

Abstract
All mammalian species depend upon the placenta, a transient organ, for exchanges of gases, nutrients, and waste between the mother and conceptus. Besides serving as a conduit for such exchanges, the placenta produces hormones and other factors that influence maternal physiology and fetal development. To meet all of these adaptations, the placenta has evolved to become the most structurally diverse organ within all mammalian taxa. However, commonalities exist as to how placental responses promote survival of the fetus against in utero threats and can alter the trajectory of fetal development, in particular the brain. Increasing evidence suggests that placenta reactions to various in utero stressors may lead to longstanding health outcomes, otherwise considered developmental origin of health and disease (DOHaD) effects. Besides transferring nutrients and gases, the placenta produces neurotransmitters, including serotonin (5-HT), dopamine, norepinephrine/epinephrine, that may circulate and influence brain development. Neurobehavioral disorders, such as autism spectrum disorders (ASD), likely trace their origins back to placental disturbances. This intimate relationship between the placenta and brain has led to coinage of the term, the placenta-brain-axis. This axis will be the focus herein, including how conceptus sex might influence it and technologies employed to parse out effects of placental-specific transcript changes on later neurobehavioral disorders. Ultimately, the placenta might provide a historical record of in utero threats the fetus confronted and a roadmap to understand how placenta responses to such encounters impacts the placental-brain-axis. Improved early diagnostic and preventative approaches may thereby be designed to mitigate such placental disruptions.
Keywords: Pregnancy, trophoblast, neurotransmitters, nutrients, inflammation, neurodevelopment, epigenetics
Introduction
All mammals and marsupials depend upon the placenta for initial survival. While ephemeral, the placenta serves throughout gestation as the primary communication organ for exchange of nutrients, gases, waste, and hormones between the dam and fetus. To meet these varying requirements, evolutionary pressures have resulted in the placenta becoming the most diverse of all organs in mammalian species (Reviewed in (Carter, 2007; Carter & Mess, 2007; Chavatte-Palmer & Guillomot, 2007; Enders & Carter, 2006; Georgiades, Ferguson-Smith, & Burton, 2002). Even though some aspects of placental anatomy and hormones/factors produced may differ across taxa, commonalities exist in the placenta of all species, such as facilitating fetal growth, mechanisms to help maintain homeostasis, and production of factors to safeguard pregnancy maintenance, otherwise termed maternal recognition of pregnancy (R. M. Roberts, Xie, & Mathialagan, 1996).
The placenta must be able to recognize even the potential slightest alteration and respond rapidly to fluctuating in utero environmental changes, such as poor maternal nutrition or obesity, maternal stress, infection/inflammation, or environmental chemicals. It is not clear if the placenta of one sex over the other possesses greater ability to adapt and thereby buffer the fetus to such changes. Should the placenta fail to respond or mount an insufficient response to in utero insults, the fetus may be at risk for developing later diseases, otherwise considered developmental origins of adult health and disease(DOHaD) effects. While many fetal organs depend upon the placenta to respond to in utero environmental challenges and to produce factors promoting fetal growth, the brain is one of the most vulnerable organs to placental disruptions. Many neurobehavioral disorders likely trace their genesis back to pathophysiological changes in the placenta (Green et al., 2015; Lesseur et al., 2014; Marsit, Lambertini, et al., 2012; Paquette et al., 2013; Rosenfeld, 2015). Because of this intimate connection between the placenta and brain, it has resulted in branding of the term, the placenta-brain-axis, which will be the focus herein. The significance of this axis is that if placental changes can guide fetal brain development, then they may also provide mechanistic understanding of the fetal origin of neurobehavioral disorders, such as autism spectrum disorders (ASD), and open up new early diagnostic and treatment avenues.
I will consider common mechanisms by which the placenta can affect fetal brain development, including through production of neurotransmitters and transfer of nutrients to the fetus. To examine how placental gene expression affects later neurobehavioral patterns, conditional knockout (KO) mice have been crucial to our understanding. Examples of such transgenic mouse models and later neurobehavioral alterations exhibited by them will be discussed. The evidence linking human placental diseases, such as fetal growth restriction (FGR) and preeclampsia (PE), and neurobehavioral disorders will be explored. Lastly, we will consider sex as a variable in affecting the placenta-brain-axis.
Placental Production of Neurotransmitters
It has long been recognized that the placenta is a steroid hormone-producing organ. However, placental production of neurotransmitters and its ability to regulate fetal brain development through these factors is assumingly less well-recognized. This is despite the fact that for some neurotransmitters the placenta is likely the sole source during the initial brain formation. In this section, we will consider three neurotransmitters produced by the placenta that can circulate and influence the fetal brain: serotonin (5-HT), dopamine (DA), and epinephrine/norepinephrine. The synthesis and metabolism of these neurotransmitters is shown in Figure 1.
Figure 1.
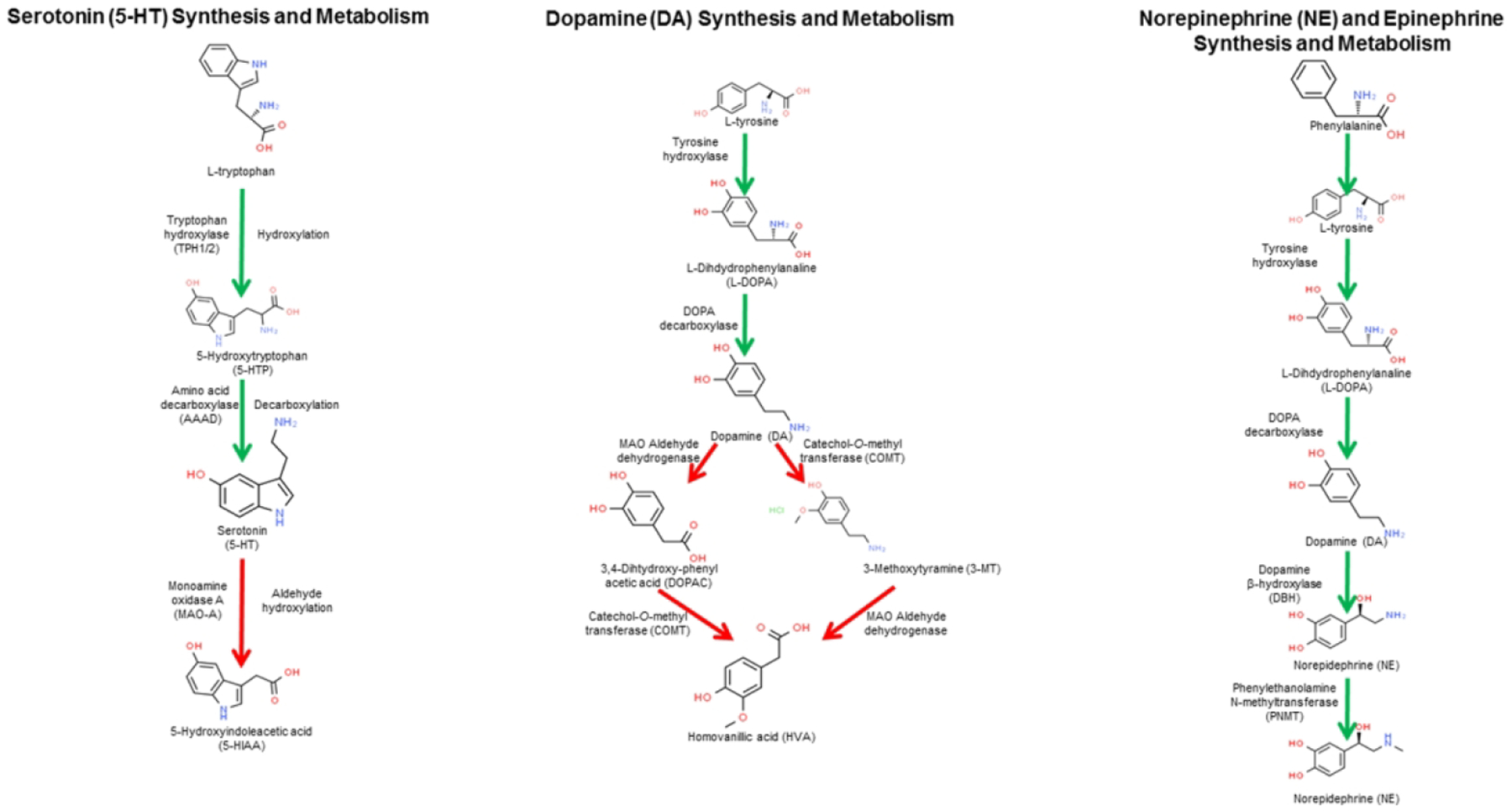
Synthesis and metabolism of 5-HT, DA, and NE/Epinephrine. All three of these neurotransmitters can be synthesized and metabolized in the placenta. In the placenta, these neurotransmitters may induce autocrine and paracrine effects. Active concentrations of these placental-derived neurotransmitters can also circulate and influence fetal brain development. Chemical structures are from www.chemspider.com/. The first panel of this figure has been modified from (Rosenfeld, 2019).
Serotonin:
Serotonin synthesis requires L-tryptophan, which is hydrolyzed by tryptophan hydroxylase (TPH1/2) to 5-hydroxytryptophan (5-HTP). Amino acid decarboxylase (AAAD) converts 5-HTP to serotonin (5-HT). 5-HT is catabolized and assumingly inactivated by monoamine oxidase A (MAO-A) stimulated conversion to hydroxyindoleacetic acid (5-HIAA).
Abundant evidence suggests that the placenta directly synthesizes 5-HT (Bonnin et al., 2011; Bonnin & Levitt, 2011; Huang, Zhang, Di, & Zhang, 1998; Laurent et al., 2017; Muller et al., 2017; Tuteja, Chung, & Bejerano, 2016). In the mouse placenta, the genetic machinery to produce 5-HT increases from embryonic (e) 7.5 to 9.5 (Tuteja et al., 2016). One study, however, suggests that the placenta relies on transport of maternal 5-HT (Kliman et al., 2018). In our recently published studies, we found that exposure to the endocrine disrupting chemicals (EDC), bisphenol A (BPA) and bisphenol S (BPS) lower the total concentrations of 5-HT in the placenta (Mao et al., In Press). Additionally, we localized 5-HT to trophoblast giant cells, which are in contact with the underlying uterine tissue, but the number of giant cells staining immunopositive for 5-HT was reduced with BPA or BPS exposure (Mao et al., In Press). As the potential sole provider of 5-HT during early brain development, placental hyposerotonemia or hyperserotonemia can disrupt early neural programming.
Autism spectrum disorders (ASD) presumably arise due to a range of intrinsic and extrinsic factors, and alterations in placental production of 5-HT has been postulated to increase the risk for ASD (Sato, 2013; Yang, Tan, & Du, 2014), FGR (Ranzil, Ellery, et al., 2019; Ranzil, Walker, et al., 2019). Changes in placental 5-HT production might also increase the likelihood of anxiogenic behaviors (Belmaker, Agam, & Bersudsky, 2008; Hendricks et al., 2003).
One reason the brain may be susceptible to fluctuations in placental 5-HT is due the fact that the placenta is apparently the sole source of this neurotransmitter during early brain development (Bonnin et al., 2011; Bonnin & Levitt, 2011). During fetal brain development, 5-HT stimulates cell division, neuronal migration, cell differentiation, and synaptogenesis (Yang et al., 2014). Hyperserotonemia at this time may cause a negative feedback loop that inhibits 5-HT signaling by potentially suppressing the expression of its cognate receptors, such as 5-HT1A, 5-HT1B, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT4, 5-HT6, and 5-HT7 receptors. Elevated fetal levels of 5-HT may also inhibit oxytocin production by the paraventricular nucleus (PVN) of the hypothalamus but enhance calcitonin gene related peptide (CGRP) in the central nucleus of the amygdala. Both oxytocin and CGRP promote social behaviors and consequently, are implicated in ASD. While elevated concentrations of 5-HT from the placenta can disrupt early brain development, hyposerotonemia may also be problematic as it can impair sensory, motor, and cognitive abilities, collectively leading to ASD or other neurobehavioral disorders (Yang et al., 2014).
Serotonin-reuptake inhibitors (SSRI) are increasingly being prescribed to combat depression in pregnant women (Huybrechts et al., 2013; Mitchell et al., 2011). Besides altering 5-HT concentrations available in the synaptic terminals of the maternal brain, such drugs care likely to be transferred across the placenta and increase extracellular concentrations of 5-HT within the placenta that subsequently circulates to the fetal brain. However, more work is needed to determine the pharmokinetics and potential concentrations of SSRI in the placenta of women prescribed these drugs. Moreover, the full range of placental effects of SSRI remains uncertain but highly relevant given the mounting usage of these drugs to treat depression in pregnant women.
Catecholamines- Dopamine, epinephrine, norepinephrine
Dopamine (DA) is another neurotransmitter that may be produced by the placenta where it can induce direct effects through various DA receptors, including D1 and D2 receptors (Ben-Jonathan & Munsick, 1980; H. J. Kim, Koh, Kang, Paik, & Choi, 2001; M. O. Kim et al., 1997; Mao et al., In Press; Tomogane, Arbogast, Soares, Robertson, & Voogt, 1993; Vaillancourt et al., 1994; Y. Zhu, Zhang, Chen, Liu, & Guo, 2002). Several lines of evidence support the placental as a dopaminergic/adrenergic organ. Copious amounts of DA are found in human amniotic fluid (Ben-Jonathan & Munsick, 1980). One of the primary actions of DA is to inhibit prolactin secretion (Fitzgerald & Dinan, 2008; Grattan, 2015). A factor, presumably DA, produced by a rat trophoblast line inhibits prolactin secretion both in vitro and in vivo (Tomogane et al., 1993). Immunohistochemical analysis of human placenta at various points throughout gestation reveals that during early pregnancy DA and norepinephrine (NE) are localized to placenta cytotrophoblasts but then shift to being expressed primarily by synctiotrophoblast cells during mid and late stages of pregnancy (Y. Zhu et al., 2002). This study also found that the level of NE escalated in cases of pregnancy-induced hypertension syndrome. We recently used to immunohistochemistry to show that trophoblast giant cells of the mouse placenta are positive for DA, but developmental exposure to the EDC, BPA or BPS increases DA immunoreactivity in these cells (Mao et al., In Press). Our recent results found the total amount of DA concentration in the placenta increases with developmental exposure to BPA and BPS (Mao et al., In Press).
Analogous to SERT binding to 5-HT and causing transport and internalization of this neurotransmitter into the trophoblast cells, similar receptors exist for the catecholamines. These include norepinephrine transporter (NET) and extraneuronal monoamine transporter (MET). Both of these receptors are expressed by the placenta (Bottalico et al., 2004). Placentae from PE pregnancies show reduced mRNA expression for NET and MET (Bottalico et al., 2004). The prevailing notion is that such receptors may prevent catecholamine-induced vasoconstriction of placental vasculature, which ensures continuous blood flow to the fetus. Higher levels of noradrenaline are fond in the bloodstream of PE relative to normotensive women (Manyonda et al., 1998). The likely source of this catecholamine is the placenta as greater activity of the rate limiting enzyme, tyrosine hydroxylase, occurs in placental tissue from PE pregnancies. Whether this is a maladaptive or beneficial response by the placenta in these cases remain unclear.
Within the placenta, catecholamines have been suggested to regulate several other factors and down-stream processes. In the sheep placenta, NE and/or DA regulate synthesis of polyamines, secretion of the maternal recognition factor in ruminants, interferon-τ (IFNT), and expression of pro-apoptotic genes (Elmetwally, Lenis, Tang, Wu, & Bazer, 2018). In the human placenta, NE appears to increase hCG and progesterone production by first trimester tissue (Shi & Zhuang, 1993a, 1993b). Two human choriocarcinoma cell lines, BeWo and JEG-3, express β-adrenergic receptors coupled to catecholamine-sensitive adenylate cyclase (Moore & Whitsett, 1982; Moore, Workman, & Whitsett, 1982), which may be one signaling pathway by which these catecholamines affect placental metabolism and hormone production.
The current data thus provide robust support that the placenta can synthesize and respond to catecholamines. It is not clear the extent these placental-derived neurotransmitters transits to the fetal brain. Injection of radioactive precursors into the placenta and then measurement of resulting radiolabeled catecholamines in the placenta and fetal brain may help answer this question. Another method to address this outstanding question is the usage of conditional knockout mice. While such transgenic mice have not been created for any of the enzymes regulating synthesis and metabolism for 5-HT, DA, NE, or epinephrine, they have been generated for other placental genes that in turn seemingly affects neural function, which will be considered in the next section.
Usage of Placental-Specific Transgenic Mice to Examine Linkages Between the Placenta and Brain
Cre/loxP transgenic mice have been invaluable in understanding how conditional deletion of a certain gene in the placenta leads to later neurobehavioral disturbances. To create mice lacking a select placental genes, current reports used mice with a Cre recombinase gene under control of an introduced human CYP19 (exon I.1) promoter (Wenzel & Leone, 2007). A 501 base pair region within the first exon of CYP19 (exon I.1) induces specific expression of a reporter transgene in mouse synctiotrophoblast cells (Kamat, Graves, Smith, Richardson, & Mendelson, 1999). Analysis of mouse placenta at various stages reveals that the CYP19-Cre transgene is active by E6.5 only in diploid trophoblast cells committed to differentiation but inactive in early undifferentiated trophoblastic stem cells (Kamat et al., 1999). Moreover, expression in precursor cells giving rise to spongiotrophoblast cells or giant cells is not complete until E11.5-E13.5, suggesting that any gene targeted for deletion in trophoblast cells will be expressed prior to this embryonic period. Whether such early trophoblast expression of the gene of interest affects fetal brain development remains ambiguous. Even with this caveat, the current data with such conditional knockout mice have yielded important mechanistic insight into how the expression of select placental genes affects neurobehavioral functions.
Example genes that have been conditionally deleted in this manner include those associated with inflammation and metabolism. Conditional deletion of interleukin 6 (IL6) signaling in the placenta was done through generation of mice that were Cy19Cre+;Il6 receptor (r)fl/fl (Wu, Hsiao, Yan, Mazmanian, & Patterson, 2017). Maternal immune activation (MIA) causes acute inflammatory responses in the fetal brain. However, absence of IL-6 signaling in placental trophoblasts blocks such responses in the placenta and fetal brain. Behavioral abnormalities and cerebellar pathologies observed in control MIA offspring are mitigated in Cy19Cre+;Il6 rfl/fl offspring.
In mice, maternal stress suppresses O-GlcNAc transferase (Ogt, an X-linked gene) in the placenta of males but not females (Howerton, Morgan, Fischer, & Bale, 2013). Hemizygous mice that conditionally lack Ogt in the placenta (Pl-OGT) show similar gene expression patterns in the placenta as males exposed to early prenatal stress (Howerton & Bale, 2014). Functional enrichment analyses on differentially expressed genes suggest that mitochondrial function is impaired in Pl-OGT and EPS males. ChIP-Seq for the O-GlcNAc mark reveals17β-hydroxysteroid dehydrogenase (Hsd17b3) locus to be affected and reduced in early prenatal stressed males who have associated reductions in testosterone conversion. Pl-OGT adult offspring exhibit reduced body weight and enhanced responsivity of the hypothalamic-pituitary-adrenal (HPA) axis.
Disruptions in placental insulin receptor (InsR) signaling can occur in response to a variety of pregnancy complications, including gestational diabetes, intrauterine growth restriction, and PE (Colomiere, Permezel, Riley, Desoye, & Lappas, 2009; Desoye, Hofmann, & Weiss, 1992; Li et al., 2014; Petropoulos et al., 2015; Rademacher, Gumaa, & Scioscia, 2007; Street et al., 2011), and deficits in this placental signaling pathway may serve as the bridge between maternal metabolic disorders and offspring neurodevelopmental disorders. To test this hypothesis, transgenic mice with conditional deletion of InsR in fetal trophoblasts were generated (Bronson, Chan, & Bale, 2017). The HPA stress response is enhanced in males but not females lacking trophoblast InsR. These males also show deficits in sensorimotor gating. Gene expression changes in the placenta of these males correlates with vasculature function, amino acid transport, 5-HT homeostasis, and mitochondrial function. The brain of these males have transcriptomic differences that collectively suggest impaired cortical development.
Placental specific deletion of the Igf2 P0 transcript in mice results in intrauterine growth restriction, an imbalance between fetal requirement and placental delivery of nutrients, and notably, offspring become more anxious as they mature (Mikaelsson, Constancia, Dent, Wilkinson, & Humby, 2013). The findings provide further support for a linkage between placental responses and later offspring behavioral changes, but potential sex differences in placental responses and later offspring behavioral disruptions were not considered. Epigenetic modifications discussed in the next section may underpin such gene expression changes in the placenta.
Placental Epigenetic Changes and Neurobehavioral Outcomes
Epigenetic changes are those that affect transcription or translation of a gene to the protein it encodes but without altering the DNA sequence. Two main types of epigenetic changes, DNA methylation and microRNA (miR) expression patterns, within the placenta have been examined for potential associations with later neurobehavioral disruptions. While it is not clear based on current reports if such placental alterations affect fetal brain development, they may at least serve as biomarkers to identify those children at risk of developing such disorders. Thus, in this section, we will consider the studies to date linking such epigenetic modifications and neurobehavioral deficits.
There are several cohort studies examining a variety of environmental, genetic, and other factors that correlate with risk for ASD. One of the most longstanding cohorts is the Markers of Autism Risk in Babies Learning Early Signs (MARBLES). A prospective study of high-risk pregnancies as part of this cohort reveals that 400 differentially methylated regions (DMR) distinguish placentas from offspring later diagnosed with ASD relative to those not diagnosed with such disorders (Y. Zhu et al., 2019). DMR in CYP2E1 and IRS2 from placenta of autistic children reach genome-wide significance with methylation at CYP2E1 positively correlating with ASD diagnosis and genotype within the DMR. An earlier study done with the MARBLES cohort discovered that pesticide exposure significantly influences placental DNA methylation patterns (Schmidt et al., 2016), suggesting that pesticides might affect fetal brain development by altering the placental epigenome. Genome bisulfite sequencing of human placentas from the MARBLES study provides strong evidence that DNA methylation changes in the placenta might serve as barometers to identify those children at risk of developing ASD (Schroeder et al., 2016).
Maternal stress and effects on the placenta-brain-axis have of been longstanding interest. Glucocorticoids are considered one of the primary stress hormones. The placenta possesses high concentrations of 11β-Hydroxysteroid dehydrogenase 2 (11β-HSD2) to shield the fetus from maternal glucocorticoids by metabolizing cortisol to the inactive metabolite, cortisone (P. Zhu, Wang, Zuo, & Sun, 2019). Select amounts of cortisol, however, might escape and penetrate into the placenta whereupon they can bind and activates glucocorticoid receptors (GR), encoded by the NR3C1 gene. With this pathway in mind, several studies have examined how extrinsic factors affect methylation of NR3C1 and HSD11B2. Variance in placental NR3C1 methylation is associated with infant crying parameters, such as energy variation and frequency in cry utterances (Sheinkopf, Righi, Marsit, & Lester, 2016). Increased placental NR3C1 methylation correlates with increased infant attention and self-attention but reduced lethargy and need for emotional soothing during the first postnatal month (Stroud et al., 2016). Varying NR3C1 methylation is also associated with infant quality of movement and attention (Bromer, Marsit, Armstrong, Padbury, & Lester, 2013). Further analysis into DMR within NR3C1 establishes that CpG sites 5–13 is related to increased cortisol activity and infant self-regulation (Conradt et al., 2015). Increasing amounts of placental methylation of HSD11B2 coincides with low birth weight and reduced scores for quality of movement (Marsit, Maccani, Padbury, & Lester, 2012). Examination of the joint contribution of placental NR3C1 and HSD11B2 methylation shows that infants with low NR3C1 methylation but high HSD11B2 methylation have low excitability scores (Appleton, Lester, Armstrong, Lesseur, & Marsit, 2015). Those with high NR3C1 methylation but low HSD11B2 methylation demonstrated increased asymmetrical reflexes, and infants with elevated methylation for both cortisol pathway genes exhibit higher habituation scores. These differential effects are likely due to varying glucocorticoid exposure and activity within the placenta during gestation.
Besides placental 5-HT affecting neurobehavioral development, methylation of placental HTR2A, the receptor mediating the effects of 5-HT, may also be involved. HTR2A methylation associates inversely with infant quality of movement but positively with infant attention (Paquette et al., 2013). Placental leptin (LEP) is another gene whose methylation pattern is linked strongly with infant neurobehavioral outcomes. A 10% increase in LEP DNA methylation correlates with a pattern of infant neurobehavior typified by increased lethargy and hypotonicity (Lesseur et al., 2014). When considered together, methylation patterns for 10 imprinted genes, DLX5, DHCR24, VTRNA2–1, PHLDA2, NPA1, FAM50B, GNAS-AS1, PAX8-AS1, SHANK2, and COPG2IT1 stratifies infants into those characterized by reduced quality of movement, elevated indices of asymmetrical and non-optimal reflexes, and increased likelihood of physiological stress (Green et al., 2015).
Alterations in miR expression profiles represent another type of epigenetic change that can occur in the placenta and may modulate infant neurobehavioral patterns. One report that has examined such epigenetic modifications reports that increased placental miR-16 expression is negatively associated with attention score (Maccani, Padbury, Lester, Knopik, & Marsit, 2013). In contrast, miR-146a and miR-182 are positively related to quality of movement score. Notably, miR-16, along with miR-15a, regulates SERT expression in human placental and rat brain raphe cells (Moya, Wendland, Salemme, Fried, & Murphy, 2013).
Placental Histopathological Changes, Diseases and Neurobehavioral Disorders
A handful of studies have considered the potential link between histopathological changes in the placenta and later neurobehavioral disorders. Inclusion bodies with trophoblast cells may serve as a good predictor of those children showing poor neurodevelopment and at potential risk for ASD (Anderson, Jacobs-Stannard, Chawarska, Volkmar, & Kliman, 2007; Firestein, Abellar, Myers, & Welch, 2017; Walker et al., 2013). A study examining 55 ASD cases and 199 neurotypical controls presenting at New York Methodist Hospital determined that inflammation within the chorionic plate vessels and maternal vascular malperfusion are strongly associated with increased risk for ASD in male but not female offspring (Straughen et al., 2017). Inexplicably, placental villous edema relates to decreased risk of ASD in males. Reductions in overall placental shape variability have been noted in high-risk ASD siblings, which may suggest limited ability to adapt to in utero changes (Park et al., 2018).
In CD1 mice, maternal exposure to the inflammatory factor, IL-1β, causes pronounced pathological changes in the placenta and fetal brain (Novak et al., 2019). Both maternal and fetal placenta of treated dams is hypoplastic, and correspondingly, cortical atrophy is evident in the fetal brain. Maternal exposure to IL-1β upregulates mRNA expression for Nfkb2 and Cxcl11. CD4+ and CD8+ infiltrates into these same placenta. IL-1β leads to maternal sub-chronic and widespread inflammation with decrease pup survival and perinatal brain injury, and such morbidities and mortalities may be due to resulting placentitis.
Besides maternal inflammation affecting the placenta-brain-axis, maternal starvation and/or underperfusion to the placenta may also deleteriously affect both organs. To study how placental underperfusion affectes fetal offspring brain development and birth weight, ligation of the uteroplacental vessel was performed in rabbit does at day 25 of gestation (Illa et al., 2017), whereas to examine effects of maternal undernutrition, does were subjected to a 70% reduction in basal intake at 22 days of pregnancy (Illa et al., 2017). Both approaches result in reduced birth weight with the placental underperfusion model also increasing incidence of stillbirths. Offspring from both models exhibit compromised neurobehavioral performance.
One of the most common placental diseases is PE. PE is characterized by gestational hypertension and proteinuria with diagnoses most commonly after 20 weeks of gestation in women who were previously symptom-free (Lisonkova & Joseph, 2013). The pathogenesis of PE remains enigmatic as it is seemingly influenced by both genetic and extrinsic factors (Oudejans, van Dijk, Oosterkamp, Lachmeijer, & Blankenstein, 2007; J. M. Roberts & Cooper, 2001). Upon delivery or removal of the placenta, the disease symptoms resolve, suggesting the placenta is the initiating organ. Early onset PE has been ascribed to improper remodeling of the uterine spiral arteries by the invasive extravillous trophoblasts (Pijnenborg, Vercruysse, Hanssens, & Brosens, 2011). With the overall PE incidence rate at approximately 3.1% (Lisonkova & Joseph, 2013), increasing numbers of studies have examined for potential linkages between PE and offspring neurobehavioral disorders, especially ASD. The majority of the studies support an association between PE and increased risk for ASD, as detailed below.
A linkage between ASD and PE was examined in children from 20 California counties with 517 being afflicted with ASD and 194 diagnosed with developmental delay (Walker et al., 2015). Children were recruited through the California Department of Developmental Services, the Medical Investigation of Neurodevelopmental Disorders (MIND) Institute, and referrals. This retrospective study determined that children with ASD are two-times as likely to have been exposed in utero to PE. Analysis of approximately 88,000 births from 1996 through 2002 issued by the South Carolina Medicaid Program revealed that maternal PE/eclampsia is associated with a significantly greater odds ratio of ASD in offspring with reduced birth weight likely mediating some of these effects (Mann, McDermott, Bao, Hardin, & Gregg, 2010). Comparison of 323 probands with ASD to 257 unaffected siblings and 1504 neurotypical controls reveals that PE, polyhydramnios (too much amniotic fluid), oligoamnios (insufficient amniotic fluid), placenta previa, umbilical cord knot, and gestational diabetes correlate with autistic symptoms, in particular stereotyped behaviors and socio-communication deficits (Chien et al., 2019). A retrospective population-based cohort study with approximately 254,000 singletons from 1991 through 2014 exposed to PE and those derived from healthy pregnancies found that children exposed prenatally to PE have a significantly higher risk of developing a neurophychiatric morbidity during childhood (Nahum Sacks et al., 2019), suggesting PE can lead to other neurobehavioral diseases besides ASD. Meta-analysis based on 37,634 autistic and 12,081,416 non-autistic children reported that prenatal factors that raise the likelihood of ASD include PE, spontaneous labor, induced labor, no labor, breech presentation, and fetal distress, but it is not clear how these factors may inter-relate to each other and whether they a play a causal or secondary role (Wang, Geng, Liu, & Zhang, 2017). Analysis of singleton live births in Sweden from 1982 through 2010 that included 2,842,230 children with 54,071 diagnosed with ASD determined that PE in absence of small for gestational age is linked with a 25% increase in the likelihood of developing ASD. An increased association exists though for developing ASD in offspring who experience a combination of PE and small for gestational age (Maher et al., 2019). The combined data provide robust evidence that pregnancies complications, such as PE, which are associated with placental abnormalities, may increase the likelihood of offspring developing ASD.
Sexually Dimorphic Differences in Placental Responses and Potential Risk for Neurobehavioral Disorders
How the placenta responds to in utero environmental challenges with examples provided previously may differ based on the conceptus and correspondingly, one sex over the other may be more vulnerable to programmed effects of an adverse gestational environment. The preceding sections alluded to some cases where such sex differences in placental responses have been noted. Sexually dimorphic differences in the occurrence of PE may also exist with some studies suggesting that preterm PE tending to be more prevalent in women gestating a female fetus (Liu et al., 2019; Schalekamp-Timmermans et al., 2017; Taylor et al., 2018), whereas an extensive meta-analysis spanning studies from 1950 to 2015 comprised of non-Asian individuals indicate this disease is more prevalent with mothers carrying male fetuses (Jaskolka, Retnakaran, Zinman, & Kramer, 2017). It is clear though that males have a greater likelihood of developing ASD with the reported odds ratio varying from 3 to 4:1 (Fombonne, 2008; Loomes, Hull, & Mandy, 2017; “Prevalence of autism spectrum disorders--Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008,” 2012). In this section, we will focus on how sexually dimorphic responses in the placenta may occur and examine the evidence to date that later neurobehavioral deficits may originate due to such placental sex differences.
A comprehensive discussion of how such sex differences in placental responses might occur and studies providing support for the various mechanisms is provided in (Rosenfeld, 2015). Briefly, several underpinning mechanisms may modulate sex differences in placental responses. Differences in DNA methylation or other epigenetic changes can cause such differences (Gabory et al., 2012; Gabory, Roseboom, Moore, Moore, & Junien, 2013; Gallou-Kabani et al., 2010; Rosenfeld, 2012; Tarrade, Panchenko, Junien, & Gabory, 2015; Tekola-Ayele et al., 2019). Changes in the expression of miR represent another type of epigenetic change, as detailed above. In newborn girls, placenta miR-34a, miR-146a, and miR-222 are positively associated with placental relative telomere length (Tsamou et al., 2018). Telomeres represents repetitive nucleotide sequences that protects the end of the chromosome from deterioration or fusion with other chromosomes, and this region of the chromosome diminishes with age (Bernadotte, Mikhelson, & Spivak, 2016). Conversely, elevated expression of placental miR-21 in newborn boys is only weakly associated with shorter placental telomere length.
While the paternal X-chromosome is presumed to undergo X-inactivation exclusively in the placenta, some genes on this X chromosome may escape such inactivation resulting in the placenta of females expressing twice the number of gene copies as males (Gabory et al., 2013). Several of the genes on the X-chromosome encode those involved in nutrient metabolism, which has considerable relevance should the conceptus experience a surfeit of nutrients on undernutrition.
In support of this notion, we and others have reported that the mid-gestational placenta of female mice mounts a more robust response to a maternal high fat diet, HFD (Gabory et al., 2012; Gallou-Kabani et al., 2010; Mao et al., 2010). A recent study that examined the rat placenta at day 21 of gestation in dams fed a HFD vs. a control diet also found sex differences in the placental transcriptome with those of males demonstrating increased plasticity relative to females (Lin et al., 2019). Pups of both sexes born to dams on a HFD exhibited fetal growth restriction (FGR), and activation of the placental renin-angiotensin system by this maternal diet was found to be associated with this disorder in both sexes. A maternal HFD to mice results in increased placental macrophage activation and cytokine gene expression that is enhanced in males but both sexes show reduced labyrinth thickness and decrease trophoblast proliferation (D. W. Kim, Young, Grattan, & Jasoni, 2014). Placental lipidomic profiles in rabbits also show sexually-dimorphic differences in response to a maternal diet enriched in fat and cholesterol with stored fatty acid concentrations increasingin the placenta of females relative to males (Tarrade et al., 2013). Triacylgycerol is elevated, however, in these males (Tarrade et al., 2013). A systemic review of animal studies further suggests that maternal obesity is associated with offspring neurobehavioral disruptions as evidenced by elevated locomotor activity and anxiogenic behaviors (Menting et al., 2019). Rhesus macaque offspring of mothers with increased baseline adiposity or gestational weight gain display poor adaptability with exaggerated emotional responses and reduced interest in novel stimuli (Walker, VandeVoort, Li, Chaffin, & Capitanio, 2018). Correspondingly, they have greater suppression of cortisol following dexamethasone administration. Whether such behavioral and other phenotypic changes are due to maternal obesity acting on the fetal brain or through the placenta is uncertain.
In humans, the placenta of males but not females from overweight and obese women is typified by increased activation of autophagosomal formation and autophagosome-lysosome fusion (Muralimanoharan, Gao, Weintraub, Myatt, & Maloyan, 2016). Another study by this group notably showed that maternal obesity associates with increased miR-210 but decreased brain derived neural factor (BDNF) expression in female placenta, whereas proBDNF and mature BDNF are reduced in male vs. female placenta in conceptuses gestated by obese mothers (Prince, Maloyan, & Myatt, 2017). miR210 expression is inversely correlated with mature BDNF protein. Tropomyosin receptor kinase B (TRKB, BDNF receptor) phosphorylation at tyrosine 817 is elevated in male and female placenta from obese women, but phosphorylation of mitogen-activated protein kinase (MAPK p38) is solely increased in female placenta from these women (Prince et al., 2017). Collectively, these data show that maternal obesity alters in a sex-dependent manner BDNF/TRKB/MAPK p38 signaling in the placenta. Placenta of lean women with a male fetus have higher supraoxide dismutase (SOD) activity and total antioxidant capacity (Evans & Myatt, 2017). On the other hand, placenta from obese women with a male fetus contain increased amounts of carbonyls and nitrotyrosine, and as a potential compensatory mechanism, these placentas have elevations in glutathione peroxidase and thioredoxin reductase activity. The collective findings from this latter study reveal that the placenta of males derived from lean mothers possess the greatest antioxidant activity, but such protective responses are abolished with maternal obesity, which may account for later adverse outcomes in sons, in particular those born to obese mothers (Bridgman et al., 2018; Greene-Cramer et al., 2018).
The above studies suggest that a maternal HFD results in sex differences in placental anatomy and physiology. However, the current data do not establish causation that such placental changes increase the risk of neurobehavioral disorders in one sex over the other. Maternal stress has been another extrinsic factor extensively examined as to how it affects placental function and later neurobehavioral outcomes and will thus be considered.
As detailed above, maternal stress suppresses Ogt (an X-linked gene) in the placenta of male but not female mice (Howerton et al., 2013). Coding genes and miR expression patterns are disrupted in the hypothalami of placental-specific hemizygous OGT mice. Another study showed that maternal corticosterone exposure in mice increases Akt-O-GlcNacylation that associates with decreased phosphorylation in male placentas only (Pantaleon, Steane, McMahon, Cuffe, & Moritz, 2017). In contrast, control female placenta have an upsurge in basal OGT and OGT/GR complex relative to control male placenta. Maternal corticosterone exposure does affect these levels, but female placenta from this group demonstrate increased global O-GlcNacylation, whereas corticosterone exposure raises OGT and OGT/GR complex with no alteration in O-GlcNacylation.
Maternal stress in mice induces expression of Pppara, Igfbp1, Hifa, and Glut4 in male but not female placentae (Mueller & Bale, 2008). Males subjected to maternal stress during gestation develop several maladaptive stress responses, namely anhedonia (inability to experience pleasure), and deficits in the hypothalamic-pituitary axis (HPA). Other placental-neurobehavioral comorbidities are observed in prenatal stressed male mice, such as up-regulation of proinflammatory cytokines (Il6 and Il1b) in the placentae, stress-induced locomotor hyperactivity, and alteration of neural expression of DA D1 and D2 receptors (Bronson & Bale, 2014). Maternal exposure to dexamethasone stimulates sex-specific transcriptome changes in the mouse placenta at E18.5 based on RNA-Seq analysis with male placenta showing activation of various inflammatory response-related genes, chemokines, and their receptors (Lee et al., 2017). Another study testing the effects of maternal dexamethasone exposure on placental protein expression patterns found that placenta of control females have increased expression of glucocorticoid receptor (GR) (α-A and P variants) in the cytoplasm than control male placenta, whereas GRα-C is elevated in the nucleus of control female relative to control male placenta (Cuffe, Saif, Perkins, Moritz, & Clifton, 2017). Maternal dexamethasone triggers the cytoplasmic expression of GRα-A in the cytoplasm but suppresses the expression of GRα-C in male placenta. Increased expression of GRα-C-regulated genes, Sgk1 and Bcl2l11 occurs in females placenta exposed to dexamethasone. Effects of dexamethasone in spiny mouse have also been tested (O’Connell, Moritz, Walker, & Dickinson, 2013). Female placenta of this species normally have greater amounts of glycogen at day 25 of gestation with parturition occurring at approximately 39 days. Dexamethasone treatment to the dams initially decreases expression of Gsk3b, Gys1, Gbe1, Foxo1, and Ugp2 in male and female placenta, but only females from this group have reductions in placental glycogen amounts. In male placenta, reductions in Gsk3b and Ugp2 persist through to day 37 of gestation.
In rats, advanced maternal age leas to differences in placental responses and structure relative to placenta derived from pregnancies with younger dams, and such differences may also be sex-dependent (Napso, Hung, Davidge, Care, & Sferruzzi-Perri, 2019). In female placenta of aged dams, placental transport and endocrine regions are enlarged with elevations in insulin-like growth factor 2 (Igf2) and placental lactogen (Prl3b1). Female placenta from aged dams also display heightened glucocorticoid metabolism with elevations in 11bhsd2 expression, whereas, the opposite is the case with male placenta from these same dams who show reductions in this gene. Both male and female placenta of aged dams are noted for increase oxidative stress, but apoptosis is only prominent in the male placenta from these dams. In contrast to the sex differences observed in the placenta, advanced maternal age causes FGR in both males and females.
11β-HSD1 and 11β-HSD2 were examined in the placenta of sons and daughters for pregnant women treated with a single course of betamethasone between 23–34 weeks of gestation (Braun et al., 2018). Males exposed to this glucocorticoid, especially those born prematurely, have increased 11β-HSD2 protein concentrations in the placenta. Such a response may serve as a protective mechanism as females whose placenta show reduced concentrations of this enzyme following betamethasone exposure exhibit increased amounts of maternal cortisol transferred across the placenta, and these daughters have an associated reduction in head circumference and risk for heightened stress responses in adulthood.
Taken together, the data suggest that maternal stress due to intrinsic factors or pharmacological induction leads to sex-dependent differences in placental responses with the placenta in male rodent models in general being more vulnerable to such maternal stressors that may in turn increase the risk for later neurobehavioral deficits. Additional retrospective studies with humans and non-human primate models are needed to determine if in contrast to rodents, the female placenta is more vulnerable to maternal stress in these species. If so, it is not clear how well the maternal stress-induced rodent studies may actually mirror those resulting in the human placenta.
Conclusions
While the placenta is an emphermal organ, how it responds to in utero environmental challenges can lead to long term consequences on offspring health. Nowhere is this truer than in the brain where early on in development it depends upon the placenta for nutrient delivery, oxygen, and as it is increasingly becoming recognized, neurotransmitters. The primary neurotransmitters conveyed from the placenta to the brain include 5-HT, DA, and NE/epinephrine. Anything that compromises the delivery of these factors from the placenta to the brain can lead to deleterious health consequences, including risk for neurobehavioral disorders. Increasing usage of SSRI by pregnant womenplagued with depression is thus of major concern. While some studies have begun to examine how these drugs affect the placenta (Clabault, Cohen, Vaillancourt, & Sanderson, 2018; Clabault, Flipo, et al., 2018; Hudon Thibeault et al., 2017; Laurent et al., 2016), more experiments are needed to determine what concentrations of these drugs are reached in the placenta and whether such drugs affect the placental-brain-axis.
Conditional transgenic mice that lack select metabolic, growth-dependent, and inflammatory genes in trophoblast cells have been invaluable in teasing apart their role in the placenta and later neurobehavioral outcomes, including those that may be sex-dependent (Bronson et al., 2017; Howerton & Bale, 2014; Mikaelsson et al., 2013; Wu et al., 2017). Conceivably, the usage of lentiviral vectors and CRISPR/Cas9 system will allow for greater ability to manipulate placental gene expression patterns (Tobita, Kiyozumi, & Ikawa, 2017) and then study resulting neurobehavioral outcomes. This procedure could also be used to perform therapeutic genome editing in the placenta and thereby prevent later neurobehavioral disorders whose genesis traces its origins back to placental dysfunction. Thus, a better understanding of how transcriptome changes in the placenta affects such diseases may pidentify potential biomarkers for early diagnosis and be exploited for therapeutic purposes.
Epigenetic changes in the placenta that include DNA methylation alterations and changes in miR expression are other potential barometers of later neurobehavioral disorders, especially ASD. As with transcript changes, such epigenetic modifications may provide both early biomarkers for detection of such diseases and serve as the basis for interventionary approaches. Accumulative histopathological changes in the placenta may provide valuable insights into stressors the placenta was exposed to during gestation and potential diseases that may be of concern as the infant matures. Based on the studies to date, clinicians should consider routinely performing histological examination of term placenta from newborn infants. While there are clear linkages between PE and ASD and other neurobehavioral disorders, even subtle changes in the placental architecture might provide valuable clues as to the in utero challenges that sculptedd both the placenta and developing fetus and ensuing DOHaD effects.
When considering how maternal stressors impact the placental-brain-axis, the placenta should be regarded as a sexually dimorphic organ with one sex possibly more vulnerable than the other. Such sex differences may arise due to improper inactivation of the X-chromosome, epigenetic changes, or other mechanisms. However, the net result is that changes in maternal diet, stress, and exposure to other extrinsic factors are likely not handled in the same manner in male vs. female placenta. Depending on the stressor and species, the placenta of one sex may be better equipped to adapt and confront such in utero fluctuations. In this sense, the placenta might serve as the gatekeeper to prevent pernicious factors from reaching the fetus. A muted or maladaptive placental response to such in utero changes might causes a permanent stamp on the fetus with the offspring developing neurological and other disorders with age. Thus, the cumulative placenta responses in each sex are important variables to consider in trying to understand fetal programming of later diseases.
In sum, the placenta and fetal brain are inextricably linked. A better understanding of how the placenta responds to in utero stressors it encounters will likely yield mechanistic insight into neurobehavioral and other disorders with a DOHaD-basis and potential avenues to thereby diagnosis and treat such diseases at the outset.
Acknowledgements:
CSR is supported by NIEHS 1R01ES025547.
References
- Anderson GM, Jacobs-Stannard A, Chawarska K, Volkmar FR, & Kliman HJ (2007). Placental trophoblast inclusions in autism spectrum disorder. Biological Psychiatry, 61(4), 487–491. doi: 10.1016/j.biopsych.2006.03.068 [DOI] [PubMed] [Google Scholar]
- Appleton AA, Lester BM, Armstrong DA, Lesseur C, & Marsit CJ (2015). Examining the joint contribution of placental NR3C1 and HSD11B2 methylation for infant neurobehavior. Psychoneuroendocrinology, 52, 32–42. doi: 10.1016/j.psyneuen.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmaker RH, Agam G, & Bersudsky Y (2008). Role of GSK3beta in behavioral abnormalities induced by serotonin deficiency. Proceedings of the National Academy of Sciences of the United States of America, 105(20), E23; author reply E24. doi: 10.1073/pnas.0801168105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Jonathan N, & Munsick RA (1980). Dopamine and prolactin in human pregnancy. Journal of Clinical Endocrinology and Metabolism, 51(5), 1019–1025. doi: 10.1210/jcem-51-5-1019 [DOI] [PubMed] [Google Scholar]
- Bernadotte A, Mikhelson VM, & Spivak IM (2016). Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY), 8(1), 3–11. doi: 10.18632/aging.100871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, … Levitt P (2011). A transient placental source of serotonin for the fetal forebrain. Nature, 472(7343), 347–350. doi: 10.1038/nature09972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A, & Levitt P (2011). Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience, 197, 1–7. doi: 10.1016/j.neuroscience.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottalico B, Larsson I, Brodszki J, Hernandez-Andrade E, Casslen B, Marsal K, & Hansson SR (2004). Norepinephrine transporter (NET), serotonin transporter (SERT), vesicular monoamine transporter (VMAT2) and organic cation transporters (OCT1, 2 and EMT) in human placenta from pre-eclamptic and normotensive pregnancies. Placenta, 25(6), 518–529. doi: 10.1016/j.placenta.2003.10.017 [DOI] [PubMed] [Google Scholar]
- Braun F, Hardt AK, Ehrlich L, Sloboda DM, Challis JRG, Plagemann A, … Braun T (2018). Sex-specific and lasting effects of a single course of antenatal betamethasone treatment on human placental 11beta-HSD2. Placenta, 69, 9–19. doi: 10.1016/j.placenta.2018.07.007 [DOI] [PubMed] [Google Scholar]
- Bridgman SL, Azad MB, Persaud RR, Chari RS, Becker AB, Sears MR, … Kozyrskyj AL (2018). Impact of maternal pre-pregnancy overweight on infant overweight at 1 year of age: associations and sex-specific differences. Pediatric Obesity, 13(10), 579–589. doi: 10.1111/ijpo.12291 [DOI] [PubMed] [Google Scholar]
- Bromer C, Marsit CJ, Armstrong DA, Padbury JF, & Lester B (2013). Genetic and epigenetic variation of the glucocorticoid receptor (NR3C1) in placenta and infant neurobehavior. Developmental Psychobiology, 55(7), 673–683. doi: 10.1002/dev.21061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson SL, & Bale TL (2014). Prenatal stress-induced increases in placental inflammation and offspring hyperactivity are male-specific and ameliorated by maternal antiinflammatory treatment. Endocrinology, 155(7), 2635–2646. doi: 10.1210/en.2014-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson SL, Chan JC, & Bale TL (2017). Sex-Specific Neurodevelopmental Programming by Placental Insulin Receptors on Stress Reactivity and Sensorimotor Gating. Biological Psychiatry, 82(2), 127–138. doi: 10.1016/j.biopsych.2016.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AM (2007). Animal models of human placentation--a review. Placenta, 28 Suppl A, S41–47. doi: 10.1016/j.placenta.2006.11.002 [DOI] [PubMed] [Google Scholar]
- Carter AM, & Mess A (2007). Evolution of the placenta in eutherian mammals. Placenta, 28(4), 259–262. doi: 10.1016/j.placenta.2006.04.010 [DOI] [PubMed] [Google Scholar]
- Chavatte-Palmer P, & Guillomot M (2007). Comparative implantation and placentation. Gynecologic and Obstetric Investigation, 64(3), 166–174. doi: 10.1159/000101742 [DOI] [PubMed] [Google Scholar]
- Chien YL, Chou MC, Chou WJ, Wu YY, Tsai WC, Chiu YN, & Gau SS (2019). Prenatal and perinatal risk factors and the clinical implications on autism spectrum disorder. Autism, 23(3), 783–791. doi: 10.1177/1362361318772813 [DOI] [PubMed] [Google Scholar]
- Clabault H, Cohen M, Vaillancourt C, & Sanderson JT (2018). Effects of selective serotonin-reuptake inhibitors (SSRIs) in JEG-3 and HIPEC cell models of the extravillous trophoblast. Placenta, 72–73, 62–73. doi: 10.1016/j.placenta.2018.10.007 [DOI] [PubMed] [Google Scholar]
- Clabault H, Flipo D, Guibourdenche J, Fournier T, Sanderson JT, & Vaillancourt C (2018). Effects of selective serotonin-reuptake inhibitors (SSRIs) on human villous trophoblasts syncytialization. Toxicology and Applied Pharmacology, 349, 8–20. doi: 10.1016/j.taap.2018.04.018 [DOI] [PubMed] [Google Scholar]
- Colomiere M, Permezel M, Riley C, Desoye G, & Lappas M (2009). Defective insulin signaling in placenta from pregnancies complicated by gestational diabetes mellitus. European Journal of Endocrinology of the European Federation of Endocrine Societies, 160(4), 567–578. doi: 10.1530/eje-09-0031 [DOI] [PubMed] [Google Scholar]
- Conradt E, Fei M, LaGasse L, Tronick E, Guerin D, Gorman D, … Lester BM (2015). Prenatal predictors of infant self-regulation: the contributions of placental DNA methylation of NR3C1 and neuroendocrine activity. Frontiers in Behavioral Neuroscience, 9, 130. doi: 10.3389/fnbeh.2015.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuffe JSM, Saif Z, Perkins AV, Moritz KM, & Clifton VL (2017). Dexamethasone and sex regulate placental glucocorticoid receptor isoforms in mice. Journal of Endocrinology, 234(2), 89–100. doi: 10.1530/joe-17-0171 [DOI] [PubMed] [Google Scholar]
- Desoye G, Hofmann HH, & Weiss PA (1992). Insulin binding to trophoblast plasma membranes and placental glycogen content in well-controlled gestational diabetic women treated with diet or insulin, in well-controlled overt diabetic patients and in healthy control subjects. Diabetologia, 35(1), 45–55. doi: 10.1007/bf00400851 [DOI] [PubMed] [Google Scholar]
- Elmetwally MA, Lenis Y, Tang W, Wu G, & Bazer FW (2018). Effects of catecholamines on secretion of interferon tau and expression of genes for synthesis of polyamines and apoptosis by ovine trophectoderm. Biology of Reproduction, 99(3), 611–628. doi: 10.1093/biolre/ioy085 [DOI] [PubMed] [Google Scholar]
- Enders AC, & Carter AM (2006). Comparative placentation: some interesting modifications for histotrophic nutrition -- a review. Placenta, 27 Suppl A, S11–16. doi: 10.1016/j.placenta.2005.10.013 [DOI] [PubMed] [Google Scholar]
- Evans L, & Myatt L (2017). Sexual dimorphism in the effect of maternal obesity on antioxidant defense mechanisms in the human placenta. Placenta, 51, 64–69. doi: 10.1016/j.placenta.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein MR, Abellar R, Myers MM, & Welch MG (2017). Increased trophoblast inclusions in placentas from prematurely born infants: A potential marker of risk for preterm neurodevelopmental outcomes. Placenta, 60, 61–63. doi: 10.1016/j.placenta.2017.10.006 [DOI] [PubMed] [Google Scholar]
- Fitzgerald P, & Dinan TG (2008). Prolactin and dopamine: what is the connection? A review article. J Psychopharmacol, 22(2 Suppl), 12–19. doi: 10.1177/0269216307087148 [DOI] [PubMed] [Google Scholar]
- Fombonne E (2008). Is autism getting commoner? British Journal of Psychiatry, 193(1), 59. doi: 10.1192/bjp.193.1.59 [DOI] [PubMed] [Google Scholar]
- Gabory A, Ferry L, Fajardy I, Jouneau L, Gothie JD, Vige A, … Junien C (2012). Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PloS One, 7(11), e47986. doi: 10.1371/journal.pone.0047986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabory A, Roseboom TJ, Moore T, Moore LG, & Junien C (2013). Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biology of Sex Differences, 4(1), 5. doi: 10.1186/2042-6410-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallou-Kabani C, Gabory A, Tost J, Karimi M, Mayeur S, Lesage J, … Junien C (2010). Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PloS One, 5(12), e14398. doi: 10.1371/journal.pone.0014398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades P, Ferguson-Smith AC, & Burton GJ (2002). Comparative developmental anatomy of the murine and human definitive placentae. Placenta, 23(1), 3–19. doi: 10.1053/plac.2001.0738 [DOI] [PubMed] [Google Scholar]
- Grattan DR (2015). 60 YEARS OF NEUROENDOCRINOLOGY: The hypothalamo-prolactin axis. Journal of Endocrinology, 226(2), T101–122. doi: 10.1530/joe-15-0213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green BB, Kappil M, Lambertini L, Armstrong DA, Guerin DJ, Sharp AJ, … Marsit CJ (2015). Expression of imprinted genes in placenta is associated with infant neurobehavioral development. Epigenetics, 10(9), 834–841. doi: 10.1080/15592294.2015.1073880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene-Cramer B, Harrell MB, Hoelscher DM, Sharma S, Ranjit N, Gupta V, … Arora M (2018). Association between parent and child weight status among private school children in Delhi, India. Glob Health Promot, 25(2), 67–74. doi: 10.1177/1757975916658002 [DOI] [PubMed] [Google Scholar]
- Hendricks TJ, Fyodorov DV, Wegman LJ, Lelutiu NB, Pehek EA, Yamamoto B, … Deneris ES (2003). Pet-1 ETS gene plays a critical role in 5-HT neuron development and is required for normal anxiety-like and aggressive behavior. Neuron, 37(2), 233–247. [DOI] [PubMed] [Google Scholar]
- Howerton CL, & Bale TL (2014). Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proceedings of the National Academy of Sciences of the United States of America, 111(26), 9639–9644. doi: 10.1073/pnas.1401203111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerton CL, Morgan CP, Fischer DB, & Bale TL (2013). O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proceedings of the National Academy of Sciences of the United States of America, 110(13), 5169–5174. doi: 10.1073/pnas.1300065110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WQ, Zhang CL, Di XY, & Zhang RQ (1998). Studies on the localization of 5-hydroxytryptamine and its receptors in human placenta. Placenta, 19(8), 655–661. [DOI] [PubMed] [Google Scholar]
- Hudon Thibeault AA, Laurent L, Vo Duy S, Sauve S, Caron P, Guillemette C, … Vaillancourt C (2017). Fluoxetine and its active metabolite norfluoxetine disrupt estrogen synthesis in a co-culture model of the feto-placental unit. Molecular and Cellular Endocrinology, 442, 32–39. doi: 10.1016/j.mce.2016.11.021 [DOI] [PubMed] [Google Scholar]
- Huybrechts KF, Palmsten K, Mogun H, Kowal M, Avorn J, Setoguchi-Iwata S, & Hernandez-Diaz S (2013). National trends in antidepressant medication treatment among publicly insured pregnant women. General Hospital Psychiatry, 35(3), 265–271. doi: 10.1016/j.genhosppsych.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illa M, Eixarch E, Munoz-Moreno E, Batalle D, Leal-Campanario R, Gruart A, … Gratacos E (2017). Neurodevelopmental effects of undernutrition and placental underperfusion in fetal growth restriction rabbit models. Fetal Diagnosis and Therapy, 42(3), 189–197. doi: 10.1159/000454859 [DOI] [PubMed] [Google Scholar]
- Jaskolka D, Retnakaran R, Zinman B, & Kramer CK (2017). Fetal sex and maternal risk of pre-eclampsia/eclampsia: a systematic review and meta-analysis. BJOG: An International Journal of Obstetrics and Gynaecology, 124(4), 553–560. doi: 10.1111/1471-0528.14163 [DOI] [PubMed] [Google Scholar]
- Kamat A, Graves KH, Smith ME, Richardson JA, & Mendelson CR (1999). A 500-bp region, approximately 40 kb upstream of the human CYP19 (aromatase) gene, mediates placenta-specific expression in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America, 96(8), 4575–4580. doi: 10.1073/pnas.96.8.4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Young SL, Grattan DR, & Jasoni CL (2014). Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biology of Reproduction, 90(6), 130. doi: 10.1095/biolreprod.113.117259 [DOI] [PubMed] [Google Scholar]
- Kim HJ, Koh PO, Kang SS, Paik WY, & Choi WS (2001). The localization of dopamine D2 receptor mRNA in the human placenta and the anti-angiogenic effect of apomorphine in the chorioallantoic membrane. Life Sciences, 68(9), 1031–1040. doi: 10.1016/s0024-3205(00)01006-7 [DOI] [PubMed] [Google Scholar]
- Kim MO, Kim JH, Choi WS, Lee BH, Cho GJ, Roh SM, … Baik SH (1997). Colocalization of dopamine D1 and D2 receptor mRNAs in rat placenta. Molecules and Cells, 7(6), 710–714. [PubMed] [Google Scholar]
- Kliman HJ, Quaratella SB, Setaro AC, Siegman EC, Subha ZT, Tal R, … Steck TL (2018). Pathway of maternal serotonin to the human embryo and fetus. Endocrinology, 159(4), 1609–1629. doi: 10.1210/en.2017-03025 [DOI] [PubMed] [Google Scholar]
- Laurent L, Deroy K, St-Pierre J, Cote F, Sanderson JT, & Vaillancourt C (2017). Human placenta expresses both peripheral and neuronal isoform of tryptophan hydroxylase. Biochimie, 140, 159–165. doi: 10.1016/j.biochi.2017.07.008 [DOI] [PubMed] [Google Scholar]
- Laurent L, Huang C, Ernest SR, Berard A, Vaillancourt C, & Hales BF (2016). In utero exposure to venlafaxine, a serotonin-norepinephrine reuptake inhibitor, increases cardiac anomalies and alters placental and heart serotonin signaling in the rat. Birth Defects Research. Part A: Clinical and Molecular Teratology, 106(12), 1044–1055. doi: 10.1002/bdra.23537 [DOI] [PubMed] [Google Scholar]
- Lee JY, Yun HJ, Kim CY, Cho YW, Lee Y, & Kim MH (2017). Prenatal exposure to dexamethasone in the mouse induces sex-specific differences in placental gene expression. Development Growth and Differentiation, 59(6), 515–525. doi: 10.1111/dgd.12376 [DOI] [PubMed] [Google Scholar]
- Lesseur C, Armstrong DA, Murphy MA, Appleton AA, Koestler DC, Paquette AG, … Marsit CJ (2014). Sex-specific associations between placental leptin promoter DNA methylation and infant neurobehavior. Psychoneuroendocrinology, 40, 1–9. doi: 10.1016/j.psyneuen.2013.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hadden C, Singh P, Mercado CP, Murphy P, Dajani NK, … Kilic F (2014). GDM-associated insulin deficiency hinders the dissociation of SERT from ERp44 and down-regulates placental 5-HT uptake. Proceedings of the National Academy of Sciences of the United States of America, 111(52), E5697–5705. doi: 10.1073/pnas.1416675112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YJ, Huang LT, Tsai CC, Sheen JM, Tiao MM, Yu HR, … Tain YL (2019). Maternal high-fat diet sex-specifically alters placental morphology and transcriptome in rats: Assessment by next-generation sequencing. Placenta, 78, 44–53. doi: 10.1016/j.placenta.2019.03.004 [DOI] [PubMed] [Google Scholar]
- Lisonkova S, & Joseph KS (2013). Incidence of preeclampsia: risk factors and outcomes associated with early- versus late-onset disease. American Journal of Obstetrics and Gynecology, 209(6), 544.e541–544.e512. doi: 10.1016/j.ajog.2013.08.019 [DOI] [PubMed] [Google Scholar]
- Liu Y, Li N, Li Z, Zhang L, Li H, Zhang Y, … Ye R (2019). Impact of gestational hypertension and preeclampsia on fetal gender: A large prospective cohort study in China. Pregnancy Hypertension, 18, 132–136. doi: 10.1016/j.preghy.2019.09.020 [DOI] [PubMed] [Google Scholar]
- Loomes R, Hull L, & Mandy WPL (2017). What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. Journal of the American Academy of Child and Adolescent Psychiatry, 56(6), 466–474. doi: 10.1016/j.jaac.2017.03.013 [DOI] [PubMed] [Google Scholar]
- Maccani MA, Padbury JF, Lester BM, Knopik VS, & Marsit CJ (2013). Placental miRNA expression profiles are associated with measures of infant neurobehavioral outcomes. Pediatric Research, 74(3), 272–278. doi: 10.1038/pr.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher GM, O’Keeffe GW, Dalman C, Kearney PM, McCarthy FP, Kenny LC, & Khashan AS (2019). Association between preeclampsia and autism spectrum disorder: a population-based study. Journal of Child Psychology and Psychiatry and Allied Disciplines. doi: 10.1111/jcpp.13127 [DOI] [PubMed] [Google Scholar]
- Mann JR, McDermott S, Bao H, Hardin J, & Gregg A (2010). Pre-eclampsia, birth weight, and autism spectrum disorders. Journal of Autism and Developmental Disorders, 40(5), 548–554. doi: 10.1007/s10803-009-0903-4 [DOI] [PubMed] [Google Scholar]
- Manyonda IT, Slater DM, Fenske C, Hole D, Choy MY, & Wilson C (1998). A role for noradrenaline in pre-eclampsia: towards a unifying hypothesis for the pathophysiology. British Journal of Obstetrics and Gynaecology, 105(6), 641–648. doi: 10.1111/j.1471-0528.1998.tb10179.x [DOI] [PubMed] [Google Scholar]
- Mao J, Jain A, Denslow ND, Nouri M-Z, Chen S, Wang T, … Rosenfeld CS (In Press). Bisphenol A and bisphenol S disruptions of the mouse placenta and potential effects on the placenta-brain axis. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Zhang X, Sieli PT, Falduto MT, Torres KE, & Rosenfeld CS (2010). Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proceedings of the National Academy of Sciences of the United States of America, 107(12), 5557–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit CJ, Lambertini L, Maccani MA, Koestler DC, Houseman EA, Padbury JF, … Chen J (2012). Placenta-imprinted gene expression association of infant neurobehavior. Journal of Pediatrics, 160(5), 854–860.e852. doi: 10.1016/j.jpeds.2011.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit CJ, Maccani MA, Padbury JF, & Lester BM (2012). Placental 11-beta hydroxysteroid dehydrogenase methylation is associated with newborn growth and a measure of neurobehavioral outcome. PloS One, 7(3), e33794. doi: 10.1371/journal.pone.0033794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menting MD, van de Beek C, Mintjens S, Wever KE, Korosi A, Ozanne SE, … Painter RC (2019). The link between maternal obesity and offspring neurobehavior: A systematic review of animal experiments. Neuroscience and Biobehavioral Reviews, 98, 107–121. doi: 10.1016/j.neubiorev.2018.12.023 [DOI] [PubMed] [Google Scholar]
- Mikaelsson MA, Constancia M, Dent CL, Wilkinson LS, & Humby T (2013). Placental programming of anxiety in adulthood revealed by Igf2-null models. Nat Commun, 4, 2311. doi: 10.1038/ncomms3311 [DOI] [PubMed] [Google Scholar]
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, & Hernandez-Diaz S (2011). Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. American Journal of Obstetrics and Gynecology, 205(1), 51.e51–58. doi: 10.1016/j.ajog.2011.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JJ Jr., & Whitsett JA (1982). The beta-adrenergic receptor in mammalian placenta: species differences and ontogeny. Placenta, 3(3), 257–268. doi: 10.1016/s0143-4004(82)80003-9 [DOI] [PubMed] [Google Scholar]
- Moore JJ Jr., Workman L, & Whitsett JA (1982). Trophoblastic cells of the hydatidiform mole contain a beta 1-subtype adrenergic receptor. Journal of Clinical Endocrinology and Metabolism, 55(2), 341–346. doi: 10.1210/jcem-55-2-341 [DOI] [PubMed] [Google Scholar]
- Moya PR, Wendland JR, Salemme J, Fried RL, & Murphy DL (2013). miR-15a and miR-16 regulate serotonin transporter expression in human placental and rat brain raphe cells. International Journal of Neuropsychopharmacology, 16(3), 621–629. doi: 10.1017/s1461145712000454 [DOI] [PubMed] [Google Scholar]
- Mueller BR, & Bale TL (2008). Sex-specific programming of offspring emotionality after stress early in pregnancy. Journal of Neuroscience, 28(36), 9055–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CL, Anacker AM, Rogers TD, Goeden N, Keller EH, Forsberg CG, … Veenstra-VanderWeele J (2017). Impact of maternal serotonin transporter genotype on placental serotonin, fetal forebrain srotonin, and neurodevelopment. Neuropsychopharmacology, 42(2), 427–436. doi: 10.1038/npp.2016.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralimanoharan S, Gao X, Weintraub S, Myatt L, & Maloyan A (2016). Sexual dimorphism in activation of placental autophagy in obese women with evidence for fetal programming from a placenta-specific mouse model. Autophagy, 12(5), 752–769. doi: 10.1080/15548627.2016.1156822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahum Sacks K, Friger M, Shoham-Vardi I, Sergienko R, Spiegel E, Landau D, & Sheiner E (2019). Long-term neuropsychiatric morbidity in children exposed prenatally to preeclampsia. Early Human Development, 130, 96–100. doi: 10.1016/j.earlhumdev.2019.01.016 [DOI] [PubMed] [Google Scholar]
- Napso T, Hung YP, Davidge ST, Care AS, & Sferruzzi-Perri AN (2019). Advanced maternal age compromises fetal growth and induces sex-specific changes in placental phenotype in rats. Scientific Reports, 9(1), 16916. doi: 10.1038/s41598-019-53199-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell BA, Moritz KM, Walker DW, & Dickinson H (2013). Treatment of pregnant spiny mice at mid gestation with a synthetic glucocorticoid has sex-dependent effects on placental glycogen stores. Placenta, 34(10), 932–940. doi: 10.1016/j.placenta.2013.06.310 [DOI] [PubMed] [Google Scholar]
- Oudejans CB, van Dijk M, Oosterkamp M, Lachmeijer A, & Blankenstein MA (2007). Genetics of preeclampsia: paradigm shifts. Human Genetics, 120(5), 607–612. doi: 10.1007/s00439-006-0259-1 [DOI] [PubMed] [Google Scholar]
- Pantaleon M, Steane SE, McMahon K, Cuffe JSM, & Moritz KM (2017). Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Scientific Reports, 7(1), 2017. doi: 10.1038/s41598-017-01666-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette AG, Lesseur C, Armstrong DA, Koestler DC, Appleton AA, Lester BM, & Marsit CJ (2013). Placental HTR2A methylation is associated with infant neurobehavioral outcomes. Epigenetics, 8(8), 796–801. doi: 10.4161/epi.25358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BY, Misra DP, Moye J, Miller RK, Croen L, Fallin MD, … Salafia CM (2018). Placental gross shape differences in a high autism risk cohort and the general population. PloS One, 13(8), e0191276. doi: 10.1371/journal.pone.0191276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos S, Guillemin C, Ergaz Z, Dimov S, Suderman M, Weinstein-Fudim L, … Szyf M (2015). Gestational dabetes alters offspring DNA methylation profiles in human and rat: identification of key pathways involved in endocrine system disorders, insulin signaling, diabetes signaling, and ILK signaling. Endocrinology, 156(6), 2222–2238. doi: 10.1210/en.2014-1643 [DOI] [PubMed] [Google Scholar]
- Pijnenborg R, Vercruysse L, Hanssens M, & Brosens I (2011). Endovascular trophoblast and preeclampsia: A reassessment. Pregnancy Hypertension, 1(1), 66–71. doi: 10.1016/j.preghy.2010.10.010 [DOI] [PubMed] [Google Scholar]
- Prevalence of autism spectrum disorders--Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. (2012). MMWR: Surveillance Summaries, 61(3), 1–19. [PubMed] [Google Scholar]
- Prince CS, Maloyan A, & Myatt L (2017). Maternal obesity alters brain derived neurotrophic factor (BDNF) signaling in the placenta in a sexually dimorphic manner. Placenta, 49, 55–63. doi: 10.1016/j.placenta.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademacher TW, Gumaa K, & Scioscia M (2007). Preeclampsia, insulin signalling and immunological dysfunction: a fetal, maternal or placental disorder? Journal of Reproductive Immunology, 76(1–2), 78–84. doi: 10.1016/j.jri.2007.03.019 [DOI] [PubMed] [Google Scholar]
- Ranzil S, Ellery S, Walker DW, Vaillancourt C, Alfaidy N, Bonnin A, … Murthi P (2019). Disrupted placental serotonin synthetic pathway and increased placental serotonin: Potential implications in the pathogenesis of human fetal growth restriction. Placenta. doi: 10.1016/j.placenta.2019.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzil S, Walker DW, Borg AJ, Wallace EM, Ebeling PR, & Murthi P (2019). The relationship between the placental serotonin pathway and fetal growth restriction. Biochimie, 161, 80–87. doi: 10.1016/j.biochi.2018.12.016 [DOI] [PubMed] [Google Scholar]
- Roberts JM, & Cooper DW (2001). Pathogenesis and genetics of pre-eclampsia. Lancet, 357(9249), 53–56. doi: 10.1016/s0140-6736(00)03577-7 [DOI] [PubMed] [Google Scholar]
- Roberts RM, Xie S, & Mathialagan N (1996). Maternal recognition of pregnancy. Biology of Reproduction, 54(2), 294–302. [DOI] [PubMed] [Google Scholar]
- Rosenfeld CS (2012). Effects of maternal diet and exposure to bisphenol A on sexually dimorphic responses in conceptuses and offspring. Reprod Domest Anim, 47 Suppl 4, 23–30. doi: 10.1111/j.1439-0531.2012.02051.x [DOI] [PubMed] [Google Scholar]
- Rosenfeld CS (2015). Sex-specific placental responses in fetal development. Endocrinology, 156(10), 3422–3434. doi: 10.1210/en.2015-1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CS (2019). Placental serotonin signaling, pregnancy outcomes, and regulation of fetal brain development. Biology of Reproduction. doi: 10.1093/biolre/ioz204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K (2013). Placenta-derived hypo-serotonin situations in the developing forebrain cause autism. Medical Hypotheses, 80(4), 368–372. doi: 10.1016/j.mehy.2013.01.002 [DOI] [PubMed] [Google Scholar]
- Schalekamp-Timmermans S, Arends LR, Alsaker E, Chappell L, Hansson S, Harsem NK, … Steegers EA (2017). Fetal sex-specific differences in gestational age at delivery in pre-eclampsia: a meta-analysis. International Journal of Epidemiology, 46(2), 632–642. doi: 10.1093/ije/dyw178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RJ, Schroeder DI, Crary-Dooley FK, Barkoski JM, Tancredi DJ, Walker CK, … LaSalle JM (2016). Self-reported pregnancy exposures and placental DNA methylation in the MARBLES prospective autism sibling study. Environ Epigenet, 2(4). doi: 10.1093/eep/dvw024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder DI, Schmidt RJ, Crary-Dooley FK, Walker CK, Ozonoff S, Tancredi DJ, … LaSalle JM (2016). Placental methylome analysis from a prospective autism study. Molecular Autism, 7, 51. doi: 10.1186/s13229-016-0114-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinkopf SJ, Righi G, Marsit CJ, & Lester BM (2016). Methylation of the Glucocorticoid Receptor (NR3C1) in Placenta Is Associated with Infant Cry Acoustics. Frontiers in Behavioral Neuroscience, 10, 100. doi: 10.3389/fnbeh.2016.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CZ, & Zhuang LZ (1993a). Norepinephrine regulates human chorionic gonadotrophin production by first trimester trophoblast tissue in vitro. Placenta, 14(6), 683–693. doi: 10.1016/s0143-4004(05)80385-6 [DOI] [PubMed] [Google Scholar]
- Shi CZ, & Zhuang LZ (1993b). Stimulatory effect of norepinephrine on progesterone production by human first trimester placenta explants in vitro. Life Sciences, 52(20), 1657–1665. doi: 10.1016/0024-3205(93)90048-8 [DOI] [PubMed] [Google Scholar]
- Straughen JK, Misra DP, Divine G, Shah R, Perez G, VanHorn S, … Salafia CM (2017). The association between placental histopathology and autism spectrum disorder. Placenta, 57, 183–188. doi: 10.1016/j.placenta.2017.07.006 [DOI] [PubMed] [Google Scholar]
- Street ME, Viani I, Ziveri MA, Volta C, Smerieri A, & Bernasconi S (2011). Impairment of insulin receptor signal transduction in placentas of intra-uterine growth-restricted newborns and its relationship with fetal growth. European Journal of Endocrinology of the European Federation of Endocrine Societies, 164(1), 45–52. doi: 10.1530/eje-10-0752 [DOI] [PubMed] [Google Scholar]
- Stroud LR, Papandonatos GD, Salisbury AL, Phipps MG, Huestis MA, Niaura R, … Lester BM (2016). Epigenetic regulation of placental NR3C1: mechanism underlying prenatal programming of infant neurobehavior by maternal smoking? Child Development, 87(1), 49–60. doi: 10.1111/cdev.12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrade A, Panchenko P, Junien C, & Gabory A (2015). Placental contribution to nutritional programming of health and diseases: epigenetics and sexual dimorphism. Journal of Experimental Biology, 218(Pt 1), 50–58. doi: 10.1242/jeb.110320 [DOI] [PubMed] [Google Scholar]
- Tarrade A, Rousseau-Ralliard D, Aubriere MC, Peynot N, Dahirel M, Bertrand-Michel J, … Chavatte-Palmer P (2013). Sexual dimorphism of the feto-placental phenotype in response to a high fat and control maternal diets in a rabbit model. PloS One, 8(12), e83458. doi: 10.1371/journal.pone.0083458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BD, Ness RB, Klebanoff MA, Tang G, Roberts JM, Hougaard DM, … Haggerty CL (2018). The impact of female fetal sex on preeclampsia and the maternal immune milieu. Pregnancy Hypertension, 12, 53–57. doi: 10.1016/j.preghy.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekola-Ayele F, Workalemahu T, Gorfu G, Shrestha D, Tycko B, Wapner R, … Louis GMB (2019). Sex differences in the associations of placental epigenetic aging with fetal growth. Aging (Albany NY), 11(15), 5412–5432. doi: 10.18632/aging.102124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobita T, Kiyozumi D, & Ikawa M (2017). Placenta-specific gene manipulation using lentiviral vector and its application. Placenta, 59 Suppl 1, S37–s43. doi: 10.1016/j.placenta.2017.09.012 [DOI] [PubMed] [Google Scholar]
- Tomogane H, Arbogast LA, Soares MJ, Robertson MC, & Voogt JL (1993). A factor(s) from a rat trophoblast cell line inhibits prolactin secretion in vitro and in vivo. Biology of Reproduction, 48(2), 325–332. doi: 10.1095/biolreprod48.2.325 [DOI] [PubMed] [Google Scholar]
- Tsamou M, Martens DS, Cox B, Madhloum N, Vrijens K, & Nawrot TS (2018). Sex-specific associations between telomere length and candidate miRNA expression in placenta. Journal of Translational Medicine, 16(1), 254. doi: 10.1186/s12967-018-1627-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja G, Chung T, & Bejerano G (2016). Changes in the enhancer landscape during early placental development uncover a trophoblast invasion gene-enhancer network. Placenta, 37, 45–55. doi: 10.1016/j.placenta.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillancourt C, Petit A, Gallo-Payet N, Bellabarba D, Lehoux JG, & Belisle S (1994). Labelling of D2-dopaminergic and 5-HT2-serotonergic binding sites in human trophoblastic cells using [3H]-spiperone. Journal of Receptor Research, 14(1), 11–22. [DOI] [PubMed] [Google Scholar]
- Walker CK, Anderson KW, Milano KM, Ye S, Tancredi DJ, Pessah IN, … Kliman HJ (2013). Trophoblast inclusions are significantly increased in the placentas of children in families at risk for autism. Biological Psychiatry, 74(3), 204–211. doi: 10.1016/j.biopsych.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CK, Krakowiak P, Baker A, Hansen RL, Ozonoff S, & Hertz-Picciotto I (2015). Preeclampsia, placental insufficiency, and autism spectrum disorder or developmental delay. JAMA Pediatrics, 169(2), 154–162. doi: 10.1001/jamapediatrics.2014.2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CK, VandeVoort CA, Li CS, Chaffin CL, & Capitanio JP (2018). Adiposity and weight gain during pregnancy associate independently with behavior of infant rhesus monkeys (Macaca mulatta). Developmental Psychobiology, 60(6), 629–638. doi: 10.1002/dev.21744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Geng H, Liu W, & Zhang G (2017). Prenatal, perinatal, and postnatal factors associated with autism: A meta-analysis. Medicine (Baltimore), 96(18), e6696. doi: 10.1097/md.0000000000006696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel PL, & Leone G (2007). Expression of Cre recombinase in early diploid trophoblast cells of the mouse placenta. Genesis, 45(3), 129–134. doi: 10.1002/dvg.20276 [DOI] [PubMed] [Google Scholar]
- Wu WL, Hsiao EY, Yan Z, Mazmanian SK, & Patterson PH (2017). The placental interleukin-6 signaling controls fetal brain development and behavior. Brain, Behavior, and Immunity, 62, 11–23. doi: 10.1016/j.bbi.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CJ, Tan HP, & Du YJ (2014). The developmental disruptions of serotonin signaling may involved in autism during early brain development. Neuroscience, 267, 1–10. doi: 10.1016/j.neuroscience.2014.02.021 [DOI] [PubMed] [Google Scholar]
- Zhu P, Wang W, Zuo R, & Sun K (2019). Mechanisms for establishment of the placental glucocorticoid barrier, a guard for life. Cellular and Molecular Life Sciences, 76(1), 13–26. doi: 10.1007/s00018-018-2918-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Mordaunt CE, Yasui DH, Marathe R, Coulson RL, Dunaway KW, … LaSalle JM (2019). Placental DNA methylation levels at CYP2E1 and IRS2 are associated with child outcome in a prospective autism study. Human Molecular Genetics, 28(16), 2659–2674. doi: 10.1093/hmg/ddz084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhang W, Chen M, Liu N, & Guo J (2002). [Study on expression of norepinephrine and dopamine placental tissues of normal pregnancy and pregnancy induced hypertension syndrome]. Zhonghua Fu Chan Ke Za Zhi, 37(3), 142–145. [PubMed] [Google Scholar]