

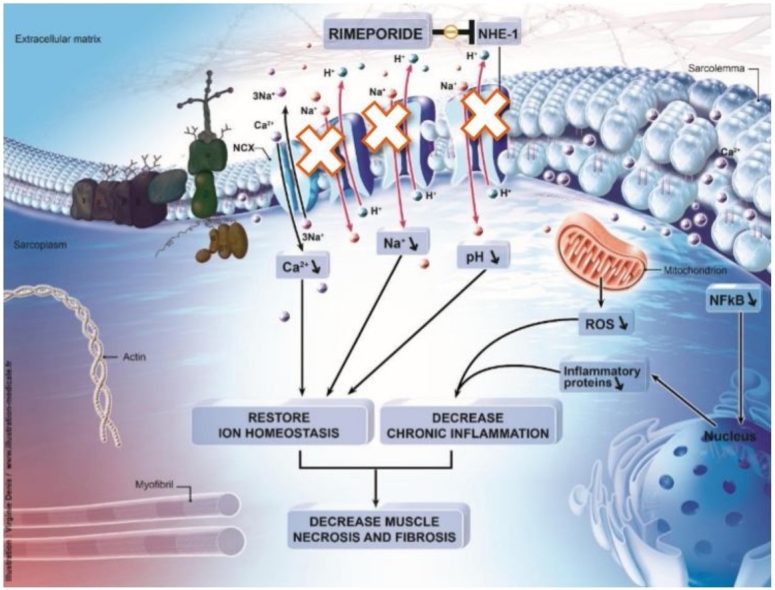
Graphical abstract
Keywords: Duchenne Muscular Dystrophy, NHE-1, Rimeporide, Safety, Pharmacokinetic, Cardiomyopathy
Abstract
Rimeporide, a first-in-class sodium/proton exchanger Type 1 inhibitor (NHE-1 inhibitor) is repositioned by EspeRare for patients with Duchenne Muscular Dystrophy (DMD). Historically, NHE-1 inhibitors were developed for cardiac therapeutic interventions. There is considerable overlap in the pathophysiological mechanisms in Congestive Heart Failure (CHF) and in cardiomyopathy in DMD, therefore NHE-1 inhibition could be a promising pharmacological approach to the cardiac dysfunctions observed in DMD. Extensive preclinical data was collected in various animal models including dystrophin-deficient (mdx) mice to characterise Rimeporide’s anti-fibrotic and anti-inflammatory properties and there is evidence that NHE-1 inhibitors could play a significant role in modifying DMD cardiac and also skeletal pathologies, as the NHE-1 isoform is ubiquitous. We report here the first study with Rimeporide in DMD patients. This 4-week treatment, open label phase Ib, multiple oral ascending dose study, enrolled 20 ambulant boys with DMD (6–11 years), with outcomes including safety, pharmacokinetic (PK) and pharmacodynamic (PD) biomarkers. Rimeporide was safe and well-tolerated at all doses. PK evaluations showed that Rimeporide was well absorbed orally reaching pharmacological concentrations from the lowest dose, with exposure increasing linearly with dose and with no evidence of accumulation upon repeated dosing. Exploratory PD biomarkers showed positive effect upon a 4-week treatment, supporting its therapeutic potential in patients with DMD, primarily as a cardioprotective treatment, and provide rationale for further efficacy studies.
1. Introduction
1.1. Disease’s overview
Duchenne Muscular Dystrophy (DMD) is a paediatric orphan disease with an incidence of approximately 1 in 5000 male births worldwide [1,2]. It is a rapidly progressing and severe form of muscular dystrophy where children typically display skeletal muscle weakness by the age of 2–6 years, followed by progressive loss of their ability to walk between the ages of 7–13 years. By the age of 15–18 years, most patients start to develop progressive respiratory muscle weakness leading to respiratory insufficiency requiring ventilator support. While the widespread use of corticosteroids improves muscle function and delay age at loss of ambulation, there continues to be a high unmet need for treatments for cardiorespiratory complications in these patients. The improved respiratory intervention has also improved the respiratory outcomes for affected patients. However, cardiac disease (cardiomyopathy leading to heart failure and arrhythmias) has become an increasingly important cause of morbidity and mortality [3]. Pathological cellular hallmarks of DMD include, downstream the lack of dystrophin, abnormal ionic homeostasis. Cytoplasmic sodium overload was shown to cause severe osmotic oedema in DMD patients [4]. The dual overload in sodium ion [5] and water precedes the dystrophic process and persists until fatty muscle degeneration is complete. In addition, intracellular overload of calcium is well described as a major cause of muscle cell dysfunction in DMD [6] and necrosis.
1.2. Scientific rationale of Rimeporide repositioning in DMD
Rimeporide (initial code EMD 87580), is a benzoyl-guanidine derivative. It is a potent and selective inhibitor of the sodium-proton exchanger isoform 1 (NHE-1) which is found ubiquitously in mammalian cell membranes throughout the body [7]. There are ten NHE isoforms but NHE-1 is the predominant isoform in the heart and in skeletal muscles [8]. This ubiquitous protein, encoded by the SLC9A1 gene, has several important physiological and pathological influences on mammalian cells as a result of its activity. In normal conditions, NHE-1 maintains intracellular pH (pHi) and volume by removing one intracellular proton (H+) ion in exchange for a single extracellular sodium (Na+) ion [9]. In certain pathological conditions, NHE-1 is activated, leading to a rapid accumulation of sodium in cells [8] and an acidification of the extracellular space. The high sodium concentration drives an increase in calcium (Ca2+) via direct interaction and reversal of the Na+/Ca2+ exchanger (NCX). The resulting accumulation of calcium triggers various pathways leading to cell death. As one example, NHE-1 is known to contribute to cardiac hypertrophy [10]. The concept of NHE-1 involvement in cardiac pathology has been adopted for decades and is supported by a plethora of experimental studies demonstrating effective NHE-1 inhibition in protecting the myocardium against ischemic and reperfusion injury as well as attenuating myocardial remodeling and heart failure [11].
The cardioprotective effects of NHE-1 inhibitors, including Rimeporide, have been extensively studied in various animal models of myocardial infarction and dystrophic cardiomyopathy including DMD. A recently completed study in Golden Retriever muscular dystrophy (GRMD) dogs has confirmed the cardioprotective role of Rimeporide after a treatment in a preventing setting [12]. Other preclinical experiments [13,14] have underlined the significance of myocardial necrosis, due to pH abnormalities as well as calcium and sodium imbalances in the pathophysiology of heart failure and have demonstrated the beneficial effects of NHE-1 inhibition using Rimeporide in preventing the deleterious effects of Ca2+ and Na + overload [14].
There is also clear scientific rationale supporting the concept that NHE-1 could play an important role in several pathophysiological processes involved in DMD. In skeletal muscles of DMD patients, due to the lack of dystrophin, mechanical stress during contraction leads to skeletal and cardiac muscle fibers fragility, tears in the sarcolemma, influx of extracellular Ca2+ [15], subsequent inflammation and Na + overload [16], and alkalization [17]. Na + is presumed to accumulate in dystrophic muscle fibers through microtears and channels leading to enhanced Ca2+ entry through the NCX. Cytosolic Na + levels have been described to be elevated in myofibers from dystrophin-deficient (mdx) mice, the most used animal model of DMD [18] and in skeletal muscles in DMD patients [4,19].
Also, pH is increased in resting muscle tissue of patients with DMD [20]. Interestingly, pH increase is seen in descending order in DMD, Becker muscular dystrophy, and female carriers, when compared to healthy controls [21], i.e. pH increases with the severity of the genetic alteration. An increase of pH in muscle tissue has also been reported in mdx mice [18]. In addition, Na + influx via the NHE-1 is increased in mdx myocytes compared to wild-type myocytes [22].
NHE-1 inhibition by Cariporide, another selective Na+/H + exchange inhibitor discontinued in late stage clinical development for safety reasons, has been shown to have significant beneficial effects on skeletal muscle function, intracellular ion homeostasis and pH in mdx mice [22]. In these mice, treatment with Cariporide markedly reduced creatine kinase (CK) levels, inflammation, fibrosis and histological skeletal muscle damage, whilst preserving muscle performance. In the BIO 14.6 hamster (delta sarcoglycan deficiency related dystrophy, also relevant to dystrophinopathies), Cariporide normalised intracellular Ca2+ levels and Na + influx and decreased stretch induced CK release, strongly supporting that inhibition of NHE-1 can lead to regulation of Ca2+ via normalisation of NCX activity [22].
Rimeporide, another NHE-1 inhibitor which showed a good safety and tolerability profile so far in adults (tested in 166 subjects in 7 phase I studies) proved to provide potent anti-inflammatory and anti-fibrotic effects in short and long term studies in mdx mice both in skeletal and cardiac muscles activity [23]. In UMX 101 hamsters, also a delta sarcoglycan deficiency related dystrophy and a well-established and accepted model of dilated cardiomyopathy activity [14], lifelong preventive treatment with Rimeporide led to a dramatic reduction in mortality and prevented myocardial necrosis and hypertrophy, as well as Ca2+ accumulation [14,24]. Present preclinical findings suggest that Rimeporide has a potential to be a successful pharmacological approach to the cardiac and skeletal dysfunction observed in DMD.
1.3. Summary of clinical data with Rimeporide in adults
Rimeporide was developed up to clinical Phase I for the treatment of advanced congestive heart failure (CHF) by Merck KGaA (Germany). Development was discontinued for strategic reasons by Merck KGaA despite a data package including preclinical pharmacology studies, 7 clinical pharmacology studies involving 145 healthy adults and a clinical study in 21 patients with CHF, with oral and/or intravenous (IV) dosing. In 2013, EspeRare entered in a license agreement with Merck KGaA for the repositioning and development of Rimeporide.
Rimeporide was well tolerated in healthy male subjects as a single oral dose up to 600 mg, as multiple oral doses three times a day (TID) up to 600 mg daily for seven days, and as a single IV dose of 350 mg. No dose limiting toxicity was observed in these studies and a maximum tolerated dose could not be defined. Adverse events (AEs) included chest discomfort, abdominal discomfort, postural dizziness, vaso-vagal episodes, dizziness, headache, light-headedness, paraesthesia of mouth, tongue, hands, rash, respiratory tract disorders, diarrhoea, and elevated liver enzymes (in IV study only) and were equally reported in placebo and treated patients. The intensity of the AEs was mild to moderate. No serious AEs were reported. In general, vital signs and laboratory investigations did not reveal any dose-limiting effects of Rimeporide.
Rimeporide was also well tolerated in patients with CHF when administered at 300 mg daily for 8 days on top of standard heart failure medications. One serious AE of worsening heart failure was reported, which was not related to the study drug but to the fact that this patient had stopped his heart failure medication.
Based on an extensive data package including anti-inflammatory and antifibrotic effects in mdx mice, improved survival in cardiomyopathic hamsters and cardioprotection in GRMD dogs [12, 13, 14, 23] and good safety and tolerability in adults, EspeRare has commenced the clinical development of Rimeporide for boys with DMD, with the main goal of preventing or slowing the progression of myocardial damage in these children.
This Phase 1b open label study (Esperare RIM4DMD, clinicaltrials.gov NCT02710591, EudraCT number 2015-002530-50) aimed to evaluate the safety, tolerability, pharmacokinetics and to explore biomarkers of efficacy of multiple ascending oral doses of Rimeporide in young ambulant patients with DMD.
2. Materials and methods
2.1. Investigational medicinal product
The active drug substance was manufactured by Merck KGaA. The investigational medicinal product (IMP) Rimeporide was provided as hard gelatine 25 mg or 50 mg capsules, in bottles of 50 capsules each. Rimeporide capsules were manufactured by GLATT Pharmaceutical Services GmbH. Rimeporide hard gelatine capsules were then supplied to study sites by Clinical Supplies Management Holdings, Inc. (CSM). The capsules are immediate release formulations. The capsules contain mannitol, hydroxypropyl methylcellulose, low-substituted hydroxypropyl cellulose, and magnesium stearate as excipients.
2.2. Study design and conduct
The study was designed as an open-label, multiple dose study with 4 ascending dose cohorts. Scientific Advice was received from the European Medicines Agency (EMA) on the study design before study initiation. It was a multicenter European study (UK, France, Italy, Spain).
The study protocol was approved by the Ethics Committees at each participating site and National Competent Authority from each country prior to the study initiation. The legal guardian for each study participant signed informed consent prior to any study procedures. Main criteria for inclusion were male subjects between 6–14 years of age at study entry, genetically confirmed DMD, able to walk at least 75 m independently and on a stable dose of corticosteroids for at least 6 months prior to baseline.
Oral doses of Rimeporide from 50 mg to 300 mg three times daily (TID) were administered. Each subject participated in only 1 dose cohort and subsequently received Rimeporide for a total of 4 weeks as shown in Fig. 1.
Fig. 1.

Dose escalation process in RIM4DMD phase I trial.
Abbreviation: kg: kilograms; mg: milligrams TID: three time a day, 4w: 4weeks, N: number of subjects, PK: Pharmacokinetic data.
The primary objective of this study was to evaluate the safety of Rimeporide in ambulant DMD boys. Secondary objective was to evaluate PK profile of Rimeporide. Exploratory objectives aimed to quantify biomarkers of cardiac and skeletal muscle inflammation, metabolism or injury, as well as other markers of efficacy mechanistically related to Rimeporide mode of action or to DMD pathophysiology. These biomarkers included serum/plasma and imaging assessments.
2.3. Selection of doses in the study
Given the rarity and morbidity of DMD, it was aimed to start dosing at a safe, yet pharmacologically effective dose. Dose finding studies in animal models indicated that the exposure/concentration range that would have an efficacious response were ranging from 500 to 2500 ng/mL [13]. Thus, simulated plasma concentrations for all cohorts were selected to fall within this range.
The dose-escalation procedure also included the fewest dose levels without escalating up to exposures of overt toxicity. It was intended to proceed cautiously to take into account both the safety profile known to date and emerging safety data in this young and vulnerable patient population. Further dose escalation commenced only after safety and PK data from all patients of the previous cohort had been reviewed by the independent Safety Monitoring Committee (SMC). The recommendation to escalate to the next cohort was given by the SMC.
2.4. Study assessments
Safety assessments were conducted at the screening visit and after one, two and four weeks of treatment, as well as within 1–2 weeks following the end of treatment period. The clinical safety in these young patients was carefully evaluated throughout the study by experienced clinical staff. The safety assessments included: physical examinations, AEs/serious AEs, vital signs, diastolic and systolic blood pressure in supine and standing positions, heart rate in same position (supine and standing) and respiratory rate; 12-lead electrocardiogram (ECG) including QT (reviewed both locally by a cardiologist and centrally by an independent cardiologist). Laboratory parameters (from blood and urine samples) included haematology and biochemistry panel. Gastrin was also measured in serum as it was shown in some animal studies to increase and anecdotal and moderate elevations in blood gastrin were observed in humans in one of the previous phase I study. Finally, the number of patients withdrawn for safety issues was also captured.
For the PK assessments, as advised by the EMA Guideline on the clinical investigation of medicinal products for the treatment of Duchenne and Becker muscular dystrophy [25], a population PK analysis model developed by Calvagone Sarl (France) was used based on prior PK data obtained with Rimeporide in adults. A 3-compartment disposition model with an absorption phase and a first order linear elimination from the central compartment was designed. Using this 3-compartment distribution population PK model, a sparse sampling approach including 6 time points over the study was derived from simulated PK profiles in children to best fit to the model. On day 1 of treatment, one time point in the absorption phase, one time point near the maximum plasma concentration (Cmax) and two time points in the biphasic decay were deemed appropriate to document the PK model in DMD boys while minimizing burden of blood sampling. At the end of the 4-week treatment, only 2 points were needed to confirm the lack of time dependency of Rimeporide PK profile. Given that 20 patients were planned to be enrolled, the amount of data for validation of the population PK model was 120 concentration time points. On Study Day 1, samples were taken pre-dose and within the following time intervals (0.5–1 h), (1–2 h), (2.5–3.5 h) and approximately 6 h after the first or the second dose. On study day 28, 2 samples were taken at the following time interval (0.5–1 h) post dose and approximately 6 h after the last dose. For the purpose of the population PK model, real sampling times were considered for the analysis.
For the PD imaging, all patients were proposed to have a Nuclear Magnetic Resonance Imaging (NMRI) at baseline and week 4 to explore changes in NMRI indices in skeletal muscle (water T2, fat fraction). The acquisitions were performed locally in both lower limbs of the patients.
For the PD biomarker analysis, two serum/plasma samples were analysed for each subject: baseline (pre-treatment), and after four weeks of daily treatment with Rimeporide at the indicated dose level (post treatment). The selection of biomarkers was based on a semi-targeted and targeted analysis of circulating proteins according to Rimeporide’s mode of action, DMD pathological processes (inflammation, fibrosis and muscle injury) and already established biomarkers in DMD patients.
2.5. Sample management and analysis
Collection kits, shipment material and documentation, and a laboratory manual were provided by the Sponsor to each clinical site before any patient inclusion in the study. Safety blood samples were handled and analysed locally. Remaining plasma/serum aliquots were stored on site in secured and controlled freezers until shipment by an authorised carrier to Eurofins central laboratory in the Netherlands. Temperature was monitored during shipment.
PK plasma samples were transferred to Nuvisan GmbH, Germany for the analysis of plasma concentrations of Rimeporide using a previously validated LC–MS/MS method.
PD samples (serum and plasma) were transferred to the Serum bank of Geneva University Hospital or to KTH Royal Institute of Technology, Sweden or to Olink for analysis on behalf of the Department of Human Genetics, Leiden University Medical Center, The Netherlands. Two-hundred and thirty protein biomarkers were measured with four different technologies: Olink’s Proximity Extension Assay (PEA) [26], Electrophoresis, ELISA and bead based immuno-Luminex assay [27] in two types of biological matrix (serum and plasma). The Olink PEA immunoassay technology was used to measure 2 cardiometabolic panels of 92 protein biomarkers each. Another 48 biomarkers were selected and analysed at KTH by suspension bead array using antibodies validated by western blot, immunoassay and immunochemistry. Active TGF-β1, MMP-9, and PINP were measured from serum by enzyme-linked immunosorbent assay ELISA kits (R&D Systems, Abingdon, UK) using the protocol recommended by the manufacturer. For the measure of active TGF-β1 (free + latent TGF-β1), serum samples were activated with reagents as recommended by manufacturer.
TNFα, IL-6, IL-34 and CXCL10/IP-10 were measured by a commercially available multiplex beads immunoassay (Luminex Performance assay, Human Obesity Panel, R&D Systems, Minneapolis, USA) according to supplier’s instruction.
Creatine kinase total (CK T) and isoenzymes (CK MM and MB) were measured by the central laboratory Eurofins. CK total and isoenzymes to monitor muscle damage were measured from serum by Roche Imidazole buffered NADPH rate reaction for CK total and by Sebia Agarose electrophoresis for CK isoenzymes.
2.6. Quality assurance
Quality assurance and quality control systems were implemented and maintained using written standard operating procedures to ensure that the trial was conducted and data were generated, documented (recorded), and reported in compliance with the protocol, Good Clinical Practice, and the applicable regulatory requirement(s).
2.7. Statistical methods and analysis
Since this study was descriptive and exploratory in nature, no formal sample size calculation was performed. A sample size of 20 evaluable patients receiving active drug was proposed for this phase 1b study with no placebo control. This was discussed and deemed acceptable by the Committee for Medicinal Products for Human Use during the EMA scientific advice meeting (EMA/CHMP/SAWP/376863/2015).
The Intention to treat (ITT) population consisted of all patients who received at least one dose of study drug. This population was defined also as the Safety population. The PK and exploratory PD variables were analysed on the ITT population.
For the safety data, descriptive statistics were provided for all variables in summary tables by cohort according to the type of variable summarised. AEs were tabulated by body system and by preferred term for each dose level. AEs were also tabulated by severity and relationship to the study medication. Summaries were produced for Serious AEs (SAEs). For each clinical laboratory test, individual patient values were listed and changes from baseline were calculated and summarised.
2.7.1. Population PK analysis
Using population PK (PPK), PK data were acquired by analyzing data pooled from healthy volunteers, CHF patients and DMD patients. Model development, model diagnostics and simulations were performed using NONMEM 7.3 and PsN 4.6.0 by Calvagone Sarl [28]. Primary PK parameters like clearance and volume of distribution were directly obtained from the final NONMEM run. The following PK parameters such as the Cmax, the area under the curve at 24 h (AUC24), the time at which Cmax is observed (Tmax), the drug elimination half-life (T1/2) and the time spent above the pharmacological targeted concentration (defined as >500 ng/mL) over 24 h (TAT) were obtained by a non-compartmental analysis (NCA) from simulated PK profiles of the first day and the last day of administration. NCA was performed using SAS software (version 9.4). An empirical Bayesian feedback estimation (MAXEVAL = 0) was performed on the simulated dataset (on data points every 0.05 h) using the final PK parameter estimates from the final Rimeporide PK model and the actual dosing schedule for each DMD patient. In order to evaluate the predictive performance of the final model, a prediction corrected visual predictive check (VPC) was performed on the concentration-time data of the different studies [29].
The PPK model explored the influence of the following covariates: alanine transaminase (ALT), aspartate transaminase (AST), bilirubin, body mass index (BMI), bodyweight (BW), glomerular filtration rate (GFR), serum creatinine, and lean body mass (LBM).
Descriptive statistics were also used for the main PK parameters and presented by dose cohorts and by day of administration.
2.7.2. PD exploratory analysis
Descriptive statistics were calculated for all parameters, separately for each cohort. Graphical and tabular presentations were produced when appropriate. An additional exploratory statistical analysis was performed for the plasma/serum biomarkers with the aim of identifying candidate biomarkers associated with Rimeporide treatment in a dose-dependent or dose-independent manner.
For treatment association analyses, the following linear mixed effect models [30] were fitted to data:
| B ∼ TRT + Age + BMI + (1|SUBJID) |
Where B is the biomarker expression (at Day 0 or 28), TRT is a dummy variable equal to 0 at Day 0 and 1 at Day 28, Age and BMI (Body Mass Index) are covariates, and SUBJID is a subject-specific random intercept that accounts for correlation between repeated measurements from the same patient. A type II analysis of deviance was performed. Wald chi-square p-values of the TRT terms were corrected using the Benjamini-Hochberg method [31] and biomarkers with False Discovery Rate (FDR) ≤10 % were reported.
3. Results
3.1. Disposition of subjects and demographics
A total of 24 patients were screened and 20 enrolled in 4 clinical centres (Fig. 2): one patient was a screened failure as he was previously involved in another research study (less than 4 weeks before IMP administration) and the 3 other patients did not receive the study drug within 1 month of screening due to the unavailability of the IMP.
Fig. 2.

Disposition of subjects.
Abbreviation: N: number of subjects: TID: three time a day.
All enrolled patients completed the study. The analysis set consisted of 20 DMD patients, aged from 6–11 years with a confirmed diagnosis of DMD. The demographic and clinical information for the subjects enrolled are reported in Table 1.
Table 1.
Patient demographic and characteristics.
| Characteristics | Cohort 1 N = 5 |
Cohort 2 N = 5 |
Cohort 3 N = 5 |
Cohort 4 N = 5 |
All N = 20 |
|---|---|---|---|---|---|
| Age (years) | |||||
| Mean (SD) | 8.4 (1.7) | 8.2 (1.5) | 8.8 (1.6) | 9.2 (0.4) | 8.7 (1.3) |
| Median | 8.0 | 8.0 | 8.0 | 9.0 | 9.0 |
| Range | 6; 10 | 6; 10 | 7; 11 | 9; 10 | 6; 11 |
| Height (cm) | |||||
| Mean (SD) | 126.4 (6.9) | 127.6 (2.3) | 125.4 (6.8) | 121.2 (9.3) | 125.2 (6.7) |
| Median | 127.0 | 127.0 | 125.0 | 120.0 | 125.5 |
| Range | 119; 135 | 125; 130 | 117; 134 | 110; 135 | 110; 135 |
| Weight (kg) | |||||
| Mean (SD) | 28.8 (9.0) | 29.0 (5.2) | 29.0 (4.9) | 25.8 (6.9) | 28.1 (6.3) |
| Median | 24.6 | 27.0 | 27.7 | 23.5 | 25.7 |
| Range | 20.4; 39.0 | 24.5; 37.6 | 24.0; 35.9 | 21.0; 37.9 | 20.4; 39.0 |
| BMI (kg/m2) | |||||
| Mean (SD) | 17.7 (3.9) | 17.8 (3.2) | 18.3 (1.9) | 17.3 (2.2) | 17.8 (2.7) |
| Median | 17.1 | 17.3 | 17.7 | 17.2 | 17.4 |
| Range | 13.6; 22.2 | 15.4; 23.3 | 16.3; 21.3 | 14.6; 20.8 | 13.6; 23.3 |
| Time since DMD diagnosis (years) | |||||
| Mean (SD) | 4.5 (2.7) | 4.9 (1.5) | 5.5 (2.5) | 6.6 (2.7) | 5.4 (2.3) |
| Median | 3.4 | 4.6 | 4.8 | 6.4 | 4.7 |
| Range | 2.9; 9.1 | 3.2; 7.2 | 2.2; 8.3 | 3.2; 9.6 | 2.2; 9.6 |
Abbreviation: BMI: body mass index; cm: centimetres; DMD: Duchenne muscular dystrophy; kg: kilograms; mg: milligrams; N: number of subjects; SD: standard deviation.
A focus was given to concomitant medications that were relevant for DMD throughout the study duration as shown in Table 2. All patients but one in cohort 3 (no corticosteroid at screening and during the entire study duration) were on a stable dose of corticosteroids. In addition, another patient in cohort 3 received omeprazole for the treatment of heartburn at a stable dose of 10 mg daily. Treatment was started three months before inclusion in the study and continued throughout the study period.
Table 2.
Relevant concomitant medications for DMD by patients throughout the study duration.
| Anatomical Main Group Chemical Subgroup Preferred Name |
Cohort 1 N = 5 |
Cohort 2 N = 5 |
Cohort 3 N = 5 |
Cohort 4 N = 5 |
All patients N = 20 |
|---|---|---|---|---|---|
| Systemic hormonal preparations, excl. sex hormones and insulins | |||||
| Glucocorticoids | 5 (100 %) | 5 (100 %) | 4 (80 %) | 5 (100 %) | 19 (95 %) |
| Deflazacort | 0 | 3 (60 %) | 2 (40 %) | 5 (100 %) | 10 (50 %) |
| Prednisone | 5 (100 %) | 1 (20 %) | 0 | 0 | 6 (30 %) |
| Prednisolone | 0 | 1 (20 %) | 3 (60 %) | 0 | 4 (20 %) |
| Alimentary tract and metabolism | |||||
| Proton pump inhibitors | 0 | 0 | 1 (20 %) | 0 | 1 (5 %) |
| Omeprazole | 0 | 0 | 1 (20 %) | 0 | 1 (5 %) |
Abbreviations: %: percentage; N: number of subjects. In cohort 3, one patient did not receive any glucocorticoid while another one received prednisolone and deflazacort.
Finally, the overall compliance and adherence to the dosing schedule of Rimeporide was excellent: mean compliance was 97.1 %.
3.2. Primary objective: safety of Rimeporide after repeated dosing in young DMD boys
As already established in previous studies in healthy adults, Rimeporide had a good safety profile in young boys with DMD over a wide range of doses. No SAE related to study medication was reported and no withdrawal due to adverse events or for any other reasons was observed.
Twelve patients reported a total of 37 treatment emergent adverse events (TEAEs). Thirty-five TEAEs were of mild intensity and 2 were reported as severe. All were self-limiting. There was no evidence for an increase in frequency or severity of AEs with increasing dose. The most frequently reported TEAEs were headache (6 events reported in 5 patients); diarrhoea (4 events reported in 3 patients); and nasopharyngitis (4 events reported in 4 patients) as shown in Table 3.
Table 3.
Treatment-Emergent Adverse Events classified by System Organ Class and Preferred Term defined in term of number of patients and of events.
| System Organ Class Preferred Term |
Cohort 1 N = 5 |
Cohort 2 N = 5 |
Cohort 3 N = 5 |
Cohort 4 N = 5 |
All patients N = 20 |
|---|---|---|---|---|---|
| Any treatment-emergent adverse event | 2 (6 events) | 1 (3 events) | 5 (15 events) | 4 (13 events) | 12 (37 events) |
| Nervous system disorders | 1 (1 event) | 0 | 3 (3 events) | 3 (4 events) | 7 (8 events) |
| Headache | 1 (1 event) | 0 | 2 (2 events) | 2 (3 events) | 5 (6 events) |
| Dizziness | 0 | 0 | 1 (1 event) | 0 | 1 (1 event) |
| Presyncope | 0 | 0 | 0 | 1 (1 event) | 1 (1 event) |
| Gastrointestinal disorders | 0 | 1 (1 event) | 4 (8 events) | 1 (1 event) | 6 (10 events) |
| Diarrhoea | 0 | 0 | 3 (4 events) | 0 | 3 (4 events) |
| Abdominal pain upper | 0 | 0 | 1 (2 events) | 1 (1 event) | 2 (3 events) |
| Vomiting | 0 | 0 | 2 (2 events) | 0 | 2 (2 events) |
| Mouth ulceration | 0 | 1 (1 event) | 0 | 0 | 1 (1 event) |
| Infections and infestations | 0 | 1 (1 event) | 2 (2 events) | 1 (1 event) | 4 (4 events) |
| Nasopharyngitis | 0 | 1 (1 event) | 2 (2 events) | 1 (1 event) | 4 (4 events) |
| Respiratory, thoracic and mediastinal disorders | 0 | 1 (1 event) | 1 (1 event) | 2 (2 events) | 4 (4 events) |
| Cough | 0 | 1 (1 event) | 1 (1 event) | 1 (1 event) | 3 (3 events) |
| Oropharyngeal pain | 0 | 0 | 0 | 1 (1 event) | 1 (1 event) |
| General disorders and administration site conditions | 1 (2 events) | 0 | 0 | 1 (1 event) | 2 (3 events) |
| Chills | 1 (2 events) | 0 | 0 | 0 | 1 (2 events) |
| Pyrexia | 0 | 0 | 0 | 1 (1 event) | 1 (1 event) |
| Vascular disorders | 0 | 0 | 1 (1 event) | 1 (2 events) | 2 (3 events) |
| Flushing | 0 | 0 | 1 (1 event) | 1 (2 events) | 2 (3 events) |
| Ear and labyrinth disorders | 0 | 0 | 0 | 1 (1 event) | 1 (1 event) |
| Tympanic membrane perforation |
0 | 0 | 0 | 1 (1 event) | 1 (1 event) |
| Injury, poisoning and procedural complications | 1 (1 event) | 0 | 0 | 1 (1 event) | |
| Fall | 1 (1 event) | 0 | 0 | 1 (1 event) |
Abbreviation: N: number of subjects.
The only SAEs reported were diarrhoea and vomiting, in one patient in Cohort 3 not reported as related to study drug. The symptoms occurred simultaneously in a single episode considered to be due to food poisoning. This was corroborated by the fact that the patient ´s mother, experienced similar symptoms on the same day after eating the same (fish) meal.
The only events which were reported as adverse drug reactions (ADRs) were an event of urticaria in one subject in Cohort 2, two events of flushing in one subject in Cohort 4 and one episode of nasopharyngitis in the same subject. All these events were reported as possibly related to the study drug and none as probably related. Therefore, apart from transient flushing which was observed in one subject in Cohort 3 on one occasion and in one subject in Cohort 4 on two occasions, there was no emergent pattern of AEs.
There were no consistent or clinically significant effects on vital signs (data not shown). Standing and supine systolic and diastolic blood pressure as well as heart rate appeared to be unaffected by the treatment. Clinical laboratory parameters (biochemistry and haematology parameters) revealed no clinically significant changes over treatment period. No modifications of gastrin levels, a surrogate marker for signs of adverse effect on gastric parietal cells [32], were observed after a four-week treatment. In general, vital signs and laboratory investigations did not reveal any adverse effects of Rimeporide. Most notably, Rimeporide was devoid of any blood pressure or heart rate lowering effects.
There were no clinically important changes in 12-lead ECGs. All values for QTcB (corrected QT interval based on Bazett formula) or QTcF (corrected QT interval based on Fridericia formula) were below 450 msec, apart from two values in two subjects, for whom the elevations were not confirmed by the central reading. Some increases in QTcB intervals above 30 msec were seen in a few patients, but these were either not present on both the local and central readings or were not accompanied by changes in QTcF, except for one value at week 2 in a subject in cohort 3 receiving 450 mg/day.
3.3. Secondary objective: pharmacokinetic profile of Rimeporide in young DMD boys
The objectives of this PK analysis were 1) to investigate the PK profile of Rimeporide after single- and multiple-dose, and 2) to explore the linearity of the different doses administered in boys ages 6–11 years with DMD. The PK full dataset consisted in a pool of over 3400 Rimeporide concentrations, among healthy adults, CHF patients and 20 DMD children. The model qualification on the final model through the goodness of fit (GOF) plots showed a good agreement between the observed and predicted Rimeporide concentrations. Epsilon shrinkage was acceptable, indicating that the GOF plots are reliable. A prediction-corrected visual predictive check for the final PPK model showed that most of the observed concentrations fell within the 95 % confidence interval of the simulated data. The final PPK model described the data well and was able to explain part of the variability in exposure on the basis of some covariates (Glomerular filtration rate, serum creatinine and food status). Finally, the PK model had an adequate predictive performance to simulate PK profiles in DMD children and to derive individual PK parameters by NCA.
Overall, Rimeporide PK in DMD patients was dose linear with no sign of any accumulation with time. Rate of absorption was slowed down under fed conditions without changing the drug exposure (AUC). Based on the identified covariates, dose adjustment did not seem to be necessary according to body weight. Oral dosing with immediate release capsules resulted in a quick and extensive absorption from the gastrointestinal tract. Cmax occurred within 1–2 h post dose on both Days 1 and 28 as shown in Table 4. The drug elimination half-life across all doses was short and unchanged between Day 1 and Day 28, averaging about 3–4 h and justifying a three times daily administration to maintain plasma levels above pharmacological levels. Cmax and AUC24 values were generally comparable on Days 1 and 28 at all dose levels suggesting no accumulation occurs upon multiple dosing. PK parameters increased linearly with increasing doses. When administered under fed condition, a decrease of Cmax and a shift of Tmax are observed while no impact on AUC24 is noticed. Regarding the time spent above targeted pharmacological concentration (e.g. > 500 ng/mL) over 24 h, TAT in cohort 1 was around 6 h suggesting that doses in cohort 1 might be too low to maintain plasma levels above the pharmacological target concentration throughout the day. In cohorts 2, 3 and 4, TAT increased to 17 h suggesting that doses above 100 mg TID were more suitable to lead to a sustained 24h-exposure to Rimeporide and potentially to a sustained pharmacological effect. Finally, PK profiles in children were compared to previous PK studies in healthy adults. It was observed that PK profiles in children result in a slightly higher exposure than in adults.
Table 4.
PK parameters after oral administration of Rimeporide from 150 to 900 mg daily obtained with non-compartmental analysis at Day 1 and Day 28 by dose group.
| Cohort/ Dose (mg) | Day | Statistics | T1/2 | Cmax | AUC24 | TAT |
|---|---|---|---|---|---|---|
| (h) | (ng/mL) | (ng h/mL) | (h) | |||
| Cohort 1 150 to 225 |
1 | N | 5 | 5 | 5 | 5 |
| Mean | 3.3 | 1286 | 8857 | 6.6 | ||
| SD | 0.5 | 568 | 3868 | 4 | ||
| 28 | N | 4 | 4 | 4 | 4 | |
| Mean | 3.4 | 1361 | 10423 | 8.4 | ||
| SD | 0.6 | 431 | 1208 | 1.7 | ||
| Cohort 2 300 to 450 |
1 | N | 5 | 5 | 5 | 5 |
| Mean | 4.6 | 1987 | 19996 | 17 | ||
| SD | 1.4 | 650 | 4903 | 3 | ||
| 28 | N | 5 | 5 | 5 | 5 | |
| Mean | 4.66 | 2077 | 18667 | 15 | ||
| SD | 1.46 | 974 | 5669 | 3 | ||
| Cohort 3 450 to 600 |
1 | N | 5 | 5 | 5 | 5 |
| Mean | 3.9 | 3013 | 29390 | 18 | ||
| SD | 0.9 | 730 | 4510 | 1 | ||
| 28 | N | 5 | 5 | 5 | 5 | |
| Mean | 3.76 | 2817 | 26332 | 16 | ||
| SD | 0.991 | 675 | 3026 | 2.3 | ||
| Cohort 4 600 to 900 |
1 | N | 5 | 5 | 5 | 5 |
| Mean | 3.9 | 3819 | 36179 | 19.0 | ||
| SD | 1 | 743 | 7647 | 2 | ||
| 28 | N | 5 | 5 | 5 | 5 | |
| Mean | 3.75 | 3909 | 35987 | 18 | ||
| SD | 0.910 | 846 | 6981 | 1.1 |
Abbreviations: AUC24: area under the curve at 24 h; Cmax: maximal concentration over 24 h after the first administration of the day; N: number; SD: standard deviation; TAT: time spent above the target concentration of 500 ng/ml over 24 h; T1/2: terminal elimination half-life.
3.4. Exploratory pharmacodynamic biomarkers
3.4.1. Imaging biomarkers
No notable change (worsening or improvement) in the NMRI indices was observed following Rimeporide dose escalation and during the 4-week treatment. There was however no worsening of skeletal muscles indices over the treatment period (data not shown).
3.4.2. Serum biomarkers
From the total of 230 biomarkers that were assessed in this study, 10 proteins were analysed with two or more assays. Cross-correlations across technology platforms showed lack of correlation, probably due to the use of different antibodies and sample matrices (serum and plasma).
Twelve proteins were significantly modulated by 4-week treatment with Rimeporide, as assessed by the comparison of baseline and 4-week follow-up samples. There was no association between biomarkers levels and dose, age and BMI, which was not surprising considering the small range of variations in these covariates. These 12 biomarkers, listed in Table 5, represent potential new candidate biomarkers to evaluate the biological efficacy of Rimeporide non-invasively.
Table 5.
Circulating biomarkers associated with Rimeporide treatment.
| Rank | Biomarker | Biomarker name | Source | Term | p-value | FDR |
|---|---|---|---|---|---|---|
| 1 | IGFBP1 | Insulin Growth Factor Binding Protein 1 | OLink | −2.7123 | 1.7e-8 | 4e-6 |
| 2 | TR | Transferrin receptor (also called TFRC) | OLink | 0.5252 | 3.0e-6 | 0.0004 |
| 3 | SCGB3A2 | Secretoglobulin family 3A-member-2 | OLink | −0.5466 | 1.3e-5 | 0.001 |
| 4 | CCL15 | C-C motif chemokine ligand 15 | OLink | −0.4489 | 0.0003 | 0.17 |
| 5 | KLK6 | Kallikrein 6 | OLink | −0.2612 | 0.0004 | 0.20 |
| 6 | FAS | Fas Ligand | OLink | −0.2823 | 0.0007 | 0.24 |
| 7 | CK T | Creatine Kinase total | Electrophoresis | −0.3851 | 0.0007 | 0.24 |
| 8 | TNFα | Tumor Necrosis Factor α | Luminex | −0.1726 | 0.0009 | 0.24 |
| 9 | CK MM | Creatine Kinase MM isoform | Electrophoresis | −0.3886 | 0.0009 | 0.24 |
| 10 | MCP1 | Monocyte Chemoattractant Protein | OLink | −0.5675 | 0.0016 | 0.39 |
| 11 | MB | Myoglobin | OLink | −0.6763 | 0.0036 | 0.79 |
| 12 | IGFBP6 | Insulin Growth Factor Binding Protein 6 | OLink | −0.3144 | 0.0044 | 0.89 |
Abbreviations: FDR (False discovery rate): p-value corrected for multiple testing.
All proteins showed down-regulation upon treatment with Rimeporide with the exception of TR showing an upregulation. Among modulated biomarkers, IGFBP1 (p = 1.7e-8; FDR = 4e-6) is of particular statistical relevance (Fig. 3). Of note, the concentration of the well-known CK T (p = 0,0007; FDR = 0,24) and its CK MM (p = 0,0009; FDR = 0,24) were reduced by the treatment.
Fig. 3.

Evolution of key biomarkers after a 4-week treatment with Rimeporide.
Dot plot showing the reduction of selected individual biomarkers in the 4 cohorts between Week 0 (baseline) and Week4 (end of treatment period). Log transformed concentration data are plotted on the y-axis.
From these proteins significantly associated to Rimeporide treatment, 8 were already studied in large scale protein biomarker discovery longitudinal studies in DMD patients using SOMAscan and described in case control comparisons between DMD patients and aged-matched controls [33,34]. These already studied biomarkers are CK MM, IGFBP6, IGFBP1, MB, KLK6, CCL15, FAS and TNFα. In these longitudinal studies, all these proteins plus TR in Spitali’s study [34] were shown to be stable over time in DMD patients except for CK, which is known to be variable in such a population.
4. Discussion
Seven phase I studies have been conducted in healthy adults with Rimeporide during its initial development for patients with congestive heart failure and have shown no safety concern, a good tolerability and a good PK profile. No safety and PK data were available in pediatrics nor in young DMD boys. The objectives of this phase I study were to provide complementary data to the existing data package on Rimeporide safety, tolerability, PK and biomarker(s) of biological response in pediatric patients with DMD.
The safety and tolerability profile of Rimeporide in children was good and similar to that observed in healthy adults. No treatment-related adverse events, nor dose limiting toxicity were observed as well as no consistent pattern of non-serious adverse events. There was also no significant modification in the laboratory values. ECGs and QT intervals did not present any significant change from baseline as confirmed by a central reading. No clinically relevant change in blood pressure was observed.
A PPK model was applied and showed that the PK profiles in children were increasing linearly with rising doses, with no sign of accumulation upon multiple dosing and that interaction with food did not significantly impact exposure. Concentrations above targeted pharmacological exposure (500 ng/mL) were achieved in all cohorts although in cohort 1, TAT was significantly shorter than in the other cohorts.
Particular efforts were also dedicated to the identification of imaging and serum candidate biomarkers. While skeletal NMRI indices did not change significantly over the 4-week treatment period (no improvement and no worsening), this was expected given the short treatment duration. Prior studies [35] showed that therapeutic effect of corticosteroids can be observed no earlier than 3 months of treatment by an initiation of a decrease in muscle T2, consistent with the anti-inflammatory action of corticosteroids.
There was however, preliminary evidence for biological effect on several serum/plasma biomarkers relevant for Rimeporide mode of action including inflammation, muscular repair and injury. The observed biomarker changes provide encouraging evidence of the biological activity of Rimeporide at all doses tested over a 4-week period in children with DMD. Although the biomarker changes were not dose-dependent, it is notable that even at the lowest dose, Rimeporide plasma peak concentrations were within the expected range of pharmacological concentration. However, TAT in cohort 1 was much shorter (6 h) than that in cohorts 2, 3 and 4 where TAT was around 17 h. It is not known at this stage whether a continuous inhibition of NHE-1 is needed to achieve an optimal efficacy.
For this study, we selected 230 candidate serum biomarkers based on the disease’s hallmarks and Rimeporide mode of action. 12 protein biomarkers were found to be significantly correlated with treatment. For example, a significant reduction in circulating levels of IGFBP1 and IGFBP6 was observed. The levels of circulating IGFBP1 & 6 are not known to be modulated over time in natural history studies in DMD patients [33,34], suggesting that the decrease of IGFBPs level observed in DMD patients may be associated to Rimeporide and may reflect positive changes in muscle regeneration by different mechanisms. IGFBP1 binding to endogenous insulin-like growth factor-1 (IGF1) was reported to prevent its regenerative action in dystrophic muscles [36]. IGFBP1 knockout mice show increased muscle mass and force [37]. Decreased levels of IGF binding proteins are expected to lead to increased levels of free IGF1 which appeared to be effective in mdx mice in increasing muscle strength and preventing muscle necrosis [36,38,39]. IGF1 has also been shown to have cardioprotective effect on hypertensive rats by inhibiting NHE-1 activity necrosis [40]. Such biomarker signature nicely correlates with the recent cardioprotective effect observed in GRMD dogs treated with Rimeporide [12].
A significant reduction in total Creatine Kinase CK T (p < 0,0007; FDR 0.24) and its isoform CK MM (p < 0,0009; FDR 0.24) were also reported. Both enzymes are known to be increased in boys with DMD owing to muscle damage [27]. Downregulation of both enzymes in such young patients (6–11 years old) could be suggestive of a muscle protective effect by Rimeporide.
Although still exploratory, these biomarkers results could prove to be helpful in a confirmatory phase II study for dose/dosing regimen(s) adjustment. Positive findings on these biomarkers may help to predict clinical benefit non-invasively at early stage of clinical development, and may also confirm the pharmacological efficacy of Rimeporide while further understanding its mechanism of action.
5. Conclusions
This phase Ib study establishes the safety of Rimeporide in young boys with DMD along with encouraging early changes in PD markers of efficacy and biological effects.
Cardiac disease is one of the leading causes of death in DMD patients. There is today no drug that specifically treats the cardiac pathogenesis related to DMD. Heart Failure drugs such as ACE inhibitors, beta blockers, angiotensin II receptor blockers and eplerenone are prescribed despite the certainty of evidence being very low. Interestingly, Rimeporide working through NHE-1 inhibition, is a first in class treatment and addresses some of the pathways that likely are unique and target a portion of the DMD pathology, regardless of the mutation, attributable to its more directly related mechanism of action.
A larger and longer clinical study with a placebo control is under preparation to confirm the beneficial effect of Rimeporide to prevent the cardiac and skeletal degeneration. The rationale for future studies is strongly underpinned both by the positive effects on function and survival consistently seen in dystrophic animal models and the good tolerability and the encouraging comprehensive biomarkers efficacy profile in patients, suggesting that Rimeporide has potential to provide a much-needed therapeutic approach for young boys with DMD.
Funding
This work was supported by EspeRare as well as AFM Telethon (grants 19163 & 17724), SwissLife, Innosuisse grant (25946.1 PFLS-LS), PPMD Spain, PPMD Italy and Duchenne UK. This work was also supported in part by The Muscular Dystrophy UK Central Grant to the DNC and by the NIHR Great Ormond Street Hospital Biomedical Research Centre.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
F. Porte Thomé and D. Labolle are employees and receive remuneration from EspeRare. Dr J. Gray is employed as consultant for EspeRare.
P. Spitali and M. Signorelli are employed and receive remuneration by the Leiden University Medical Center. P. Spitali has served as ad-hoc consultant for Summit PLC, Bioclinica BC, Marathon Pharmaceuticals and Solid Bio; all payments were directed to the Leiden University Medical Center and P. Spitali did not receive remuneration. P. Spitali has received grants from the AFM Telethon, the Dutch Duchenne Parent Project, the Prinses Beatrix Spierfonds, Parent Project Muscular Dystrophy and Spieren voor Spieren.
C. Al-Khalili Szigyarto and C. Johansson are employed by KTH, the Royal Institute of Technology. C. Al-Khalili Szigyarto has received research grants from the AFM Telethon and the EU commission.
Dr. F. Muntoni reports grants from AFM Telethon, during the conduct of the study; personal fees from Pfizer, Roche, Biogen, Avexis, Sarepta as well as grants from Sarepta, outside the submitted work.
The other authors declared to have no conflict of interest.
Acknowledgements
The study was designed by the sponsor, EspeRare with the support of investigators. Trial execution was supervised by the sponsor in collaboration with the investigators and the contract research organizations (Orphan Reach, Cros NT, Leon Research, Ricerche Nuove Srl, Kasaconsult).
We wish to acknowledge of patients, families and care givers as well as the SMC members, e.g. Professor Rudolf Korinthenberg, Professor Denis Duboc and Professor Behrouz Kassaï for their contributions.
We also wish to acknowledge Merck KgAa for its support.
References
- 1.Mendel J. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012;71(3):304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 2.Moat S.J., Bradley D.M., Salmon R., Clarke A., Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK) Eur. J. Hum. Genet. 2013;21(10):1049–1053. doi: 10.1038/ejhg.2012.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNally E.M. Contemporary cardiac issues in duchenne muscular dystrophy. Circulation. 2015;131(18):1590–1598. doi: 10.1161/CIRCULATIONAHA.114.015151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber M.A. Permanent muscular sodium overload and persistent muscle edema in Duchenne muscular dystrophy: a possible contributor of progressive muscle degeneration. J. Neurol. 2012;259(11):2385–2392. doi: 10.1007/s00415-012-6512-8. [DOI] [PubMed] [Google Scholar]
- 5.Fanchaouy M., Polakova E., Jung C., Ogrodnik J., Shirokova N., Niggli E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2020;46(2):114–121. doi: 10.1016/j.ceca.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen D.G., Gervasio O.L., Yeung E.W., Whitehead N.P. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 2010;88:83–91. doi: 10.1139/Y09-058. [DOI] [PubMed] [Google Scholar]
- 7.Numata M., Petrecca K., Norma Lake N., Orlowski J. Identification of a mitochondrial Na+/H+ exchanger. J. Biol. Chem. 1998;273(12):6951–6959. doi: 10.1074/jbc.273.12.6951. [DOI] [PubMed] [Google Scholar]
- 8.Fliegel L. Structural and functional changes in the Na+/H+ exchanger isoform 1, induced by Erk1/2 phosphorylation. Int. J. Mol. Sci. 2019;20(10):2378. doi: 10.3390/ijms20102378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fliegel L. The Na+/H+ exchanger (NHE) isoform 1. Int. J. Biochem. Cell Biol. 2005;37(1):33–37. doi: 10.1016/j.biocel.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Odunewu-Aderibigbe A., Fliegel L. The Na(+) /H(+) exchanger and pH regulation in the heart. IUBMB Life. 2014;66(10):679–685. doi: 10.1002/iub.1323. [DOI] [PubMed] [Google Scholar]
- 11.Evans N.P., Misyakb S.A., Robertsonc J.L., Bassaganya-Rierab J., Grangea R.W. Immune mediated mechanisms potentially regulate the disease time course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R. 2009;1(8):755–768. doi: 10.1016/j.pmrj.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghaleh B. Protective effects of rimeporide on left ventricular function in golden retriever muscular dystrophy dogs. Int. J. Cardiol. 2020;S0167–5273(19) doi: 10.1016/j.ijcard.2020.03.031. 35776-6. [DOI] [PubMed] [Google Scholar]
- 13.Chahine M. NHE-1 dependent intracellular sodium overload in hypertrophic hereditary cardiomyopathy: prevention by NHE-1 inhibitor. J. Mol. Cell. Cardiol. 2005;38:571–582. doi: 10.1016/j.yjmcc.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Bkaily G., Jacques D. Na+-H+ exchanger and proton channel in heart failure associated with Becker and Duchenne muscular dystrophies. Can. J. Physiol. Pharmacol. 2017;95:1213–1223. doi: 10.1139/cjpp-2017-0265. [DOI] [PubMed] [Google Scholar]
- 15.Deconinck N., Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr. Neurol. 2007;36(1):1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 16.Hirn C., Shapovalov G., Petermann O., Roulet E., Ruegg U.T. Nav1.4 deregulation in dystrophic skeletal muscle leads to Na+ overload and enhanced cell death. J. Gen. Physiol. 2008;132(2):199–208. doi: 10.1085/jgp.200810024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paoletti M. Advances in quantitative imaging of genetic and acquired myopathies: clinical applications and perspectives. Front. Neurol. 2019;10(78):1–21. doi: 10.3389/fneur.2019.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunn J.F., Tracey I., Radda G.K. Exercise metabolism in Duchenne muscular dystrophy: a biochemical and (31P)-nuclear magnetic resonance study of mdx mice. Proc. Biol. Sci. 1993;251(1332):201–206. doi: 10.1098/rspb.1993.0030. [DOI] [PubMed] [Google Scholar]
- 19.Weber M.A., Nagel A.M., Jurkat-Rott K., Lehmann-Horn F. (23Na) MRI detects elevated muscular sodium concentration in Duchenne muscular dystrophy. Neurology. 2011;77:2017–2024. doi: 10.1212/WNL.0b013e31823b9c78. [DOI] [PubMed] [Google Scholar]
- 20.Newman R.J., Bore P.J., Chan L., Gadian D.G., Styles P., Taylor D., Radda G.K. Nuclear magnetic resonance studies of forearm muscle in Duchenne dystrophy. Br. Med. J. 1982;284:1072–1074. doi: 10.1136/bmj.284.6322.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kemp G.J., Taylor D.J., Dunn J.F., Frostick S.P., Radda G.K. Cellular energetics of dystrophic muscle. J. Neurol. Sci. 1993;116:201–206. doi: 10.1016/0022-510x(93)90326-t. [DOI] [PubMed] [Google Scholar]
- 22.Iwata Y., Katanosaka Y., Hisamitsu T., Wakabayashi S. Enhanced Na/H exchange activity contributes to the pathogenesis of muscular dystrophy via involvement of P2 receptors. Am. J. Pathol. 2007;71(5):1576–1587. doi: 10.2353/ajpath.2007.070452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porte-Thomé F. Development of Rimeporide, a sodium-hydrogen exchanger (NHE-1) inhibitor, for patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2020;25(2):S259–S260. doi: 10.1016/j.nmd.2015.06.269. [DOI] [Google Scholar]
- 24.Bkaily G. Na+-H+ exchanger inhibitor prevents early death in hereditary cardiomyopathy. Can. J. Physiol. Pharmacol. 2015;93(11):923–934. doi: 10.1139/cjpp-2015-0107. [DOI] [PubMed] [Google Scholar]
- 25.Guideline on the clinical investigation of medicinal products for the treatment of Duchenne and Becker muscular dystrophy 17 December 2015 1 EMA/CHMP/236981/2011, Corr. 1.
- 26.Assarsson E. Homogenous 96-Plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9(4) doi: 10.1371/journal.pone.009519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayoglu B. Affinity proteomics within rare diseases: a BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol. Med. 2014;6(7):918–936. doi: 10.15252/emmm.201303724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beal S.L., Sheiner L.B., Boeckmann A.J., Bauer R.J. Icon Development Solutions; Ellicott City Maryland, USA: 2020. NONMEM Users Guides. (Eds. 1989-2011) [Google Scholar]
- 29.Bergstrand M., Hooker A.C., Wallin J.E., Karlsson M.O.K. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;(2):143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCulloch C.E., Searle S.R., Neuhaus J.M. John Wiley & Sons; 2008. Generalized, Linear, and Mixed Models. [DOI] [Google Scholar]
- 31.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995;57(1):289–300. [Google Scholar]
- 32.Creutzfeldt W., Lamberts R. Is hypergastrinaemia dangerous to man? Scand. J. Gastroenterol. 1991;26(suppl. 180):179–191. doi: 10.3109/00365529109093198. [DOI] [PubMed] [Google Scholar]
- 33.Hathout Y. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. PNAS. 2015;112(23):7153–7158. doi: 10.1073/pnas.1507719112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spitali P. Tracking disease progression non‐invasively in Duchenne and Becker muscular dystrophies. J. Cachexia Sarcopenia Muscle. 2020;9(4):715–726. doi: 10.1002/jcsm.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arpan I. Examination of effects of corticosteroids on skeletal muscles of boys with DMD using MRI and MRS. Neurology. 2014;83(11) doi: 10.1212/WNL.0000000000000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barton E.R., Morris L., Musaro A., Rosenthal N., Sweeney H.L. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J. Cell Biol. 2002;157(1):137–148. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eilers W., Crossey P., Foster K. Insulin-like growth factor binding protein 1 knockout increases free serum IGF-1 and enhances skeletal muscle mass and force in vivo. Neuromuscul. Disord. 2020;24(5):S184–S316. doi: 10.1016/j.nmd.2015.06.124. [DOI] [Google Scholar]
- 38.Gehrig S.M., Ryall J.G., Schertzer J.D., Lynch G.S. Insulin-like growth factor-I analogue protects muscles of dystrophic mdx mice from contraction-mediated damage. Exp. Physiol. 2008;93(11):1190–1198. doi: 10.1113/expphysiol.2008.042838. [DOI] [PubMed] [Google Scholar]
- 39.Schertzer J., Ryall J., Lynch G. Systemic administration of IGF-I enhances oxidative status and reduces contraction-induced injury in skeletal muscles of mdx dystrophic mice. Am. J. Physiol. Endocrinol. Metab. 2006;291(3):499–505. doi: 10.1152/ajpendo.00101.2006. [DOI] [PubMed] [Google Scholar]
- 40.Yeves A.M., Burgos J.I., Medina A.J., Villa‐Abrille M.C., Ennis I.L. Cardioprotective role of IGF1 in the hypertrophied myocardium of the spontaneously hypertensive rats: a key effect on NHE‐1 activity. Acta Physiol. 2018;224(2) doi: 10.1111/apha.13092. [DOI] [PubMed] [Google Scholar]