

Abstract
Cardiac paraganglioma (PGL) is a rare neuroendocrine tumour causing significant morbidity primarily due to norepinephrine secretion potentially causing severe hypertension, palpitations, lethal tachyarrhythmias, stroke and syncope. Cardiologists are faced with two clinical scenarios. The first is the elevated norepinephrine, whose actions must be properly counteracted by adrenoceptor blockade to avoid catastrophic consequences. The second is to evaluate the precise location of a cardiac PGL and its spread since compression of cardiovascular structures may result in ischaemia, angina, non-noradrenergic-induced arrhythmia, cardiac dysfunction or failure. Thus, appropriate assessment of elevated norepinephrine by its metabolite normetanephrine is a gold biochemical standard at present. Furthermore, dedicated cardiac CT, MRI and transthoracic echocardiogram are necessary for the precise anatomic information of cardiac PGL. Moreover, a cardiologist needs to be aware of advanced functional imaging using 68Ga-DOTA(0)-Tyr(3)-octreotide positron emission tomography/CT, which offers the best cardiac PGL-specific diagnostic accuracy and helps to stage and rule out metastasis, determining the next therapeutic strategies. Patients should also undergo genetic testing, especially for mutations in genes encoding succinate dehydrogenase enzyme subunits that are most commonly present as a genetic cause of these tumours. Curative surgical resection after appropriate α-adrenoceptor and β-adrenoceptor blockade in norepinephrine-secreting tumours is the primary therapeutic strategy. Therefore, appropriate and up-to-date knowledge about early diagnosis and management of cardiac PGLs is paramount for optimal outcomes in patients where a cardiologist is an essential team member of a multidisciplinary team in its management.
INTRODUCTION
Paragangliomas (PGLs) are rare neuroendocrine tumours that arise from chromaffin cells located outside of the adrenal gland. Cardiac PGLs can receive parasympathetic innervation that may affect the function including the catecholamines content or catecholamine turnover with implications on cardiac physiology. PGLs are broadly classified into sympathetic and parasympathetic nervous system tumours.1 Sympathetic PGLs produce catecholamines (particularly norepinephrine or epinephrine) while parasympathetic PGLs are usually non-functional and mainly located in the head and neck. Cardiac PGLs are rare, accounting for <0.3% of mediastinal tumours and 1%–3% of primary cardiac tumours.2,3 Cardiac PGLs arise from the visceral or branchiomeric autonomic paraganglia resulting in left atrial and aortic body tumours, respectively. Although cardiac PGLs are commonly described in all cardiac chambers, left atrial PGLs are the most common, followed by aortic body tumours. The objective of this comprehensive review is to provide most recent advancements and future directions in the molecular origins, genetic background, evaluation and contemporary management of cardiac PGLs.
CURRENT VIEW ON MOLECULAR ORIGINS OF CARDIAC PARAGANGLIOMAS
Currently, 22 genes are linked to the pathogenesis of PGLs. Among these, approximately 27%–35% are germline mutations, whereas 30%–39% are somatic mutations and 7% are fusion genes.4 A previous case series of 10 patients with mediastinal PGLs showed that all patients (100%) had germline mutations in either SDHB or SDHD.5 Moreover, it was found that underlying mutations in SDHB/C/D (SDHx, presenting with pseudohypoxia) were implicated in approximately 77% of cardiac PGLs in a multi-institutional case series.2 Figure 1 summarises the anatomic distribution of cardiac PGLs based on the SDHx mutations that were reported in journals indexed to PubMed between 2000 and 2019.2,6–18 A total of 38 patients (39 tumours) had undergone testing for germline genetic mutation and out of these, 12, 6 and 21 tumours were identified to carry underlying SDHB, SDHC and SDHD mutations, respectively. However, it is important to note that cardiac PGLs can occur in non-SDHx or apparently sporadic patients as well. The solid evidence of PGL hypoxiom supports the notion that there may be undetected hypoxia-related genes or other cell signalling/regulating genes that are implicated in the pathogenesis of cardiac PGLs.
Figure 1.
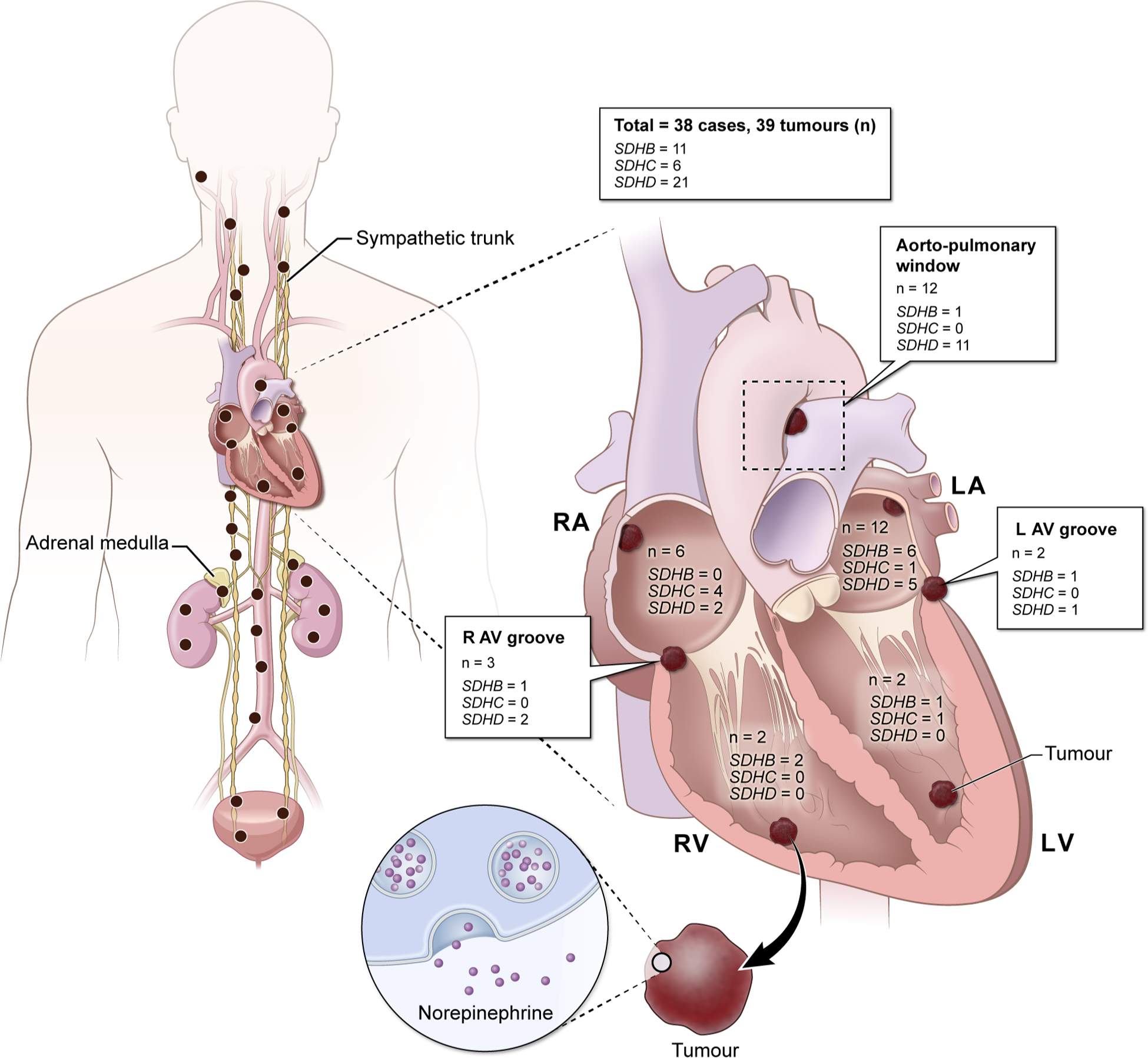
Schematic representation of mutation-wise distribution of cardiac paragangliomas in genes encoding subunits of succinate dehydrogenase enzyme that were reported to PubMed between the years 2000 and 2019.
CLINICAL PRESENTATION AND DIAGNOSIS
Clinical presentation
Clinical manifestations of PGLs are highly determined by the biochemical profile of the tumour. In general, PGLs are broadly classified to three phenotypical biochemical categories: noradrenergic, adrenergic and dopaminergic as they predominantly produce norepinephrine, epinephrine or dopamine and produce their metabolites, normetanephrine, metanephrine and methoxytyramine, respectively.19 As cardiac PGLs are extra-adrenal and predominantly associated with SDHx mutations, these PGLs present with the noradrenergic biochemical phenotype (with or without elevation of dopamine/methoxytyramine), very rarely with only a dopaminergic biochemical phenotype or biochemically silent.20,21 Cardiac PGLs can also be clinically silent and patients may present with non-specific symptoms such as weight loss, fatigue or fever, which may be attributed to desensitisation of catecholamine receptors or due to sole dopamine secretion. In fact, a meta-analysis on cardiac PGLs (n=150) have shown that approximately 77% of the study cohort had norepinephrine-related symptoms such as hypertension, sweating, diaphoresis, palpitations, headache and dizziness.22 Chest pain or distress was not quite common and were only seen in approximately 18% of the patients. Additionally, other constitutional symptoms such as hypertensive crisis, heart failure, mitral insufficiency, embolisation and compressive or obstructive symptoms were reported. These symptoms and signs are more related to a specific location of cardiac PGL rather than its secretory profile. It is yet to be determined if secretion of catecholamines from cardiac PGLs may have unique local paracrine or autocrine actions on surrounding cardiac tissue. Table 1 summarises the typical clinical manifestations in patients with cardiac PGL based on their biochemical profile.
Table 1.
Biochemical phenotypes, associated clinical presentations and therapeutic options of paragangliomas
| Biochemical phenotype | Typical signs and symptoms | Therapeutic options |
|---|---|---|
| Noradrenergic (norepinephrine/normetanephrine) | Sustained hypertension, constipation, diaphoresis, headache, nervousness/anxiety, nausea/vomiting, paleness, organ ischaemia |
|
| Adrenergic (epinephrine/metanephrine) | Episodic hypertension, episodic palpitations/tachycardia; headache, nervousness/anxiety, hyperglycaemic, hyperlipidaemia, anxiety, diaphoresis, rarely flushing episodes | |
| Dopaminergic | Asymptomatic, hypotension, diarrhoea (only if dopamine levels are very high) | Patients presenting with hypotension should have adequate volume repletion including 1–2 L of 0.9% normal saline on the day before surgery (to prevent postsurgical hypotension). |
| Biochemically silent (not producing any catecholamines) | Asymptomatic/non-specific symptoms | None. |
Mild orthostatic hypotension is the most common side effect, which can be minimised by starting the medications at a low dose at night and titrate to blood pressure as a patient tolerates.
Potential risk of causing postoperative hypotension.
Typically used in normotensive and borderline hypertension patients and on the day of the surgical resection or the night before surgery (long-acting α-adrenoceptor blockers are not typically used on the day of surgery to avoid postoperative hypotension).
Pathology and metastatic behaviour
Although a tissue diagnosis plays an important role for an accurate diagnosis of cardiac tumours, extensive vascularity of cardiac PGLs renders the biopsy hazardous. Biopsy is not recommended, especially in biochemically positive PGLs with a typical positivity of 68Ga-DOTA(0)-Tyr(3)-octreotide (68Ga-DOTATATE) or 18F-dihydroxyphenylalanine (18F-FDOPA) positron emission tomography/CT (PET/CT), which all point specifically to the presence of PGL.23–25
There are no specific histological features, immunohistochemical stains or molecular criteria that point towards the diagnosis of malignancy, except for the presence of distant metastases.1 However, some studies have elucidated the role of Grading System for Adrenal Pheochromocytoma and Paraganglioma (GAPP), modified-GAPP, lack of SDHB staining and ASES score (age, size, extra-adrenal location), vascular/lymphatic/capsular invasion, the presence of necrosis and secretory-type score(s) in determining the metastatic potential of PGLs.26,27 These factors/scores, SDHB mutations, a large size (>5–6 cm) and noradrenergic/dopaminergic biochemical phenotypes are the only reliable predictors of a high risk for metastasis.28,29
Current views on biochemical diagnosis of cardiac PGLs
Missing a cardiac PGL can have a detrimental outcome due to excessive catecholamine levels, which is why highly sensitive biochemical tests should be used to exclude a cardiac PGL. Given the episodic nature of catecholamine release, the use of catecholamine metabolites, released continuously and independently of catecholamines has a superior diagnostic sensitivity over the simple measurement of catecholamines.30 The diagnostic pitfalls and methods of collecting the biochemical specimens are discussed in detail elsewhere.23 A recent meta-analysis showed a superior diagnostic accuracy of appropriately collected plasma metanephrines as compared with that of 24 hours urine specimens.31 Alternatively, 24 hours urine metanephrines and creatinine may be measured to identify the biochemical nature of the tumour. A detailed list of medications that may lead to false-positive metanephrines is discussed in figure 2.23 A twofold elevation in metanephrines above the upper reference limit almost always favours the diagnosis of PGLs. However, this elevation in metanephrines may not be evident all the time due to a very small tumour size or its low tumoural catecholamine contents that is typical for SDHx-related PGLs. The major problem with the biochemical evaluation is the borderline positivity (less than two-times above the upper reference limit), which may or may not be related to the presence of PGL. Thus, if a patient has cardiac tumour with no clinical manifestations of PGLs but with borderline positive biochemical evaluation, patient may be further evaluated by getting the clonidine suppression test, which is almost 100% specific and 97% sensitive for the diagnosis of PGLs presenting with the noradrenergic phenotype.32 However, to proceed with the test, the patient’s cardiac status should be stable without any underlying hypotension (<110/60 mm Hg) or hypovolaemia at the baseline. In addition to a noradrenergic phenotype, cardiac PGLs can also present with the dopaminergic phenotype. In a report, about 60% (8/15) of cardiac PGLs had dopamine elevations.2 Out of these eight patients, seven had both normetanephrine and dopamine elevation and the remaining one had only dopamine elevation. The dopaminergic phenotype has been well documented in tumours harbouring SDHx mutations.2 This signifies the importance of adding dopamine and its metabolite, methoxytyramine in the biochemical evaluation of cardiac PGLs.33 Also, urine dopamine level is not recommended in the diagnostic workup of cardiac PGL as most of the dopamine present in the urine is formed in renal cells.34 Nevertheless, when there is a high suspicion of cardiac PGL, despite normal plasma norepinephrine/normetanephrine/dopamine/methoxytyramine levels (so-called non-functional PGLs), elevated chromogranin A can be a good biomarker to prove that such a tumour belongs to neuroendocrine ones.
Figure 2.
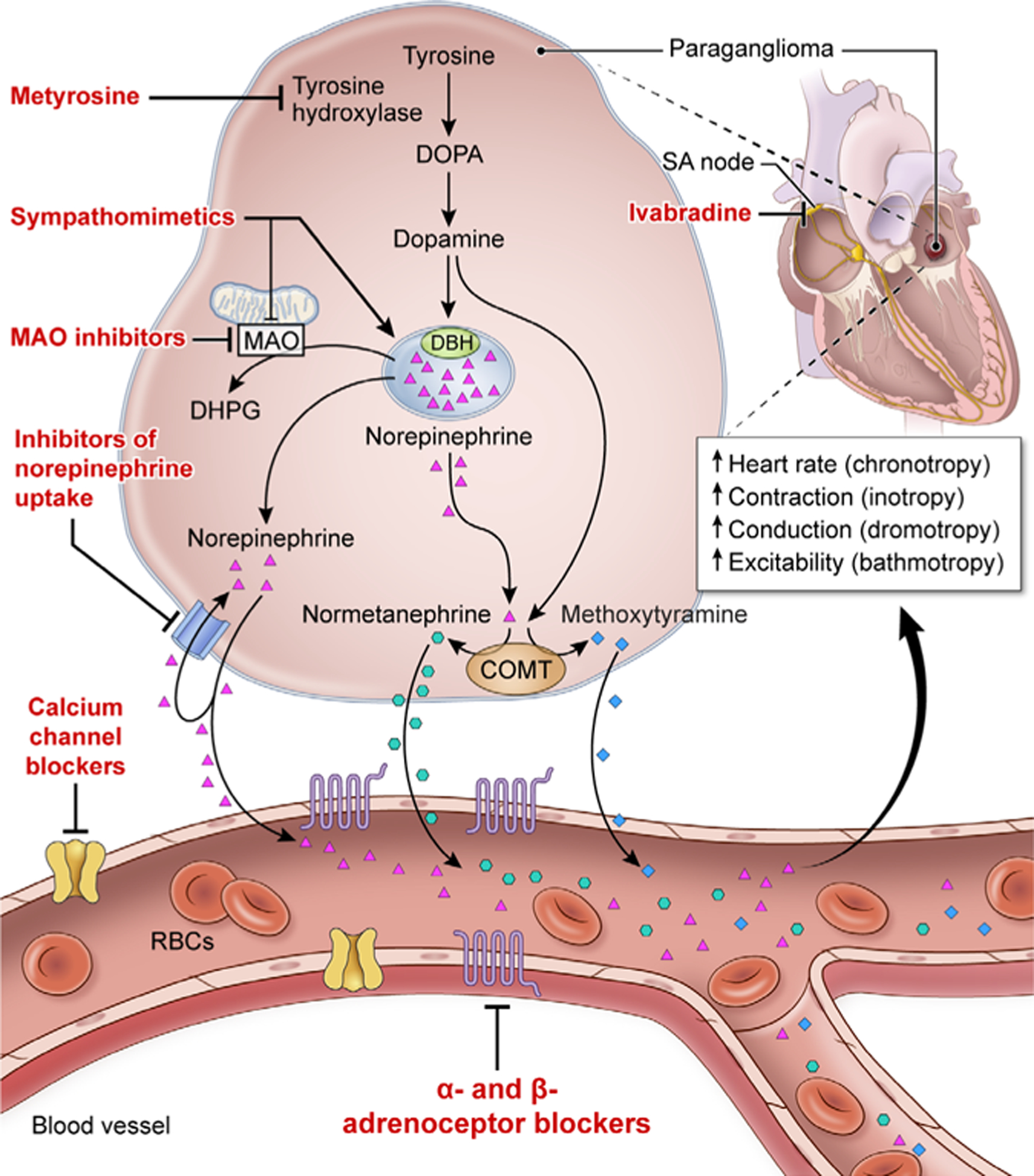
Schematic representation of synthesis, release and action of catecholamines and the respective metabolites released from cardiac paraganglioma and drugs that act on the catecholamine pathway. The α-adrenoceptor and β-adrenoceptor blockers reducethe effects of catecholamines on end organs such as blood vessels, heart and others that harbour adrenoceptors. Metyrosine blocks the rate-limiting step in catecholamine synthesis by inhibiting tyrosine hydroxylase. Calcium channel blockers cause smooth muscle relaxation in the blood vessel. Ivabradine, an If current inhibitor, acts on the sinoatrial node (SA node), thereby targeting the chronotropic effect of catecholamines at SA node. Agents such as sympathomimetic drugs (ephedrine, caffeine, amphetamine and nicotine) cause displacement of norepinephrine from the stores (vesicular sequestration, dominant mechanism indicated by bold arrow) and partly by inhibiting monoamine oxidase (MAO). MAO inhibitors block the conversion of catecholamines to dihydroxyphenylglycol (DHPG), thereby increasing the concentrations of norepinephrine. Antidepressants such as selective-norepinephrine re-uptake inhibitors, and tricyclic antidepressants that inhibit norepinephrine re-uptake leading to increased concentration of norepinephrine. RBC, red blood cell.
Richter et al extended the utilisation of liquid chromatography with mass spectrometry technique to identify the underlying metabolomics by quantifying the metabolites such as succinate-to-fumarate ratio. This helps in identifying the presence of SDHx mutations, and new underlying pathogenic variants that have led to the presence of PGL.35 Notably, succinate-to-fumarate ratio can also be detected by nuclear magnetic resonance (MR) spectroscopy.36 MR spectroscopy proposed by Varoquax et al has novel implications in localising and identifying the culprit tumour(s), their underlying genetic defect and therapeutic response without the need for a tumour sample, which is an enormous advantage in cardiac PGLs that cannot undergo simple biopsy in most cases.37
Role of anatomical imaging in localising cardiac PGLs
Anatomical imaging including contrast-enhanced CT scan, dedicated cardiac MRI and transthoracic echocardiogram (TTE) play a significant role in the localisation of the lesion and to evaluate the key characteristic features of cardiac PGL.38 These features include tumour attenuation, contrast enhancement, detailed understanding of the feeding vessel (if any) and close proximity to surrounding blood vessels and vital structures.
On cardiac MRI, cardiac PGLs are well-circumscribed ovoid/spherical lesions and are typically hyperintense on T2 imaging sequences.38 In contrast, angiosarcomas of the heart usually have low-intensity on T2 MRI sequences and are typically irregular nodules.38 Cardiac PGLs should also be high in the differential diagnosis if a mass is located in the left atrial area and inter-atrial septum beneath the great vessels-ascending aorta and pulmonary artery (visceral autonomic and branchiomeric PGL, respectively).22 This higher sensitivity and specificity of cardiac MRI over CT scans has been well documented in multiple case series.2,22,38 However, one must consider the limitations and contraindications of cardiac MRI such as the presence of pacemaker and/or surgical clips in place in which contrast-enhanced CT would be the anatomical imaging modality of choice (in patients with normal renal function) (figure 3).
Figure 3.
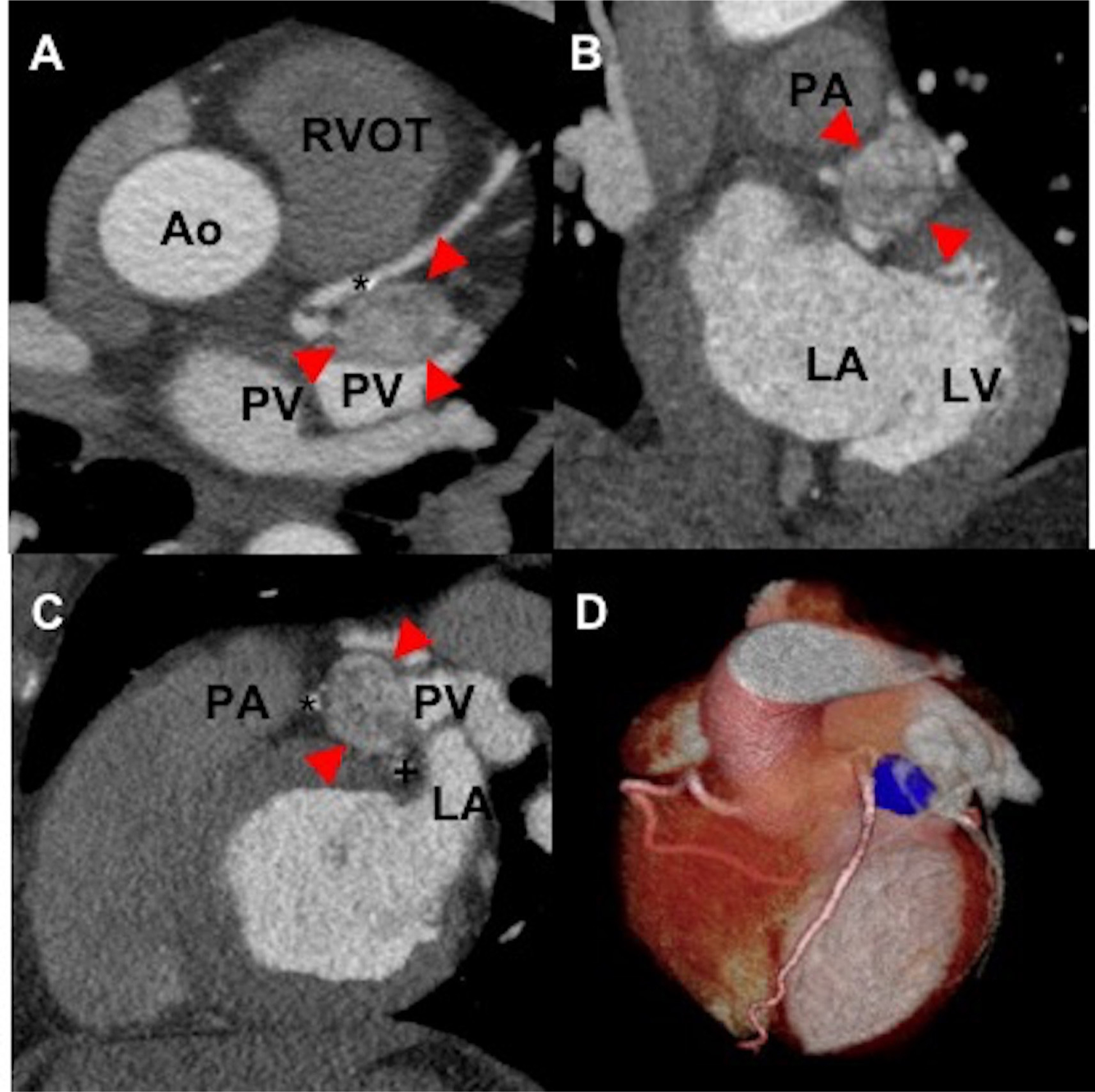
The axial (A), coronal (B), sagittal (C) and reformatted three-dimensional images (D) of a contrast-enhanced cardiac-gated CT of a woman aged 50 years without mutation in genes encoding subunits of succinate dehydrogenase enzyme demonstrates a well-circumscribed cardiac mass (arrows) measuring 3×2×2 cm. This mass is located superior to left atrium (B, C) with the inferior border extending to the left atrioventricular groove. The mass is bordered laterally by the left atrial appendage (C) and medially by the main pulmonary artery (superiorly, (B, C)) and proximal left anterior descending artery (*, (C)). The proximal left circumflex courses just inferior to the mass (+, (D)). There is no evidence of luminal compression in the left anterior descending or left circumflex arteries by the mass. Ao, aortic root; LA, left atrium; LV, left ventricle; PA, pulmonary artery; PV, pulmonary vein; RVOT, right ventricular outflow tract; asterisk (*), left anterior descending artery; plus (+), left circumflex.
On TTE, cardiac PGLs look like echogenic well-defined tumours/masses and are often misdiagnosed as atrial myxomas.39 One key differentiating feature between the two tumours is that myxomas have broad bases, whereas cardiac PGLs have narrow origin bases.39
Role of functional imaging in localising cardiac PGLs
Due to the extreme rarity of cardiac PGLs, patients with symptoms of PGL may not routinely undergo cardiac imaging.2 Therefore, functional imaging aids with initial thoracic localisation (figures 4 and 5) and encouraging more cardiac-specific imaging. Furthermore, it can help to stage and rule out multifocality/metastasis in a patient (figures 4 and 5).2
Figure 4.
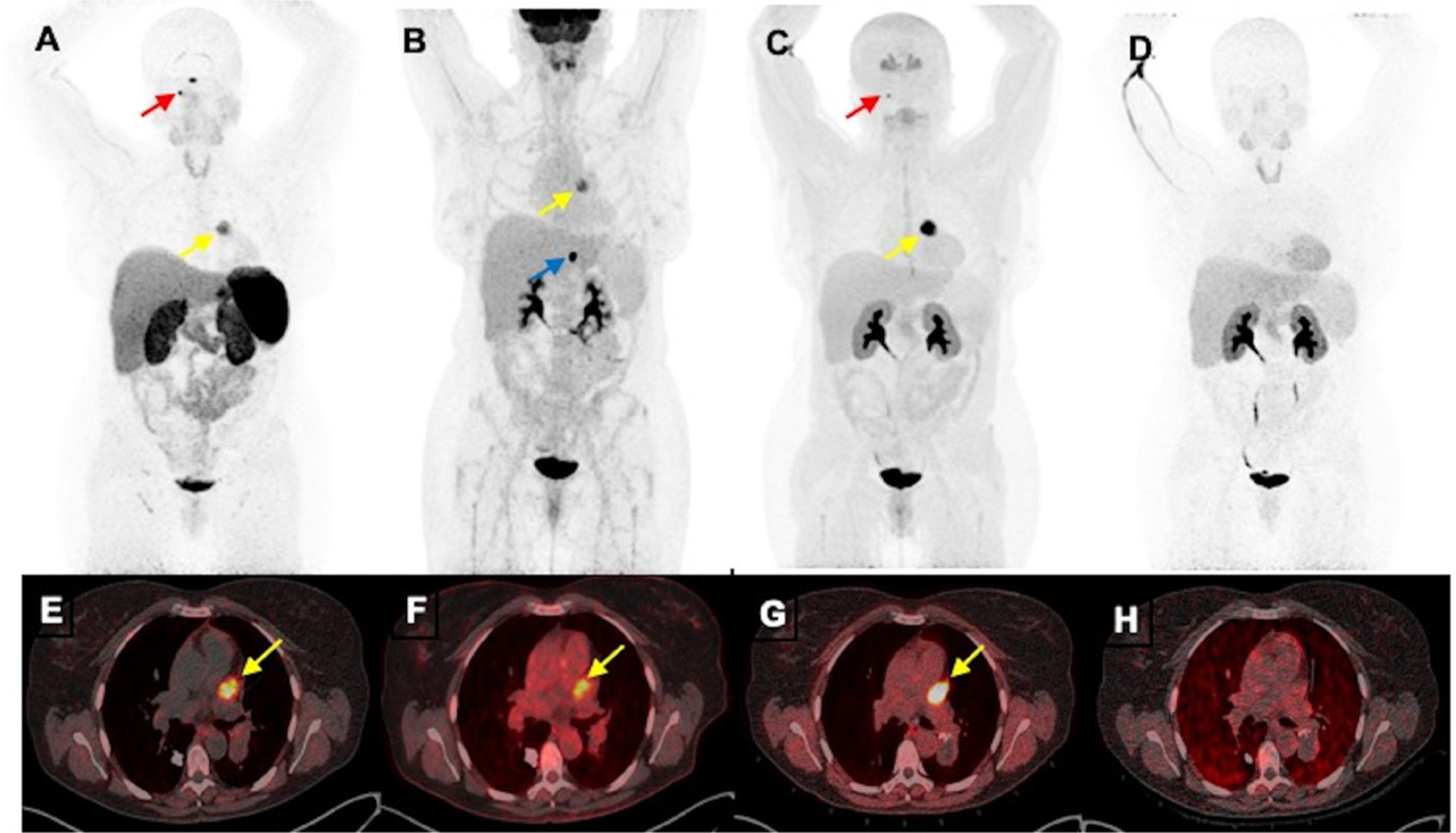
Functional positron emission tomography/CT (PET/CT) imaging of a cardiac paraganglioma of a woman aged 50 years without mutation in genes encoding subunits of succinate dehydrogenase enzyme. The anterior maximum intensity projection images (A–D) and fused axial PET/CT images (E–H) of 68Ga-DOTA(0)-Tyr(3)-octreotide PET/CT (A, E), 18F-fluorodeoxyglucose (B, F) and 18F-dihydroxyphenylalanine (C, G) PET/CT scans demonstrate a cardiac paraganglioma (yellow arrows) located in between pulmonary trunk and left atrium and superior aspect of left ventricle. However, this lesion lacks avidity on 18F-fluorodopamine (D, H). Furthermore, 68Ga-DOTA(0)-Tyr(3)-octreotide and 18F-dihydroxyphenylalanine PET/CT demonstrate a small right glomus jugulare paraganglioma (red arrows; A, B) and 18F-fluorodeoxyglucose PET/CT alone demonstrates a gastrohepatic nodule (blue arrow, (B)).
Figure 5.
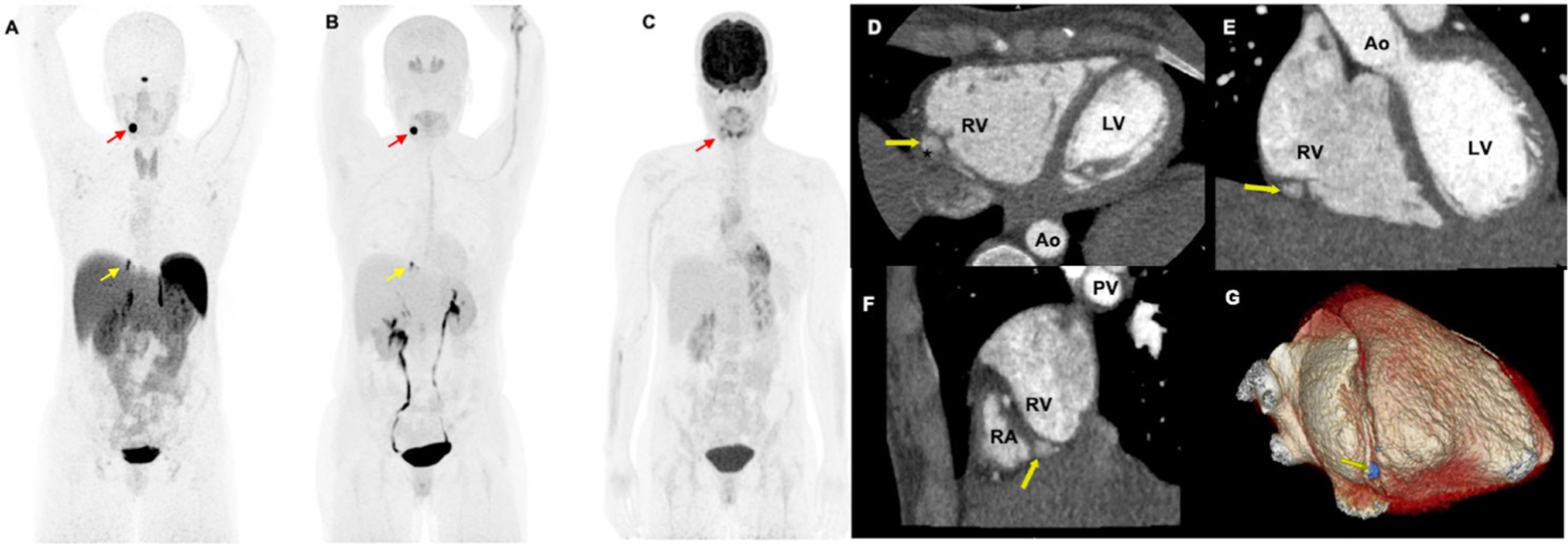
Functional positron emission tomography/CT (PET/CT) and contrast-enhanced cardiac-gated CT imaging of a woman aged 34 years with mutation in succinate dehydrogenase D gene. The anterior maximum intensity projection images (A–C) of 68Ga-DOTA(0)-Tyr(3)-octreotide PET/CT (A) and 18F-dihydroxyphenylalanine demonstrates a pericardiac paraganglioma (yellow arrows; A, B) as well as a left carotid body paraganglioma (red arrows; A, B). However, 18F-fluorodeoxyglucose (C) PET/CT scan demonstrates faint uptake in the left carotid body paraganglioma (red arrow, (C)) but lacks avidity in the pericardiac paraganglioma. This pericardiac paraganglioma was not localised on contrast-enhanced chest CT. The patient further underwent a contrast-enhanced cardiac-gated CT imaging to further characterise this pericardiac mass. The axial (D), coronal (E), sagittal (F) and reformatted three-dimensional images (G) demonstrate a well-circumscribed pericardiac mass (yellow arrows; D–G measuring 0.6×0.7×1.1 cm. This mass is located adjacent to mid-distal right carotid artery (*, (D)) without impingement of vessel lumen. The mass is outside the right ventricular free wall (yellow arrows, (D–F)) in the basal portion of the right atrial-ventricular groove (F) and within the pericardium. Ao, aortic root; LV, left ventricle; PV, pulmonary vein; RA, right atrium; RV, right ventricle; asterisk (*), right carotid artery.
Ruling out multiplicity/metastasis of the tumours and invasion to the surrounding structures are crucial steps as they guide the final decision on proceeding with surgery or systemic therapy.
The various functional imaging radiopharmaceuticals target different mechanisms of tumourigenesis in PGLs. The patient’s genetic mutation has an important influence on the radiopharmaceutical utilisation.24,25
Somatostatin receptors (SSTR) are overexpressed in PGLs, especially SSTR2 subtype40 and 68Ga-DOTATATE demonstrates higher affinity for SSTR2, whereas 18F-FDOPA targets the tumour via the large amino acid transporter system and 123/131I-metaiodobenzylguanidine (123/131I-MIBG) or 18F-fluorodopamine (18F-FDA) targets norepinephrine transporter system found in PGLs.24,25 However, 18F-fluorodeoxyglucose (18F-FDG) is a non-specific radiopharmaceutical that enters the tumour through glucose transporters.41
In the detection of cardiac PGLs, the sensitivity of 18F-FDOPA and 18F-FDG PET/CT have been reported as 100%.2,22 However, given the high proportion of SDHx mutations in cardiac PGLs (~80%), 68Ga-DOTATATE PET/CT scan should be the imaging modality of choice due to their documented higher sensitivity in SDHx mutations.24,25,42 Besides its diagnostic value, 68Ga-DOTATATE PET/CT should be a method of a choice for cardiac PGL staging as it has higher sensitivity to detect metastatic disease.24,25,42 This is particularly important when deciding how to treat these tumours (discussed in ‘Contemporary management’ section). Furthermore, it can help in radio-guided surgery using 68Ga-DOTATATE-based hand-held gamma probes.43
Additionally, other radiopharmaceuticals have been used to image cardiac PGLs and have shown inferior detection rates ranging from 54.5% to 75% with 123/131I-MIBG scintigraphy and 57.1% with 18F-FDA PET/CT.22 However, 123/131I-MIBG-avid cardiac PGLs can be treated with conventional 131I-MIBG or high-specific activity 131I-MIBG for metastatic or inoperable disease.44 Furthermore, PET/MRI can provide a comprehensive imaging evaluation of cardiac PGL.45
CONTEMPORARY MANAGEMENT
Preoperative medical therapy (adrenoceptor blockade)
Since cardiac PGLs produce catecholamines, patients are prone to life-threatening peri-operative complications such as hypertensive crisis, cardiac arrhythmias or even myocardial infarction. To prevent such peri-operative complications, these patients should be placed on alpha (α)-adrenoceptor and beta (β)-adrenoceptor blockers at least 2 weeks prior to the surgery (table 1, figure 6).23
Figure 6.
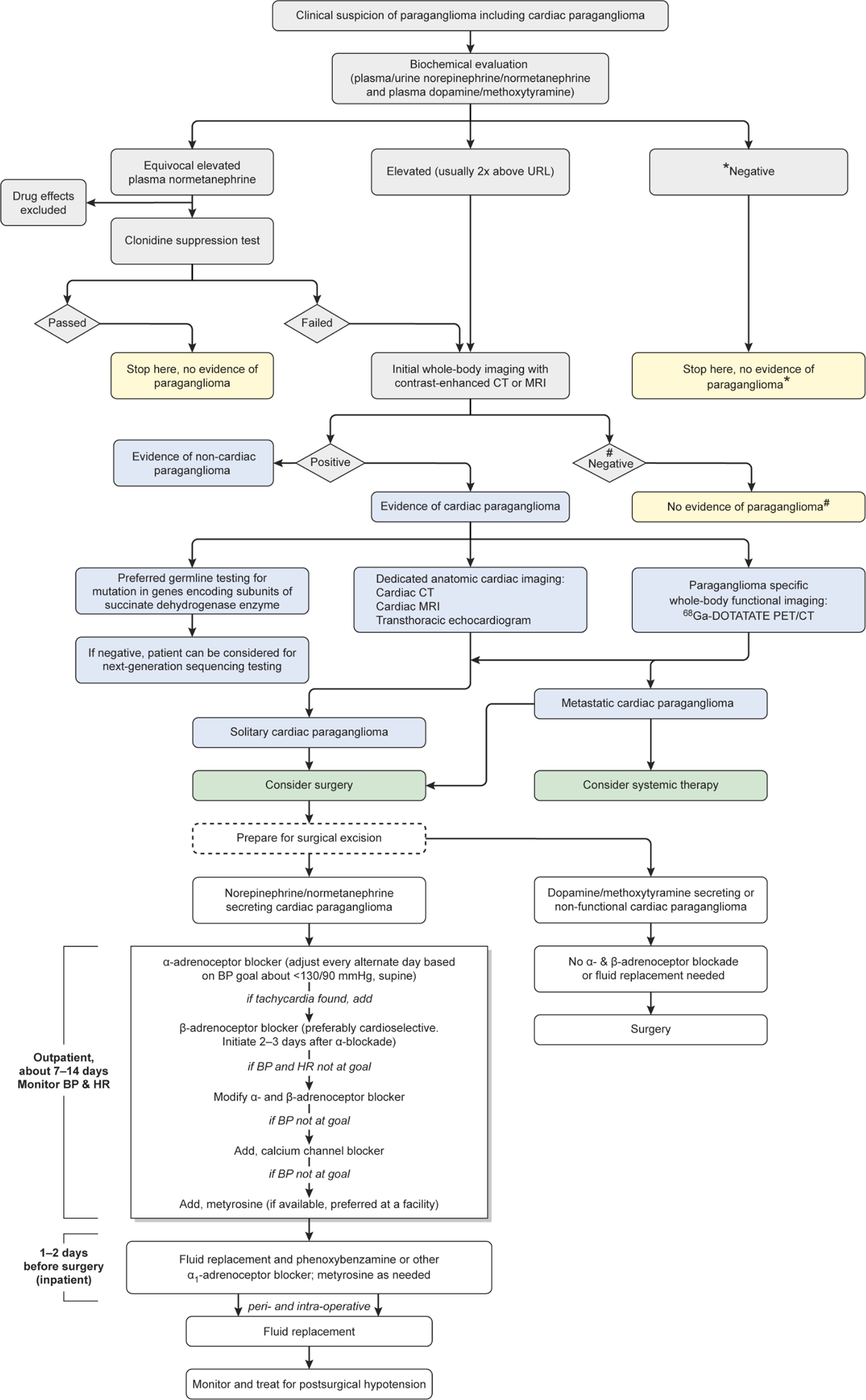
Flow chart enumerating the diagnosis and management of cardiac paraganglioma. *Results are to be interpreted with caution that biochemical evaluation may be negative in non-functioning paraganglioma. #Results are to be interpreted with caution that anatomical imaging may miss small paragangliomas and functional imaging can act as complimentary diagnostic tool. BP, blood pressure; HR, heart rate, 68Ga-DOTATATE, 68Ga-DOTA(0)-Tyr(3)-octreotide; PET/CT, positron emission tomography/CT.
Therapy should be initiated with an α-adrenoceptor blocker first followed by a β-adrenoceptor blocker 2–3 days later if tachycardia is present. This sequential therapy is primarily based on the fact that using β-adrenoceptor blockers leads to a loss of β-adrenoceptor receptor-mediated vasodilatation with unopposed catecholamine-induced vasoconstriction (due to unopposed action of catecholamines on α-adrenoceptors), which can potentially lead to a hypertensive crisis.
Phenoxybenzamine is the commonly preferred long-acting α-adrenoceptor blocker, but alternative options include short-acting α-blockers (prazosin, terazosin and doxazosin).46 To minimise hypotension caused by the α-adrenoceptor blockers, adequate hydration needs to be maintained and these medications should be administered at bedtime.
Metyrosine (α-methyl-p-tyrosine), a tyrosine hydroxylase inhibitor blocking the final step in the catecholamine pathway, may be used in patients in whom α-adrenoceptor and β-adrenoceptor blockers failed to normalise the blood pressures at their maximum doses.47
Patients who harbour biochemically silent PGLs are not placed on adrenoceptor blockers. On the day of surgery, the patients should have adequate volume repletion with 1–2 L of 0.9% normal saline to prevent hypotension and may receive a short-acting α-adrenoceptor blocker to prevent severe postoperative hypotension.23
In patients with refractory tachycardia due to catecholamine β-adrenoceptor stimulating activity, ivabradine (If current inhibitor) may be a good option (figure 2).48 Patients who harbour non-functional PGLs (ie, tumours that do not synthesise catecholamines) or dopamine-producing cardiac PGLs are not placed on adrenoceptor blockers. The day before surgery, patients should have adequate volume repletion with 1–2 L of 0.9% normal saline (most often used) to prevent postoperative hypotension (but not for those with non-functional or only dopamine-producing PGLs).46
Surgical management
The initial question regarding the surgical management of cardiac PGLs is whether the tumour is resectable. The topography of PGL tumours and their relationship with cardiac structures should be taken into account when assessing the surgical treatment of cardiac PGLs.
PGLs are frequently close to great vessels and can sometimes involve cardiac cavities. Moreover, these tumours infiltrate cardiac tissues; hence, resection may be challenging and at times require reconstruction of cardiac structures. However, in some situations such as infiltration of an atrioventricular groove or more rarely, involvement of extracardiac structures, resection becomes almost impossible. Since reconstruction after tumour resection may require revascularisation of the coronary arteries and reconstruction of great vessels, preoperative evaluation of the coronary arteries may be required by cardiac-gated CT scan or coronary angiography. Most commonly, PGLs are fed by coronary arteries, which can be ascertained by preoperative angiography and preparation for concomitant coronary artery bypass grafting should be planned.
Additionally, one must be cautious and prepared to tackle the peri-operative complications such as cardiac arrhythmias and hypertensive crisis.38 Given the cumbersome nature of the procedure, an accurate assessment of myocardial and valvular function is mandatory. In patients with poor general condition, surveillance and medical management should be advocated.
Intraoperatively, given the predilection for PGLs to arise in the atrioventricular groove and the base of the great vessels, cardiopulmonary bypass is typically required for complete resection and reconstruction.22 Therefore, these operations should be conducted in centres with established cardiac surgery programmes in the hands of teams experienced with complex cardiac surgical reconstruction. With the problematic locations of these tumours, occasionally surgical treatment can include complete removal of the heart and resection of the tumour followed by auto-transplantation of the reconstructed heart.49 Fortunately, technical improvements in the use of the heart-lung machine have obviated the need for auto-transplantation and affords the ability to completely excise masses that may breach the walls of the cardiac chambers while allowing for isolation of the tumour from the circulation. This permits better haemodynamic control if manipulation of the mass leads to catecholamine release.
Once on cardiopulmonary bypass, the masses can be easily identified and resected. They may be locally invasive and may also have a thin covering which facilitates removal. Defects created by removal can be repaired either primarily or with autologous, xenogeneic or synthetic materials. These resections and concomitant reconstructions can be quite extensive but are generally successful.50 Given the potential extensive removal of tissue to achieve complete excision, in very rare cases, cardiac-transplantation may be required as a last result. Haemorrhage as a result of reconstruction can occur. Particular care and inspection of the suture lines is necessary particularly once the heart is reloaded with volume and contracting but prior to completely disengaging from cardiopulmonary bypass support. As noted, if a main coronary artery is involved, coronary artery bypass may be required and either an autologous arterial or venous conduit would be harvested to perform grafting. Additional anaesthetic management includes being prepared to deal with the residual effects of the significant preoperative catecholamine blockade and the resulting downregulation of adrenoceptors, initial pressor requirements once weaned from cardiopulmonary bypass may be significant.
Postoperatively, recurrence can occur but is rare and the majority of surgically treated patients do not experience hyper-secretion of catecholamines. Regardless, lifelong surveillance is usually required. Survival following surgical resection is similar to that of normal population unless the tumour is metastatic, in which case the ‘5-year survival is <50% of age-matched controls’.38
Management of metastatic disease
Potential therapeutic options for metastatic disease include conventional chemotherapy with cyclophosphamide, vincristine and dacarbazine (CVD regimen); temozolomide monotherapy; radiopharmaceuticals such as conventional or high-specific activity 131I-MIBG in 123/131I-MIBG-positive tumours; peptide receptor radionuclide therapy (177Lu/90Y-DOTA peptides) either alone or in combination with octreotide, lanreotide or low-dose capecitabine in 68Ga-DOTATATE-positive tumours and molecular targeted therapies such as tyrosine kinase inhibitors (sunitinib, cabozantinib).51,52 In patients with rapidly progressive disease, especially in the tumours with SDHB mutations, combination chemotherapy with CVD followed by temozolomide monotherapy would be a reasonable first-line option.52 In the patients who have failed temozolomide monotherapy (172 mg/m2/day for 5 days/month), long-term low-dose temozolomide (metronomic regimen) (75 mg/m2/day with 3 weeks on and 1 week off treatment) in combination with lanreotide may be tried.53 Alternatively, in patients with slowly progressive disease, systemic radiotherapy (131I-MIBG or 177Lu/90Y-DOTA peptides) should be considered.
Furthermore, patients harbouring metastatic disease should be highly encouraged to participate in clinical trials that evaluate the role of targeted therapies such as tyrosine kinase inhibitors, Poly (ADP-ribose) polymerase inhibitors, hypoxia-inducible factor-2α inhibitors, hypomethylating agents and topoisomerase inhibitors.52 It is important to note that these patients are prone to hypertensive crisis and should be placed on adequate α-adrenoceptor and β-adrenoceptor blockade along with metyrosine.
CONCLUSION AND FUTURE DIRECTIONS
Cardiac PGLs are exceptionally rare tumours. We emphasise the importance of biochemical evaluation and functional imaging for establishing a definitive diagnosis (figure 6). Simultaneously, patients with confirmed PGLs should be screened for the presence of predisposing germline mutations, especially SDHx. Curative surgical resection after appropriate α-adrenoceptor and β-adrenoceptor blockade remains the main treatment option. Referral to a tertiary care centre that has multidisciplinary team approach involving cardiologists, endocrinologists, nuclear medicine physicians, cardiothoracic surgeons and oncologists (in the case of metastatic disease) is essential for optimal outcomes. The availability of metabolomics (succinate-to-fumarate ratio) and proton MR spectroscopy may play an important role in management of cardiac PGLs, especially in difficult tumour locations or inoperable tumours, and 68Ga-DOTATATE-PET/MRI can provide a comprehensive evaluation of cardiac PGL. Going forward, further research on the genetic basis and metabolomics of cardiac PGLs will undoubtedly help in developing effective forms of therapies.
Acknowledgements
Alan Hoofring assisted with medical illustration of the figures 1, 2 and 6. Dr Alexander Ling proofread the figure legends 3 and 5. Dr Babak Saboury helped with the figure 5.
Funding This study was funded by the National Institutes of Health (grant number Z1AHD008735) awarded to KP. This work was supported by the Intramural Research Program of the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Footnotes
Competing interests None declared.
REFERENCES
- 1.Lam AK-Y, Lam AK. Update on adrenal tumours in 2017 World Health organization (who) of endocrine tumours. Endocr Pathol 2017;28:213–27. [DOI] [PubMed] [Google Scholar]
- 2.Martucci VL, Emaminia A, del Rivero J, et al. Succinate dehydrogenase gene mutations in cardiac paragangliomas. Am J Cardiol 2015;115:1753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel J, Sheppard MN. Pathological study of primary cardiac and pericardial tumours in a specialist UK centre: surgical and autopsy series. Cardiovasc Pathol 2010;19:343–52. [DOI] [PubMed] [Google Scholar]
- 4.Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017;31:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghayee HK, Havekes B, Corssmit EPM, et al. Mediastinal paragangliomas: association with mutations in the succinate dehydrogenase genes and aggressive behavior. Endocr Relat Cancer 2009;16:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michałowska I, Ćwikła J, Prejbisz A, et al. Mediastinal paragangliomas related to SDHx gene mutations. Kitp 2016;3:276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romano S, Fava C, Minuz P, et al. Succinate dehydrogenase gene mutation with cardiac paraganglioma: multimodality imaging and pathological correlation. Eur Heart J 2017;38:1853–4. [DOI] [PubMed] [Google Scholar]
- 8.Abdelhady K, Durgam S, Orza D, et al. Left atrial and carotid body paraganglioma. Ann Thorac Surg 2017;103:e323–5. [DOI] [PubMed] [Google Scholar]
- 9.Degrauwe S, Monney P, Qanadli SD, et al. Intrapericardial paraganglioma: the role of integrated advanced multi-modality cardiac imaging for the assessment and management of rare primary cardiac tumors. Cardiol J 2017;24:447–9. [DOI] [PubMed] [Google Scholar]
- 10.Otani N, Sugano K, Inami S, et al. Cardiac paraganglioma with a novel germline mutation of succinate dehydrogenase gene D. Jpn J Clin Oncol 2017;47:1193–7. [DOI] [PubMed] [Google Scholar]
- 11.Hardegree EL, Patel SM, Maleszewski JJ, et al. The heart of the matter. Am J Med 2012;125:873–5. [DOI] [PubMed] [Google Scholar]
- 12.González López MT, González SG, García ES, et al. Surgical excision with left atrial reconstruction of a primary functioning retrocardiac paraganglioma. J Cardiothorac Surg 2013;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miraldi F, Taffon C, Toscano M, et al. Black cardiac paraganglioma in a multiple paraganglioma syndrome. Eur J Cardiothorac Surg 2007;32:940–2. [DOI] [PubMed] [Google Scholar]
- 14.Beroukhim RS, del Nido P, Teot LA, et al. Cardiac paraganglioma in an adolescent. Circulation 2012;125:e322–4. [DOI] [PubMed] [Google Scholar]
- 15.Dhanasopon AP, Shemin RJ, Yeh MW. Cardiac paraganglioma presenting as gestational hypertension. Surgery 2010;147:459–61. [DOI] [PubMed] [Google Scholar]
- 16.Illouz F, Pinaud F, De Brux J-L, et al. Long-Delayed Localization of a Cardiac Functional Paraganglioma With SDHC Mutation. Ann Intern Med 2012;157:222–3. [DOI] [PubMed] [Google Scholar]
- 17.Millar AC, Mete O, Cusimano RJ, et al. Functional cardiac paraganglioma associated with a rare SdhC mutation. Endocr Pathol 2014;25:315–20. [DOI] [PubMed] [Google Scholar]
- 18.Siejka DA, Vittorio AF, Thakur S, et al. Resection of a functioning intrapericardial paraganglioma associated with succinate dehydrogenase B mutation. SAGE Open Medical Case Reports 2019;7:2050313X1983953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Berkel A, Lenders JWM, Timmers HJLM. Diagnosis of endocrine disease: biochemical diagnosis of phaeochromocytoma and paraganglioma. Eur J Endocrinol 2014;170:R109–19. [DOI] [PubMed] [Google Scholar]
- 20.Timmers HJLM Gimenez-Roqueplo A-P, Mannelli M, et al. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 2009;16:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenhofer G, Lenders JWM, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2012;48:1739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J-G, Han J, Jiang T, et al. Cardiac paragangliomas. J Card Surg 2015;30:55–60. [DOI] [PubMed] [Google Scholar]
- 23.Lenders JWM, Duh Q-Y, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014;99:1915–42. [DOI] [PubMed] [Google Scholar]
- 24.Taieb D, Jha A, Treglia G, et al. Molecular imaging and radionuclide therapy of paraganglioma and pheochromocytoma. Endocr Relat Cancer 2019;26:R627–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taïeb D, Hicks RJ, Hindié E, et al. European association of nuclear medicine practice Guideline/Society of nuclear medicine and molecular imaging procedure standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2019;46:2112–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho YY, Kwak MK, Lee S-E, et al. A clinical prediction model to estimate the metastatic potential of pheochromocytoma/paraganglioma: AsES score. Surgery 2018;164:511–7. [DOI] [PubMed] [Google Scholar]
- 27.Koh J-M, Ahn SH, Kim H, et al. Validation of pathological grading systems for predicting metastatic potential in pheochromocytoma and paraganglioma. PLoS One 2017;12:e0187398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crona J, Lamarca A, Ghosal S, et al. Genotype-Phenotype correlations in pheochromocytoma and paraganglioma. Endocr Relat Cancer 2019;26:539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hescot S, Leboulleux S, Amar L, et al. One-Year progression-free survival of therapy-naive patients with malignant pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2013;98:4006–12. [DOI] [PubMed] [Google Scholar]
- 30.Eisenhofer G, Lenders JWM, Linehan WM, et al. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel–Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med 1999;340:1872–9. [DOI] [PubMed] [Google Scholar]
- 31.Därr R, Kuhn M, Bode C, et al. Accuracy of recommended sampling and assay methods for the determination of plasma-free and urinary fractionated metanephrines in the diagnosis of pheochromocytoma and paraganglioma: a systematic review. Endocrine 2017;56:495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisenhofer G, Goldstein DS, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true-from false-positive test results. J Clin Endocrinol Metab 2003;88:2656–66. [DOI] [PubMed] [Google Scholar]
- 33.Rao D, Peitzsch M, Prejbisz A, et al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol 2017;177:103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenhofer G, Goldstein DS, Sullivan P, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab 2005;90:2068–75. [DOI] [PubMed] [Google Scholar]
- 35.Richter S, Gieldon L, Pang Y, et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med 2019;21:705–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lussey-Lepoutre C, Bellucci A, Burnichon N, et al. Succinate detection using in vivo 1H-MR spectroscopy identifies germline and somatic SDHx mutations in paragangliomas. Eur J Nucl Med Mol Imaging 2019;104. [DOI] [PubMed] [Google Scholar]
- 37.Varoquaux A, le Fur Y, Imperiale A, et al. Magnetic resonance spectroscopy of paragangliomas: new insights into in vivo metabolomics. Endocr Relat Cancer 2015;22:M1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav PK, Baquero GA, Malysz J, et al. Cardiac paraganglioma. Circulation 2014;7:851–6. [DOI] [PubMed] [Google Scholar]
- 39.Araoz PA, Mulvagh SL, Tazelaar HD, et al. Ct and MR imaging of benign primary cardiac neoplasms with echocardiographic correlation. RadioGraphics 2000;20:1303–19. [DOI] [PubMed] [Google Scholar]
- 40.Reubi J, Waser B, Schaer J-C, et al. Somatostatin receptor sst1–sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur J Nucl Med 2001;28:836–46. [DOI] [PubMed] [Google Scholar]
- 41.Belhocine T, Spaepen K, Dusart M, et al. 18Fdg PET in oncology: the best and the worst (review). Int J Oncol 2006;28:1249–61. [PubMed] [Google Scholar]
- 42.Janssen I, Blanchet EM, Adams K, et al. Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clinical Cancer Research 2015;21:3888–95. 10.1158/1078-0432.CCR-14-2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El Lakis M, Gianakou A, Nockel P, et al. Radioguided surgery with gallium 68 Dotatate for patients with neuroendocrine tumors. JAMA Surg 2019;154:40–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pryma DA, Chin BB, Noto RB, et al. Efficacy and Safety of High-Specific-Activity 131 I-MIBG Therapy in Patients with Advanced Pheochromocytoma or Paraganglioma. J Nucl Med 2019;60:623–30. 10.2967/jnumed.118.217463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhojwani N, Huang J, Garg V, et al. Utility of (18)F-Fluorodeoxyglucose Positron Emission Tomography/Magnetic Resonance Imaging in the Diagnosis of Cardiac Paraganglioma. Indian J Nucl Med 2017;32:380–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pacak K Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab 2007;92:4069–79. [DOI] [PubMed] [Google Scholar]
- 47.Steinsapir J, Carr AA, Prisant LM, et al. Metyrosine and pheochromocytoma. Arch Intern Med 1997;157:901–6. [PubMed] [Google Scholar]
- 48.Malaza G, Brofferio A, Lin F, et al. Ivabradine in catecholamine-induced tachycardia in a patient with paraganglioma. N Engl J Med 2019;380:1284–6. 10.1056/NEJMc1817267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J 2002;29:105–8. [PMC free article] [PubMed] [Google Scholar]
- 50.Lupinski RW, Shankar S, Agasthian T, et al. Primary cardiac paraganglioma. Ann Thorac Surg 2004;78:e43–4. 10.1016/j.athoracsur.2004.01.015 [DOI] [PubMed] [Google Scholar]
- 51.Kong G, Grozinsky-Glasberg S, Hofman MS, et al. Efficacy of peptide receptor radionuclide therapy for functional metastatic paraganglioma and pheochromocytoma. J Clin Endocrinol Metab 2017;102:3278–87. 10.1210/jc.2017-00816 [DOI] [PubMed] [Google Scholar]
- 52.Nölting S, Grossman A, Pacak K. Metastatic phaeochromocytoma: spinning towards more promising treatment options. Exp Clin Endocrinol Diabetes 2019;127:117–28. 10.1055/a-0715-1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tena I, Gupta G, Tajahuerce M, et al. Successful second-line metronomic temozolomide in metastatic paraganglioma: case reports and review of the literature. Clin Med Insights Oncol 2018;12:117955491876336–67. [DOI] [PMC free article] [PubMed] [Google Scholar]