

Abstract
Amplicons are helper-dependent herpes simplex virus type 1 (HSV-1)-based vectors that can deliver very large, foreign DNA sequences and, as such, are good candidates for both gene delivery and vaccine development. However, many studies have shown that innate immune responses induced by virus vectors can play a significant role in the control of transgenic expression and in the induction of inflammatory responses. Furthermore, amplicons are very interesting tools to study innate cellular responses elicited by entry of HSV-1 particles in the absence of any virus gene expression. For these reasons, in this study we characterized the innate antiviral response established in human fibroblasts of limited passage (HFFF-2) infected by amplicons. Our results indicate that infection with amplicons triggered an interferon (IFN)-regulatory factors 3 and 7 (IRF3/7)-dependent antiviral response, rendered the cells resistant to vesicular stomatitis virus infection and induced significant changes in the pattern of cellular gene expression, including the upregulation of Toll-like receptor 3 (TLR3), IRF7 and IFN-stimulated genes (ISGs). In contrast, we observed only a mild and contained type I IFN response in infected cells. Amplicon infection induced nuclear translocation and subsequent degradation of IRF3, without hyperphosphorylation of the protein. Inhibition of endosome-resident TLR signalling by blocking lysosome maturation or the knockdown of TLR3 and 4 did not abolish the cellular response to amplicons, whereas knockdown of IRF3 and 7 inhibited ISG and IFN-β expression severely. Therefore, our results confirm the existence of TLR-independent, IRF3/7-dependent activation pathways triggered by HSV-1 particles in human fibroblasts.
INTRODUCTION
Herpes simplex virus type 1 (HSV-1)-based amplicons are helper-dependent vectors (Spaete & Frenkel, 1982) that can convey up to 150 kbp of foreign DNA to a wide range of cell types, making these amplicons very promising gene-delivery tools (Wade-Martins et al., 2003). It is possible to prepare amplicon stocks that are free of helper particles (Saeki et al., 2001) or that bear <1 % replication-defective helper particles (Zaupa et al., 2003), which are candidates for gene therapy and vaccine applications (reviewed by Cuchet et al., 2007). However, previous studies have shown that innate antiviral responses induced by these vectors can reduce the efficacy of transgenic expression and cause adverse inflammatory reactions in mice (Olschowka et al., 2003; Suzuki et al., 2007). In addition to their applications as gene-delivery vectors, amplicons are powerful tools to investigate HSV-1 functions (Hodge & Stow, 2001; Porter & Stow, 2004; Sourvinos & Everett, 2002). As amplicons carry no virus genes, they can be used to study very early interactions between HSV-1 particles and host cells, including the innate cellular responses elicited by virus entry, thus avoiding the requirement to use virus mutants, chemicals or UV inactivation of virus particles. UV treatment of the virus may disrupt the protein structure of the particles, affecting entry, and may cross-link virus proteins and nucleic acids, affecting nuclear release of the genome. Amplicons, in contrast, are able to penetrate cells normally and target their genome to the nucleus, and are transcriptionally active. Furthermore, the amplicon genome can be engineered to express selected virus or cellular proteins, potentially affecting early virus–host interactions as we have shown previously (Cuchet et al., 2005), thereby allowing analysis of how these functions modulate the cellular responses. In this study, we characterized the innate immune response induced by green fluorescent protein (GFP)-expressing amplicons in human fibroblasts.
Type I interferon (IFN-α/β), IFN-stimulated genes (ISGs) and pro-inflammatory cytokines represent a critical first barrier that limits virus expression and replication and provides signals for the development of the adaptive branch of the immune response (Kadowaki et al., 2000; Randall & Goodbourn, 2008). This innate response is initiated by pattern-recognition receptors (PRRs), among which are the family of membrane-bound Toll-like receptors (TLRs) and the RNA helicases retinoic acid-inducible gene (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) (Kawai & Akira, 2006a). The cytoplasmic B-DNA sensor DAI was identified as an important molecule in the induction of innate responses to DNA viruses (Takaoka et al., 2007), although recent studies argue against a role of DAI in the recognition of bacterial or cytosolic DNA (Lippmann et al., 2008). PRRs are activated by molecular structures presented on virus particles or by virus gene products, leading to the activation of IFN-regulatory factors (IRFs), NF-κB and AP-1 transcription factors (Kawai & Akira, 2006b; Mogensen & Paludan, 2005). Activated IRF3 and/or IRF7 dimers form a complex with the transcriptional co-activators CBP/p300, bind IFN-stimulated response elements at the promoters of some ISGs (Grandvaux et al., 2002) and, together with the NF-κB and AP-1 transcription factors, bind the promoters of the IFN-α and -β genes, resulting in induction of the immediate-early phase of the IFN pathway (Guo et al., 2000; Schafer et al., 1998; Servant et al., 2003; Yang et al., 2004). Binding of secreted type I IFNs to their cognate receptor (IFNAR) activates the Jak/STAT pathway, resulting in the transcription of a multitude of ISGs that are involved in the inhibition of virus replication, such as PKR, oligo(A) synthetase/RNase L and Mx (Imada & Leonard, 2000; Leonard, 2001; Samuel, 2001).
HSV-1 overcomes this cellular response through the inhibition of IFN and ISG expression by the immediate-early proteins ICP0 (Eidson et al., 2002; Everett et al., 2008; Lin et al., 2004; Melroe et al., 2004; Mossman et al., 2001) and ICP27 (Johnson et al., 2008; Melchjorsen et al., 2006), besides the virus host shutoff protein Vhs (Chee & Roizman, 2004; Smiley, 2004; Yokota et al., 2001). Other virus proteins like γ34.5 and US11 also confer resistance to the IFN-mediated cellular response (He et al., 1997; Mossman & Smiley, 2002; Mulvey et al., 2004). In addition, recent studies suggested that HSV-1 mutants lacking the γ34.5 gene are unable to inhibit the autophagy pathway of cellular recognition of viruses (Orvedahl et al., 2007).
HSV-1 induces the innate immune response through TLR-dependent and -independent pathways, involving TLR2 and 9, as well as the mitochondrial antiviral signalling protein (MAVS)/IFN-β promoter stimulator 1 (IPS-1) and member(s) of the phosphoinositide 3-kinase family (Hochrein et al., 2004; Noyce et al., 2006; Rasmussen et al., 2007, 2009). Two independent groups have shown that UV inactivation of HSV-1 fails to activate fully the type I IFN pathway in murine embryo fibroblasts (MEFs) and in vivo (Paladino et al., 2006; Rasmussen et al., 2007). We extended these studies by examining the signal-transduction pathways that are activated following entry of amplicons carrying no virus genes into human fibroblasts. Our results demonstrated that infection by amplicons, but not by helper or wild-type virus, resulted in the establishment of an IRF3/7-dependent antiviral state in these fibroblasts. Amplicon infection induced nuclear translocation and subsequent degradation of IRF3, without the hyperphosphorylation of the protein. These data confirm that IRF3 activation by HSV-1 particles results in robust ISG mRNA induction without the hyperphosphorylation of IRF3 (Collins et al., 2004). In addition, we concluded that TLRs were not involved in the initiation of the antiviral response to amplicons in human fibroblasts, as demonstrated (i) by the lack of effect of the inhibition of endosome-resident TLR signalling by blocking the maturation of endosomes to lysosomes and (ii) through knockdown of TLR3 and 4 expression by RNA interference (RNAi), thus supporting and extending previous studies using MEFs derived from transgenic mice (Paladino et al., 2006).
METHODS
Cells and viruses.
Vero 7b (Krisky et al., 1998), TE-CRE-GRINA129 (Zaupa et al., 2003) and Gli36 (Kashima et al., 1995) cells and human fetal foreskin fibroblasts (HFFF-2) (European Collection of Cell Cultures) were propagated in Dulbecco's minimum essential medium (Invitrogen) supplemented with 10 % fetal bovine serum (FBS) (Invitrogen), penicillin (100 U ml−1) and streptomycin (100 μg ml−1) (Invitrogen). Cells were infected at indicated m.o.i.s in 199 medium (Lonza) supplemented with 1 % FBS. HSV-1 17 syn+ and HSV-1 17 syn+-derived ΔICP4 and Cre recombinase-sensitive HSV-1-LaLΔJ virus were grown and titrated in Vero and Vero 7b cells, respectively. Vesicular stomatitis virus (VSV) and Newcastle disease virus (NDV) strain LaSota were kind gifts from from Fabian Wild (Institut Pasteur de Lyon, Lyon, France) and Eliette Bonnefoy (CNRS-UPR 2228, Paris, France), respectively.
Amplicon vector preparation.
Amplicon plasmid pA-(GFP), in which GFP expression is driven by the HSV-1 IE4/5 promoter (Cuchet et al., 2005), was used for amplicon preparation as described by Zaupa et al. (2003), using Vero 7b and TE-CRE-GRINA129 cells. Amplicon vectors were concentrated from supernatant of infected TE-CRE-GRINA129 cells, washed with PBS and resuspended in Optimem Plus Glutamax medium (Invitrogen). Amplicon GFP-transducing units were titrated in Gli36 cells and helper virus (p.f.u.) was titrated in Vero 7b cells. The ratio of amplicon to helper virus ranged from 1000 : 1 to 300 : 1.
Western blot analysis.
For preparation of whole-cell extracts, cells were washed twice and harvested in cold PBS, followed by centrifugation at 200 g for 3 min at 4 °C. Cell pellets were resuspended in whole-cell extract buffer [20 mM HEPES (pH 7.4), 100 mM NaCl, 10 mM β-glycerolphosphate, 0.2 % Triton X-100, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM PMSF, 2 mM dithiothreitol and 1× protease-inhibitor cocktail (Sigma)], lysed on ice for 15 min and centrifuged for 10 min at 13 000 g at 4 °C. Extract concentrations were determined with a Bradford assay (Bio-Rad) and 20 μg total cell proteins was separated on 7.5 % polyacrylamide gels, transferred to nitrocellulose and probed with antibodies specific to IRF3 (BD Biosciences, cat. no. 550428), ICP0 (obtained from Dr Roger Everett, Institute of Virology, University of Glasgow, Glasgow, UK) and β-actin (Sigma, cat. no. A5060).
Indirect immune fluorescence.
Cells seeded on glass coverslips were washed twice with PBS, fixed [paraformaldehyde 4 % (v/v), sucrose 2 % (w/v) in PBS], permeabilized (PBS, 0.2 % Triton X-100) and probed with antibodies specific for IRF3 (BD Biosciences, cat. no. 550428), EEA1 (Abcam, ab 2900) or Rab7 (Sigma, cat. no. R4779), followed by Alexa 555-conjugated goat anti-mouse or anti-rabbit (Invitrogen) in antibody dilution buffer (PBS, 0.5 % FBS, 3 mg BSA ml−1, 0.1 % Triton X-100). The coverslips were washed with washing buffer (PBS, 0.5 % FBS, 0.1 % Triton X-100), followed by PBS and distilled water, dried and mounted with Vectashield H-1500 (Vector Laboratories).
RNA extraction and real-time quantitative RT-PCR (qRT-PCR).
Total cellular RNA was harvested with RNAII Nucleospin (Macherey-Nagel), followed by a DNase I step using DNAfree (Ambion). One microgram of total RNA was reverse-transcribed by using random primers (Promega) and RevertAid H− Moloney murine leukemia virus reverse transcriptase (Fermentas). PCR was subsequently performed using primers (see Supplementary Table S1, available in JGV Online). Real-time PCR was performed with Quantitect SYBR green PCR mix (Qiagen) according to the manufacturer, with a Roche LightCycler. Results were analysed by using the internal standard-curve method. The expression values for each gene were normalized to hypoxanthine phosphoribosyltransferase 1 (HPRT) or 18S rRNA expression values quantified in parallel. The bars represent sd of at least three independent experiments.
pI : C, CpG and NH4Cl treatment.
Cells were treated with polyinosinic : polycytidilic acid (pI : C; Sigma) added to culture medium to a final concentration of 100 μg ml−1, or fluorescein isothiocyanate (FITC)–ODN 2006 (InvivoGen) at a final concentration of 1 μM for the times indicated. For the inhibition of endosome maturation, NH4Cl (Sigma) to a final concentration of 20 mM was added to cells 1 h prior to infection.
RNAi treatment.
For the knockdown of specific RNA targets, cells were transfected with 100 pmol Stealth RNAi (Invitrogen) (see Supplementary Table S2, available in JGV Online) in Optimem Plus Glutamax medium (Invitrogen), using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's protocol. Seventy-two hours later, cells were infected by amplicons at an m.o.i. of 5 and, 18 h post-infection (p.i.), total RNA was extracted.
RESULTS
Infection with HSV-1 amplicons results in the establishment of an antiviral state in HFFF-2 cells, showing ISG expression and IFN-β secretion
Amplicon particles are HSV-1 particles containing, in the place of the virus genome, a concatemeric, head-to-tail, double-stranded (ds) DNA molecule derived from the ‘amplicon plasmid’. This plasmid contains the origin of virus DNA replication (OriS) and ‘a’ packaging sequences of HSV-1, the essential bacterial sequences for propagation in Escherichia coli, and desired transgene cassettes. The replication and packaging of the amplicon plasmid in HSV-1 particles rely on virus proteins provided in trans by the replication-defective HSV-1-LaLΔJ virus, which does not express the ICP4, γ34.5 or gC proteins (Zaupa et al., 2003). In addition, this virus contains a single ‘a’ packaging sequence, flanked by loxP sites, inserted in the gC coding region. This single packaging sequence is excised during the production of the amplicon vectors in a cell line expressing both ICP4 and Cre recombinase (Zaupa et al., 2003), allowing high-titre amplicon production, contaminated only with 0.1–1.0 % defective helper particles.
We investigated the innate antiviral response established in human fibroblasts of limited passage (HFFF-2) infected by the amplicon vector system developed in our laboratory (Zaupa et al., 2003). Infection with 10 and 1 transducing units (TU) of amplicons per cell conferred resistance to VSV infection [Fig. 1a(i, ii)], similarly to IFN-β pretreatment [Fig. 1a(iv)]. In contrast, amplicon infection at low m.o.i. (0.1 TU per cell) did not protect cells from the VSV-induced cytopathic effect [Fig. 1a(iii)]. The inhibition of VSV replication in the amplicon pre-infected cells suggested that an m.o.i.-dependent antiviral response was in place, indicating that amplicons, similarly to UV-inactivated HSV-1, trigger innate antiviral responses. Amplicon infection also resulted in cytokine secretion, as demonstrated by VSV plaque-reduction assays on Vero cells conditioned with the culture supernatants of infected HFFF-2 cells. Cytokine secretion was detectable only in the culture supernatants of the cells infected by amplicons at high m.o.i.s (Fig. 1b). Parallel infection with NDV stimulated a much more robust IFN-β response, earlier than the amplicon infection and at lower m.o.i. (Fig. 1b).
Fig. 1.

Amplicon infection of HFFF-2 cells inhibits VSV replication and induces cytokine secretion and IFN-β and ISG expression. (a) HFFF-2 cells were infected with 10 (i), 1 (ii) or 0.1 (iii) TU amplicons per cell, treated by 1000 U IFN-β ml−1 (iv) or left untreated (vi) and, 8 h later, infected by 500 p.f.u. VSV or mock-infected (NI; v). (b) Supernatants of HFFF-2 cells infected with 10 and 0.1 TU per cell or p.f.u. per cell of amplicons or NDV, respectively, collected at 8 and 24 h p.i., were added to Vero cells for 16 h, followed by infection with 200 p.f.u. VSV. Vero cells were treated with 1000 U IFN-β ml−1 and supernatant of mock-infected HFFF-2 cells as positive and negative controls, respectively. (c) ISG54 and ISG56 (i), IFN-β (ii) and IRF7 (iii) mRNA levels were quantified by qRT-PCR from HFFF-2 cells infected with 10 TU amplicons per cell or 10 p.f.u. NDV per cell for 18 h, normalized to HPRT and represented as percentages relative to NDV.
The levels of ISG54, ISG56, IRF7 and IFN-β mRNAs, relative to the HPRT mRNA, were compared by qRT-PCR in cells infected with amplicon vectors or NDV. Amplicon infection induced the ISG54 and 56 mRNAs, which reached approximately 40 and 10 %, respectively, of the levels induced by NDV infection [Fig. 1c(i)]. On the other hand, NDV infection induced 10 000× more IFN-β mRNA than in the mock-infected cells, whereas amplicon infection increased IFN-β mRNA by approximately 100-fold [Fig. 1c(ii)]. This difference in IFN-β mRNA levels was confirmed with IFN-β ELISA (data not shown) and was consistent with the results of the VSV plaque-reduction experiments presented above.
Comparison of the levels of IRF7 mRNA in amplicon- and NDV-infected HFFF-2 cells showed that NDV and amplicons increased IRF7 expression by 100- and 50-fold, respectively, whereas in non-infected HFFF-2 cells, IRF7 showed limited expression [Fig. 1c(iii)]. IRF7 participates in the amplification phase of IFN-β expression and is important for IFN-β secretion in fibroblasts (Yang et al., 2004). Therefore, our results suggest that the low IFN-β expression by the amplicon-infected cells is not due to impaired induction of IRF7 expression.
As the amplicons used in this study contained 0.1–1.0 % defective helper particles, we tested the effect of small levels of HSV-1-LaLΔJ in the antiviral response. To this end, HFFF-2 cells were infected with 0.1 p.f.u. HSV-1-LaLΔJ per cell, which corresponded to the p.f.u. of HSV-1-LaLΔJ added to the cells during infection with 10 TU amplicon vectors per cell. We found neither significant induction of ISGs (Supplementary Fig. S1, available in JGV Online) nor IFN-β expression and inhibition of VSV replication (data not shown) in HFFF-2 cells infected with 0.1 p.f.u. HSV-1-LaLΔJ per cell. Therefore, the response of HFFF-2 cells to amplicon infection demonstrated high induction of ISG mRNAs and very low levels of IFN-β secretion, and this response was not due to the low levels of helper contamination.
IRF3 activation in amplicon- and helper-infected HFFF-2 cells
The low levels of IFN-β expression induced by amplicon infection, compared with NDV and VSV infection, raised the question of the state of IRF3 activation in amplicon-infected cells. The activation of IRF3 was therefore examined in the infected cells. VSV and NDV infections served as positive controls for the detection of IRF3 nuclear translocation. The monoclonal antibody used in this study did not recognize IRF3 in the untreated cells and, upon VSV or NDV infections, nuclear staining became intense in the majority of the cells [Fig. 2a(ii); Fig. 6a(iv)]. Amplicon infection at an m.o.i. of 10 TU per cell resulted in the nuclear accumulation of IRF3 in 30–50 % of cells [Fig. 2a(i); Fig. 6a(ii)]. Interestingly, infection with the replication-defective HSV-1-LaLΔJ helper virus at the same m.o.i. also induced the nuclear accumulation of IRF3 [Fig. 2a(iii)]. Infection with 0.1 p.f.u. HSV-1-LaLΔJ per cell resulted in the nuclear accumulation of IRF3 in <5 % of cells. However, as expected, HSV-1-LaLΔJ, like wild-type HSV-1 infections, did not stimulate ISG expression (Supplementary Fig. S1). This suggested that HSV-1-LaLΔJ induced IRF3 activation, but that IRF3 transcriptional activity was inhibited, as described previously (Lin et al., 2004).
Fig. 2.

IRF3 activation in infected HFFF-2 cells. (a) IRF3 localization in HFFF-2 cells infected with 10 TU amplicons per cell (i), 10 p.f.u. VSV per cell (ii), 10 p.f.u. HSV-1-LaLΔJ per cell (iii) or not infected (NI; iv) for 7 h. (b) IRF3, β-actin and ICP0 Western blot analysis of HFFF-2 cells infected as described in (a) and collected at 3, 8 and 24 h p.i.
Examination of IRF3 by SDS-PAGE for the detection of slower-migrating phosphorylated forms showed clear differences in the IRF3 electrophoretic profile between the amplicon- and VSV-infected cells. VSV infection resulted in a shift of the IRF3 protein to a slower-migrating form (Fig. 2b, VSV, 8 h), followed by the disappearance of the activated protein (Fig. 2b, VSV, 24 h). In contrast, in amplicon-infected cells, only a fraction of IRF3 shifted to moieties of higher molecular mass (Fig. 2b, Amp, 8 h). However, the levels of IRF3 decreased significantly by 24 h p.i. (Fig. 2b, Amp, 24 h). HSV-1-LaLΔJ infection also failed to induce hyperphosphorylation of IRF3 (Fig. 2b, LaLΔJ, 8 and 24 h). Therefore, amplicon infection resulted in the activation of IRF3, as demonstrated by nuclear translocation and subsequent degradation, but without full hyperphosphorylation. The lack of IRF3 hyperphosphorylation could explain the low levels of IFN-β expression by the amplicon infection compared with NDV and VSV infections.
HSV-1 amplicon infection results in an IRF3/7-dependent induction of ISGs and IFN-β
Our results showed that amplicons induced a distinct IRF3 activation, followed by high expression of IRF7, but low levels of IFN-β. Therefore, we tested whether ISG induction by the amplicons was IRF3- and/or IRF7-dependent, as observed previously for HSV-1. Transfection with IRF3-specific small interfering (si) RNA resulted in >50 % downregulation of the basal levels of IRF3 mRNA in uninfected HFFF-2 cells and, upon infection, a 60 % drop in IRF3 mRNA relative to the cells treated with control siRNA (Fig. 3a). IRF3 mRNA downregulation resulted in a 60 % reduction of ISG54 and IFN-β mRNAs and only a 30 % reduction of IRF7 mRNA. As our transient siRNA knockdown protocol did not result in 100 % silencing of the targeted gene, the remaining IRF3 could still cause enough IRF7 induction, possibly allowing IRF7 to substitute for IRF3 in the induction of ISGs. For this reason, we proceeded to the double knockdown of IRF3 and IRF7, which resulted in >70 % downregulation of basal IRF3 and IRF7 mRNA levels in the uninfected cells, in comparison to transfection with a negative siRNA control (Fig. 3c). IRF3 and IRF7 mRNA levels decreased by >80 and >90 %, respectively, in the amplicon-infected cells. The knockdown of IRF7 alone did not affect the induction of ISG54, ISG56 and IFN-β mRNAs by amplicon infection (Fig. 3b). The double knockdown of IRF3/7 resulted in a greater inhibition of the induction of ISG54, ISG56 and IFN-β mRNAs by amplicon infection, compared with single knockdown of IRF3 in HFFF-2 cells (Fig. 3c). Hence, we confirmed that the ISG upregulation in HSV-1 amplicon-infected HFFF-2 cells was IRF3/7-dependent.
Fig. 3.

IRF3/7 knockdown abolishes ISG and IFN-β induction by amplicons. HFFF-2 cells were transfected by siRNAs specific for IRF3 (a), IRF7 (b) or both (c) and infected with 5 TU amplicons per cell for 18 h or mock-infected (NI). IRF3, IRF7, ISG54, ISG56 and IFN-β mRNA levels were quantified by qRT-PCR, normalized to 18S rRNA and represented as percentages relative to the control siRNA (csi)-treated/amplicon-infected sample (csi-Amp).
Induction of ISGs and IFN-β by HSV-1 amplicons in human fibroblasts is independent of TLRs 3, 4, 7 and 9
Previous studies with UV-inactivated HSV-1 in human fibroblasts showed that ISG56 expression was independent of TLR or RIG-I signalling (Paladino et al., 2006). However, in those studies, UV-inactivated HSV-1 did not induce IRF3 phosphorylation, IRF3 nuclear accumulation was not demonstrated and there was no IFN-β expression, in contrast to our results. Therefore, due to the differences between amplicons and UV-inactivated HSV-1, we decided to test whether the innate response induced by amplicons was TLR-independent. Initially, we analysed HFFF-2 cells by RT-PCR for the expression of TLRs 3 and 4, which activate IRF3, and TLRs 7 and 9, which signal through IRF7. We detected expression of TLR3 and 4, but not of TLR7 or 9, by RT-PCR (Fig. 4a). TLR3 expression was inducible following amplicon infection, whereas the levels of TLR4 were high in both untreated and infected cells (Fig. 4b). In order to verify the RT-PCR results, we also tested the response of HFFF-2 cells to TLR9- and TLR3-specific ligands, CpG oligodeoxynucleotides and pI : C, respectively. FITC-labelled CpG–DNA added extracellularly in the culture medium co-localized with early endosome antigen 1 (EEA1) and later with Rab7 protein, confirming the entry of CpG–DNA into the early and late endosome compartments [Fig. 5a(i, ii)]. However, no induction of ISG54 mRNA (Fig. 4c) or IRF3 activation (data not shown) was detectable. In contrast, extracellular addition of pI : C to the cells caused IRF3 nuclear translocation [Fig. 5b(i)] and upregulation of ISG 54 mRNA (Fig. 4c). Therefore, these results confirmed the absence of TLR9 and the presence of TLR3 in HFFF-2 cells.
Fig. 4.

TLR3 and 4 are highly expressed in HFFF-2 cells, although not involved in the recognition of amplicons. (a) TLR3, 4, 7 and 9 were detected by RT-PCR from genomic DNA (lanes 1), mRNA (lanes 2) or negative cDNA control (lanes 3). (b) TLR3, 4 and β-actin mRNA levels were quantified by qRT-PCR from HFFF-2 cells infected with 10 TU amplicons per cell or mock-infected (NI) for 18 h, normalized to HPRT and represented as percentages relative to the amplicon-infected sample. (c) ISG54 mRNA levels were quantified from HFFF-2 cells treated with pI : C and CpG oligodeoxynucleotides for 18 h by qRT-PCR, normalized to HPRT and represented as percentages relative to the pI : C-treated sample. (d, e) HFFF-2 cells were transfected by siRNAs specific for TLR3 or TLR4 and infected with 5 TU amplicons per cell for 18 h or mock-infected (NI). TLR3, 4, ISG54, ISG56 and IFN-β mRNA levels were quantified by qRT-PCR, normalized to 18S rRNA and represented as percentages relative to the control siRNA (csi)-treated/amplicon-infected sample.
Fig. 5.
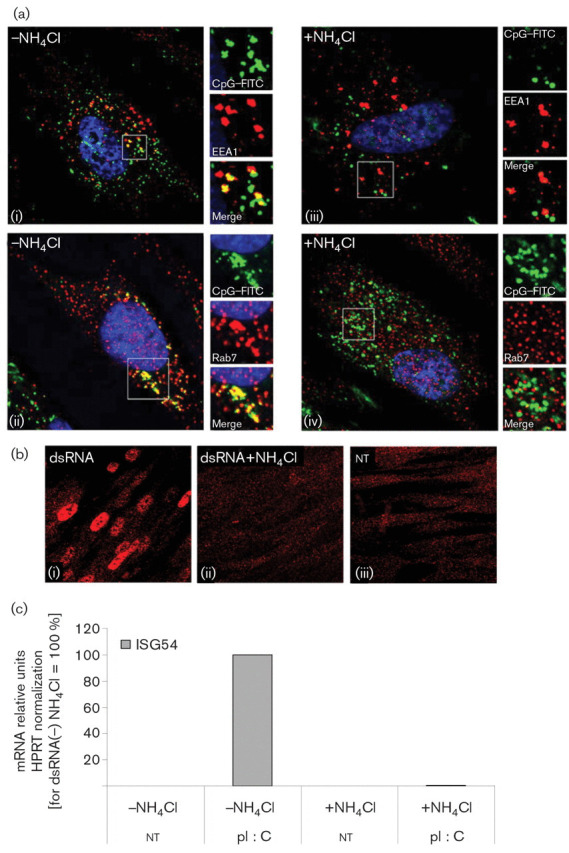
HFFF-2 cells do not respond to CpG oligodeoxynucleotides and the response to pI : C is dependent on endosome maturation. (a) Co-localization of CpG–FITC added extracellularly with EEA1 (i, iii; 30 min) and Rab7 (ii, iv; 2 h) with or without NH4Cl treatment. (b) IRF3 localization in HFFF-2 cells following treatment with pI : C for 6 h, with or without NH4Cl. nt, Not treated with pI : C. (c) ISG54 mRNA levels following treatment with pI : C for 18 h were quantified by qRT-PCR, normalized to HPRT and represented as percentages relative to pI : C (−) NH4Cl.
Next, we tested the effect of TLR3 and 4 knockdowns on the induction of ISG54, 56 and IFN-β mRNAs by amplicon infection. TLR3- and -4-specific siRNAs reduced the respective basal mRNA levels by >50 % relative to the control siRNA in uninfected HFFF-2 cells. The TLR3 and 4 reductions were more pronounced in the amplicon-infected cells, where it reached 80 % relative to the control (Fig. 4d, e). However, neither TLR3 nor 4 RNAi silencing inhibited the induction of ISG54, 56 or IFN-β mRNAs following amplicon infection (Fig. 4d, e). Therefore, our results showed a TLR3-, TLR4- and TLR9-independent induction of the innate antiviral response following HSV-1 amplicon infection.
Inhibition of endosome maturation into lysosomes does not inhibit IRF3 activation and ISG mRNA upregulation by HSV-1 amplicon infection
Finally, we investigated the possible involvement of lysosome-related signalling that could lead to the activation of IRF7 and the induction of ISGs following amplicon infection. In addition, recent studies suggested that HSV-1 mutants lacking the γ34.5 gene could undergo degradation in the autolysosome (Orvedahl et al., 2007). We inhibited endosome acidification and maturation into lysosomes, which consequently resulted in the inhibition of endosome-associated TLR3, 7, 8 and 9 signalling (de Bouteiller et al., 2005; Rutz et al., 2004). NH4Cl treatment inhibited endocytosis, as demonstrated by the lack of co-localization of FITC-labelled CpG–DNA with the EEA1- and Rab7-positive early and late endosomes, respectively [Fig. 5a(iii, iv)]. In addition, treatment of the HFFF-2 cells with NH4Cl inhibited the TLR3-dependent activation of IRF3 and induction of ISG54 following treatment with pI : C (Fig. 5b, c).
We next examined the response of HFFF-2 cells to amplicons in the presence of NH4Cl in comparison to VSV and NDV. Both viruses signal through TLR-dependent and -independent pathways. However, VSV enters cells in a pH-dependent mechanism and, therefore, NH4Cl treatment inhibits VSV entry and replication, in contrast to NDV and HSV-1, which enter mainly through a pH-independent mechanism. IRF3 nuclear translocation and ISG54, 56 and IFN-β upregulation by amplicon infection were not inhibited by NH4Cl [Fig. 6a(ii, vi); Fig. 6b]. IRF3 nuclear translocation by HSV-1-LaLΔJ was also unaffected by the acidification of the endosomes (data not shown), suggesting that amplicons, as well as HSV-1, did not activate IRF3 through the intracellular TLRs residing in endosomes. Similarly to amplicons, IRF3 nuclear translocation induced by NDV was not sensitive to NH4Cl treatment [Fig. 6a(iv, viii); Fig. 6b]. In contrast, NH4Cl clearly inhibited IRF3 activation and ISG and IFN-β mRNA induction by VSV [Fig. 6a(iii, vii); Fig. 6b]. Therefore, inhibition of pH-dependent endocytosis and lysosome maturation did not play any role in the induction of the innate antiviral response to the HSV-1 particle in human fibroblasts, indicating that lysosome-dependent virus-recognition systems such as the TLR-dependent and autophagy-dependent pathways are not responsible for the recognition of HSV-1 particles in the absence of virus gene expression and replication.
Fig. 6.

IRF3 activation and ISG expression are independent of endosome maturation in amplicon-infected HFFF-2 cells in contrast to VSV. (a) IRF3 localization in HFFF-2 cells infected for 6 h with 10 TU amplicons per cell (ii, vi), 10 p.f.u. VSV per cell (iii, vii) or NDV (iv, viii) or not infected (NI; i, v), with or without NH4Cl. (b) ISG54, ISG56 and IFN-β mRNAs in HFFF-2 cells, infected as in (a) for 18 h were quantified by qRT-PCR, normalized to HPRT and represented as percentages relative to NDV −NH4Cl.
DISCUSSION
In this study, we investigated the innate antiviral response established in human fibroblasts of limited passage (HFFF-2) infected by amplicons, both because these vectors are a good model to study very early virus–host interactions and because innate immune responses induced by virus vectors play a significant role in the control of transgenic expression and in the induction of adverse inflammatory responses in vivo (Suzuki et al., 2007). Our results demonstrated that infection by amplicons resulted in the expression of several ISGs and the establishment of an antiviral state in HFFF-2 cells that was mainly mediated through IRF3 and 7. Furthermore, despite the significant upregulation of TLR3 and IRF7 mRNAs, the response to amplicons was independent of TLR signalling. The establishment of the antiviral response observed in the infected fibroblasts was clearly due to the amplicon particles and not to the low amounts of contaminating helper particles, as we demonstrated that the helper virus actually inhibited the induction of ISGs.
Several studies characterizing the induction of innate immune responses by HSV-1, UV-inactivated HSV-1 or HSV-1 mutants with the immediate-early genes deleted in mouse and human fibroblasts agree that IFN-independent ISG expression induced by virus entry is inhibited by certain virus proteins (Eidson et al., 2002; Mossman et al., 2001; Mulvey et al., 2004; Nicholl et al., 2000). There are, however, minor differences regarding the expression of type I IFN. Infection with UV-inactivated HSV-1 resulted in ISG expression, but not the secretion of IFN-β (Paladino et al., 2006; Rasmussen et al., 2007). The lack of type I IFN induction in the above studies was accounted for by the absence of NF-κB activation and the lack of IRF3 nuclear accumulation and hyperphosphorylation (Collins et al., 2004; Paladino et al., 2006). On the other hand, infection with HSV-1 induced low levels of type I IFN expression in a foreskin fibroblast lineage, but not in fetal lung fibroblasts (Preston et al., 2001). In our study, IFN-β secretion from the amplicon-infected fibroblasts was low even at high m.o.i. and correlated with the absence of hyperphosphorylation of IRF3. According to previous studies, the lack of virus replication could account for low IFN-β secretion, due to the absence of recognition of virus replication products and the full activation of IRF3 (Paladino et al., 2006; Rasmussen et al., 2007); however, we cannot exclude the possibility that amplicon particles may also intrinsically inhibit IRF3 hyperphosphorylation by tegument proteins, which may in turn inhibit the induction of high levels of IFN-β.
Interestingly, the ICP4− HSV-1-LaLΔJ virus induced the nuclear accumulation of IRF3 despite the expression of ICP0, which was shown previously to cause the inhibition of IRF3 nuclear accumulation and the faster degradation of activated IRF3 in Sendai virus-infected cells (Melroe et al., 2004, 2007). However, despite the activation of IRF3, ISG mRNA induction was inhibited following HSV-1-LaLΔJ infection, in agreement with previous studies showing that ICP0 and ICP27 expression inhibits IFN-dependent and -independent ISG expression (Eidson et al., 2002; Johnson et al., 2008; Lin et al., 2004).
HSV-1 induces innate immune responses through TLR-dependent and -independent pathways (Hochrein et al., 2004; Noyce et al., 2006; Rasmussen et al., 2007, 2009). TLR9 has been already proposed as a sensor of HSV-1, due to the unmethylated CpG motifs contained in the HSV-1 genome (Hemmi et al., 2000; Hochrein et al., 2004; Krug et al., 2004; Lundberg et al., 2003). Virus glycoproteins or other protein components of the HSV-1 particle could also induce the innate response through interaction with TLR2 (Compton et al., 2003; Kurt-Jones et al., 2004). However, TLR2 activation alone would not activate IRF3 and it was recently shown that dendritic cell-induced maturation by HSV-1 was TLR2-independent (Reske et al., 2008). HSV-1 particles contain small levels of cellular and virus RNAs (Sciortino et al., 2001), and dsRNA molecules are generated during the virus life cycle (Weber et al., 2006), which could also be responsible for IRF3 activation through TLR3-dependent or -independent pathways. TLR4, typically assigned to lipopolysaccharide signalling, which results in IRF3 activation, has also been implicated in the recognition of glycoprotein components of certain viruses, and therefore cannot be excluded (Kopp & Medzhitov, 2003). However, previous studies with UV-inactivated HSV-1 in murine fibroblasts indicated that ISG56 expression was independent of TLR or RIG-I signalling (Paladino et al., 2006).
We examined the expression profile of TLR3, 4, 7 and 9, which may be involved in the activation of IRF3 and IRF7 in HFFF-2 cells. TLR3 mRNA was detected in uninfected fibroblasts and showed a considerable increase following amplicon infection, whereas treatment of HFFF-2 cells with synthetic pI : C dsRNA resulted in IRF3 activation and the increase in expression of ISG54. However, TLR3 knockdown had no effect on the ISG and IFN-β expression induced by amplicon infection, overall suggesting that HSV-1 particles were not recognized by TLR3 in HFFF-2 cells. It has been reported that, although the TLR3 and 4 signalling pathways converge with regard to IRF3 and NF-κB activation (Doyle et al., 2002), there are differences in the mechanism of IRF3 activation (Wietek et al., 2003) resulting, in the case of TLR4, in a less important IFN-β production. TLR4 knockdown in HFFF-2 cells did not inhibit the amplicon-induced ISG expression, indicating that activation of IRF3 by amplicon particles was TLR4-independent. The innate response to amplicons was also independent of the endosomal TLR7, 8 and 9. Therefore, our results based on human primary cells agree with recent data based on mouse fibroblasts, which suggested a TLR-independent pathway of IRF3 activation following HSV-1 entry in the absence of virus gene expression and replication (Noyce et al., 2006; Paladino et al., 2006; Rasmussen et al., 2007, 2009). Furthermore, the results from the prevention of endosome TLR signalling and the inhibition of the endosome maturation to lysosomes also suggest that the recognition of the amplicon vector particles does not occur through an autophagy-related pathway.
There are recent data regarding cytosolic recognition of CpG–DNA that is independent of TLR9, which results in IRF3 and NF-κB activation and a robust induction of type I IFN response in dendritic cells (Stetson & Medzhitov, 2006; Takaoka et al., 2007). However, we have preliminary observations suggesting that the cytosolic DNA sensor DAI (DLM-1/ZBP1) is not expressed in quiescent HFFF-2 cells, but is induced upon infection with amplicons (E. Tsitoura, unpublished data). However, according to the current models of HSV-1 infection, following entry, virus capsids are transported to the nuclear pores from where the virus DNA is released into the nucleus. Amplicon or HSV-1 DNA is thus not expected to be exposed to the cytoplasm. This raises the possibility of a nuclear sensor of incoming DNA that could result in IRF3 activation.
To conclude, our results show clearly that amplicons rendered infected HFFF-2 cells resistant to virus infection and induced significant changes in the pattern of cellular gene expression, including the upregulation of TLR3, IRF7 and ISGs. Overall, however, we observed a mild antiviral response. These observations provide new insights into the interaction of HSV-1 amplicons with human cells, important for the development of more efficient and safer vectors. Regarding the molecular basis underlying the innate responses elicited by HSV-1 particles in human fibroblasts, our results present further experimental support for the notion that the HSV-1-induced antiviral response in fibroblasts is largely TLR-independent.
Supplementary Data
Acknowledgments
This work was supported by grants from the French institutions Association pour la Recherche contre le Cancer (ARC) (no. 3271) and Association Française contre les Myopathies (AFM) (no. 10713). E. T. was supported by a European Commission grant (LSHB-CT-2004-005246). D. C. was supported by Bioviron S. A. R. L. (France).
Footnotes
Supplementary tables showing primers and RNAi reagents used in this study and a supplementary figure are available with the online version of this paper.
†Present address: Centre de Génétique Moléculaire et Cellulaire, CNRS-UMR 5534, Université Claude Bernard Lyon 1, 2nd floor, 16 rue Raphaël Dubois, F-69622 Villeurbanne, France.
References
- Chee, A. V. & Roizman, B. ( 2004. ). Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J Virol 78, 4185–4196.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, S. E., Noyce, R. S. & Mossman, K. L. ( 2004. ). Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 78, 1706–1717.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton, T., Kurt-Jones, E. A., Boehme, K. W., Belko, J., Latz, E., Golenbock, D. T. & Finberg, R. W. ( 2003. ). Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol 77, 4588–4596.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet, D., Ferrera, R., Lomonte, P. & Epstein, A. L. ( 2005. ). Characterization of antiproliferative and cytotoxic properties of the HSV-1 immediate-early ICP0 protein. J Gene Med 7, 1187–1199.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Cuchet, D., Potel, C., Thomas, J. & Epstein, A. L. ( 2007. ). HSV-1 amplicon vectors: a promising and versatile tool for gene delivery. Expert Opin Biol Ther 7, 975–995.[CrossRef] [DOI] [PubMed] [Google Scholar]
- de Bouteiller, O., Merck, E., Hasan, U. A., Hubac, S., Benguigui, B., Trinchieri, G., Bates, E. E. & Caux, C. ( 2005. ). Recognition of double-stranded RNA by human Toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem 280, 38133–38145.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Doyle, S., Vaidya, S., O'Connell, R., Dadgostar, H., Dempsey, P., Wu, T., Rao, G., Sun, R., Haberland, M. & other authors ( 2002. ). IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17, 251–263.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Eidson, K. M., Hobbs, W. E., Manning, B. J., Carlson, P. & DeLuca, N. A. ( 2002. ). Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J Virol 76, 2180–2191.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett, R. D., Parada, C., Gripon, P., Sirma, H. & Orr, A. ( 2008. ). Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82, 2661–2672.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandvaux, N., Servant, M. J., tenOever, B., Sen, G. C., Balachandran, S., Barber, G. N., Lin, R. & Hiscott, J. ( 2002. ). Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol 76, 5532–5539.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J., Peters, K. L. & Sen, G. C. ( 2000. ). Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology 267, 209–219.[CrossRef] [DOI] [PubMed] [Google Scholar]
- He, B., Gross, M. & Roizman, B. ( 1997. ). The γ 134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94, 843–848.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H., Matsumoto, M., Hoshino, K., Wagner, H. & other authors ( 2000. ). A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Hochrein, H., Schlatter, B., O'Keeffe, M., Wagner, C., Schmitz, F., Schiemann, M., Bauer, S., Suter, M. & Wagner, H. ( 2004. ). Herpes simplex virus type-1 induces IFN-α production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A 101, 11416–11421.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge, P. D. & Stow, N. D. ( 2001. ). Effects of mutations within the herpes simplex virus type 1 DNA encapsidation signal on packaging efficiency. J Virol 75, 8977–8986.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imada, K. & Leonard, W. J. ( 2000. ). The Jak-STAT pathway. Mol Immunol 37, 1–11.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Johnson, K. E., Song, B. & Knipe, D. M. ( 2008. ). Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 374, 487–494.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki, N., Antonenko, S., Lau, J. Y. & Liu, Y. J. ( 2000. ). Natural interferon α/β-producing cells link innate and adaptive immunity. J Exp Med 192, 219–226.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima, T., Vinters, H. V. & Campagnoni, A. T. ( 1995. ). Unexpected expression of intermediate filament protein genes in human oligodendroglioma cell lines. J Neuropathol Exp Neurol 54, 23–31.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Kawai, T. & Akira, S. ( 2006a. ). Innate immune recognition of viral infection. Nat Immunol 7, 131–137. [DOI] [PubMed] [Google Scholar]
- Kawai, T. & Akira, S. ( 2006b. ). TLR signaling. Cell Death Differ 13, 816–825.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Kopp, E. & Medzhitov, R. ( 2003. ). Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol 15, 396–401.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Krisky, D. M., Marconi, P. C., Oligino, T. J., Rouse, R. J., Fink, D. J., Cohen, J. B., Watkins, S. C. & Glorioso, J. C. ( 1998. ). Development of herpes simplex virus replication-defective multigene vectors for combination gene therapy applications. Gene Ther 5, 1517–1530.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Krug, A., Luker, G. D., Barchet, W., Leib, D. A., Akira, S. & Colonna, M. ( 2004. ). Herpes simplex virus type 1 activates murine natural interferon-producing cells through Toll-like receptor 9. Blood 103, 1433–1437. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones, E. A., Chan, M., Zhou, S., Wang, J., Reed, G., Bronson, R., Arnold, M. M., Knipe, D. M. & Finberg, R. W. ( 2004. ). Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101, 1315–1320.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard, W. J. ( 2001. ). Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol 73, 271–277.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Lin, R., Noyce, R. S., Collins, S. E., Everett, R. D. & Mossman, K. L. ( 2004. ). The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol 78, 1675–1684.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann, J., Rothenburg, S., Deigendesch, N., Eitel, J., Meixenberger, K., van Laak, V., Slevogt, H., N'Guessan, P. D., Hippenstiel, S. & other authors ( 2008. ). IFNβ responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell Microbiol 10, 2579–2588.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Lundberg, P., Welander, P., Han, X. & Cantin, E. ( 2003. ). Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J Virol 77, 11158–11169.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchjorsen, J., Siren, J., Julkunen, I., Paludan, S. R. & Matikainen, S. ( 2006. ). Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-κB and IRF-3. J Gen Virol 87, 1099–1108.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Melroe, G. T., DeLuca, N. A. & Knipe, D. M. ( 2004. ). Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J Virol 78, 8411–8420.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melroe, G. T., Silva, L., Schaffer, P. A. & Knipe, D. M. ( 2007. ). Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-β induction. Virology 360, 305–321.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen, T. H. & Paludan, S. R. ( 2005. ). Reading the viral signature by Toll-like receptors and other pattern recognition receptors. J Mol Med 83, 180–192.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Mossman, K. L. & Smiley, J. R. ( 2002. ). Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J Virol 76, 1995–1998.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman, K. L., Macgregor, P. F., Rozmus, J. J., Goryachev, A. B., Edwards, A. M. & Smiley, J. R. ( 2001. ). Herpes simplex virus triggers and then disarms a host antiviral response. J Virol 75, 750–758.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey, M., Camarena, V. & Mohr, I. ( 2004. ). Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and γ 134.5 gene products. J Virol 78, 10193–10196.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholl, M. J., Robinson, L. H. & Preston, C. M. ( 2000. ). Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J Gen Virol 81, 2215–2218. [DOI] [PubMed] [Google Scholar]
- Noyce, R. S., Collins, S. E. & Mossman, K. L. ( 2006. ). Identification of a novel pathway essential for the immediate-early, interferon-independent antiviral response to enveloped virions. J Virol 80, 226–235.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olschowka, J. A., Bowers, W. J., Hurley, S. D., Mastrangelo, M. A. & Federoff, H. J. ( 2003. ). Helper-free HSV-1 amplicons elicit a markedly less robust innate immune response in the CNS. Mol Ther 7, 218–227.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Orvedahl, A., Alexander, D., Talloczy, Z., Sun, Q., Wei, Y., Zhang, W., Burns, D., Leib, D. A. & Levine, B. ( 2007. ). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Paladino, P., Cummings, D. T., Noyce, R. S. & Mossman, K. L. ( 2006. ). The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol 177, 8008–8016.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Porter, I. M. & Stow, N. D. ( 2004. ). Replication, recombination and packaging of amplicon DNA in cells infected with the herpes simplex virus type 1 alkaline nuclease null mutant ambUL12. J Gen Virol 85, 3501–3510.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Preston, C. M., Harman, A. N. & Nicholl, M. J. ( 2001. ). Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J Virol 75, 8909–8916.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall, R. E. & Goodbourn, S. ( 2008. ). Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89, 1–47.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Rasmussen, S. B., Sorensen, L. N., Malmgaard, L., Ank, N., Baines, J. D., Chen, Z. J. & Paludan, S. R. ( 2007. ). Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol 81, 13315–13324.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, S. B., Jensen, S. B., Nielsen, C., Quartin, E., Kato, H., Chen, Z. J., Silverman, R. H., Akira, S. & Paludan, S. R. ( 2009. ). Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J Gen Virol 90, 74–78.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reske, A., Pollara, G., Krummenacher, C., Katz, D. R. & Chain, B. M. ( 2008. ). Glycoprotein-dependent and TLR2-independent innate immune recognition of herpes simplex virus-1 by dendritic cells. J Immunol 180, 7525–7536.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Rutz, M., Metzger, J., Gellert, T., Luppa, P., Lipford, G. B., Wagner, H. & Bauer, S. ( 2004. ). Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur J Immunol 34, 2541–2550.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Saeki, Y., Fraefel, C., Ichikawa, T., Breakefield, X. O. & Chiocca, E. A. ( 2001. ). Improved helper virus-free packaging system for HSV amplicon vectors using an ICP27-deleted, oversized HSV-1 DNA in a bacterial artificial chromosome. Mol Ther 3, 591–601.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Samuel, C. E. ( 2001. ). Antiviral actions of interferons. Clin Microbiol Rev 14, 778–809.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, S. L., Lin, R., Moore, P. A., Hiscott, J. & Pitha, P. M. ( 1998. ). Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem 273, 2714–2720.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Sciortino, M. T., Suzuki, M., Taddeo, B. & Roizman, B. ( 2001. ). RNAs extracted from herpes simplex virus 1 virions: apparent selectivity of viral but not cellular RNAs packaged in virions. J Virol 75, 8105–8116.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant, M. J., Grandvaux, N., tenOever, B. R., Duguay, D., Lin, R. & Hiscott, J. ( 2003. ). Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem 278, 9441–9447.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Smiley, J. R. ( 2004. ). Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol 78, 1063–1068.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourvinos, G. & Everett, R. D. ( 2002. ). Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J 21, 4989–4997.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaete, R. R. & Frenkel, N. ( 1982. ). The herpes simplex virus amplicon: a new eucaryotic defective-virus cloning-amplifying vector. Cell 30, 295–304.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Stetson, D. B. & Medzhitov, R. ( 2006. ). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Suzuki, M., Chiocca, E. A. & Saeki, Y. ( 2007. ). Early STAT1 activation after systemic delivery of HSV amplicon vectors suppresses transcription of the vector-encoded transgene. Mol Ther 15, 2017–2026.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Takaoka, A., Wang, Z., Choi, M. K., Yanai, H., Negishi, H., Ban, T., Lu, Y., Miyagishi, M., Kodama, T. & other authors ( 2007. ). DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Wade-Martins, R., Saeki, Y. & Chiocca, E. A. ( 2003. ). Infectious delivery of a 135-kb LDLR genomic locus leads to regulated complementation of low-density lipoprotein receptor deficiency in human cells. Mol Ther 7, 604–612.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Weber, F., Wagner, V., Rasmussen, S. B., Hartmann, R. & Paludan, S. R. ( 2006. ). Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80, 5059–5064.[CrossRef] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietek, C., Miggin, S. M., Jefferies, C. A. & O'Neill, L. A. ( 2003. ). Interferon regulatory factor-3-mediated activation of the interferon-sensitive response element by Toll-like receptor (TLR) 4 but not TLR3 requires the p65 subunit of NF-kappa. J Biol Chem 278, 50923–50931.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Yang, H., Ma, G., Lin, C. H., Orr, M. & Wathelet, M. G. ( 2004. ). Mechanism for transcriptional synergy between interferon regulatory factor (IRF)-3 and IRF-7 in activation of the interferon-β gene promoter. Eur J Biochem 271, 3693–3703.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Yokota, S., Yokosawa, N., Kubota, T., Suzutani, T., Yoshida, I., Miura, S., Jimbow, K. & Fujii, N. ( 2001. ). Herpes simplex virus type 1 suppresses the interferon signaling pathway by inhibiting phosphorylation of STATs and Janus kinases during an early infection stage. Virology 286, 119–124.[CrossRef] [DOI] [PubMed] [Google Scholar]
- Zaupa, C., Revol-Guyot, V. & Epstein, A. L. ( 2003. ). Improved packaging system for generation of high-level noncytotoxic HSV-1 amplicon vectors using Cre-loxP site-specific recombination to delete the packaging signals of defective helper genomes. Hum Gene Ther 14, 1049–1063.[CrossRef] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.