

Abstract
Here we report a novel poly(2-oxazoline)-based block copolymer with the aromatic heterocyclic side chains in one block, poly(2-methyl-2-oxazoline)-b-poly(2-N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl-2-oxazoline) (PMeOx-PcBOx), and demonstrate its potential application as a drug delivery platform. The copolymer was synthesized via the condensation of N,N-dimethylbiguanide with the methyl ester side chain in poly(2-methoxycarboxyethyl-2-oxazoline) block (PMestOx) of the PMeOx-PMestOx diblock copolymer. We confirmed the N,N-dimethylbiguanide condensation with PMestOx and the complete conversion of the side chain to the N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl moiety by NMR spectroscopy, MALDI-TOF mass spectroscopy, UV–Vis spectroscopy, and titration analysis. The PMeOx-PcBOx copolymer self-assemble into polymeric micelles in aqueous solution. Successful encapsulation into these micelles has been demonstrated for 1) several poorly soluble drugs, such as bruceantin and LY2109761, and 2) dichloro(1,2-diaminocyclohexane)platinum(II) (DachPt). The first class of drugs is incorporated possibly via hydrogen bonding and pi-pi interactions with the PcBOx side groups, while the second one is likely forms coordination bonds with the same side groups. The capability of this new copolymer to solubilize a uniquely diverse set of active pharmaceutical ingredients suggests potential applications in drug delivery.
Keywords: Biguanide, Condensation, Coordination, Hydrogen bonding, Solubilization, Poly(2-oxaozoline), Polymeric micelles
1. Introduction
With an increasing number of approved drugs, the design of novel drug molecules has become more complicated. The challenge in drug design is the concurrent demand for achieving higher therapeutic potency with proper physicochemical properties and lowering toxicity. Large pharmaceutical companies employ high throughput screening (HTS) to find lead compounds, but the structure optimization that takes place after HTS often results in therapeutic failure. It is well-known that the increased structural complexity of therapeutic compounds is positively correlated to drug development success rate [1]. Therefore, the use of additional strategies, such as rational drug design, is a necessity to help simplify this complex, failure-prone process [2]. One potential approach to simplify drug development is to take advantage of drug delivery systems. Drug delivery systems can enhance the therapeutic efficacy and expand the viable drugs by compensating for undesirable physicochemical properties of therapeutic agents and modulating their pharmacokinetics, biodistribution, and cellular uptake [3–5].
Among a variety of drug delivery systems, the most successful approach thus far is the encapsulation of physicochemically challenging active pharmaceutical ingredients (APIs) in nanoparticles [6,7]. Currently, several drug delivery-based products are approved for the clinic, and many clinical and preclinical studies are in progress [8]. For example, the liposomal formulation of Amphotericin B dramatically decreased the risk of systemic side effects of the compound [9]. Abraxane®, a nanoparticle albumin–bound paclitaxel, has eliminated the use of toxic excipients [10], and lipid nanoparticle formulations can transform siRNA into a useful therapeutic which acts on a previously undruggable target [11].
However, the majority of reported nanoformulations are limited by the API’s physicochemical properties. For example, liposomes are suitable carriers for hydrophilic, slightly basic compounds [12,13]. On the other hand, previously reported polymeric micelles are most suitable to load hydrophobic drugs, because the driving force for drug encapsulation is overwhelmingly due to hydrophobic interactions [3].
N,N-dimethylbiguanide, known more commonly as metformin, has largely been used as an anti-diabetic medication. It has a strong capacity to form hydrogen bonds due to its nitrogen-rich structure [14]. It was also reported that N,N-dimethylbiguanide can condense with alkyl ester compounds to form heteroaromatic (triazine) ring structures which exhibit strong pi-pi interactions [15]. We designed a novel polymer functionalized with the condensed form of N,N-dimethylbiguanide based on poly(2-oxazoline)s which takes advantage of hydrogen bonding and pi-pi stacking interactions inherent in this structure. These new interactions may assist in the solubilization of otherwise incompatible small molecules. The novel polymer self-assembles into nanoparticles in aqueous medium and shows the ability to encapsulate a diverse space of therapeutic agents. Taken together, this novel polymer may increase the success rate of drug development.
2. Materials and methods
2.1. Chemicals
All chemicals for polymer synthesis and dichloro(1,2-diaminocyclohexane)platinum(II) (DachPt) were purchased from Sigma-Aldrich (St. Louis, MO). Oxaliplatin was purchased from LC Laboratories (Woburn, MA). Metformin hydrochloride (N,N-dimethylbiguanide hydrochloride) was purchased from Tokyo Chemical Industry Co., Ltd. (Portland, OR). N,N, 6-trimethyl-1,3,5-triazine-2,4-diamine (cBG) was purchased from Santa Cruz Biotechnology (Dallas, TX). 2-methoxycarboxyethyl-2-oxazoline (MestOx) was synthesized as previously reported [16]. For synthesis of the polymers, the reagents (methyl tri-fluoromethanesulfonate (MeOTf), 2-methyl-2-oxazoline (MeOx), MestOx, 2-n-butyl-2-oxazoline (BuOx)) and solvent (acetonitrile (ACN)) and others were dried by refluxing over calcium hydride (CaH2) under inert nitrogen gas and subsequently distilled prior to use [17]. P [MeOx35-b-BuOx20-b-MeOx35] (P2) was synthesized as described previously [17]. Cell counting kit (CCK-8) was purchased from Dojindo Molecular Technologies (Rockville, MD).
2.2. Polymer characterization
NMR spectra were recorded on an INOVA 400 at room temperature (RT). The spectra were calibrated using the solvent signals (CDCl3 7.26 ppm, (CD3)2SO 2.50 ppm, D2O 4.80 ppm). Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) was performed on a Sciex 5800 MALDI-TOF/TOF mass spectrometer and 2-(4′-Hydroxybenzeneazo)benzoic acid (HABA) (20 mg/mL in acetonitrile) was used as the matrix. Gel permeation chromatography (GPC) was performed on a GPCmax VE-2001 system (Viscotek) (RI detector mode, PSS SEC column (GRAM 100 Å 8 × 300 mm, SDV 5 μm) with N,N-Dimethylformamide (DMF) (25 mM LiBr, 1 mL/min) as eluent and calibrated against polymethylmethacrylate (PMMA) standards.
2.3. Synthesis of methyl-P[MeOx60-b-MestOx30]-piperidine (PMeOx-PMestOx)
Under dry and inert conditions, 32.2 mg (0.2 mmol, 1 eq) of initiator (MeOTf) and 1015 mg (11.93 mmol, 60 eq) of MeOx monomer were dissolved in 4 mL dry acetonitrile at RT. The mixture was stirred at 80 °C for 4 h. After cooling to RT, the monomer for the second block, MestOx (942 mg, 6.01 mmol, 30 eq), was added and the mixture was stirred at 80 °C overnight. The polymer was terminated by addition of 0.1 mL piperidine (1.01 mmol, 5 eq) and the mixture was stirred overnight at RT. An excess of K2CO3 was added to the mixture, and then the mixture was allowed to stir for 12 h. After filteration of the mixture, 5 mL of chloroform-methanol mixture (1:1) was added to the filtrate containing the product (PMeOx-PMestOx). After precipitation of the polymer by ice-cold diethyl ether (approximately 50 times the volume of polymer solution of diethyl ether was added), the product was isolated by centrifugation and organic solvent was decanted. The polymer product was dissolved in ~50 mL of DI water and dialyzed against DI water (3.5 kDa membrane) for 3 days, changing the water every day, to remove organic solvent and any remaining monomers. The resulting solution was lyophilized, and the polymer was obtained as a white powder (1428 mg, 73%). GPC (DMF (25 mM LiBr)): Mn = 13.4 kg/mol (PDI 1.038); 1H NMR (D2O, 298 K): 2.4–2.7 (4H, CO–CH2–CH2–CO–OCH3); 3.2–3.7 (7H, –N–CH2–CH2, –O–O–CH3); 1.8–2.0 (3H, –CO–CH3).
2.4. Synthesis of Methyl-P[MeOx60-b-(2-N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl-2-oxazoline)30]-piperidine (PMeOx-PcBOx)
N,N-dimethylbiguanide (Metformin) free base was prepared as previously reported [18]. Briefly, metformin hydrochloride (4.51 g, 27.27 mmol) was suspended in isopropyl alcohol (i-PrOH) (40 mL) and potassium hydroxide (1.83 g, 32.62 mmol) was added to the suspension at 50 °C. The white slurry was stirred at 50 °C for 2 h, and then the mixture was cooled to RT. The resulting mixture was filtered and the filter-cake was washed with acetone (2 × 10 mL). The combined filtrates were concentrated under reduced pressure yielding a white solid (metformin free base). Yield: 98% (3.45 g); 1H NMR (400 MHz, D2O) δ 3.07 (s, -N-(CH3)2). Subsequently, N,N-dimethylbiguanide free base (1.55 g, 12.02 mmol (20-fold excess amount of MestOx unit in polymer)) was added to a solution of PMeOx-PMestOx (200 mg, 22.2 μmol (0.6 mmol of total MestOx unit)) in dimethylformamide (DMF) (15 mL). The mixture was stirred at 75 °C for 48 h to form PcBOx. The reaction mixture was then diluted with DI water and dialyzed against DI water to completely remove unreacted free N,N-dimethylbiguanide and organic solvent. The polymer was obtained as a white powder (162 mg, 81% yield) after lyophilization from water. 1H NMR (D2O, 298 K): 2.2–2.7 (4H, CO–CH2–CH2–C3N3(NH2)(N(CH3)2); 2.6–3.0 (6H, C3N3(NH2)(N(CH3)2); 3.1–3.7 (4H, –N–CH2–CH2–); 1.8–2.0 (3H, –CO–CH3).
2.5. Analysis of PMeOx-PcBOx structure
UV–Vis spectroscopic analysis was performed to investigate the formation of poly(2-N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl-2-oxazoline) (PcBOx) structure in PMeOx-PcBOx. Samples (PMeOx-PcBOx, PMeOx-PMestOx, and cBG, and N,N-dimethylbiguanide) were dissolved (equimolar N,N-dimethylbiguanide or cBG units) in DI water and UV absorption spectra were measured over the wavelength range of 200 to 300 nm in 1 nm steps. (SpectraMax M5, Molecular Devices). In order to investigate the mass shift of PMestOx after conjugation with N,N-dimethylbiguanide and confirm the formation of the defined structure using MALDI-TOF/TOF analysis, we synthesized a PcBOx homopolymer from MestOx homopolymer with a degree of polymerization (DP) of 10 as described above. MALDI-TOF/TOF was performed on a Sciex 5800 MALDI-TOF/TOF mass spectrometer and 2-(4′-Hydroxybenzeneazo)benzoic acid (HABA) (20 mg/mL in acetonitrile) was used as the matrix. Polymer samples were dissolved in acetonitrile (2 mg/mL). Analytes were prepared by mixing 10 μL of matrix and 5 μL of polymer samples. Samples were applied using the dried droplet method.
2.6. Titration analysis
PMeOx-PcBOx (12 mg) was dissolved in 10 mL 0.01 N HCl solution and titrated with 0.1 N NaOH solution added in increments of 0.020 mL after the pH values were stabilized. Titration assay for control samples (saline, cBG and PMeOx-PMestOx) was performed at the same molar concentration of cBG units with the same volume of titrant. Derivative ΔOH/ΔpH was plotted from the obtained titration curves to investigate the buffering-capacity of samples.
2.7. Self-assembly of PMeOx-PcBOx in aqueous solution and particle formation
Briefly, the sample was diluted with DI water to yield 1 mg/mL final polymer concentration before the measurement. Particle z-average effective diameter and polydispersity index (PDI) were measured by dynamic light scattering (DLS) (Nano-ZS, Malvern Instruments, UK). Results are the mean of three independent sample measurements. The morphology of PMeOx-PcBOx particle in aqueous media was determined using a LEO EM910 TEM operating at 80 kV (Carl Zeiss SMT Inc., Peabody, MA). One drop of PMeOx-PcBOx solution (1 mg/mL final polymer concentration) was deposited on a copper grid/carbon film for 5 min and excess solution was wiped off using fine filter paper. Then one drop of negative staining solution (1% uranyl acetate) was added and allowed to dry for 10 s prior to the TEM imaging. Digital images were obtained using a Gatan Orius SC1000 CCD Digital Camera in combination with Digital Micrograph 3.11.0 software (Gatan Inc., Pleasanton, CA). Two dimensional nuclear Overhauser effect spectroscopy (2D NOESY) NMR measurement were performed for both PMeOx-PMestOx and PMeOx-PcBOx in D2O at 1 mg/mL polymer concentration to investigate the molecular interactions of the polymers in aqueous media. Critical micelle concentration (CMC) of PMeOx-PcBOx was determined by DLS [19]. PMeOx-PcBOx solution in DI water (1 mg/mL) was gradually diluted with DI water and derived count rates (kcps) were measured in triplicate after mixing. The CMC was taken as the polymer concentration at wich a significant and consistent increase in derived count rate was observed [19].
2.8. Drug encapsulation
2.8.1. Encapsulation of water-insoluble drugs and in vitro drug release
Drug-encapsulated polymeric nanoparticle formulations were prepared by the thin film hydration method as previously described [17]. For bruceantin, LY2109761, imiquimod, paclitaxel, SN-38, LY364947, GDC-0941, aclacinomycin A, wortmannin, and GW788388 stock solutions of PMeOx-PcBOx and drugs in chloroform:methanol (9:1) solution were mixed together at the pre-determined ratios (3:10 drug to polymer w/w ratio (LY2109761), 2:10 w/w ratio (bruceantin and imiquimod), 1:10 w/w ratio (paclitaxel, SN-38, wortmannin, LY364947, GDC-0941, aclacinomycin A, GW788388)). All samples were first tested at the 10:1 polymer to drug feeding ratio. If encapsulation was successful, we proceeded to test 10:2 and other feeding ratios. The reported data for maximal dispersion is the formulation which achieved the highest LC and absolute solubility of the hydrophobic drugs. This was determined by screening through the feeding ratios.
The organic solvent was evaporated at 58 °C under a stream of inert gas to form a thin-film of drug-polymer homogenous mixture. Next, the thin films were hydrated with saline and then incubated for 5–10 min at 45 °C to form drug-encapsulated polymeric micelle solutions. All samples were prepared with 2 mg of polymer and hydrated with 200 uL of saline. The formed micelle solutions were centrifuged at 10,000 rpm for 3 min (Sorvall Legend Micro 21R Centrifuge, Thermo Scientific) to remove any precipitate of unloaded drug or polymer. The final concentration of drugs in PMeOx-PcBOx micelles was analyzed by HPLC (Agilent Technologies 1200 series) using a mixture of acetonitrile/water (70%/30% v/v for bruceantin, imiquimod, wortmannin, GW788388 and paclitaxel; 50%/50% v/v for LY2109761, GDC-0941, aclacinomycin A, LY364947, and SN-38) as the mobile phase. The samples were diluted with mobile phase to final concentration of ~100 μg/mL of drugs and injected (10 μL) into the HPLC system. The flow rate was 1.0 mL/min, and column temperature was 40 °C. Detection wavelength were 227 nm for bruceantin, paclitaxel, LY2109761, GDC-0941, GW788388, aclacinomycin A, LY364947 and SN-38 and 254 nm for imiquimod and wortmannin. Drug concentration was quantified against free drug calibration curves.
The drug release from PMeOx-PcBOx nanoparticle was studied using the membrane dialysis method against phosphate buffered saline (PBS), pH 7.4 at 37 °C. Briefly, the drug loaded PMeOx-PcBOx nanoparticle formulations (paclitaxel and bruceantin) were diluted with saline to obtain solution of approximately 0.1 mg/mL of polymer. Then the diluted nanoparticle formulations (100 μL) were placed in 100 μL floatble Slide-A-Lyzer MINI dialysis devices with a MWCO of 3.5 kDa (Thermo Scientific) and suspended in 20 mL of PBS to comply with sink conditions. Three devices were used for every time point. At each time point, the samples were withdrawn from the dialysis device and the remaining drug amount of sample were quantified by HPLC as described above. Stability of both nanoparticle formulations (paclitaxel and bruceantin) was investigated via monitoring drug loading (HPLC) and size distribution (DLS) for 8 days at 4 °C.
2.8.2. Accounting for molecular characteristics of insoluble drugs
The number of hydrogen bond acceptors and donors in each drug substance were counted by Chem3D. The number of rotatable bonds in each drug substance was counted as previously reported [20]. Briefly, any single bond not in ring structures, except for the bonds connected to the terminal atom and the bonds present in amide bonding, were counted as 1. The Log P values of polymer unit structures and each drug substance were predicted by Chem3D. The definition of electron deficient aromatic ring herein is an aromatic ring, which has more electron withdrawing atoms or groups than electron donating atoms or groups. The score of each drug substance is the number of the parameters which meet our criteria: 1) electron deficient aromatic ring is present, 2,3) the number of hydrogen bonding acceptors and donors are more than 6 and 2, respectively, 4) LogP is less than 3, and 5) the number of rotatable bonds is more than 5.
2.8.3. Encapsulation of platinum-based drug (DachPt)
DachPt powder (5 mg) was suspended in PMeOx-PcBOx (10 mg) aqueous solution (3 mL) and the reaction mixture was refluxed for 48 h. After that the mixture was cooled to RT and centrifuged to precipitate free DachPt. Clear supernatant solution was then dialyzed against DI water for 3 days in 3.5 kDa dialysis membrane and lyophilized to obtain powder form of DachPt-loaded PMeOx-PcBOx (DachPt-PMeOx-PcBOx). Actual Pt loading was measured via Inductively coupled plasma mass spectrometry (ICP-MS) analysis using NexION 300D inductively coupled plasma mass spectrometer (PerkinElmer, USA) equipped with SC4-DX autosamplers (ESI, USA). Decomposition of the samples matrix was performed by heating at 70 °C for 20 h after the addition of trace metal grade nitric acid (20% aqueous solution) and hydrochloric acid (5% aqueous solution). Digested samples were diluted (1:10) with DI water, a second dilution (1:100) was performed with 2% nitric acid. ICP-MS was operated in standard mode, all operating parameters were optimized to meet requirements as defined by the manufacturer prior to method calibration and analysis. Calibration curves were constructed using a zero point standard and a six point calibration curve in a range 1–100 ppb. Five replicates were analyzed per sample. Quantification was performed with Bi as an internal standard.
2.8.4. Loading efficiency (LE) and loading capacity (LC) calculations
The following equations were used to calculate LE and LC of drug in PMeOx-PcBOx micelles:
| (1) |
| (2) |
where Mdrug and Mexcipient are the mass of the solubilized drug and polymer in the solution respectively, while Mdrug added is the weight amount of the drug added to the dispersion during the preparation of the micelle formulation.
2.9. Cell culture and cytotoxicity
In vitro cytotoxicity of DachPt-PMeOx-PcBOx, oxaliplatin, and PMeOx-PcBOx was evaluated in MDA-MB-231 and 344SQ cancer cell lines by following previously reported method [21] using [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] (WST-8) (Cell-Counting Kit-8 assay). Briefly, cells were seeded in 96-well plates at a density of 5000 cells/well 24 h prior to the treatment. Subsequently, cells were treated with DachPt-PMeOx-PcBOx, oxaliplatin, or PMeOx-PcBOx in full medium at the same concentration of platinum. Following 72 h, the incubation medium was removed, and 100 μL of fresh medium with WST-8 (10 μL/well) was added and incubated for 4 h at 37 °C. Subsequently, absorbance was read at 450 nm using a plate reader (SpectraMax M5, Molecular Devices). Cell survival rates were calculated and normalized to the control untreated wells. Data represents average of six replicates in means ± standard deviation (SD). The mean drug concentration required for 50% growth inhibition (IC50) was determined using Graphpad Prism 7 software.
3. Results
3.1. Polymer synthesis and post-polymerization modification
We first synthesized PMeOx-PMestOx diblock copolymer as the starting material via living cationic ring-opening polymerization [16] and then converted it to N,N-dimethylbiguanide derivative by the polymer analogous condensation reaction of the MestOx block with N,N-dimethylbiguanide (Scheme 1). The full conversion of PMestOx was confirmed by 1H NMR spectra (1H NMR (CDCl3, 298 K)) of the reaction mixture as the methyl ester signal of the polymer (at δ 3.5 ppm) disappeared after the condensation with N,N-dimethylbiguanide (Fig. S1). The block length and the molecular weight of the PMeOx-PMestOx precursor were confirmed via 1H NMR spectroscopy (1H NMR (D2O, 298 K)) (Fig. S2) (PMeOx = 60, PMestOx = 30, Mn = 9.9 kg/mol) and GPC (Mn = 13.4 kg/mol, PDI = 1.038).
Scheme 1.
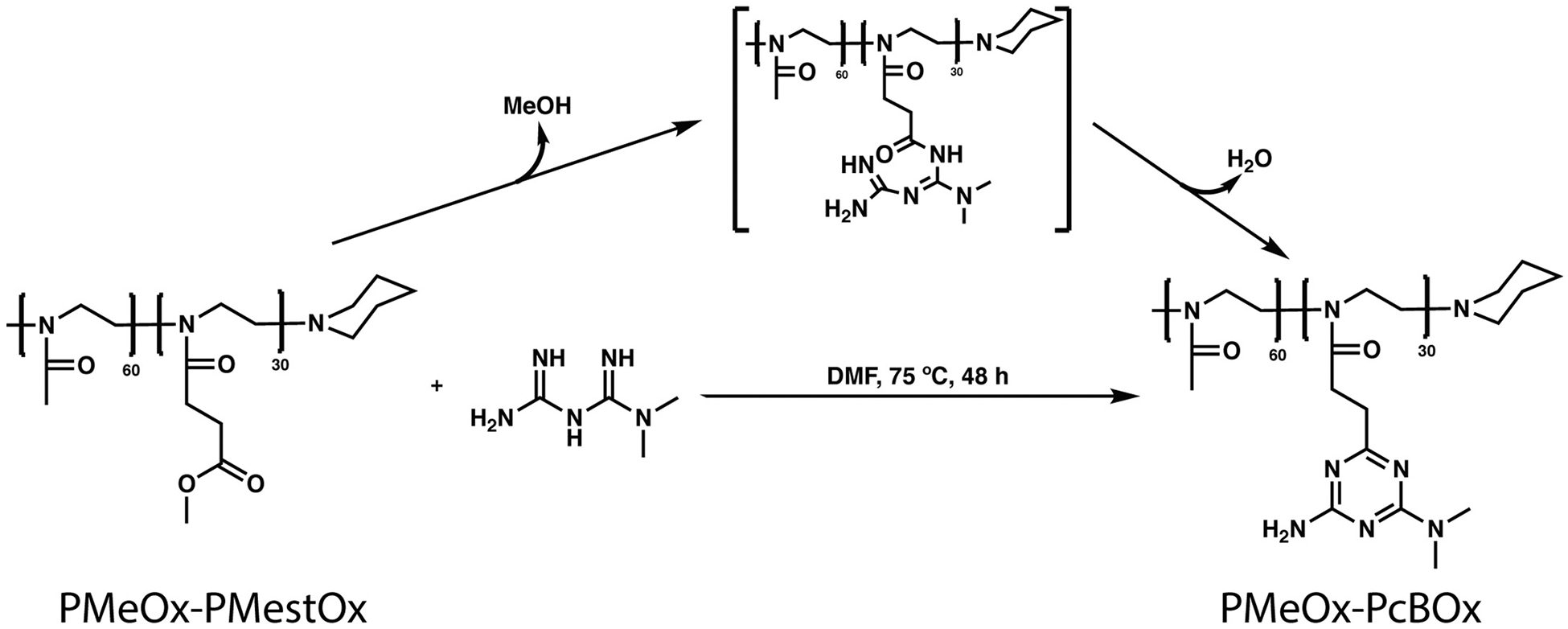
Synthesis of PMeOx-PcBOx via N,N-dimethylbiguanide condensation.
After the purification of PMeOx-PcBOx, 1H NMR spectrum of precursor (PMeOx-PMestOx), cBG, and PMeOx-PcBOx were analyzed to confirm the conversion of cBOx structure (1H NMR ((CD3)2SO, 298 K)). The disappearance of the methyl ester signal (marked by arrow in Fig. 1A and not present in Fig. 1C) suggests that there was a complete conversion from methyl ester to a new structure. The 1H NMR spectrum of the newly synthesized side chain was nearly identical to that of the reference molecule (cBG) (Fig. 1B and C) indicating that the condensed cBG-like ring structure was formed on the side chain of the polymer, consistent with PcBOx. Also, new peaks attributed to protons g and f appeared at 3.0 ppm and 6.6 ppm, respectively. There were no changes exhibited in the NMR spectrum of the other polymer block (PMeOx) (Fig. 1A and C, proton a). This suggests the conversion of PMeOx-PMestOx to PMeOx-PcBOx was successful and proceeded without side reactions. The 13C NMR spectrum of PMeOx-PcBOx was also identical to that of the reference molecule (cBG) (Fig. S3) indicating the formation of cBG strucrue on the side chain of the polymer and the disappearance of the methyl ester signal (51 ppm) suggests a complete conversion from methyl ester to cBG structure. Also, new peaks attributed to four carbons appeared at 36 ppm, 165.4 ppm, 168.8 ppm, and 176.5 ppm. There were no changes exhibited in the NMR spectrum of the other polymer block (PMeOx) (Fig. S3). The molecular mass of PMeOx-PcBOx was estimated by 1H NMR spectroscopy (1H NMR (D2O, 298 K)) (Fig. S2) (Mn = 12.2 kg/mol). The GPC value (Mn = 12.2 kg/mol, PDI = 1.056) was relatively low when compared that of the the precursor PMeOx-PMestOx, which is possibly to be due to interaction of the cBOx (polyamine moiety) with the GPC column, resulting in the apparent decrease in the molecular weight, previously reported for GPC analysis of polyamine compounds [22]. Alhough the absolute molecular weight value can not be obtained from the GPC analysis, the PDI value of PMeOx-PcBOx is narrow and suggests that no side reaction or crosslinking of the polymer chains occured.
Fig. 1.
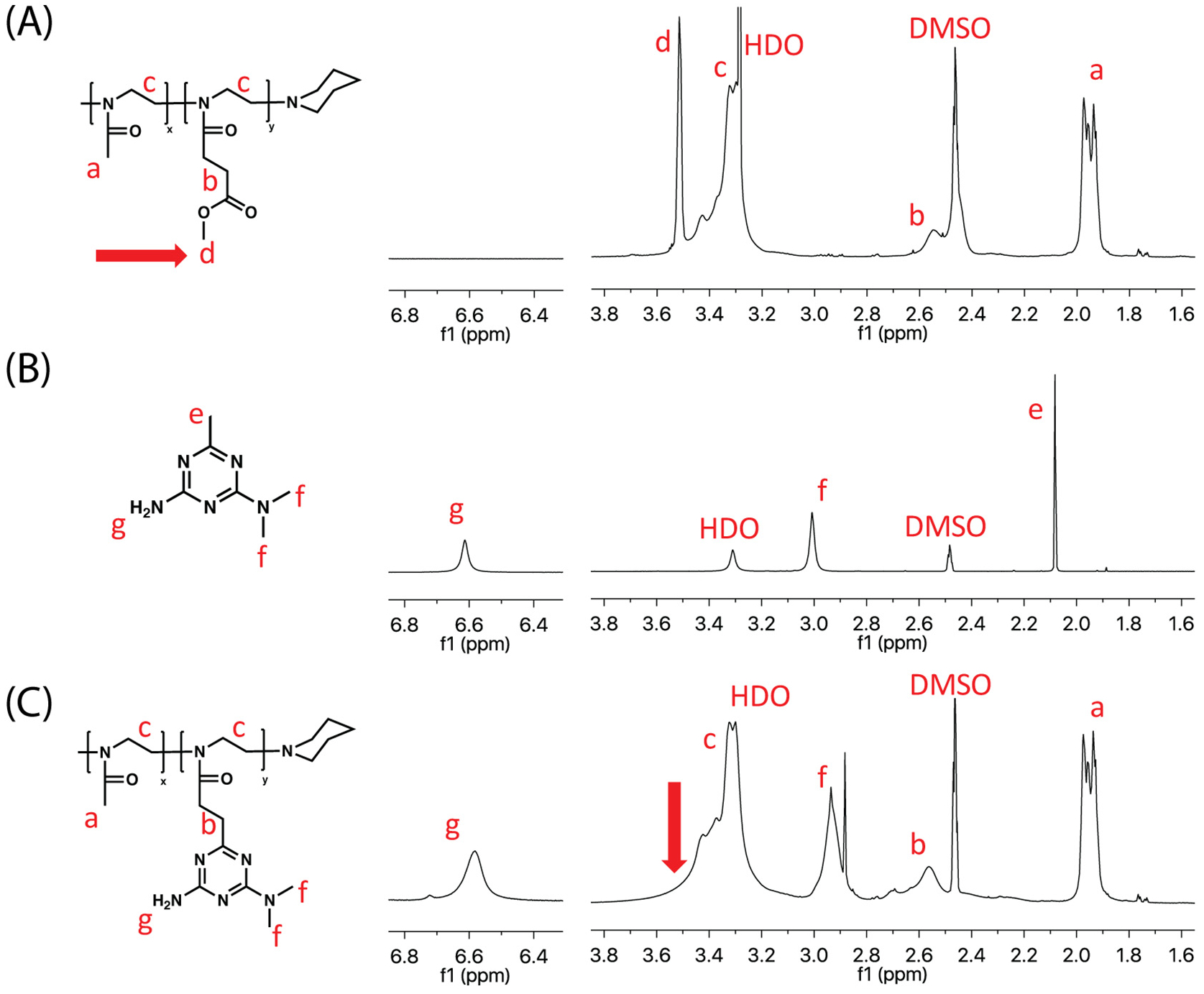
Overlay of the 1H NMR spectra of PMeOx-PMestOx, cBG and PMeOx-PcBOx (1H NMR ((CD3)2SO, 298 K).
We performed UV–Vis spectroscopic analysis to investigate the formation of the PMeOx-PcBOx structure. As shown in Fig. 2A, the UV spectrum of the PMeOx-PcBOx showed specific peak absorbance around 227 nm while uncondensed N, N-dimethylbiguanide did not show particular UV absorbance. We found only PMeOx-PcBOx exhibited a clear peak absorbance around 227 nm (PMeOx-PcBOx) confirming the formation of the PcBOx structure. These results suggest the methyl ester group in PMestOx had been condensed with N,N-dimethylbiguanide to form PcBOx. The PMeOx-PcBOx and cBG UV measurements were done with equimolar amounts of cBG molecules and PcBOX side chains, meaning we would expect nearly identical absorbance, which we see in Fig. 2A. This, along with the NMR data, indicate a full conversion of the PMestOx unit to PcBOx.
Fig. 2.
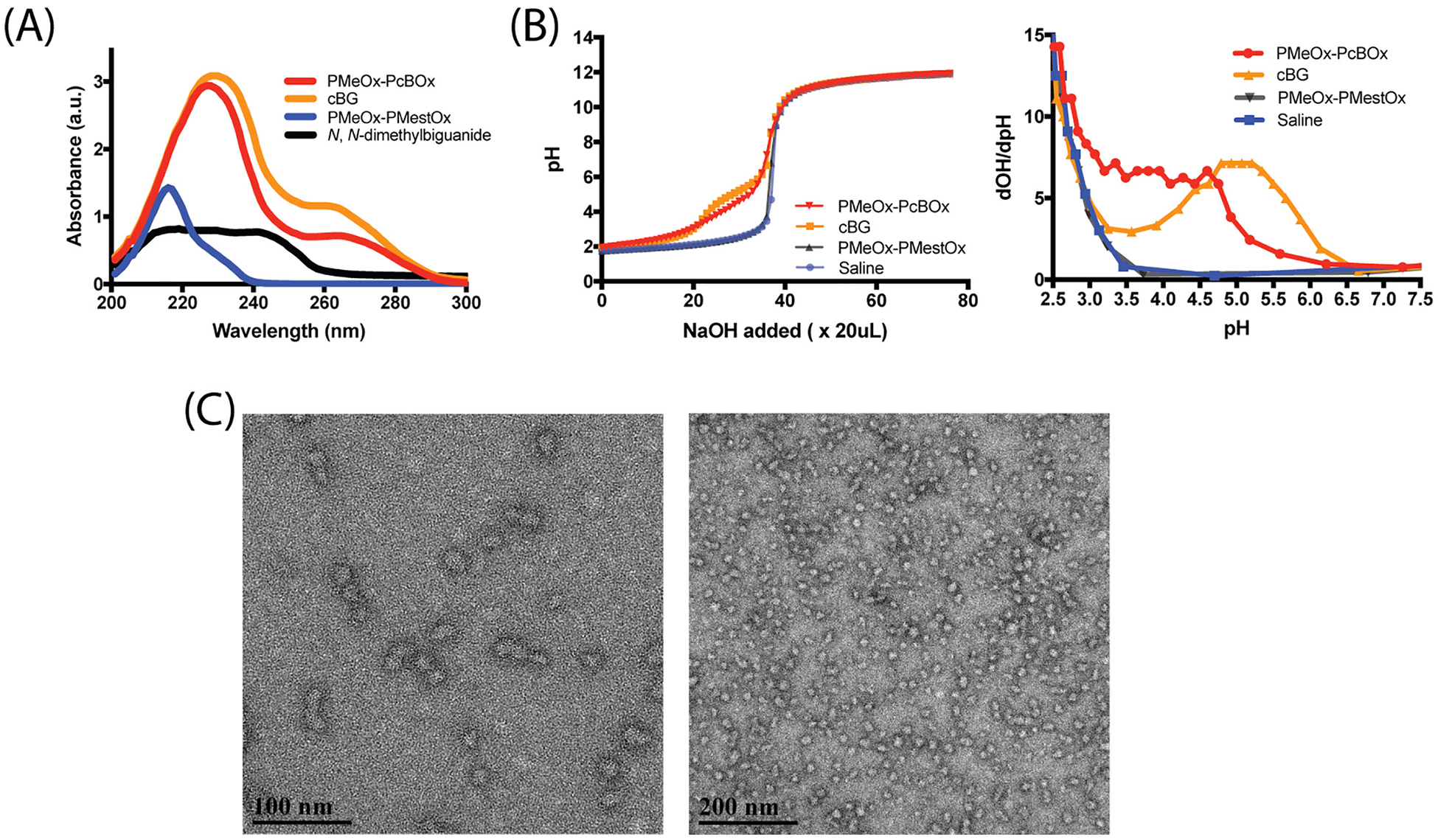
(A) UV spectra of PMeOx-PcBOx, cBG, PMeOx-PMestOx and N,N-dimethylbiguanide in the range of 200–300 nm. (B) Acid-base titration curves (left) and derivative plot dOH/dpH as a function of pH (right) of PMeOx-PcBOx, cBG, PMeOx-PMestOx and saline. (C) TEM image of self-assembled PMeOx-PcBOx. Scale bar = 200 nm (left), 50 nm (right).
To show the mass shift of PMestOx unit after conjugation with N,N-dimethylbiguanide, MALDI-TOF analysis was performed on PcBOx homopolymer with DP of 10. MALDI-TOF MS of the PMestOx revealed the expected spacing of 157.07 m/z corresponding to the MestOx monomer, while after the condensation reaction the spacing increased to 236.14 m/z. This data provides evidence supporting the condensation of N,N-dimethylbiguanide with PMestOx yielding the PcBOx structure (spacing of 236.14 m/z corresponding to repeating unit of PcBOx) (Fig. S4).
The pH-dependent protonation of PMeOx-PcBOx polymer in saline was evaluated by titration (Fig. 2B). The titration curve and dOH/dpH curve of cBG clearly showed its proton buffering profile and its pKa was estimated to be around 5. PMeOx-PcBOx copolymer also had similar but broader protonation profile, whereas PMeOx-PMestOx didn’t show any proton buffering capacity. This indicatesthe protonation profile of PMeOx-PcBOx was derived from the cBG moiety.
3.2. Self-assembly of PMeOx-PcBOx copolymer
Self-assembly of PMeOx-PcBOx copolymer and formation of particles in aqueous media was investigated by DLS and confirmed by TEM. The particles had a volume measured diameter of 28.0 nm and PDI of 0.28 as determined by DLS (Fig. S5). The particles were non-spherical, with partially elongated morphology (Fig. 2C). The dual spherical and elongated morphologies contribute to the high PDI value in the DLS measurements.
To examine the molecular interactions of PMeOx-PcBOx chains in aqueous media, 2D NOESY NMR measurements were conducted. The 2D NOESY NMR spectra (Fig. S6), in PMeOx-PcBOx dispersions display strong correlation of PcBOx protons (2.6 ppm to 3.0 ppm, 6H, C3N3(NH2)(N(CH3)2) with other PcBOx protons (2.2 ppm to 2.7 ppm, 4H, CO–CH2–CH2–C3N3(NH2)(N(CH3)2) and protons of the polymer backbone (3.1 ppm to 3.7 ppm, 3.1–3.7 (4H, –N–CH2–CH2–). This highlights some of the intramolecular and intermolecular interactions present in the PMeOx-PcBOx polymer. In the case of PMeOx-PMestOx, 2D NOESY NMR did not show any correlation of protons of PMeOx-PMestOx. Taken together, this evidence supports that the molecular interaction of PMeOx-PcBOx arises from PcBOx structure and contributes to the polymer self-assembly into particles in aqueous media.
To investigate the effect of polymer concentration on the formation of the particle in aqueous solution, the CMC of PMeOx-PcBOx was determined by DLS. Light scattering intensity and count rate of the particle was monitored over a range of PMeOx-PcBOx concentrations and the point where derived count rate becomes constant corresponds to the CMC. As shown in Fig. S7, the CMC of PMeOx-PcBOx was approximately 40 mg/L in DI water.
3.3. Solubilization of poorly soluble small molecules
We examined several poorly soluble active pharmaceutical ingredients to assess their solubilization in PMeOx-PcBOx polymeric micelles and compared it with the solubilization of these molecules in the micelles of our reference polymer P[MeOx35-b-BuOx20-b-MeOx35] (P2) [17,23–25]. The molecules examined belonged to different classes including transforming growth factor β (TGF-β) receptor inhibitors (LY2109761, GW788388 and LY364947), protein synthesis inhibitors (bruceantin), toll-like receptor (TLR) 7 agonist (imiquimod), phosphatidylinositol 3-kinase (PI3K) inhibitors (wortmannin and pictilisib (GDC-0941)), microtubule inhibitor (paclitaxel), and topoisomerase inhibitors (Aclacinomycin A and SN-38). In these experiments, we kept the polymer concentration constant at 10 mg/mL and varied the feed concentration of the drug until its maximal solubility was observed. We screened drugs at various ratios, beginning at 1:10 (drug:polymer), and increased the ratio until maximal solubility was obtained. As shown in Fig. 3, PMeOx-PcBOx micelle formulation displayed a reasonably good capacity for solubilization of several poorly soluble drug compounds (see also Table S1). In particular, we solubilized nearly ~3.28 mg/mL of LY2109761 with 24.7% LC and little if any drug precipitate (82% LE). Bruceantin also showed reasonably good maximal solubilization in this formulation: 1.87 mg/mL, 11.5% LC, 65% LE. On the other hand, paclitaxel and other drugs were not as soluble as LY209761and bruceantin (6.8% LC for aclacinomycin A, 6.3% LC for paclitaxel, 4.3% LC for GDC-0941, 3.8% LC for wortmannin), while LY364947 (0.3% LC) GW788388 (0.2% LC), Imiquimod (undetectable) and SN-38 (undetectable) were practically insoluble. Comparison of the solubilization profile of these molecules for PMeOx-PcBOx and P2 triblock copolymer micelles reveals distinct selectivity of each polymer for some of these APIs (Fig. 3, see also Table S1). With P2, we observed the ultra-high loading of bruceantin with least amount of drug precipitate (as much as 9.98 mg/mL, 50.0% LC, 99% LE). Also, as previously reported [17], paclitaxel was solubilized at maximal concentration of 6.89 mg/mL (41% LC and 86% LE). LY2109761 showed fairly high solubilization in P2 micelles (1.5 mg/mL, 14.0% LC, 78% LE) although it was distinctively less than that observed in PMeOx-PcBOx micelles. Solubilization of aclacinomycin A was low but generally comparable in both micelle systems. On the other hand, GDC-0941 and wortmannin were practically insoluble in P2 system, while theywere distinctively more soluble in PMeOx-PcBOx. The other compounds tested (GDC-0941, LY364947, SN-38, and Imiquimod) were practically insoluble in both micelle solutions. This data is briefly summarized in Supplementary Table S2 along with innate water solubilities of the drug compounds with their absolute loading (mg/mL).
Fig. 3.
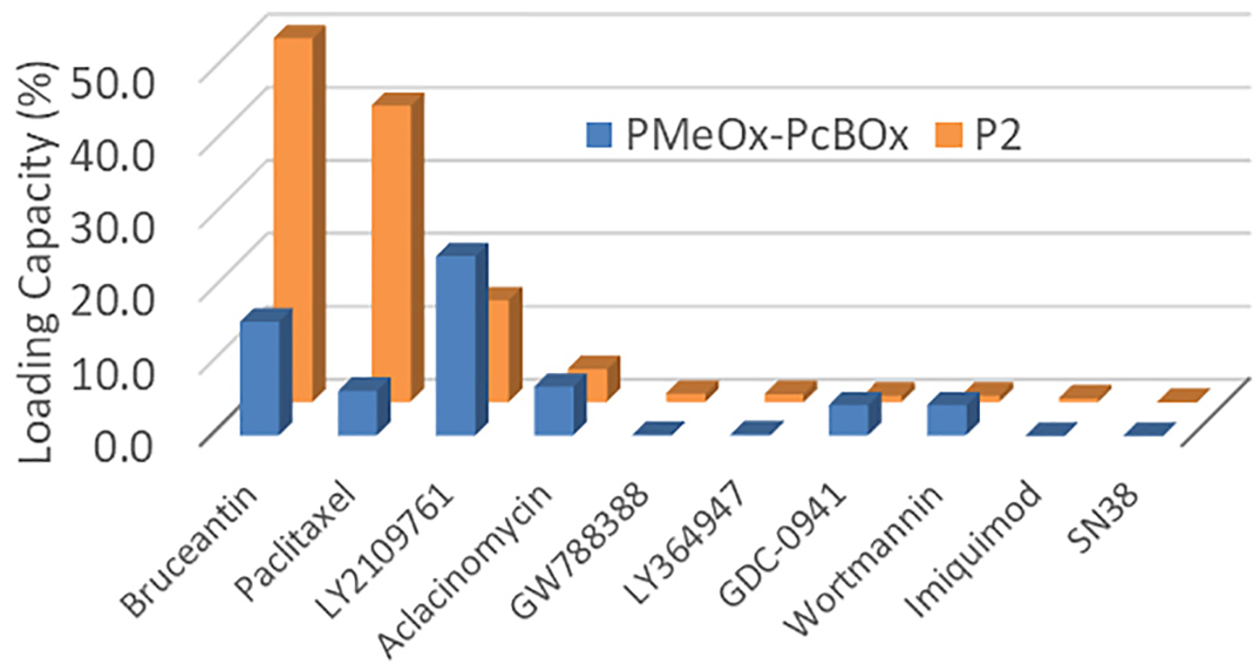
Differential solubilization of drugs indicated by their maximum LC in PMeOx-PcBOx (blue bars) and P2 (orange bars). Polymers concentration 10 mg/mL. (n = 3 for all drugs and polymers except for GW788388, GDC-0941, and LY364947 in PMeOx-PcBOx groups, which has n = 1). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
PMeOx-PcBOx micelle dispersions displayed sub-100 nm size distribution in DLS effective diameter measurements; 85 nm for paclitaxel (PDI = 0.298, Dv10 = 23.3 nm, Dv50 = 34.6 nm, Dv90 = 80.9 nm), 60 nm for bruceantin (PDI = 0.48, Dv10 = 17.2 nm, Dv50 = 25.2 nm, Dv90 = 42.9 nm), and 50 nm for LY2109761 (PDI = 0.4, Dv10 = 15.5 nm, Dv50 = 21.5 nm, Dv90 = 34.8 nm). TEM images of particles in both formulations are shown in Fig. 4A and B. The images reveal small-sized particles (approximately 30 nm) in both formulations, corresponding to the DLS measurement (volume-based). Small portions of particle aggregates were observed in both formulations and this may explain the relatively high value of PDI.
Fig. 4.
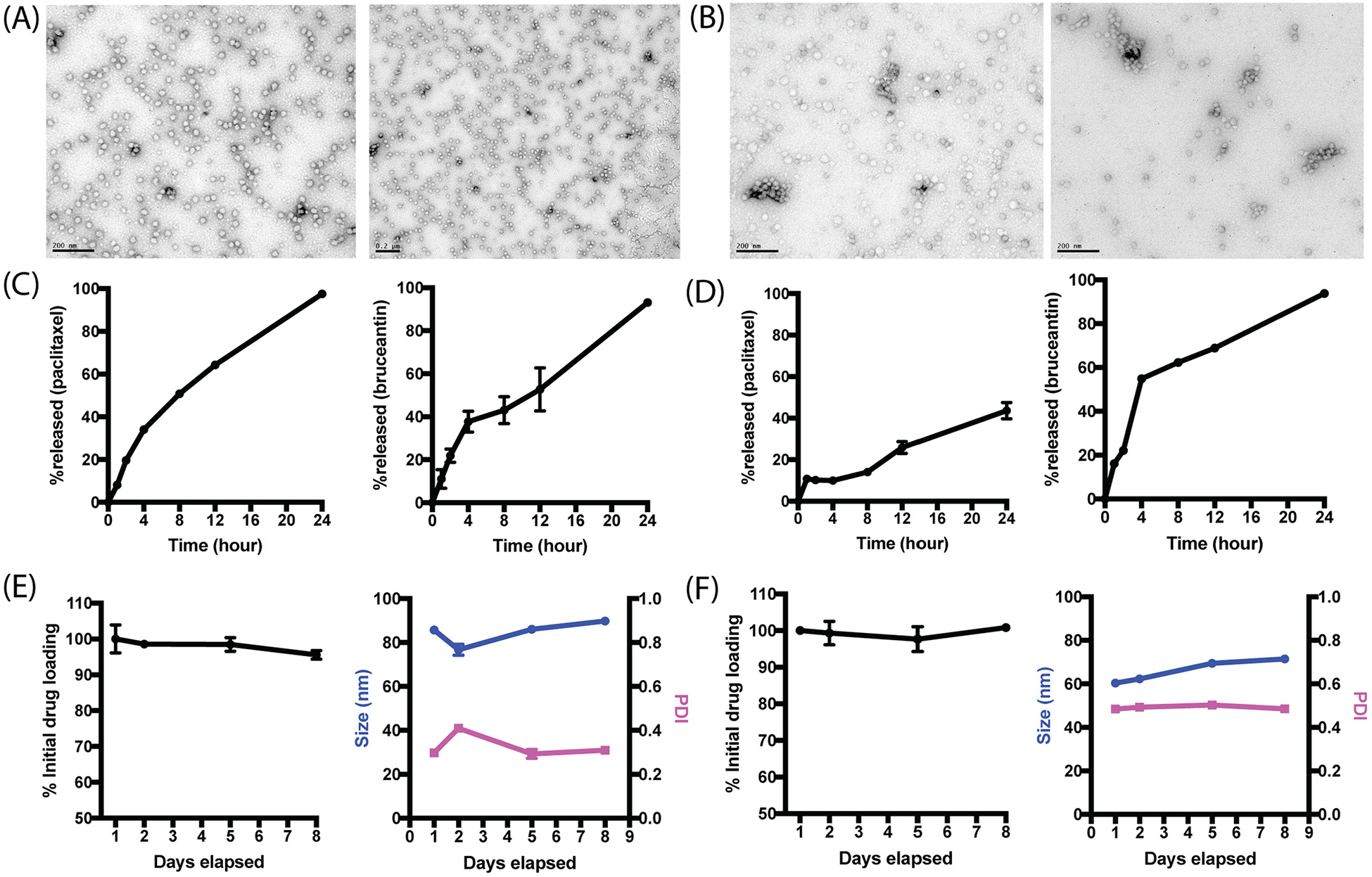
TEM image of (A) paclitaxel-PMeOx-PcBOx formulation and (B) bruceantin-PMeOx-PcBOx formulation, Release profile of (C) paclitaxel (left) and bruceantin (right) from PMeOx-PcBOx nanoparticle formulation and (D) paclitaxel (left) and bruceantin (right) from P2 polymer and stability profile of (E) paclitaxel-PMeOx-PcBOx-formulation and (F) bruceantin-PMeOx-PcBOx formulation (n = 3).
We investigated the relese profile of encapsulated drugs (paclitaxel and bruceantin) in PMeOx-PcBOx. Both paclitaxel and bruceantin were continuously released from PMeOx-PcBOx nanoparticle, with over 80% of drugs being released at 24 h time point (Fig. 4C). The release rates were similar for bruceantin in both polymers. Interestingly, the release rate was very different for paclitxael in P2 vs. PMeOx-PcBOx (Fig. 4D). After 24 h, most of the PTX was released in PcBOx whereas not even 50% was released from P2. This kind of release data is consistent with previously published data at the low (10:1) polymer:drug feeding ratio [23]. Both paclitaxel and bruceantin formulation in PMeOx-PcBOx were proven stable for 8 days at 4 °C as we monitored the drug loading and size distribution (Fig. 4E and F).
Finally, we summarized the relationship between LC and the physicochemical properties of the drug molecules that could be related to the interaction with PMeOx-PcBOx (Fig. 5). The designated potentially “influential” parameters are the presence of electron deficient aromatic structures (EDA), the numbers of hydrogen bonding acceptors (HBA) and donors (HBD), LogP, and the number of rotatable bonds (RBN).
Fig. 5.
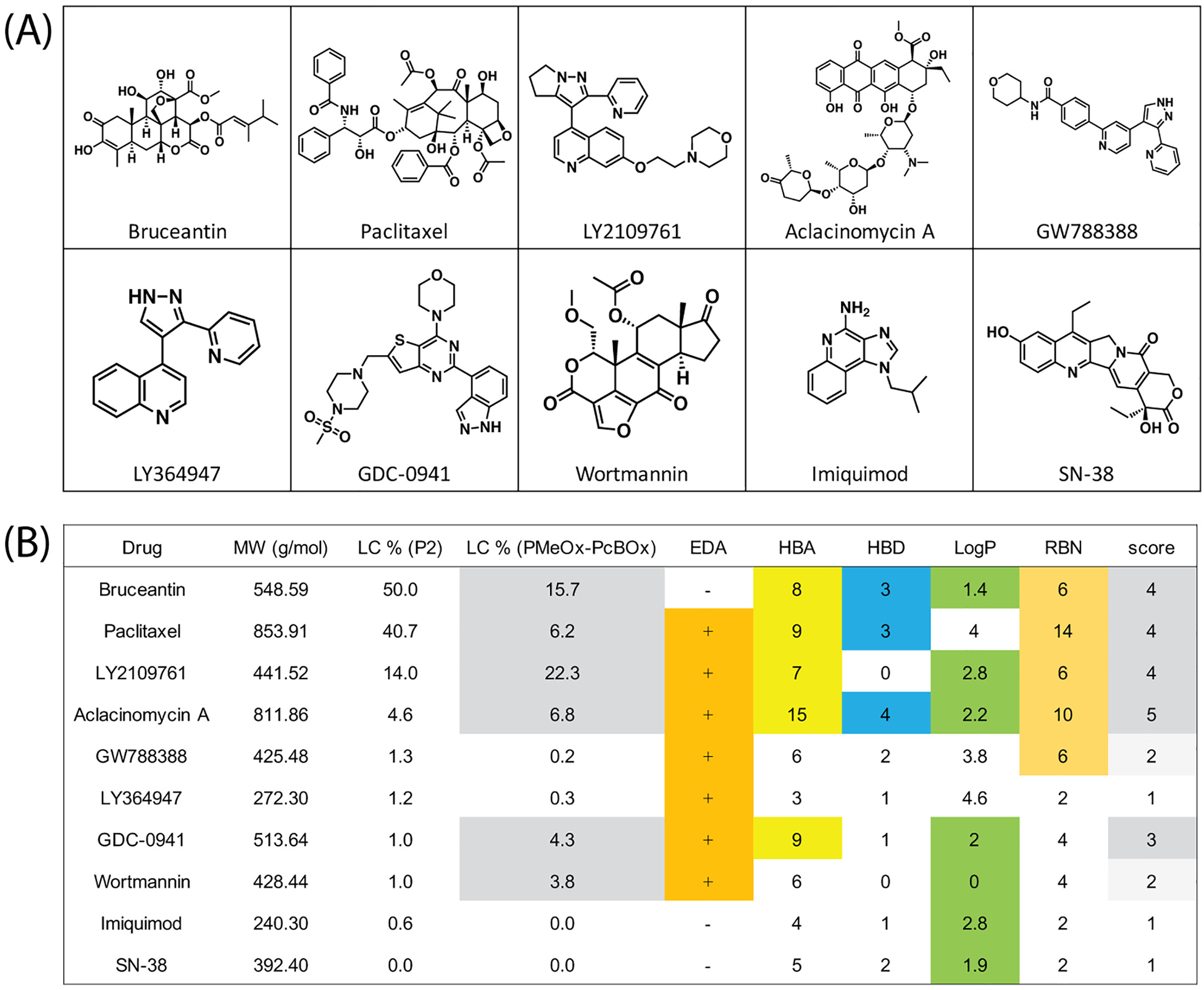
(A) Evaluated drug structures and (B) comparison of the maximal LC of each drug in PMeOx-PcBOx and P2 micelles with the molecular characteristics of these drugs. The scores correspond to the presence of electron deficient aromatic rings (EDA), the numbers of hydrogen bonding acceptors and donors (HBA and HBD, respectively), lipophilicity (LogP) and the number of rotatable bonds (RBN).
3.4. Water-soluble DachPt encapsulation
We investigated whether the cBG side chain of the PMeOx-PcBOx copolymer can facilitate incorporation of platinum drug (DachPt) in the polymeric micelles via coordination bonding. The loading was accomplished by co-incubation of the DachPt and PMeOx-PcBOx micelles in the aqueous solution followed by separation of the drug loaded micelles from unincorporated drug. The total platinum mass percent in the resulting DachPt-PMeOx-PcBOx formulation was about 17.7% as measured by ICP-MS. When converted to the equivalent content of DachPt this value results in ~34.5% LC. The analysis of DachPt-PMeOx-PcBOx formulation by DLS revealed a unimodal size distribution with an effective diameter of ~35 nm and PDI of 0.49 (Fig. S5B), The increase of the particle size and polydispersity when compared to the free PMeOx-PcBOx micelles (~28 nm, PDI = 0.28), was possibly due to conformational changes in the core-forming block and volume increase induced by DachtPt incorporation.
3.5. In vitro cytotoxicity DachPt-PMeOx-PcBOx formulation
We evaluated the in vitro cytotoxicity of DachPt-PMeOx-PcBOx in comparison with the free oxaliplatin in 344SQ murine NSCLC cells and MDA-MB-231 human breast cancer cell lines. The cytotoxicity was measured by the CCK-8 assay. Free polymer PMeOx-PcBOx was found to be non-toxic in both cell lines up to 4 mg/mL dose. On the other hand, PMeOx-PcBOx-DachPt displayed platinum concentration-dependent cytotoxicity profile in both cell lines, albeit the corresponding its IC50 exceeded those of oxaliplatin by 10 to 50 times (Fig. 6).
Fig. 6.
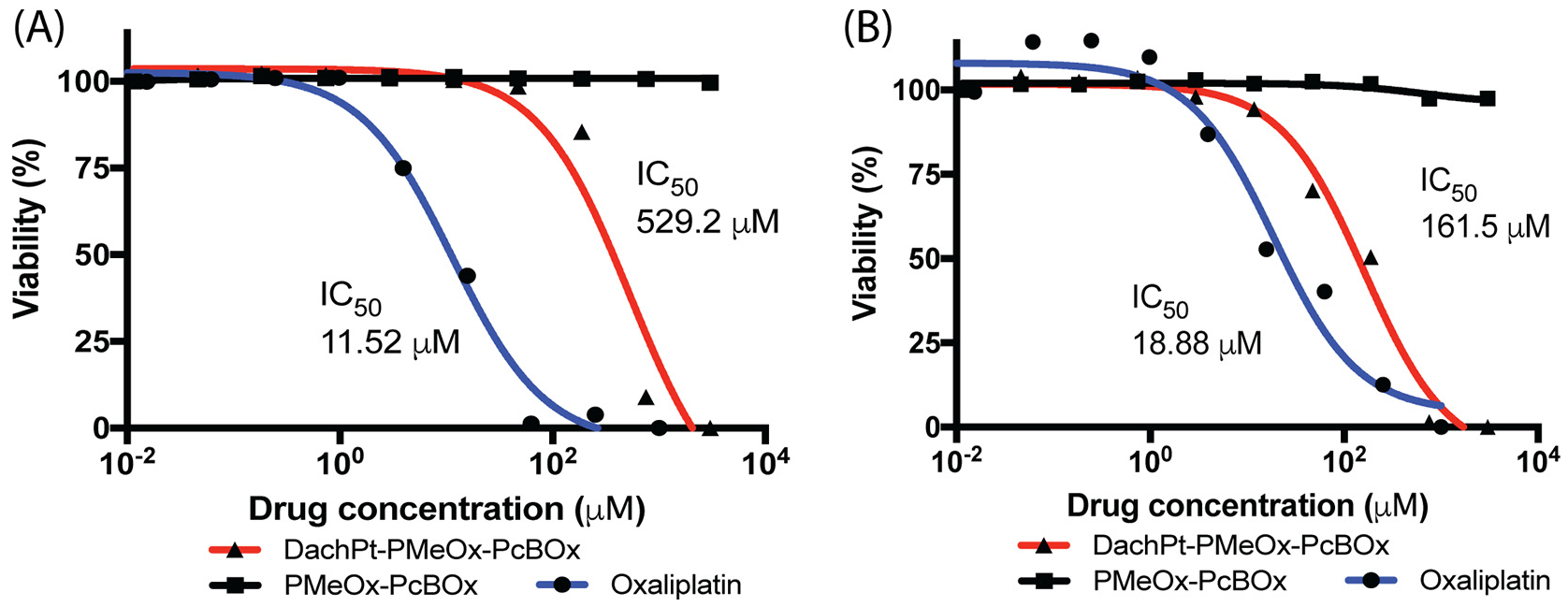
Cytotoxicity and IC50 values of PMeOx-PcBOx, DachPt-PMeOx-PcBOx and free oxaliplatin in (A) 344SQ murine NSCLC cells and (B) MDA-MB-231human breast cancer cells. DachPt-PMeOx-PcBOx concentration is presented as the oxaliplatin equivalent concentration. PMeOx-PcBOx concentration is presented as the block copolymer concentration equivalent to that in DachPt-PMeOx-PcBOx formulation.
4. Discussion
The motivation for our work was to develop an alternative polymer for drug nanoformulations which is compatible with drugs that cannot be otherwise formulated in existing drug delivery platforms. Previously, we described a poly(2-oxazoline)-based polymeric micelle platform, which is unique in its ability to incorporate large amounts of insoluble compounds, as demonstrated across many drugs and drug candidates, such as taxanes [17,26]. The hydrophobic PBuOx block in P2 (P [MeOx35-b-BuOx20-b-MeOx35]) has polar and hydrated amide functionality in each repeating unit. These micelles represent a unique dueal polar/hydrophobic environment for incorporation of drug molecules, such as paclitaxel [17,26]. These interactions are facilitated, perhaps, by the relatively small size and flexibility of the of the butyl side chain in the BuOx. In fact, substitution of the butyl group for a more hydrophobic nonyl group results in a drastic decrease in the solubility of taxanes in these poly(2-oxazoline) based micelles [17,26]. The resultant injectable aqueous micellar solutions are readily prepared and are stable for days and weeks. These formulations can contain up to 50–100 g/L of poorly soluble drugs [23,25,26]. The drug to polymer wt. ratio in these micellar formulations is also up to a hundred times better than the amounts of excipients in the current formulations. For example, for paclitaxel, the amount of P2 polymer per 1 g of drug is ~100– and ~10– times less than the amounts of excipients used in Taxol® and Abraxane®, respectively [23]. This high loading contributes strongly to the widening of the therapeutic window and allows high dose therapy as the excipient-based toxicity is drastically reduced [23].
While BuOx-based polymers, such as P2, have been extremely successful solubilizers for over two dozen drugs and drug candidates, there are many compounds which have failed to display equally good solubilization in this system [27]. This has raised a necessity for the structural modification of the block copolymers to improve solubility of the otherwise failing to be formulated drugs. Advancement in this area has been achieved recently by Luxenhofer group who modified the hydrophobic block in such polymers by replacing a substituted poly(2-oxazoline) for a substituted poly(2-oxazine), which has an additional methylene group in the backbone of the main chain [28]. The hydro-philic blocks (PMeOx) in the new copolymers remained unmodified. A novel selectivity of the new poly(2-oxazine)-based copolymers differentiating them from poly(2-oxazolines) with respect to the drugs that they can solubilize was observed [27,28]. In particular, P2 was highly effective in solubilizing paclitaxel, but much less effective with curcumin. In contrast, a copolymer containing a hydrophobic block made of 2-n-propyl-2-oxazine, which is a structural isomer of BuOx, had a clear solubilization preference for curcumin vs. paclitaxel [27,28].
In the present study, we sought to develop a novel polymeric micelle platform with differential solubilizing capacity that could bring these therapeutic advantages to a new array of insoluble drug compounds. We employed a very different approach to modification of the polymer hydrophobic block. Rather than adjusting the polymer backbone or adding a longer alkyl side chaine, we completely replaced the alkyl side chain of PBuOx for a substituted aromatic heterocyclic ring. To test this approach we designed and synthesized a novel poly(2-oxazoline) diblock copolymer, PMeOx-PcBOx, with an N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl side chain in the “hydrophobic block.” The copolymer structure has been confirmed by the 1H NMR, 2D NOESY NMR, MALDI-TOF MS, and UV–Vis spectroscopic analysis. The copolymer demonstrated the ability to self-assemble into micelle-like structures.
Next, we evaluated the drug loading capacity of this newly developed copolymer. We used drugs across the chemical space, including those molecules that have failed to be solubilized in P2 polymer. For the selected drugs that were not well formulated in P2 micelles, such as LY2109761 (and Wortmannin and GDC-0941 to lesser extents), the new block copolymer displayed a good solubilization capacitysuperior to that of P2. Despite having lower hydrophobicity of the core-forming block than P2 (the estimated LogP value of cBOx is −0.27 while that of BuOx is 1.61), PMeOx-PcBOx can still solubilize a variety of drug compounds. Interestingly, all poorly soluble compounds that show reasonably good solubilization in PMeOx-PcBOx micelles (bruceantin and LY2109761,) are moderately hydrophobic and have LogPs of 1.4 and 2.8 respectively. At the same time the more hydrophobic compounds with LogP around 4 and above (including, paclitaxel) are less soluble or practically insoluble in these micelles.
The capability of N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl side chain to engage in multiple interactions with the drug molecules is likely to be important for drug solubilization. In particular, N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl has a three-heteroatom aromatic ring, which is highly electron deficient. It is known that electron-deficient aromatic rings can form stable pi-pi stacking dimers in part due to strong interactions between pi electrons and sigma electron-deficient orbitals [29]. When considering this feature, N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl groups may enable strong pi-pi stacking interactions both with each other, resulting in the self-assembly of the PcBOx blocks in the micelle core, and with drug molecules, some of which also have electron deficient aromatic rings. Indeed, of all the studied drugs, this new polymer has a clear “preference” for LY2109761, which has highly electron deficient aromatic rings.
Additionally, the N,N-dimethyl-1,3,5-triazine-2,4-diamine-6-ethyl side chain can serve as both a hydrogen bond acceptor and a hydrogen bond donor. Since hydrogen bond formation requires strict conformational angle and distance [30], the rigid 1,3,5-triazine ring may act as a molecular “scaffold” enabling efficient hydrogen bonding with selected drugs. At the same time, flexibility of the drug structure may be important for their ability to adjust the angles and distances necessary for maximal hydrogen bonding with the cBOx units. This may explain a tendency for better solubilization observed for some drugs having higher number of rotatable bonds. Interestingly, PMeOx-PcBOx micelles appeared to have solubilization preference for some of the compounds that have higher hydrogen bonding capacity and greater number of rotatable bonds, which is known to be detrimental for drug absorption into target cells [31]. Thus, the new PMeOx-PcBOx block copolymer, and the overall class of poly(2-oxazolines) with side chains containing substituted aromatic heterocycles, may hold the potential to expand the design space of APIs and spark the development of novel polymeric micelle formulations based on multiple drug-polymer interactions.
It could be useful to quantify and correlate the physicochemical properties of drug molecules and their solubilization in PMeOx-PcBOx micelles. For this purpose we attempted to develop a simplified scoring system using the presence of electron deficient aromatic structures (EDA) as an indicator for pi-pi stacking interaction, the numbers of hydrogen bonds acceptors (HBA) and donors (HBD) as indicators for hydrogen bonding, LogP as an indicator of hydrophobicity, and rotatable bonds number (RBN) as indicator for the ease with which a drug molecule can exhibit specific interactions with the PcBOx side chains. We could not establish a clear and simple correlation between any one parameter and drug solubilization in the PMeOx-PcBOx polymeric micelles. However, when taking into account multiple parameters, this system showed some ability to rationalize the drug solubilization. For instance, bruceantin does not have an electron deficient aromatic ring whereas GW788388 does. But the solubilization of bruceantin in PMeOx-PcBOx micelles was high, whereas that of GW788388 was low. This difference could be explained by additional interactions bruceantin may have in the core of the micelles that GW788388 does not have (bruceantin Score = 4, GW788388 Score = 2). At the same time, the higher point total in this scoring system does not necessarily mean that a given drug is more soluble than another drug with a lower score. A higher score simply means that there is a higher probability of some solubility being achieved. For example, aclacinomycin A scored the highest among all analyzed compounds (Score = 5), but its solubilization was far from best and comparable to that of wortmannin that does not have a high hydrogen bonding capacity and has a relatively low number of rotatable bonds. The modest solubilization of aclacinomycin A may be connected to its relatively large volume (molecular mass 811.86 g/mol). Similarly, paclitaxel has a relatively high score (Score = 4) but shows modest solubilization, which in addition to the high molecular mass of this drug (853.90 g/mol) may be also induced by its high LogP value. With a larger data set created the relative contributions of each parameter could perhaps be, elucidated and optimized to account for differences in the drug solubilization. A prediction of the success of compounds applicable for a given polymeric micelle formulation would allow the rational design of the copolymers, or conversely, the selection of APIs, which are preferable for a given polymeric micelle system. We hope that this scoring system can provide some intuition for the design of more complex computational models to accurately predict drug solubility in these unique micelle systems.
While relying on different mechanisms for loading hydrophobic drugs, the micelles were still able to produce a similar release profile for bruceantin. Additionally, for paclitaxel, the PcBOx allowed access to a different, more complete, release profile at the given 1:10 drug to polymer ratio. Utilizing different block structures it is possible to tune the release profile of the polymeric micelles. This could be advantageous for tuning in vivo drug pharmacokinetic properties to reduce toxicity or increase therapeutic efficacy.
Heteroaromatic compounds such as substituted 1,3,5-triazine in cBOx are well known to form chelating complexes with a variety of metal species including Ag, Au, Cu, and Pt [29]. In this regard we evaluated and successfully loaded in the PMeOx-PcBOx micelles DachPt, which is an active form of the platinum chemotherapeutics, oxaliplatin and miriplatin. The resulting DachPt-PMeOx-PcBOx formulation displayed an anticancer activity in two different cancer cell lines, albeit in each cell line it had higher IC50 values than oxaliplatin. This difference in IC50 could be attributed to two factors; 1) the cell uptake mechanism is different between nanoparticle (DachPt-PMeOx-PcBOx) and small molecules (oxaliplatin), and 2) the onset of the cell damage is delayed for DachPt-PMeOx-PcBOx due to slower DachPt release mechanism compared to oxaliplatin that readily forms DachPt in cytosolic reducing conditions [32]. Further investigation is warranted to elucidate the mechanism of DachPt release in physiological conditions and cell uptake of the DachPt-PMeOx-PcBOx formulation.
Platinum drugs are widely used chemotherapies of cancer, but are known to cause peripheral neuropathy, especially sensory ataxia, due to the accumulation in the dorsal root ganglion [33]. To reduce this adverse effect, and maximize the therapeutic index, various delivery systems are explored for this drug class [34,35]. More generally, metal nanoparticle systems are interesting subjects not just for therapeutics, but also for diagnostics and imaging especially in the field of oncology [36]. For example, manganese (Mn) or gadolinium (Gd) containing nanoparticles can be used as MRI contrast agents [37,38] and gold nanoparticles are known to be useful as CT contrast agents [39]. Ruthenium (Ru) organometallic complexes were reported as photo-luminescence imaging agents that can detect hypoxic tumors [40]. While the new polymer class described in this work may not have applications in all these instances, it is truly remarkable that the same polymer excipient displays ability for incorporating both poorly soluble APIs as well as relatively well solubilized metal complexes used in chemotherapy.
5. Conclusion
In conclusion, we have designed a novel poly(2-oxazoline)-based block copolymer with a heterocyclic, aromatic side chain that can be used for 1) solubilization of a unique set of poorly soluble compounds that have previously failed in other polymeric systems, as well as 2) loading of platinum drugs through metal complexation. It is promising that the new block copolymer class can expand the application of polymeric micelle technologies to a new set of drugs and impact the drug design space by providing alternatives to hydrophobic interactions as a means of incorporation of poorly soluble APIs in the micelles. Future studies well validate this conclusion for a higher number of compounds and elucidate drug/polymer interaction mechanisms important for predictions of drug solubilization.
Supplementary Material
Acknowledgments
We thank the PhD student exchange program (great!ipid4all (group2group exchange for academic talents)) from Dresden University of Technology. We are grateful for Dr. Chaemin Lim for his consultation on polymer characterization and UNC Nanomedicines Characterization Core and Dr. Olesia Gololobova for help with ICP-MS analysis. We thank the UNC CHANL (Chapel Hill Analytical and Nanofabrication Laboratory) for the technological support for the TEM micrograph imaging. We thank the UNC Michael Hooker Proteomics Core for the technological support for the MALDI-TOF/TOF analysis.
Funding
This work was in part supported by the NCI Alliance for Nanotechnology in Cancer (U54CA198999, Carolina Center of Cancer Nanotechnology Excellence) and by the Carolina Partnership, a strategic partnership between the UNC Eshelman School of Pharmacy and The University Cancer Research Fund through the Lineberger Comprehensive Cancer Center.
Footnotes
Competing interests
AVK and RJ are listed as inventor on patents pertinent to the subject matter of the present contribution and co-founder of DelAQUA Pharmaceuticals Inc. having intent of commercial development of poly (2-oxazoline) based drug formulations. The other authors have no competing interests to report.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jconrel.2019.06.037.
References
- [1].Méndez-Lucio O, Medina-Franco JL, The many roles of molecular complexity in drug discovery, Drug Discov. Today 22 (2017) 120–126. [DOI] [PubMed] [Google Scholar]
- [2].Macalino SJY, Gosu V, Hong S, Choi S, Role of computer-aided drug design in modern drug discovery, Arch. Pharm. Res 38 (2015) 1686–1701. [DOI] [PubMed] [Google Scholar]
- [3].Kazunori K, Kwon GS, Masayuki Y, Teruo O, Yasuhisa S, Block copolymer micelles as vehicles for drug delivery, J. Control. Release 24 (1993) 119–132. [Google Scholar]
- [4].Mikhail AS, Allen C, Block copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levels, J. Control. Release 138 (2009) 214–223. [DOI] [PubMed] [Google Scholar]
- [5].Matsumura Y, Kataoka K, Preclinical and clinical studies of anticancer agent-incorporating polymer micelles, Cancer Sci. 100 (2009) 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davis ME, Chen Z, Shin DM, Nanoparticle therapeutics: an emerging treatment modality for cancer, Nanoscience and Technology: A Collection of Reviews from Nature Journals, World Scientific, 2010, pp. 239–250. [DOI] [PubMed] [Google Scholar]
- [7].Zhang L, Gu F, Chan J, Wang A, Langer R, Farokhzad O, Nanoparticles in medicine: therapeutic applications and developments, Clin. Pharmacol. Ther 83 (2008) 761–769. [DOI] [PubMed] [Google Scholar]
- [8].Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR, Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date, Pharm. Res 33 (2016) 2373–2387. [DOI] [PubMed] [Google Scholar]
- [9].Walsh TJ, Finberg RW, Arndt C, Hiemenz J, Schwartz C, Bodensteiner D, Pappas P, Seibel N, Greenberg RN, Dummer S, Liposomal amphotericin B for empirical therapy in patients with persistent fever and neutropenia, N. Engl. J. Med 340 (1999) 764–771. [DOI] [PubMed] [Google Scholar]
- [10].Green MR, Manikhas GM, Orlov S, Afanasyev B, Makhson AM, Bhar P, Hawkins MJ, Abraxane, a novel Cremophor-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer, Ann. Oncol 17 (2006) 1263–1268. [DOI] [PubMed] [Google Scholar]
- [11].Wu SY, Lopez-Berestein G, Calin GA, Sood AK, RNAi therapies: drugging the undruggable, Sci. Transl. Med 6 (2014) 240ps7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cern A, Golbraikh A, Sedykh A, Tropsha A, Barenholz Y, Goldblum A, Quantitative structure-property relationship modeling of remote liposome loading of drugs, J. Control. Release 160 (2012) 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cern A, Barenholz Y, Tropsha A, Goldblum A, Computer-aided design of liposomal drugs: in silico prediction and experimental validation of drug candidates for liposomal remote loading, J. Control. Release 173 (2014) 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mondal S, Samajdar RN, Mukherjee S, Bhattacharyya AJ, Bagchi B, Unique features of metformin: a combined experimental, theoretical, and simulation study of its structure, dynamics, and interaction energetics with DNA grooves, J. Phys. Chem. B 122 (2018) 2227–2242. [DOI] [PubMed] [Google Scholar]
- [15].Overberger C, Michelotti FW, Carabateas PM, Preparation of triazines by the reaction of biguanide and esters, J. Am. Chem. Soc 79 (1957) 941–944. [Google Scholar]
- [16].Bouten P, Lava K, van Hest J, Hoogenboom R, Thermal properties of methyl ester-containing poly (2-oxazoline) s, Polymers 7 (2015) 1998–2008. [Google Scholar]
- [17].Luxenhofer R, Schulz A, Roques C, Li S, Bronich TK, Batrakova EV, Jordan R, Kabanov AV, Doubly amphiphilic poly (2-oxazoline) s as high-capacity delivery systems for hydrophobic drugs, J. Control. Release 31 (2010) 4972–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Koh M, Lee J-C, Min C, Moon A, A novel metformin derivative, HL010183, inhibits proliferation and invasion of triple-negative breast cancer cells, Bioorg. Med. Chem 21 (2013) 2305–2313. [DOI] [PubMed] [Google Scholar]
- [19].Hu X, Lerch TF, Xu A, Efficient and selective bioconjugation using surfactants, Bioconjug. Chem 29 (2018) 3667–3676. [DOI] [PubMed] [Google Scholar]
- [20].Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem 45 (2002) 2615–2623. [DOI] [PubMed] [Google Scholar]
- [21].Oberoi HS, Nukolova NV, Bronich TK, Dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt) loaded polymer micelles with cross-linked core: preparation and characterization, PMSE Prepr. 104 (2011) 630–631. [PMC free article] [PubMed] [Google Scholar]
- [22].Mees MA, Hoogenboom R, Functional poly (2-oxazoline) s by direct amidation of methyl ester side chains, Macromolecules 48 (2015) 3531–3538. [Google Scholar]
- [23].He Z, Wan X, Schulz A, Bludau H, Dobrovolskaia MA, Stern ST, Montgomery SA, Yuan H, Li Z, Alakhova D, Sokolsky M, Darr DB, Perou CM, Jordan R, Luxenhofer R, Kabanov AV, A high capacity polymeric micelle of paclitaxel: implication of high dose drug therapy to safety and in vivo anti-cancer activity, Biomaterials 101 (2016) 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wan X, Beaudoin JJ, Vinod N, Min Y, Makita N, Bludau H, Jordan R, Wang A, Sokolsky M, Kabanov AV, Co-delivery of paclitaxel and cisplatin in poly(2-oxazoline) polymeric micelles: implications for drug loading, release, pharmacokinetics and outcome of ovarian and breast cancer treatments, Biomaterials 192 (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wan X, Min Y, Bludau H, Keith A, Sheiko SS, Jordan R, Wang AZ, Sokolsky-Papkov M, Kabanov AV, Drug combination synergy in worm-like polymeric micelles improves treatment outcome for small cell and non-small cell lung cancer, ACS Nano 12 (2018) 2426–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].He Z, Schulz A, Wan X, Seitz J, Bludau H, Alakhova DY, Darr DB, Perou CM, Jordan R, Ojima I, Kabanov AV, Luxenhofer R, Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: preparation, in vitro and in vivo evaluation, J. Control. Release 208 (2015) 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lübtow MM, Keßler L, Lorson T, Gangloff N, Kirsch M, Dahms S, Luxenhofer R, More is less: curcumin and paclitaxel formulations using poly (2-oxazoline) and poly (2-oxazine) based amphiphiles bearing linear and branched C9 side chains, Macromol. Biosci 18 (2018) 1800155. [DOI] [PubMed] [Google Scholar]
- [28].Lubtow MM, Hahn L, Haider MS, Luxenhofer R, Drug specificity, synergy and antagonism in ultrahigh capacity poly(2-oxazoline)/poly(2-oxazine) based formulations, J. Am. Chem. Soc 139 (2017) 10980–10983. [DOI] [PubMed] [Google Scholar]
- [29].Mooibroek TJ, Gamez P, The s-triazine ring, a remarkable unit to generate supramolecular interactions, Inorg. Chim. Acta 360 (2007) 381–404. [Google Scholar]
- [30].Taylor R, Kennard O, Versichel W, The geometry of the N–H⋯ O=C hydrogen bond. 3. Hydrogen-bond distances and angles, Acta Cryst 40 (1984) 280–288. [Google Scholar]
- [31].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev, 46 (2001) 3–26. [DOI] [PubMed] [Google Scholar]
- [32].Pendyala L, Creaven P, In vitro cytotoxicity, protein binding, red blood cell partitioning, and biotransformation of oxaliplatin, Cancer Res. 53 (1993) 5970–5976. [PubMed] [Google Scholar]
- [33].Kanat O, Ertas H, Caner B, Platinum-induced neurotoxicity: a review of possible mechanisms, World. J. Clin. Oncol 8 (2017) 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Marupudi NI, Han JE, Li KW, Renard VM, Tyler BM, Brem H, Paclitaxel: a review of adverse toxicities and novel delivery strategies, Expert Opin. Drug Saf 6 (2007) 609–621. [DOI] [PubMed] [Google Scholar]
- [35].Lyass O, Uziely B, Ben-Yosef R, Tzemach D, Heshing NI, Lotem M, Brufman G, Gabizon A, Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma, Cancer 89 (2000) 1037–1047. [DOI] [PubMed] [Google Scholar]
- [36].Reichert DE, Lewis JS, Anderson CJ, Metal complexes as diagnostic tools, Coord. Chem. Rev 184 (1999) 3–66. [Google Scholar]
- [37].Caravan P, Ellison JJ, McMurry TJ, Lauffer RB, Gadolinium (III) chelates as MRI contrast agents: structure, dynamics, and applications, Chem. Rev 99 (1999) 2293–2352. [DOI] [PubMed] [Google Scholar]
- [38].Mi P, Kokuryo D, Cabral H, Wu H, Terada Y, Saga T, Aoki I, Nishiyama N, Kataoka K, A pH-activatable nanoparticle with signal-amplification capabilities for non-invasive imaging of tumour malignancy, Nat. Nanotehnol 11 (2016) 724. [DOI] [PubMed] [Google Scholar]
- [39].Popovtzer R, Agrawal A, Kotov NA, Popovtzer A, Balter J, Carey TE, Kopelman R, Targeted gold nanoparticles enable molecular CT imaging of cancer, Nano Lett. 8 (2008) 4593–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang P, Huang H, Chen Y, Wang J, Ji L, Chao H, Ruthenium (II) anthraquinone complexes as two-photon luminescent probes for cycling hypoxia imaging in vivo, Biomaterials 53 (2015) 522–531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.