic receptor is associated with the cardiovascular response to metoprolol. Clin. Pharmacol. Ther 74:372–79 [DOI] [PubMed] [Google Scholar]
- 115.Sofowora GG, Dishy V, Muszkat M, Xie HG, Kim RB, et al. 2003. A common beta1-adrenergic receptor polymorphism (Arg389Gly) affects blood pressure response to beta-blockade. Clin. Pharmacol. Ther 73:366–71 [DOI] [PubMed] [Google Scholar]
- 116.Hadidi H, Zahlsen K, Idle JR, Cholerton S. 1997. A single amino acid substitution (Leu160His) in cytochrome P450 CYP2A6 causes switching from 7-hydroxylation to 3-hydroxylation of coumarin. Food Chem. Toxicol 35:903–7 [DOI] [PubMed] [Google Scholar]
- 117.Haberl M, Anwald B, Klein K, Weil R, Fuss C, et al. 2005. Three haplotypes associated with CYP2A6 phenotypes in Caucasians. Pharmacogenet. Genom 15:609–24 [DOI] [PubMed] [Google Scholar]
- 118.Lane S, Al-Zubiedi S, Hatch E, Matthews I, Jorgensen AL, et al. 2012. The population pharmacokinetics of R- and S-warfarin: effect of genetic and clinical factors. Br. J. Clin. Pharmacol 73:66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramirez C, Cavallari U, et al. 2006. Contribution of gene sequence variations of the hepatic cytochrome P450 3A4 enzyme to variability in individual responsiveness to clopidogrel. Arterioscler. Thromb. Vasc. Biol 26:1895–900 [DOI] [PubMed] [Google Scholar]
- 120.Kassimis G, Davlouros P, Xanthopoulou I, Stavrou EF, Athanassiadou A, Alexopoulos D. 2012. CYP2C19*2 and other genetic variants affecting platelet response to clopidogrel in patients undergoing percutaneous coronary intervention. Thromb. Res 129:441–46 [DOI] [PubMed] [Google Scholar]
- 121.Shuldiner AR, O’Connell JR, Bliden KP, Gandhi A, Ryan K, et al. 2009. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 302:849–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang H, Ma K, Liu W, Yang F, Liu J, Zhou H. 2016. Impact of CYP2C19 gene polymorphism on warfarin maintenance doses in patients with non-valvular atrial fibrillation. Gene 591:80–84 [DOI] [PubMed] [Google Scholar]
- 123.Sibbing D, Koch W, Gebhard D, Schuster T, Braun S, et al. 2010. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 121:512–18 [DOI] [PubMed] [Google Scholar]
- 124.Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, et al. 2009. Genetic determinants of response to clopidogrel and cardiovascular events. N. Engl. J. Med 360:363–75 [DOI] [PubMed] [Google Scholar]
- 125.Wu H, Qian J, Xu J, Sun A, Sun W, et al. 2012. Effects of CYP2C19 variant alleles on postclopidogrel platelet reactivity and clinical outcomes in an actual clinical setting in China. Pharmacogenet. Genom 22:887–90 [DOI] [PubMed] [Google Scholar]
- 126.Novkovic M, Matic D, Kusic-Tisma J, Antonijevic N, Radojkovic D, Rakicevic L. 2018. Analysis of the CYP2C19 genotype associated with bleeding in Serbian STEMI patients who have undergone primary PCI and treatment with clopidogrel. Eur. J. Clin. Pharmacol 74:443–51 [DOI] [PubMed] [Google Scholar]
- 127.Fuchshuber-Moraes M, Perini JA, Rosskopf D, Suarez-Kurtz G. 2009. Exploring warfarin pharmacogenomics with the extreme-discordant-phenotype methodology: impact of FVII polymorphisms on stable anticoagulation with warfarin. Eur. J. Clin. Pharmacol 65:789–93 [DOI] [PubMed] [Google Scholar]
- 128.Samardzija M, Topic E, Stefanovic M, Zibar L, Samardzija G, et al. 2008. Association of CYP2C9 gene polymorphism with bleeding as a complication of warfarin therapy. Coll. Antropol 32:557–64 [PubMed] [Google Scholar]
- 129.Ablin J, Cabili S, Eldor A, Lagziel A, Peretz H. 2004. Warfarin therapy is feasible in CYP2C9*3 homozygous patients. Eur. J. Intern. Med 15:22–27 [DOI] [PubMed] [Google Scholar]
- 130.Esmerian MO, Mitri Z, Habbal MZ, Geryess E, Zaatari G, et al. 2011. Influence of CYP2C9 and VKORC1 polymorphisms on warfarin and acenocoumarol in a sample of Lebanese people. J. Clin. Pharmacol 51:1418–28 [DOI] [PubMed] [Google Scholar]
- 131.Dean L 2012. Warfarin therapy and the genotypes CYP2C9 and VKORC1 In Medical Genetics Summaries, ed. Pratt V, McLeod H, Dean L, Malheiro A, Rubinstein W. Bethesda, MD: Natl. Cent. Biotechnol. Inf. [Google Scholar]
- 132.Montes R, Nantes O, Alonso A, Zozaya JM, Hermida J. 2008. The influence of polymorphisms of VKORC1 and CYP2C9 on major gastrointestinal bleeding risk in anticoagulated patients. Br. J. Haematol 143:727–33 [DOI] [PubMed] [Google Scholar]
- 133.Cavallari LH, Vaynshteyn D, Freeman KM, Wang D, Perera MA, et al. 2013. CYP2C9 promoter region single-nucleotide polymorphisms linked to the R150H polymorphism are functional suggesting their role in CYP2C9*8-mediated effects. Pharmacogenet. Genom 23:228–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cooper GM, Johnson JA, Langaee TY, Feng H, Stanaway IB, et al. 2008. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 112:1022–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rau T, Heide R, Bergmann K, Wuttke H, Werner U, et al. 2002. Effect of the CYP2D6 genotype on metoprolol metabolism persists during long-term treatment. Pharmacogenetics 12:465–72 [DOI] [PubMed] [Google Scholar]
- 136.Takekuma Y, Takenaka T, Kiyokawa M, Yamazaki K, Okamoto H, et al. 2007. Evaluation of effects of polymorphism for metabolic enzymes on pharmacokinetics of carvedilol by population pharmacokinetic analysis. Biol. Pharm. Bull 30:537–42 [DOI] [PubMed] [Google Scholar]
- 137.Rau T, Wuttke H, Michels LM, Werner U, Bergmann K, et al. 2009. Impact of the CYP2D6 genotype on the clinical effects of metoprolol: a prospective longitudinal study. Clin. Pharmacol. Ther 85:269–72 [DOI] [PubMed] [Google Scholar]
- 138.Blake CM, Kharasch ED, Schwab M, Nagele P. 2013. A meta-analysis of CYP2D6 metabolizer phenotype and metoprolol pharmacokinetics. Clin. Pharmacol. Ther 94:394–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Honda M, Ogura Y, Toyoda W, Taguchi M, Nozawa T, et al. 2006. Multiple regression analysis of pharmacogenetic variability of carvedilol disposition in 54 healthy Japanese volunteers. Biol. Pharm. Bull 29:772–78 [DOI] [PubMed] [Google Scholar]
- 140.Park KW, Park JJ, Jeon KH, Kang SH, Oh IY, et al. 2011. Enhanced clopidogrel responsiveness in smokers: Smokers’ paradox is dependent on cytochrome P450 CYP1A2 status. Arterioscler. Thromb. Vasc. Biol 31:665–71 [DOI] [PubMed] [Google Scholar]
- 141.Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, et al. 2008. CYP4F2 genetic variant alters required warfarin dose. Blood 111:4106–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Danese E, Montagnana M, Johnson JA, Rettie AE, Zambon CF, et al. 2012. Impact of the CYP4F2 p.V433M polymorphism on coumarin dose requirement: systematic review and meta-analysis. Clin. Pharmacol. Ther 92:746–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Slooter AJ, Rosendaal FR, Tanis BC, Kemmeren JM, van der Graaf Y, Algra A. 2005. Prothrombotic conditions, oral contraceptives, and the risk of ischemic stroke. J. Thromb. Haemost 3:1213–17 [DOI] [PubMed] [Google Scholar]
- 144.Newton-Cheh C, Guo CY, Larson MG, Musone SL, Surti A, et al. 2007. Common genetic variation in KCHN2 is associated with QT interval duration: the Framingham Heart Study. Circulation 116:1128–36 [DOI] [PubMed] [Google Scholar]
- 145.Yang P, Kanki H, Drolet B, Yang T, Wei J, et al. 2002. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation 105:1943–48 [DOI] [PubMed] [Google Scholar]
- 146.Sun Z, Milos PM, Thompson JF, Lloyd DB, Mank-Seymour A, et al. 2004. Role of a KCNH2 polymorphism (R1047 L) in dofetilide-induced Torsades de Pointes. J. Mol. Cell Cardiol 37:1031–39 [DOI] [PubMed] [Google Scholar]
- 147.Jiang S, Hsu YH, Xu X, Xing H, Chen C, et al. 2004. The C677T polymorphism of the methylenetetrahydrofolate reductase gene is associated with the level of decrease on diastolic blood pressure in essential hypertension patients treated by angiotensin-converting enzyme inhibitor. Thromb. Res 113:361–69 [DOI] [PubMed] [Google Scholar]
- 148.Chung JE, Chang BC, Lee KE, Kim JH, Gwak HS. 2015. Effects of NAD(P)H quinone oxidoreductase 1 polymorphisms on stable warfarin doses in Korean patients with mechanical cardiac valves. Eur. J. Clin. Pharmacol 71:1229–36 [DOI] [PubMed] [Google Scholar]
- 149.Jefferson BK, Foster JH, McCarthy JJ, Ginsburg G, Parker A, et al. 2005. Aspirin resistance and a single gene. Am. J. Cardiol 95:805–8 [DOI] [PubMed] [Google Scholar]
- 150.Wang X, Lai Y, Luo Y, Zhang X, Zhou H, et al. 2016. Relationship between clopidogrel-related polymorphisms and variable platelet reactivity at 1 year: a cohort study from Han Chinese. J. Res. Med. Sci 21:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sharma V, Kaul S, Al-Hazzani A, Alshatwi AA, Jyothy A, Munshi A. 2013. Association of COX-2 rs20417 with aspirin resistance. J. Thromb. Thrombolysis 35:95–99 [DOI] [PubMed] [Google Scholar]
- 152.Luo JQ, He FZ, Wang ZM, Sun NL, Wang LY, et al. 2015. SLCO1B1 variants and angiotensin converting enzyme inhibitor (enalapril)-induced cough: a pharmacogenetic study. Sci. Rep 5:17253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Varenhorst C, Eriksson N, Johansson A, Barratt BJ, Hagstrom E, et al. 2015. Effect of genetic variations on ticagrelor plasma levels and clinical outcomes. Eur. Heart J 36:1901–12 [DOI] [PubMed] [Google Scholar]
- 154.Shahabi P, Lamothe F, Dumas S, Rouleau-Mailloux E, Feroz Zada Y, et al. 2018. Nuclear receptor gene polymorphisms and warfarin dose requirements in the Quebec Warfarin Cohort. Pharmacogenom. J In press. 10.1038/s41397-017-0005-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Herman D, Peternel P, Stegnar M, Breskvar K, Dolzan V. 2006. The influence of sequence variations in factor VII, gamma-glutamyl carboxylase and vitamin K epoxide reductase complex genes on warfarin dose requirement. Thromb. Haemost 95:782–87 [PubMed] [Google Scholar]
- 156.D’Andrea G, D’Ambrosio RL, Di Perna P, Chetta M, Santacroce R, et al. 2005. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood 105:645–49 [DOI] [PubMed] [Google Scholar]
- 157.Pop TR, Vesa S, Trifa AP, Crisan S, Buzoianu AD. 2013. An acenocoumarol dose algorithm based on a South-Eastern European population. Eur. J. Clin. Pharmacol 69:1901–7 [DOI] [PubMed] [Google Scholar]
- 158.Kringen MK, Haug KB, Grimholt RM, Stormo C, Narum S, et al. 2011. Genetic variation of VKORC1 and CYP4F2 genes related to warfarin maintenance dose in patients with myocardial infarction. J. Biomed. Biotechnol 2011:739751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nguyen N, Anley P, Yu MY, Zhang G, Thompson AA, Jennings LJ. 2013. Genetic and clinical determinants influencing warfarin dosing in children with heart disease. Pediatr. Cardiol 34:984–90 [DOI] [PubMed] [Google Scholar]
- 160.Michaud V, Vanier MC, Brouillette D, Roy D, Verret L, et al. 2008. Combination of phenotype assessments and CYP2C9-VKORC1 polymorphisms in the determination of warfarin dose requirements in heavily medicated patients. Clin. Pharmacol. Ther 83:740–48 [DOI] [PubMed] [Google Scholar]
- 161.Chan SL, Suo C, Lee SC, Goh BC, Chia KS, Teo YY. 2012. Translational aspects of genetic factors in the prediction of drug response variability: a case study of warfarin pharmacogenomics in a multi-ethnic cohort from Asia. Pharmacogenom. J 12:312–18 [DOI] [PubMed] [Google Scholar]
- 162.Mitchell C, Gregersen N, Krause A. 2011. Novel CYP2C9 and VKORC1 gene variants associated with warfarin dosage variability in the South African black population. Pharmacogenomics 12:953–63 [DOI] [PubMed] [Google Scholar]
- 163.Saminathan R, Bai J, Sadrolodabaee L, Karthik GM, Singh O, et al. 2010. VKORC1 pharmacogenetics and pharmacoproteomics in patients on warfarin anticoagulant therapy: transthyretin precursor as a potential biomarker. PLOS ONE 5:e15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schelleman H, Brensinger CM, Chen J, Finkelman BS, Rieder MJ, Kimmel SE. 2010. New genetic variant that might improve warfarin dose prediction in African Americans. Br. J. Clin. Pharmacol 70:393–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wadelius M, Chen LY, Downes K, Ghori J, Hunt S, et al. 2005. Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharmacogenom. J 5:262–70 [DOI] [PubMed] [Google Scholar]
- 166.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, et al. 2017. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45(D1):D896–901 [DOI] [PMC free article] [PubMed] [Google Scholar]