

Abstract
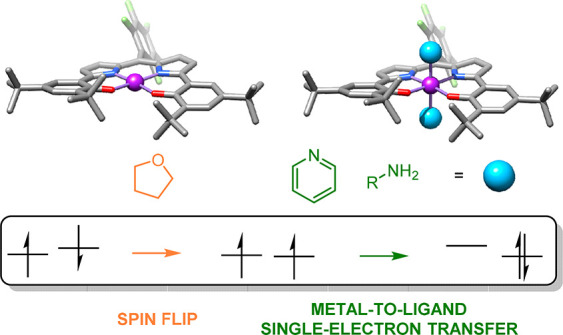
The influence of a redox-active ligand on spin-changing events induced by the coordination of exogenous donors is investigated within the cobalt complex [CoII(DPP·2–)], bearing a redox-active DPP2– ligand (DPP = dipyrrin-bis(o,p-di-tert-butylphenolato) with a pentafluorophenyl moiety on the meso-position. This square-planar complex was subjected to the coordination of tetrahydrofuran (THF), pyridine, tBuNH2, and AdNH2 (Ad = 1-adamantyl), and the resulting complexes were analyzed with a variety of experimental (X-ray diffraction, NMR, UV–visible, high-resolution mass spectrometry, superconducting quantum interference device, Evans’ method) and computational (density functional theory, NEVPT2-CASSCF) techniques to elucidate the respective structures, spin states, and orbital compositions of the corresponding octahedral bis-donor adducts, relative to [CoII(DPP·2–)]. This starting species is best described as an open-shell singlet complex containing a DPP·2– ligand radical that is antiferromagnetically coupled to a low-spin (S = 1/2) cobalt(II) center. The redox-active DPPn– ligand plays a crucial role in stabilizing this complex and in its facile conversion to the triplet THF adduct [CoII(DPP·2–)(THF)2] and closed-shell singlet pyridine and amine adducts [CoIII(DPP3–)(L)2] (L = py, tBuNH2, or AdNH2). Coordination of the weak donor THF to [CoII(DPP·2–)] changes the orbital overlap between the DPP·2– ligand radical π-orbitals and the cobalt(II) metalloradical d-orbitals, which results in a spin-flip to the triplet ground state without changing the oxidation states of the metal or DPP·2– ligand. In contrast, coordination of the stronger donors pyridine, tBuNH2, or AdNH2 induces metal-to-ligand single-electron transfer, resulting in the formation of low-spin (S = 0) cobalt(III) complexes [CoIII(DPP3–)(L)2] containing a fully reduced DPP3– ligand, thus explaining their closed-shell singlet electronic ground states.
Short abstract
A new cobalt complex bearing a redox-active dipyrrin-bisphenol ligand ([CoII(DPP·2−)]) is characterized as an open-shell singlet due to antiferromagnetic coupling between ligand- and cobalt(II)-centered unpaired electrons. Bis-coordination of THF on the axial positions results in a spin-flip to the triplet state via changing orbital overlap between the redox-active ligand and metal center. Coordination of stronger donors (pyridine and primary amines) induces metal-to-ligand single-electron transfer to yield the octahedral complexes in the closed-shell singlet state.
Introduction
Spin-state changes (spin crossover) can play an important role in chemistry and material research, among others in biochemistry (respiration, enzymatic conversions),1 development of molecular magnets2 and spintronics,3 and as a potential rate-accelerating process in organometallic chemistry and catalysis.4 Purely metal-centered spin-state changes of coordination complexes can be explained in terms of the coordination and geometry-dependent energy difference between partially filled and empty d-orbitals, as described by the ligand-field splitting parameter Δ.5
The respective roles of the metal and traditional (redox-innocent) ligands are well-understood in these cases. However, when a redox-active ligand, capable of bearing unpaired electrons, is present in the coordination sphere of the metal, the relative contributions and the roles and influence of metal and ligand to changes in the total spin state of the overall complex are far less well-understood. The main four modes of action of redox-active ligands that have been studied thoroughly can be summarized as (i) changing the Lewis acidity/basicity of the metal, (ii) acting as an electron reservoir, (iii) generation of a reactive ligand-centered radical, and (iv) radical-type activation of a substrate.6 We wish to expand upon these functions by investigating the role of a redox-active ligand in spin-changing events. Specifically, by keeping the redox-active ligand and metal center constant we set out to investigate how the coordination of different additional redox-innocent donors to the metal center influences the total spin state of the complex, which is governed by the interactions of the metal d-orbitals and the redox-active ligand orbitals of π-symmetry.
In this context, we became interested in the family of dipyrrin-bisphenol ligands (DPP, Figure 1), known since the 1970s.7 Different substitution patterns on the backbone were explored since 2009, and complexes of Al,8,9 B,8 Ga,9 In,9 Ti,10 Zr,10 Ge,10 Sn,10 and Mn11 have been reported. The DPP ligand scaffold was first described as being redox-active in 2012 after the synthesis of cobalt- and nickel-DPP complexes.12 Hereafter, the redox activity was further studied in Mn,13 Pt,14 Cu,15 and Au16 complexes. Catalytic applications have been reported for the Ti, Zr, Gn, and Sn complexes (copolymerization of epoxides with CO2) and Cu (aerobic alcohol oxidation).9,15 Contrarily, cobalt(III)-DPP complexes proved catalytically inactive in the epoxide ring-opening reaction with alcohols, which was attributed to the low Lewis acidity of the cobalt center.17
Figure 1.

General structure of a dipyrrin-bisphenol (DPP) ligand on a metal (M).
Initial studies on cobalt-DPP complexes were mainly focused on the comparison of their (redox) properties and (catalytic) reactivity with cobalt-porphyrin, -salen, and -corrole analogues. The ligand was predominantly found to coordinate as a dianionic (radical) ligand to a low-spin cobalt(II) center in neutral Co-DPP complexes.12 Density functional theory (DFT) calculations indicated that the triplet and broken-symmetry open-shell singlet (BSS) ground state (inferring (anti)ferromagnetic coupling between the metal- and ligand-centered unpaired electrons) are energetically close (∼1 kcal mol–1).12,17 Although a BSS (S = 0) spin state was inferred based on experimental data for a Co-DPP complex, the DFT calculations indicated that the triplet state was slightly favored (−1.0 kcal mol–1).18 Furthermore, monocoordination of benzonitrile, dimethyl sulfoxide (DMSO) and pyridine was observed, with conversion to the octahedral (bis-axially coordinated) complexes in neat DMSO and pyridine. Bis-coordination of two pyridine molecules to afford the octahedral complex was indicated by UV–visible (UV–vis) studies, and DFT calculations revealed orbital compositions expected for a trianionic ligand coordinated to a low-spin (B3LYP functional) or intermediate-spin (OLYP functional) cobalt(III) center. However, the exact electronic structure of the investigated species remains largely unknown at this point.
The aforementioned studies on cobalt-DPP complexes indicate that intermediate- and low-spin configurations on cobalt are energetically close and that the DPP ligand is redox-active on cobalt. Because of these properties we selected the Co-DPP system as a suitable candidate to evaluate the influence of the redox-active ligand on the total spin state of the cobalt complex in the presence and absence of axial redox-innocent donor ligands. Specifically, in this work we study the electronic configuration of a neutral [CoII(DPP·2–)] complex (with Ar = C6F5; R1 = R3 = tBu; R2 = H, Figure 1), bearing a new DPP ligand derivative, upon coordination of different axial donor ligands with experimental (X-ray diffraction (XRD), μeff, NMR, high-resolution mass spectrometry (HRMS), UV–vis) and computational (DFT, NEVPT2-CASSCF) techniques. We thereby describe how the molecular orbitals are influenced by coordination of tetrahydrofuran (THF), pyridine, and primary amines and elucidate the exact electronic structures of these complexes and the influence of the redox-active ligand on the orbital changes upon coordination of axial donors.
Results and Discussion
Synthesis and Open-Shell Singlet Electronic Ground-State Configuration of [CoII(DPP·2–)]
The dipyrrin-bisphenol ligand DPPH3, bearing two tert-butyl groups on the phenol ring and a pentafluorophenyl substituent on the meso-position, was obtained via a four-step synthesis in 65% overall isolated yield according to modified literature procedures (see Scheme S1 in the Supporting Information and Scheme 1A).8,12 Coordination of cobalt(II) and in situ oxidation to the neutral complex was achieved according to an adapted literature procedure12 by employing Co(OAc)2·4(H2O) and NEt3 under aerobic conditions to afford [CoII(DPP·2–)] as a purple powder in 88% isolated yield.
Scheme 1.

(A) Synthesis of DDPH3 and [CoII(DPP·2–)]. (B) Displacement ellipsoid plot (50% probability level) of DPPH3. (C) Displacement ellipsoid plot (50% probability level) of [CoII(DPP·2–)]. H atoms (except NH and OH) and disorder are omitted for clarity. (D) Selection of active orbitals, occupancies in parentheses, and electronic structure from a NEVPT2-CASSCF(18,14) calculation on [CoII(DPP·2–)]. Isosurface set at 80.
Slow evaporation of a concentrated solution of DPPH3 in CH2Cl2 afforded single crystals suitable for X-ray structure determination (Scheme 1B). Single crystals suitable for XRD analysis of [CoII(DPP·2–)] were also obtained in a similar manner. The molecular structure of the latter is depicted in Scheme 1C and shows a slightly distorted square planar geometry around cobalt. This distortion is most likely caused by the steric repulsion between the ortho-tert-butyl substituents on the phenolate rings. Comparison of the bond lengths in [CoII(DPP·2–)] with those found in the fully aromatic DPPH3 ligand shows alternating elongation and shortening of the C–C bond lengths (see Table S1 in the Supporting Information), consistent with the loss of aromaticity due to the oxidation of the DPP ligand in the complex. The bond lengths are similar to a previously described DPP ligand in the dianionic (radical) state on cobalt(II),12 thus supporting the proposed DPP·2– oxidation state of the ligand. The two 2-pyrrolylphenolato fragments in [CoII(DPP·2–)] have similar bond metrics, indicating a fully conjugated ligand and a delocalized ligand-centered radical coordinated to a cobalt(II) center in the neutral [CoII(DPP·2–)] complex.19
1H NMR analysis of [CoII(DPP·2–)] in CD2Cl2 showed two remarkably downfield-shifted resonances at δ = 12.82 (2H) and 4.29 (18H) ppm. Note that these signals are observed at, respectively, δ = 7.03 and 1.54 ppm in DPPH3. All other resonances are shifted ∼1 ppm relative to the free ligand. These unusual shifts are suggestive of (minor) paramagnetic contributions to the observed chemical shift in the 1H NMR spectrum, which seems to correlate with the experimentally determined bond lengths from XRD that suggest a ligand-centered radical (DDP·2–) and consequently a cobalt(II) (radical) center. However, whether these apparent paramagnetic contributions are best explained by an open-shell singlet ground state (with temperature-independent paramagnetism (TIP)) or as the result of the population of an excited higher spin-multiplicity state (either thermally or induced by weak and dynamic coordination of CD2Cl220) is unclear at this stage. Nonetheless, these shifts are noteworthy.
Measurement of the effective magnetic moment (μeff) of [CoII(DPP·2–)] in the solid state as a function of the temperature with a superconducting quantum interference device (SQUID), to investigate the coupling of the two unpaired electrons, showed no significant magnetization in the 5–290 K range (see Figure S1 in the Supporting Information). The effective magnetic moment in CD2Cl2 solution, as determined by the Evans method,21 also afforded a μeff of 0 μB. The combined XRD, NMR spectroscopic, and magnetochemical data thus indicate a diamagnetic ground state, resulting from the strong antiferromagnetic coupling of the two unpaired electrons, yielding an overall (open-shell) S = 0 singlet spin state.
To study the electronic structure, we initially performed DFT calculations at the B3LYP/def2-SVP//B3LYP/def2-TZVP level of theory, employing an m4 grid and Grimme’s version 3 dispersion corrections (see the Supporting Information for more details). The calculated bond metrics for [CoII(DPP·2–)] in the open-shell singlet state closely resemble those found in the crystal structure (see Table S1 in the Supporting Information) and show similar alternating C–C bond lengths, consistent with oxidation of the ligand to the DPP·2– redox state. The relative energies of the open-shell singlet (ΔG°298 K = +1.3 kcal mol–1), triplet (ΔG°298 K = 0.0 kcal mol–1), and closed-shell singlet (ΔG°298 K = +14.8 kcal mol–1) are consistent with the proposed open-shell (biradical) character of [CoII(DPP·2–)], but they fail to reproduce the experimentally observed (open-shell) singlet spin state being the ground state of the molecule.
Distinguishing between a triplet and a multireference broken-symmetry singlet (BSS) electronic structure is (nearly) impossible when relying on single-reference DFT calculations.22,23 We therefore investigated the electronic structure of [CoII(DPP·2–)] with multiconfigurational N-electron valence state perturbation theory (NEVPT2)-corrected complete active space self-consistent field (CASSCF) calculations (see the Supporting Information), a method we previously used successfully to study the orbital compositions of cobalt complexes bearing a redox-active ligand.24 A NEVPT2-CASSCF(18,14) calculation, employing 18 electrons in 14 active orbitals on [CoII(DPP·2–)], converged on the singlet surface and showed a dominant (>96%) contribution from a multireference open-shell singlet (OSS) electronic configuration of [CoII(DPP·2–)]. A pure triplet spin-state solution could not be found in this, nor in a reduced, active space. State averaging of the singlet and triplet state in a 50:50 mixture in the active space did afford a solution for the triplet spin state, but this triplet state was found to be +6.5 kcal mol–1 less stable than the OSS state.
A selection of the active orbitals and their occupations derived from the NEVPT2-CASSCF(18,14) calculation on [CoII(DPP·2–)] is depicted in Scheme 1D. The dxy, dyz, dxz, and a ligand (L) orbital of π symmetry are doubly filled, whereas the dx2–y2 orbital is empty. The two main contributors to the multireference OSS solution are described by the dz2 orbital, which has a bonding and antibonding combination with the π-frame of the ligand (Lπ+dz2–L′π) or is nonbonding (dz2). Specifically, 50.6% of the total wave function is described by a doubly filled Lπ+dz2–L′π orbital (and empty dz2), while 45.5% of the wave function is described by a doubly filled dz2 (and empty Lπ+dz2–L′π orbital). The electronic structure of [CoII(DPP·2–)] is thus best described as an open-shell singlet based on the combined experimental (XRD, 1H NMR, μeff) and computational (NEVPT2-CASSCF) studies. Effectively, one unpaired electron resides in the dz2 orbital on cobalt, and another unpaired electron is fully delocalized over the ligand with a small contribution from the dz2 orbital on cobalt. As such, this complex is best described as a system containing antiferromagnetically coupled cobalt(II)- and DPP·2– ligand-centered unpaired electrons.
Spin-Flip to a Triplet State upon Coordination of THF on [CoII(DPP·2–)] to Generate [CoII(DPP·2–)(THF)2]
Whereas [CoII(DPP·2–)] is purple in non-coordinating solvents (CH2Cl2, toluene) we noticed a distinct color change to green upon solvation of the complex in coordinating solvents (THF, MeOH, MeCN), indicative of solvent coordination. The UV–vis spectra of [CoII(DPP·2–)] in THF (solid green line, λmax = 318, 409, 423, 474, 632, and 833 nm) and CH2Cl2 (solid purple line, λmax = 326, 374, 556, and 755 nm) are shown in Figure 2 left. Titration of THF (guest) to a CH2Cl2 solution of [CoII(DPP·2–)] (host) afforded spectral changes in the UV–vis spectra characteristic for multiple binding events (see Figure 2 left and Figure S4 in the Supporting Information). Isosbestic points are found in two regimes; between 0 and 1.3 × 104 equiv of THF (solid purple to solid light green line, λ = 413, 488, 600, 667, 771 nm) and between 5.2 × 104 and 1.4 × 105 equiv of THF (solid orange to solid brown line, λ = 393, 489, 593, 679, 776 nm). Between these two regimes the spectral crossing points are found between the isosbestic points, suggestive of the simultaneous presence of three species.
Figure 2.

(left) UV–Vis spectra of [CoII(DPP·2–)] in pure THF ([CoII(DPP·2–)(THF)2], 32.27 μM, green solid line), in CH2Cl2 (32.27 μM, purple solid line), and titration of THF to this CH2Cl2 solution (dashed lines and solid light green, orange and brown line). (right) 1H NMR spectra of [CoII(DPP·2–)] dissolved in pure CD2Cl2 (5.89 mM, (A, F), purple), CD2Cl2 with 1.1 M THF ((B), THF/THF-d8 = 1:9), 2.0 M THF ((C), THF/THF-d8 = 1:20), 3.8 M THF ((D), THF/THF-d8 = 1:45) and pure THF-d8 ((E), green). The sample was concentrated and thoroughly dried between the measurements (E, F).
The titration data could be fitted25 to weak noncooperative host–guest–guest binding with an overall association constant (Kass) of 1.2 M–1 for binding of two THF molecules (see Figure S2 in the Supporting Information). The data are therefore consistent with the initial (predominant) formation of a mono-THF adduct (first regime), followed by the formation of a bis-THF adduct (second regime, see Figure S4 in the Supporting Information). Full conversion to this latter species is not reached at the end of the depicted titration curve (brown solid line, Figure 2 left) and is only observed in neat THF (solid green line, Figure 2 left), as indicated by the increased absorbance of various spectral bands and the shoulder at 474 nm. Similar mono- and bis-coordination of solvent has been described in the literature for related Co-DPP complexes,18 and in combination with the titration data in Figure 2 we thus propose the formation of a bis-THF adduct ([CoII(DPP·2–)(THF)2]) in neat THF (Scheme 2).
Scheme 2. Formation of [CoII(DPP·2–)(THF)2] via Coordination of THF to [CoII(DPP·2–)].

Ar = C6F5.
To further study the coordination of THF to [CoII(DPP·2–)] we followed the spectral changes in the 1H NMR spectrum upon addition of THF to a 5.89 mM CD2Cl2 solution of [CoII(DPP·2–)] (Figure 2 right). The presence of 1.1, 2.0, or 3.8 M THF (spectra B, C, D) led to signal broadening and gradual downshifting of one resonance (labeled as a red circle), while three other resonances (labeled as a blue square, orange triangle, and yellow diamond) are strongly shifted upfield, approaching the shifts observed in neat THF-d8 (spectrum E). The observed sharp paramagnetically shifted resonances in the −65 to +45 ppm region in neat THF-d8 clearly indicate conversion towards a new open-shell species. Interestingly, concentrating and thoroughly drying the sample obtained in neat THF-d8, followed by dissolution in CD2Cl2, afforded a purple solution for which the spectral data (1H NMR, UV–vis) exactly matched that of [CoII(DPP·2–)] (spectrum F). These combined data thus point to weak and reversible binding of THF to the square planar complex, consistent with the low Kass as derived from the UV–vis spectroscopic titration study.
No THF adducts were observed by HRMS, presumably due to the reversible weak binding and low boiling point of THF, which is likely easily lost in the ionization process. Attempts to crystallize a THF adduct of [CoII(DPP·2–)] were unfortunately unsuccessful. Determination of the effective magnetic moment of [CoII(DPP·2–)(THF)2] in THF-d8 by the Evans method21 afforded μeff = 2.91 μB, indicating the formation of a triplet (S = 1) complex.
DFT calculations (B3LYP/def2-SVP//B3LYP/def2-TZVP, m4 grid, Grimme’s version 3 dispersion corrections) indicated that both the square pyramidal mono-THF adduct ([CoII(DPP·2–)(THF)]) and the octahedral bis-THF complex ([CoII(DPP·2–)(THF)2]) have a triplet spin (S = 1) ground state, consistent with the experimentally determined spin state.26 To obtain more insight in the electronic structure of [CoII(DPP·2–)(THF)2] and to investigate possible multireference contributions to the ground-state wave function, we performed NEVPT2-CASSCF(18,15) calculations on the singlet, triplet, and quintet spin surfaces. The triplet state was again found to be the lowest in energy, with the (open-shell) singlet and quintet states being disfavored by +32.2 and +33.0 kcal mol–1, respectively. Dominant multireference character was observed in the triplet spin state, leading to an interesting electronic structure wherein cobalt retains the +II oxidation state and is ferromagnetically coupled to a ligand-centered radical on the DDP·2– ligand (Figure 3).
Figure 3.
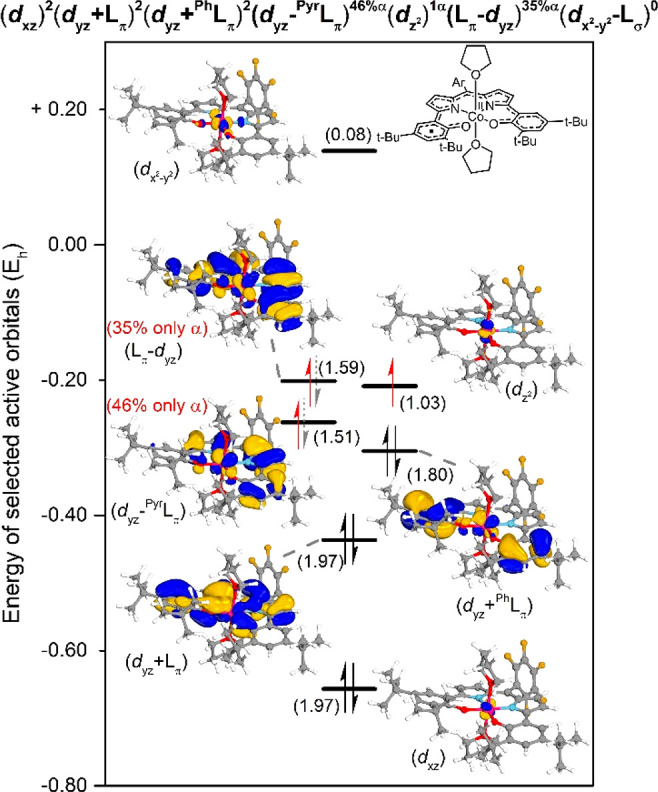
Selected active orbitals, occupancies in parentheses, and electronic structure of cobalt and the ligand from a NEVPT2-CASSCF(18,15) calculation on [CoII(DPP·2–)(THF)2]. Isosurface set at 80.
With the dxy, dxz, and dyz orbitals on cobalt doubly filled, the unpaired (and uncorrelated) α-spin electron on cobalt resides in the dz2 orbital (Figure 3). The other α-spin electron mainly resides in the strongly correlated antibonding combinations of the dyz orbital with the ligand pyrrole π-framework (dyz–PyrLπ, −0.262 54 Eh, 46.1% only α-spin, 1.51 total electron occupation) and the complete ligand π-system (Lπ–dyz, −0.208 87 Eh, 34.9% only α-spin, 1.59 total electron occupation).27 The energetically slightly higher lying Lπ–dyz orbital is more diffuse over the ligand π-system, thus leading to a smaller electron–electron repulsive interaction (i.e., pairing energy of the α- and β-spin electrons) upon filling of this orbital in comparison to the more localized (less diffuse) dyz–PyrLπ orbital. Consequently, in the multiconfigurational description of the total triplet spin state wave function, the ligand-centered unpaired electron is mainly (46.1%) localized on the least-diffuse orbital (dyz–PyrLπ).
The spin−flip in the transition from [CoII(DPP·2–)] (OSS) to [CoII(DPP·2–)(THF)2] (triplet) can be understood by looking at the composition of the SOMOs. The α- and β-spin electrons in [CoII(DPP·2–)] are located in the dz2-based orbitals (dz2 and Lπ+dz2–L′π). A triplet state would lead to severe (α) spin–spin repulsion in this cobalt-centered orbital, and consequently an OSS solution is favored. This is not the case for [CoII(DPP·2–)(THF)2], wherein the two unpaired electrons reside in spatially different orbitals (dz2 and dyz-based). In this case Hund’s rule28 applies, which states that the maximization of the total spin is favored for a given electronic configuration, thus leading to the observed triplet spin state.
Closed-Shell Singlet Spin State via Metal-to-Ligand Single-Electron Transfer Induced by Coordination of Stronger Donors
We next set out to explore the influence of replacing the weak-field ligand THF with the stronger-field ligand pyridine. Addition of excess (100 equiv) pyridine to [CoII(DPP·2–)] in CH2Cl2 afforded quantitative formation of the bis-pyridine adduct [CoIII(DPP3–)(Py)2] as a green powder after workup (Scheme 3A). The six-coordinate complex was characterized inter alia by 1H NMR spectroscopy and positive mode cold-spray ionization (CSI) HRMS. Single crystals suitable for X-ray structure determination were grown by slow evaporation of a concentrated solution of the complex in a 5:1 mixture of CH2Cl2 and MeOH. Three octahedral [CoIII(DPP3–)(Py)2] complexes are present in the unit cell (see Figure S6 in the Supporting Information), one of which (the left structure) is depicted in Scheme 3B. The bond metrics of all three [CoIII(DPP3–)(Py)2] molecules are similar, although the relative rotation of the pyridine ligands differs from nearly parallel to perpendicular (see Table S2 in the Supporting Information).
Scheme 3.

(A) Formation of [CoIII(DPP3–)(Py)2]. (B) Displacement ellipsoid plot (50% probability level) of one [CoIII(DPP3–)(Py)2] molecule. H atoms and disorder are omitted for clarity. (C) Selection of DFT-calculated orbitals and the electronic structure of cobalt and the ligand in [CoIII(DPP3–)(Py)2]. Isosurface set at 80.
The experimentally determined C–C bond lengths of the DPP moiety (see the Supporting Information) in the crystal structure of [CoIII(DPP3–)(Py)2] resemble the aromaticity that is also observed in the free DPPH3 ligand, thus suggesting a fully reduced trianionic DPP3– redox state for the ligand and consequently a cobalt(III) center in the neutral complex. The 1H NMR resonances of [CoIII(DPP3–)(Py)2] do not show any paramagnetic shifts and are found entirely in the diamagnetic region (δ = 8.04–1.25 ppm), suggesting a closed-shell singlet electronic configuration, that is, a low-spin CoIII center. The SQUID analysis of solid [CoIII(DPP3–)(Py)2] did not show significant magnetization in the 4–290 K range, again consistent with a singlet ground state (see Figure S7 in the Supporting Information).
DFT calculations (B3LYP/def2-SVP//B3LYP/def2-TZVP, m4 grid, Grimme’s version 3, dispersion corrections) indicated that formation of the closed-shell singlet octahedral bis-pyridine [CoIII(DPP3–)(Py)2] complex is more exergonic (ΔG°298 K = −14.2 kcal mol–1) than the formation of the square pyramidal monopyridine adduct [CoII(DPP·2–)(Py)] (ΔG°298 K = −9.1 kcal mol–1, S = 1). Orbital analysis clearly showed that coordination of pyridine in [CoIII(DPP3–)(Py)2] (Scheme 3C) leads to a strongly destabilized dz2 orbital (dz2–2NPy,σ), resulting in a quite large gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of 0.10346 Eh and, therefore a low-spin (CSS) configuration. The Lπ–dyz (HOMO) is doubly filled, reflecting the reduction of the ligand to the DPP3– state. Cobalt adopts the +III oxidation state in [CoIII(DPP3–)(Py)2] with doubly filled dxy, dxz, and dyz orbitals, which are stabilized due to the higher oxidation state of cobalt in comparison to [CoII(DPP·2–)]. Thus, pyridine coordination effectively results in ligand reduction via metal-to-ligand single-electron transfer. Interestingly, interaction of the pyridine-π* system with the dyz orbital is observed in the dyz–2NPy,π* orbital, reflecting at least some π-back-donation from cobalt to the pyridine ligands.
Coordination of pure σ-donors was achieved via addition of the primary amines tert-butyl amine and 1-adamantyl amine. The complex [CoIII(DPP3–)(NH2tBu)2] was obtained in quantitative yield as a green powder through addition of 100 equiv of tBuNH2 to [CoII(DPP·2–)] and subsequent concentration and drying under reduced pressure (Scheme 4A). [CoIII(DPP3–)(NH2Ad)2] (Ad = 1-adamantyl) was obtained in 43% yield as green crystals after addition of 2 equiv of AdNH2 to [CoII(DPP·2–)] and subsequent crystallization.
Scheme 4.

(A) Formation of [CoIII(DPP3–)(NH2tBu)2] and [CoIII(DPP3–)(NH2Ad)2]. Ad = 1-adamantyl. (B) Displacement ellipsoid plot (50% probability level) of [CoIII(DPP3–)(NH2Ad)2]. (C) Displacement ellipsoid plot (50% probability level) of [CoIII(DPP3–)(NH2tBu)2]. H atoms and disorder are omitted for clarity.
The 1H NMR resonances of [CoIII(DPP3–)(NH2tBu)2] and [CoIII(DPP3–)(NH2Ad)2] are similar to the bis-pyridine adduct, found within the diamagnetic region, suggesting that both complexes are most stable in the CSS spin state. Crystals suitable for X-ray structure determination of both complexes were obtained by slow evaporation of concentrated solutions in CH2Cl2 and MeOH (5:1) at room temperature. The C–C and Co–DPP bond lengths obtained from the crystal structure of [CoIII(DPP3–)(NH2Ad)2] (Scheme 4B) closely resemble those found in [CoIII(DPP3–)(Py)2] and are consistent with a fully reduced (3−) redox state of the DPP ligand and consequently a cobalt(III) center. The crystallographically independent molecules of [CoIII(DPP3–)(NH2tBu)2] found in the asymmetric unit (see Figure S9 in the Supporting Information) have mutually similar bond metrics, which are also comparable to those observed in [CoIII(DPP3–)(NH2Ad)2]. One molecule found in the crystal structure of [CoIII(DPP3–)(NH2tBu)2] is depicted in Scheme 4C.
The DFT (B3LYP/def2-SVP//B3LYP/def2-TZVP, m4 grid, Grimme’s version 3, dispersion corrections) calculated bond lengths of [CoIII(DPP3–)(NH2tBu)2] in the CSS spin state are consistent with the experimentally determined bond metrics (see Table S3 in the Supporting Information). Moreover, the corresponding triplet spin state was found to be +4.0 kcal mol–1 less stable. The DFT orbital analysis of [CoIII(DPP3–)(NH2tBu)2] (Figure 4) shows a destabilized empty dz2 orbital due to coordination of the tBuNH2 lone σ-pair (NA,σ). However, the bonding interactions of these lone pairs with the dyz and dxz orbitals are observed in the doubly filled dyz+2NA,σ–Lπ and PyrLπ–dxz+2NA,σ orbitals. The dxy, dyz, and dxz orbitals are all doubly filled, consistent with a low-spin cobalt(III) electronic configuration formed after metal-to-ligand single−electron transfer.
Figure 4.
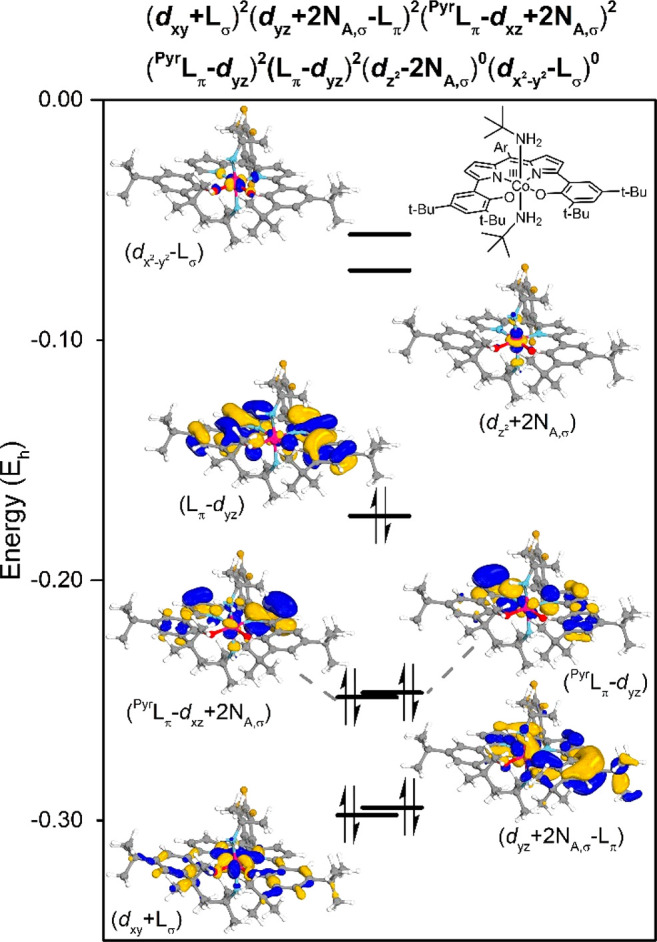
Selection of DFT calculated orbitals and electronic structure of cobalt and the ligand in [CoIII(DPP3–)(NH2tBu)2]. Isosurface set at 70.
Conclusions
The electronic ground state of [CoII(DPP·2–)] is characterized as a multiconfigurational open-shell singlet, which is best described as a system containing antiferromagnetically coupled cobalt(II)- and ligand-centered unpaired electrons. Solvation of this complex in THF (sp3-hybrid donor) affords the clean formation of a THF-adduct, [CoII(DPP·2–)(THF)2], which resides in the triplet spin state. The origin of this spin flip is the orbital overlap of the redox-active ligand π-framework with the cobalt d-orbitals, which leads to the population of two ligand-dyz orbital combinations in a multiconfigurational triplet solution to reduce spin–spin repulsion. Coordination of pyridine (σ-donor, weak π-acceptor), tBuNH2, or AdNH2 (σ-donors) afforded the closed-shell singlet octahedral complexes via metal-to-ligand single-electron transfer. The redox-active DPP ligand is reduced to the trianionic redox state, and cobalt adopts a low-spin +III oxidation state.
Concluding, we have described that a redox-active DPP ligand on cobalt can accommodate three different spin states of the complex within an integer spin system. The spin-state changes are induced via coordination of axial ligands to the square-planar complex, but the relative energy and overlap of the ligand- and cobalt-centered orbitals determines the most stable spin state. The capability of the redox-active ligand to stabilize unpaired electrons and accommodate intramolecular electron transfer was found to be crucial in this context.
Experimental Section
General Considerations
All reagents were of commercial grade and used without further purification, unless noted otherwise. All reactions were performed under an inert atmosphere in a N2-filled glovebox or by using standard Schlenk techniques (under Ar or N2), unless noted otherwise. CH2Cl2 and MeOH were distilled from CaH2; toluene was distilled from sodium, and THF was distilled from sodium benzophenone ketyl. Detailed information regarding the NMR, HRMS, UV–vis, SQUID, and XRD measurements is included in the Supporting Information. XRD- and DFT-derived bond lengths are also included in the Supporting Information. The magnetic moments in solution were determined via the Evans method.21
Synthesis and Characterization. DPPH3
Synthesized in four steps (overall isolated yield 65%) according to adapted literature procedures.8,10 Characterized by 1H and 19F NMR, HRMS-FD+, elemental analysis, and XRD (see the Supporting Information).
[CoII(DPP·2–)]
Prepared in 88% isolated yield according to a literature procedure for the insertion of cobalt in a DPP ligand.12 Characterized by 1H and 19F NMR, HRMS-FD+, UV–vis, elemental analysis, μeff (Evans method and SQUID), and XRD (see the Supporting Information).
[CoII(DPP·2–)(THF)2]
Quantitatively prepared by solvation of [CoII(DPP·2–)] in THF. Characterized by 1H NMR, μeff (Evans method), and UV–vis (see the Supporting Information).
[CoIII(DPP3–)(Py)2]
Obtained in quantitative isolated yield by the addition of pyridine (100 equiv) to a solution of [CoII(DPP·2–)] in CH2Cl2. Characterized by 1H and 19F NMR, HRMS-CSI+, UV–vis, elemental analysis, μeff (SQUID), and XRD (see the Supporting Information).
[CoIII(DPP3–)(NH2tBu)2]
Obtained in quantitative isolated yield by the addition of tBuNH2 (100 equiv) to a solution of [CoII(DPP·2–)] in CH2Cl2. Characterized by 1H and 19F NMR, HRMS-CSI+, UV–vis, and XRD (see the Supporting Information).
[CoIII(DPP3–)(NH2Ad)2]
Obtained in 43% isolated yield by addition of AdNH2 (2.0 equiv) to a solution of [CoII(DPP·2–)] in CH2Cl2. Characterized by 1H and 19F NMR, HRMS-CSI+, and XRD (see the Supporting Information).
Computational Studies
DFT
Calculations were conducted on full atomic models at the B3LYP29/def2-SVP30//B3LYP/def2-TZVP30 level of theory on an m4 grid with Grimme’s version 331 (“zero-damping”) dispersion corrections with the TURBOMOLE 7.332 software package coupled to the PQS Baker optimizer33 via the BOpt package.34 Orbital interpretation was performed by Löwdin population analysis of quasi-restricted orbitals (QRO) generated with the ORCA 4.135 software package at the B3LYP/def2-TZVP level, using the coordinates from the structures optimized in TURBOMOLE as the input and using the UNO keyword. Graphical representations of orbitals were generated using IboView.36 Energies, xyz coordinates, and more details on the calculations are included in the Supporting Information.
NEVPT2-CASSCF
The NEVPT2-corrected CASSCF calculations were performed with the ORCA 4.135 software package on the geometries optimized in TURBOMOLE. The def2-TZVP30 basis set was used together with the RIJCOSX37 approximation in conjunction with the def2-TZVP/C fitting the basis set to reduce computational cost. The single-root spin state was calculated. The NEVPT238 calculations using the RI approximation were performed on converged CASSCF wave functions. Energies, contributions to the wave functions, full representations of the active spaces, and more details on the calculations are included in the Supporting Information.
Acknowledgments
Financial support from The Netherlands Organization for Scientific Research (NWO TOP-Grant 716.015.001) to B.d.B. and the research area Sustainable Chemistry of the University of Amsterdam (RPA SuSChem, UvA) is gratefully acknowledged. Dr. E. Bill (MPI CEC) is acknowledged for the SQUID measurements and related discussions. E. Zuidinga is thanked for the HRMS measurements, and T. Bouwens is thanked for the valuable discussions regarding the UV–vis titration data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c01979.
Experimental details, synthetic procedures, NMR, HRMS, and UV–vis spectra, crystallographic refinement details, geometries (xyz coordinates) and energies of DFT-calculated structures, description of the NEVPT2-CASSCF calculations (PDF)
Accession Codes
CCDC 2012086–2012090 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Present Address
§ (J.I.v.d.V.) Bioinspired Coordination Chemistry & Homogeneous Catalysis Group, Institute of Chemistry, Carl von Ossietzky University Oldenburg, Carl-von-Ossietzky-Str. 9–11, 26129 Oldenburg, Germany.
Author Contributions
All authors have given approval to the final version of the manuscript.
This research was funded by The Netherlands Organization for Scientific Research TOP-Grant 716.015.001 to B.d.B.
The authors declare no competing financial interest.
Notes
CCDC 2012086 (DPPH3), 2012087 ([CoII(DPP·2–)]), 2012088 ([CoIII(DPP3–)(Py)2]), 2012089 ([CoIII(DPP3–)(NH2tBu)2]), and 2012090 ([CoIII(DPP3–)(NH2Ad)2]).
Supplementary Material
References
- Likhtenshtein G. I.Fundamentals, Methods, Reactions Mechanisms, Magnetic Phenomena, Structure Investigation. In Electron Spin Interactions in Chemistry and Biology ;Springer International Publishing, 2016. [Google Scholar]
- Molecular Magnets Recent Highlights ;Linert W., Verdaguer M., Eds.; Springer International Publishing, 2003. [Google Scholar]
- Wolf S. A.; Awschalom D. D.; Buhrman R. A.; Daughton J. M.; von Molnár S.; Roukes M. L.; Chtchelkanova A. Y.; Treger D. M. Spintronics: A Spin-Based Electronics Vision for the Future. Science 2001, 5546, 1488–1495. 10.1126/science.1065389. [DOI] [PubMed] [Google Scholar]
- Dzik W. I.; Böhmer W.; de Bruin B. In Spin States in Biochemistry and Inorganic Chemistry: Influence on Structure and Reactivity ;Swart M., Costas M., Eds.; Wiley, 2015; pp 103–130. [Google Scholar]
- Crabtree R. H.The Organometallic Chemistry of the Transition Metals ,5th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2009. [Google Scholar]
- a Lyaskovskyy V.; de Bruin B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. 10.1021/cs200660v. [DOI] [Google Scholar]; b van Leest N. P.; Epping R. F. J.; van Vliet K. M.; Lankelma M.; van den Heuvel E.; Heijtbrink N.; Broersen R.; de Bruin B. In Advances in Organometallic Chemistry ;Pérez P. J., Stone F. G. A., West R., Eds.; Elsevier, 2018; Vol. 70, pp 71–180. [Google Scholar]; c Broere D. L. J.; Plessius R.; van der Vlugt J. I. New avenues for ligand-mediated processes - expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. 10.1039/C5CS00161G. [DOI] [PubMed] [Google Scholar]; d van der Vlugt J. I. Radical-Type Reactivity and Catalysis by Single-Electron Transfer to or from Redox-Active Ligands. Chem. - Eur. J. 2019, 25, 2651–5662. 10.1002/chem.201802606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom S. M.; Garcia P. P. U.S. Patent 3691161, 1972.
- Ikeda C.; Ueda S.; Nabeshima T. Aluminium complexes of N2O2-type dipyrrins: the first hetero-multinuclear complexes of metallo-dipyrrins with high fluorescence quantum yields. Chem. Commun. 2009, 2544–2546. 10.1039/b820141b. [DOI] [PubMed] [Google Scholar]
- Sumiyoshi A.; Chiba Y.; Matsuoka R.; Noda T.; Nabeshima T. Efficient luminescent properties and cation recognition ability of heavy group 13 element complexes of N2O2- and N2O4-type dipyrrins. Dalton Trans. 2019, 48, 13169–13175. 10.1039/C9DT02403D. [DOI] [PubMed] [Google Scholar]
- Nakano K.; Kobayashi K.; Nozaki K. Tetravalent Metal Complexes as a New Family of Catalysts for Copolymerization of Epoxides with Carbon Dioxide. J. Am. Chem. Soc. 2011, 133, 10720–10723. 10.1021/ja203382q. [DOI] [PubMed] [Google Scholar]
- a Rausaria S.; Kamadulski A.; Rath N. P.; Bryant L.; Chen Z.; Salvemini D.; Neumann W. L. Manganese(III) Complexes of Bis(hydroxyphenyl)dipyrromethenes Are Potent Orally Active Peroxynitrite Scavengers. J. Am. Chem. Soc. 2011, 133, 4200–4203. 10.1021/ja110427e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b El Ghachtouli S.; Wójcik K.; Copey L.; Szydlo F.; Framery E.; Goux-Henry C.; Billon L.; Charlot M.-F.; Guillot R.; Andrioletti B.; Aukauloo A. Dipyrrinphenol-Mn(III) complex: synthesis, electrochemistry, spectroscopic characterisation and reactivity. Dalton. Trans. 2011, 40, 9090–9093. 10.1039/c1dt10868a. [DOI] [PubMed] [Google Scholar]
- Kochem A.; Chiang L.; Baptiste B.; Philouze C.; Leconte N.; Jarjayes O.; Storr T.; Thomas F. Ligand-Centered Redox Activity in Cobalt(II) and Nickel(II) Bis(phenolate)-Dipyrrin Complexes. Chem. - Eur. J. 2012, 18, 14590–14593. 10.1002/chem.201202882. [DOI] [PubMed] [Google Scholar]
- Lecarme L.; Chiang L.; Moutet J.; Leconte N.; Philouze C.; Jarjayes O.; Storr T.; Thomas F. The structure of a one-electron oxidized Mn(III)-bis(phenolate)dipyrrin radical complex and oxidation catalysis control via ligand-centered redox activity. Dalton Trans. 2016, 45, 16325–16334. 10.1039/C6DT02163H. [DOI] [PubMed] [Google Scholar]
- Yamamura M.; Takizawa H.; Gobo Y.; Nabeshima T. Stable neutral radicals of planar N2O2-type dipyrrin platinum complexes: hybrid radicals of the delocalized organic π-orbital and platinum d-orbital. Dalton Trans. 2016, 45, 6834–6838. 10.1039/C5DT05039A. [DOI] [PubMed] [Google Scholar]
- Lecarme L.; Kochem A.; Chiang L.; Moutet J.; Berthiol F.; Philouze C.; Leconte N.; Storr T.; Thomas F. Electronic Structure and Reactivity of One-Electron-Oxidized Copper(II) Bis(phenolate)-Dipyrrin Complexes. Inorg. Chem. 2018, 57, 9708–9719. 10.1021/acs.inorgchem.8b00044. [DOI] [PubMed] [Google Scholar]
- Thomas K. E.; Desbois N.; Conradie J.; Teat S. J.; Gros C. P.; Ghosh A. Gold dipyrrin-bisphenolates: a combined experimental and DFT study of metal-ligand interactions. RSC Adv. 2020, 10, 533–540. 10.1039/C9RA09228E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Burns L. A.; Lee L.-C.; Sherrill C. D.; Jones C. W.; Murdock C. Co(III) complexes of tetradentate X3L type ligands: Synthesis, electronic structure, and reactivity. Inorg. Chim. Acta 2015, 430, 30–35. 10.1016/j.ica.2015.01.036. [DOI] [Google Scholar]
- Shan W.; Desbois N.; Pacquelet S.; Stephane Brandes; Rousselin Y.; Conradie J.; Ghosh A.; Gros C. P.; Kadish K. M. Ligand Noninnocence in Cobalt Dipyrrin-Bisphenols: Spectroscopic, Electrochemical, and Theoretical Insights Indicating an Emerging Analogy with Corroles. Inorg. Chem. 2019, 58, 7677–7689. 10.1021/acs.inorgchem.8b03006. [DOI] [PubMed] [Google Scholar]
- Two-electron oxidation of the DPP ligand to DPP1– is highly unlikely to be stable on a cobalt(I) center. Furthermore, the DPP·2– oxidation state is also evident from 1H NMR analysis due to two downshifted resonances and was also consistent with the NEVPT2-CASSCF calculations.
- a Newbound T. D.; Colsman M. R.; Miller M. M.; Wulfsberg G. P.; Anderson O. P.; Strauss S. H. Dichloromethane is a coordinating solvent. J. Am. Chem. Soc. 1989, 111, 3762–3764. 10.1021/ja00192a052. [DOI] [Google Scholar]; b Kulawiec R. J.; Crabtree R. H. Coordination chemistry of halocarbons. Coord. Chem. Rev. 1990, 99, 89–115. 10.1016/0010-8545(90)80061-W. [DOI] [Google Scholar]
- a Evans D. F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. 10.1039/jr9590002003. [DOI] [Google Scholar]; b Deutsch J. L.; Poling S. M. The determination of paramagnetic susceptibility by NMR: A physical chemistry experiment. J. Chem. Educ. 1969, 46, 167–168. 10.1021/ed046p167. [DOI] [Google Scholar]; c Piguet C. Paramagnetic Susceptibility by NMR: The “Solvent Correction” Removed for Large Paramagnetic Molecules. J. Chem. Educ. 1997, 74, 815–816. 10.1021/ed074p815. [DOI] [Google Scholar]; d Sur S. K. Measurement of magnetic susceptibility and magnetic moment of paramagnetic molecules in solution by high-field fourier transform NMR spectroscopy. J. Magn. Reson. 1989, 82, 169–173. 10.1016/0022-2364(89)90178-9. [DOI] [Google Scholar]; e Grant D. H. Paramagnetic Susceptibility by NMR: The “Solvent Correction” Reexamined. J. Chem. Educ. 1995, 72, 39. 10.1021/ed072p39. [DOI] [Google Scholar]; f Hoppe J. I. Effective magnetic moment. J. Chem. Educ. 1972, 49, 505. 10.1021/ed049p505. [DOI] [Google Scholar]; g Bain G. A.; Berry J. F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. 10.1021/ed085p532. [DOI] [Google Scholar]
- Cramer C. J.Essentials of Computational Chemistry: Theories and Models ,2nd ed; John Wiley & Sons Ltd: West Sussex, U.K., 2004; pp 205–210. [Google Scholar]
- DFT, being a single reference method, is not suitable to accurately describe a multireference electronic structure, but it can be used to accurately calculate the molecular structure with reasonable computational costs (see the Supporting Information for comparison with experimentally determined bond lengths and geometries). Performing these geometry optimizations with multireference NEVPT2-CASSCF calculations is unfeasible in terms of computational costs. However, the NEVPT2-CASSCF calculations can be used to correctly describe the multireference electronic structure, and therefore the two computational methods are complementary.
- a van Leest N. P.; Tepaske M. A.; Oudsen J.-P. H.; Venderbosch B.; Rietdijk N. R.; Siegler M. A.; Tromp M.; van der Vlugt J. I.; de Bruin B. Ligand Redox Noninnocence in [CoIII(TAML)]0/- Complexes Affects Nitrene Formation. J. Am. Chem. Soc. 2020, 142, 552–563. 10.1021/jacs.9b11715. [DOI] [PMC free article] [PubMed] [Google Scholar]; b van Leest N. P.; Tepaske M. A.; Venderbosch B.; Oudsen J.-P. H.; Tromp M.; van der Vlugt J. I.; de Bruin B. Electronically Asynchronous Transition States for C-N Bond Formation by Electrophilic [CoIII(TAML)]-Nitrene Radical Complexes Involving Substrate-to-Ligand Single-Electron Transfer and a Cobalt-Centered Spin Shuttle. ACS Catal. 2020, 10, 7449–7463. 10.1021/acscatal.0c01343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitted with http://limhes.net/optim/ (accessed June 2, 2020) according to an HGG model with a cooperativity parameter of 1.
- See the Supporting Information for the calculated exchange coupling constant for the two unpaired electrons and the NEVPT2-CASSCF(14,10) calculations on [CoII(DPP·2–)(THF)].
- The larger occupation number for the energetically higher correlated orbital is sometimes described as a HOMO-SOMO inversion or a non-Aufbau orbital configuration. This is however, at least in our case, purely caused by the increased electron–electron repulsion (i.e., destabilization) upon double filling of the orbital. See:; a Kumar A.; Sevilla M. D. SOMO-HOMO Level Inversion in Biologically Important Radicals. J. Phys. Chem. B 2018, 122, 98–105. 10.1021/acs.jpcb.7b10002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gryn’ova G.; Coote M. L.; Corminboeuf C. Theory and practice of uncommon molecular electronic configurations. WIREs Comput. Mol. Sci. 2015, 5, 440–459. 10.1002/wcms.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins P.; Overton T.; Rourke J.; Weller M.; Armstrong F.. Shriver & Atkins’ Inorganic Chemistry ,5th ed.; Oxford University Press: Oxford, U.K., 2010; pp 18–47. [Google Scholar]
- a Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- a Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]; b Weigend F.; Haser M.; Patzelt H.; Ahlrichs R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. 10.1016/S0009-2614(98)00862-8. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- TURBOMOLE , ver. 7.3; TURBOMOLE Gmbh: Karlsruhe, Germany, 2018. [Google Scholar]
- a PQS , ver. 2.4; Parallel Quantum Solutions: Fayetteville, AR, 2001. [Google Scholar]; b Baker J. An algorithm for the location of transition states. J. Comput. Chem. 1986, 7, 385–395. 10.1002/jcc.540070402. [DOI] [Google Scholar]
- Budzelaar P. H. M. Geometry optimization using generalized, chemically meaningful constraints. J. Comput. Chem. 2007, 28, 2226–2236. 10.1002/jcc.20740. [DOI] [PubMed] [Google Scholar]
- Neese F. The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Available at; a Knizia G. Intrinsic Atomic Orbitals: An Unbiased Bridge between Quantum Theory and Chemical Concepts. J. Chem. Theory Comput. 2013, 9, 4834–4843. 10.1021/ct400687b. [DOI] [PubMed] [Google Scholar]; b Knizia G.; Klein J. E. M. N. Electron Flow in Reaction Mechanisms-Revealed from First Principles. Angew. Chem., Int. Ed. 2015, 54, 5518–5522. 10.1002/anie.201410637. [DOI] [PubMed] [Google Scholar]
- Izsak R.; Neese F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105–144111. 10.1063/1.3646921. [DOI] [PubMed] [Google Scholar]
- a Angeli C.; Cimiraglia R.; Evangelisti S.; Leininger T.; Malrieu J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. 10.1063/1.1361246. [DOI] [Google Scholar]; b Angeli C.; Cimiraglia R.; Malrieu J.-P. N-electron valence state perturbation theory: a fast implementation of the strongly contracted variant. Chem. Phys. Lett. 2001, 350, 297–305. 10.1016/S0009-2614(01)01303-3. [DOI] [Google Scholar]; c Angeli C.; Cimiraglia R.; Malrieu J.-P. N-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants. J. Chem. Phys. 2002, 117, 9138–9153. 10.1063/1.1515317. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.