

Abstract
Background
Burst suppression in mechanically ventilated intensive care unit (ICU) patients is associated with increased mortality. However, the relative contributions of propofol use and critical illness itself to burst suppression; of burst suppression, propofol, and critical illness to mortality; and whether preventing burst suppression might reduce mortality, have not been quantified.
Methods
The dataset contains 471 adults from seven ICUs, after excluding anoxic encephalopathy due to cardiac arrest or intentional burst suppression for therapeutic reasons. We used multiple prediction and causal inference methods to estimate the effects connecting burst suppression, propofol, critical illness, and in-hospital mortality in an observational retrospective study. We also estimated the effects mediated by burst suppression. Sensitivity analysis was used to assess for unmeasured confounding.
Results
The expected outcomes in a “counterfactual” Randomized Controlled Trial (cRCT) that assigned patients to mild vs. severe illness is expected to show a difference in burst suppression burden of 39%, 95% CI [8–66]%, and in mortality of 35% [29–41]%. Assigning patients to maximal (100%) burst suppression burden is expected to increase mortality by 12% [7–17]% compared to 0% burden. Burst suppression mediates 10% [2–21]% of the effect of critical illness on mortality. A high cumulative propofol dose (1316 mg/kg) is expected to increase burst suppression burden by 6% [0.8–12]% compared to a low dose (284 mg/kg). Propofol exposure has no significant direct effect on mortality; its effect is entirely mediated through burst suppression.
Conclusions
Our analysis clarifies how important factors contribute to mortality in ICU patients. Burst suppression appears to contribute to mortality but is primarily an effect of critical illness rather than iatrogenic use of propofol.
Keywords: Burst Suppression, Mortality, Critical Care, Electroencephalography
Introduction
Burst suppression is a pattern of brain activity where the electroencephalogram (EEG) is intermittently interrupted by “suppressions,” i.e., periods of reduced voltage (Figure 1). Prior studies in mechanically ventilated ICU patients found that burst suppression is associated with increased 30-day, 6-month, and one-year mortality, and with increased incidence and duration of delirium 1,2. Based on these findings, prior authors have recommended measures to reduce use of sedatives, particularly propofol, a GABA-A receptor agonist.
Figure 1.
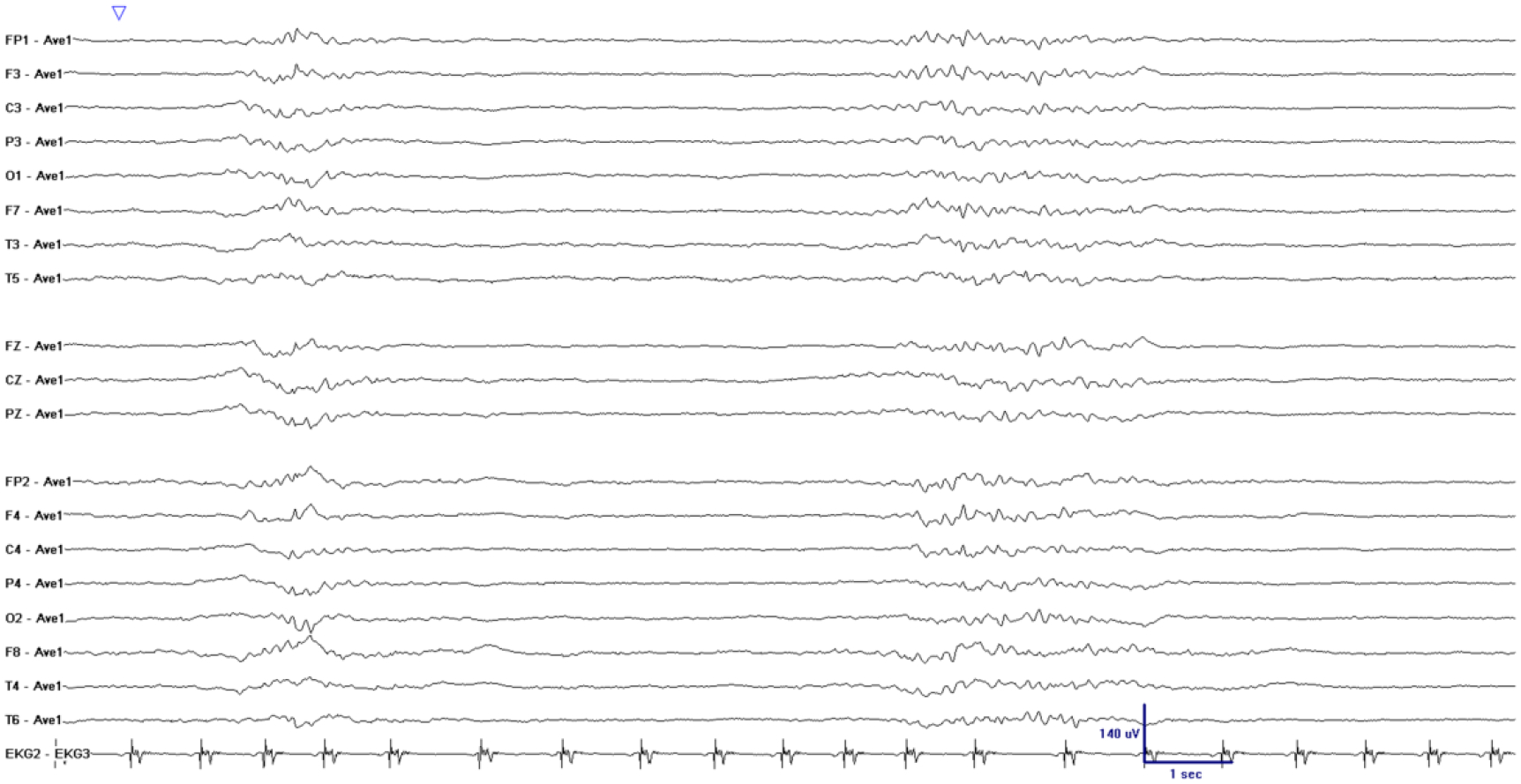
Example of unintentional burst suppression in an EEG recording from a mechanically ventilated ICU patient receiving propofol. There are two periods of higher amplitude oscillatory EEG activity (bursts), surrounded by relatively flat periods (suppressions). Electrode names refer to standard 10–20 placement, with odd-numbered electrodes placed over the left and even-numbered electrodes placed over the right hemisphere, and “Z” electrodes placed along the midline. Voltages shown are referenced to the global voltage average (labeled “Ave1”).
However, the complete picture remains unclear: among patients not receiving intentional burst suppression for therapeutic reasons or anoxic encephalopathy, how much do propofol, and critical illness itself, contribute to the occurrence of burst suppression? How important is burst suppression as a mediator in the chain of causes and effects that lead to in-hospital death? With these questions unanswered, whether reducing propofol use will reduce burst suppression and whether reducing burst suppression will save lives remains unclear.
While determining effects with a causal interpretation from observational studies is not always possible, recent work has shown that this can be accomplished under certain assumptions3,4. Here we estimate the effects: (1) of propofol dosage and patient critical illness severity on burst suppression; (2) of burst suppression on in-hospital mortality; (3) of propofol exposure on in- hospital mortality; and (4) of critical illness severity on in-hospital mortality. We also estimate the proportion mediated through burst suppression of the effects of propofol and critical illness on mortality5. Sensitivity analysis is used to quantify the robustness of the results to unmeasured or unmeasurable confounding by computing the E-value6. These approaches offer a step towards a mechanistic understanding of the causes and effects of burst suppression, where randomized trials are impossible or unethical to carry out.
Methods
Ethics Approval
This observational study was approved by the Institutional Review Board of Partners HealthCare, which waived the need for informed consent for this study. The study also complied with the World Medical Association Declaration of Helsinki regarding the ethical conduct of research involving human subjects.
Patients
Patient data were collected from seven Intensive Care Units (ICUs) at Massachusetts General Hospital (MGH) between January 2014 and December 2017. The inclusion criteria were: (1) age ≥18 years; (2) underwent continuous EEG monitoring for at least 18 hours; (3) on mechanical ventilation at the time EEG monitoring was initiated. The exclusion criteria were: (1) anoxic encephalopathy due to cardiac arrest; (2) intentional burst suppression for therapeutic reasons, such as refractory intracranial hypertension, status epilepticus, or acute respiratory distress syndrome (ARDS) / extracorporeal membrane oxygenation (ECMO) / mechanically ventilated patients that needed paralysis and paralytics for any other reason; (3) ongoing status epilepticus during EEG monitoring (patients with status epilepticus as the presenting complaint, however, were not excluded). The final cohort contained 471 patients that received anesthetics or anxiolytics purely for sedation purposes.
Patients in this dataset had a median age of 61 years with interquartile range 50 years to 72 years with almost equal number of genders (Female 233 and Male 238). 27% of the patients (128/471) died in the hospital. Half of the patients (238/471) experienced some degree of unintentional burst suppression. The histogram of the burst suppression burden (defined below) is shown in Figure S1 in the Supplementary Figures. It is notable that over half of the patients in the cohort had structural brain injuries (62.7%). Patient characteristics are described in Table 1. Potential confounding variables were selected based on clinical grounds from a list of 120 factors. Inclusion criteria for potential confounding variables were: (1) the possibility (based on pharmacologic considerations) of affecting both unintentional burst suppression and in-hospital mortality7; and (2) that data was available for more than 90% of patients.
Table 1.
Patient characteristics
| Category | Variable | Value |
|---|---|---|
| Number of patients | 471 | |
| Length of hospital stay: days, median (IQR) | 16 (9 – 26) | |
| Length of ICU stay: days, median (IQR) | 9 (5 – 16) | |
| M | In-hospital mortality: n (%) | 128 (27.2%) |
| B | Burst Suppression Burden: median % (IQR) | 9% (0% – 60%) |
| D | Propofol: mg/kg, median (IQR) | 230.5 (86.7 – 570.4) |
| Patient covariates including critical illness (C) | Age: year, median (IQR) | 61 (50 – 72) |
| Gender: n (%) | Female 233 (49.5%); Male 238 (50.5%) | |
| Smoker: n (%) | 225 (47.8%) | |
| Alcohol: n (%) | 125 (26.5%) | |
| Substance abuse: n (%) | 57 (12.1%) | |
| APACHE II: median (IQR) | 23 (20 – 27) | |
| CCI: median (IQR) | 3 (1 – 6) | |
| Initial Glasgow Coma Scale score: median (IQR) | 7 (3 – 13) | |
| Premorbid modified Rankin Score: median (IQR) | 0 (0 – 2) | |
| Structural brain injury: n (%) | 295 (62.6%) | |
| Hypertension: n (%) | 252 (53.5%) | |
| Respiratory failure: n (%) | 182 (38.6%) | |
| Seizures / status epilepticus on presentation: n (%) | 106 (22.5%) | |
| Hemorrhagic stroke: n (%) | 94 (20.0%) | |
| Cardiovascular accident: n (%) | 92 (19.5%) | |
| Subarachnoid hemorrhage: n (%) | 83 (17.6%) | |
| Septic shock: n (%) | 76 (16.1%) | |
| Toxic metabolic encephalopathy: n (%) | 74 (15.7%) | |
| History of seizure: n (%) | 72 (15.3%) | |
| Subdural hematoma: n (%) | 49 (10.4%) | |
| CNS infection or inflammation: n (%) | 48 (10.2%) | |
| Ischemic stroke: n (%) | 40 (8.5%) | |
| Cardiac failure: n (%) | 39 (8.3%) | |
| Renal failure: n (%) | 35 (7.4%) | |
| Brain tumor: n (%) | 30 (6.4%) | |
| Brain Surgery: n (%) | 29 (6.2%) | |
| Malignancy solid tumors hematologic: n (%) | 27 (5.7%) | |
| Liver failure: n (%) | 24 (5.1%) | |
| Primary psychiatric disorder: n (%) | 20 (4.2%) | |
| CNS Cancer: n (%) | 15 (3.2%) | |
| Endocrine emergency: n (%) | 10 (2.1%) | |
| Primary hematological disorder: n (%) | 9 (1.9%) | |
| Systemic hemorrhage: n (%) | 3 (0.64%) |
EEG Recording
All EEG recordings were obtained using 21 silver-silver chloride electrodes and following the standard international 10–20 system for electrode placement. Raw EEG data were reviewed by two clinical neurophysiologists, and the findings were reported in the electronic medical record every 8 to 12 hours, per institutional protocol. All EEGs were performed on clinical grounds, to evaluate suspected brain dysfunction such as subclinical seizure activity or encephalopathy.
Measures
This dataset is cross sectional, i.e. taken from a single hospitalization for each patient. For every patient, we have the following variables: burst suppression (B), in-hospital mortality (M), cumulative dose of propofol (D), and covariates (C). Note that, although the data is cross sectional, the exposure variables of interest (B, D, C) precede the outcome of interest in time (M), a necessary condition for causal inference. Beyond this requirement, to allow estimating causal effects from our cross sectional dataset, we make the assumptions encoded in the causal graph (Figure 2). These assumptions are based on domain knowledge and clinical experience. The justification of the assumptions represented by this graph is detailed in the Supplementary Methods. The estimated effects are valid to the extent that these assumptions are reasonable.
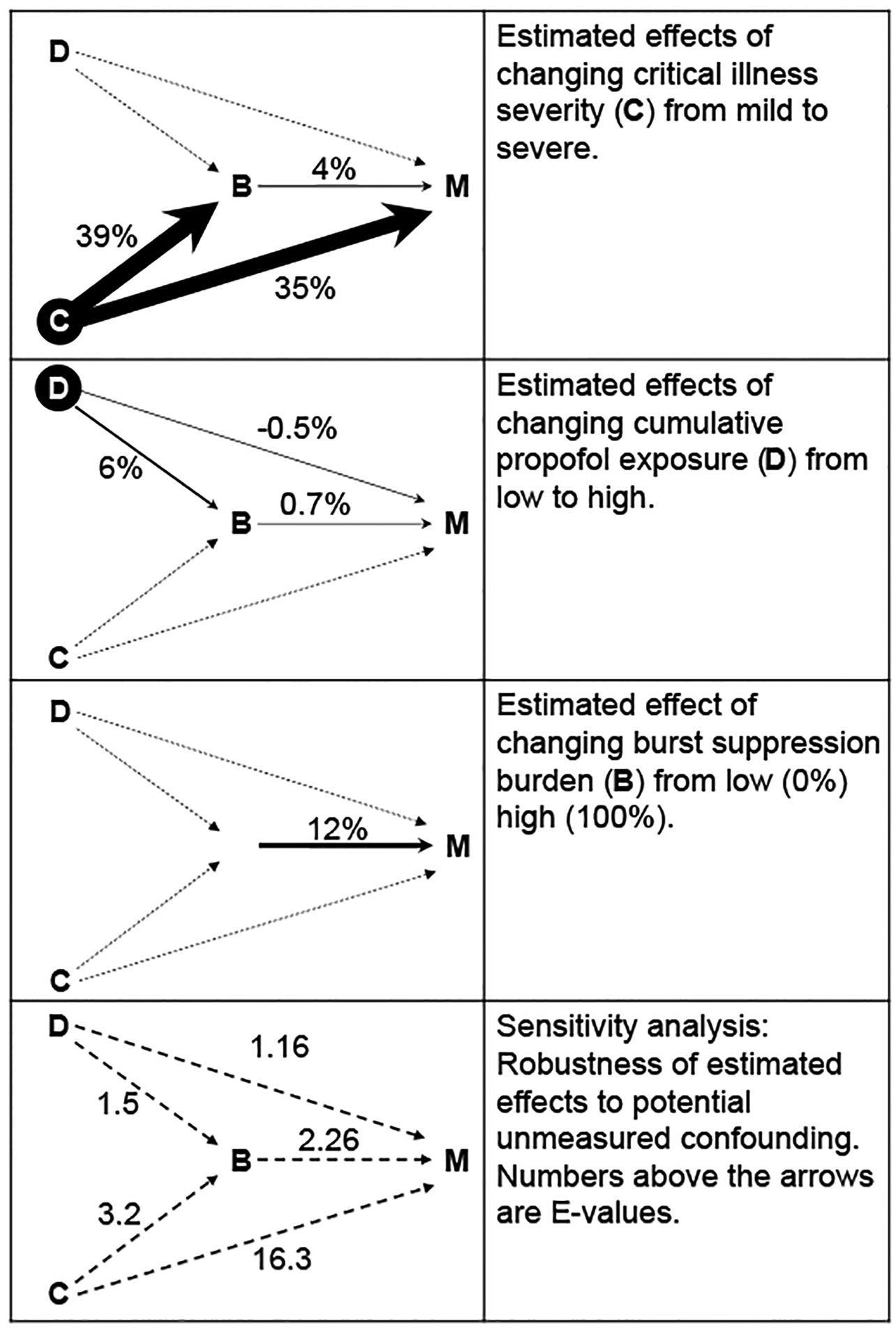
Figure 2.
Estimated direct and mediated effects on mortality of burst suppression burden, clinical condition, and drug exposure. B: burst suppression burden measured by the percentage of 8–12 hour EEG epochs with burst suppression (see methods). M: in-hospital mortality (1, deceased; 0, alive). D: drug, i.e. cumulative propofol exposure. C: patient characteristics including demographics and critical illnesses (Table 1). The estimated effect is shown above each arrow, measured on the difference scale. The width of each arrow is proportional to the estimated effect. Effects are measure as the difference in expected outcome between groups in a counterfactual randomized clinical trial (cRCT), which two groups are randomized to two different levels of B, D, or C. (A) Difference in mortality between patients randomized to 0% vs 100% burst suppression burden. (B)Difference in mortality between patients randomized to high vs. low cumulative exposure to propofol. Direct (D->M) and indirect (D->B->M) effects are shown. (C) Difference in mortality between ICU patients randomized to severe vs mild critical illness severity. Direct (C->M) an indirect (C->B->M) effects are shown. (D) Sensitivity analysis, measuring the minimum risk ratio of unmeasured confounding factor to either the exposure or the outcome that would be needed to explain away the estimated effect (E-value). These results were obtained using random forest and doubly robust estimation.
In-hospital mortality (M): This is the outcome. Mortality is 1 if the patient was deceased during hospital stay; otherwise 0.
Burst Suppression (B): Burst suppression burden (BSB) is defined as the percentage of epochs for each patient containing a generalized/global EEG pattern with intermittent periods of suppression ≤ 10 μV, with these periods of suppression occupying 50% or more of an epoch8. Burst suppression was assessed visually by clinical neurophysiologists as part of clinical care and reported in the medical record in epochs of 8–12 hours duration.
Propofol infusion (D): We computed the body-weight-normalized cumulative propofol dose (mg/kg) during ICU stay.
Patient covariates (C): Demographics and critical illnesses were included on the basis that they can potentially affect both the propensity for burst suppression and mortality (Table 1).
Effect Estimation
Effects derived from causal statistical inference can be understood as estimates of the differences (e.g. in mortality, or burst suppression burden) that one should expect to observe in various “counterfactual” randomized controlled trials (cRCTs), in which one factor is randomly assigned for each patient while other factors are held constant. These effects are counterfactual because they are estimated based on observational data, rather than directly measured in actually-performed trials.
The first step in estimating the effects is to fit two prediction models. One for mortality while adjusting for burst suppression, comorbidities, and propofol exposure, P(M|B, C, D); and another for burst suppression, adjusting for comorbidities and propofol exposure, P(B|C, D). To be robust to model misspecification, we evaluated three different nonparametric models: gradient boosted tree, random forest, and BART (Bayesian Additive Regression Trees)9 in additional to logistic regression (see Supplementary Methods regarding the rationale for considering nonparametric models). We selected the model with the best prediction performance. Each model was fit using nested 10-fold cross-validation, consisting of an external and an internal loop. The internal loop was used to select the model hyperparameters (Table S1 in the Supplementary Methods) with the best average performance on the validation folds. The external loop was used to obtain the final performance by comparing the true values and the pooled predictions from all held-out testing folds.
The next step is to define two representative patients (“base cases”) in the dataset, with respect to which we calculate patient contrasts. When estimating the effects of C->B, D->B, C->M, and D->M, we use these representative patients where the first patient representing mild illness has covariates C0 and cumulative propofol exposure D0; the second patient representing severe illness has characteristics C1 and D1. To obtain C0, C1, and D0, D1, we first compute two anchor points corresponding to the 10% (mildly ill) and 90% (severely ill) percentiles of age, initial Glasgow Coma Scale, premorbid modified Rankin Scale, APACHE II, Charlson Comorbidity Index, and normalized propofol dose. C0 and D0 are defined as the values of the patient closest to the 10% anchor point in terms of Euclidean distance after z-scoring; similarly, C0 and D0 are defined as the values closest to the 90% anchor point. The values of C0, C1, and D0, D1 are shown in Table 2.
Table 2.
The patient characteristics and propofol exposure for the representative (“base case”) patients
| Variable | Mild Illness | Severe Illness |
|---|---|---|
| Age (year) | 30.5 | 83 |
| Gender | Female | Female |
| Smoker | No | No |
| Alcohol | No | No |
| Substance abuse | No | No |
| APACHE II | 15 | 26 |
| CCI | 0 | 8 |
| Initial Glasgow Coma Scale score | 12 | 3 |
| Premorbid modified Rankin Score | 0 | 5 |
| Structural Brain Injury | No | Yes |
| Hypertension | No | No |
| Respiratory failure | Yes | No |
| Seizure or Status Epilepticus | No | Yes |
| Hemorrhagic stroke | No | No |
| Cardiovascular accident | No | No |
| Subarachnoid hemorrhage | No | No |
| Septic shock | No | No |
| Toxic metabolic encephalopathy | No | No |
| History of Seizure | No | No |
| Subdural hematoma | No | No |
| CNS infection or inflammation | Yes | No |
| Ischemic stroke | No | Yes |
| Cardiac failure | No | Yes |
| Renal failure | No | No |
| Brain tumor | No | No |
| Brain Surgery | No | No |
| Malignancy solid tumors hematologic | No | No |
| Liver failure | No | No |
| Primary psychiatric disorder | No | No |
| CNS cancer | No | No |
| Endocrine emergency | No | No |
| Primary hematological disorder | No | No |
| Systemic hemorrhage | No | No |
| Propofol (mg/kg) | 284.1 | 1315.5 |
The effect of C->B is denoted as E[B(C1)] – E[B(C0)] on the difference scale and E[B(C1)] / E[B(C0)] on the risk ratio scale, where E[] is the expectation (mean value) and B(c) is the potential outcome of BSB that would result in a cRCT in which patients were somehow randomly assigned have their covariates set to value c. The effect for D->B is similarly defined. The methods used to compute E[B(c)] or E[B(d)] are described in the Supplementary Methods.
The effect of B->M is denoted as E[M(b=100%)] – E[M(b=0%)] on the difference scale and E[M(b=100%)] / E[M(b=0%)] on the risk ratio scale. We used four different causal inference methods to compute the effect: outcome regression (OR, equivalent to adjustment), inverse propensity weighting (IPW), marginal structural modeling (MSM), and doubly robust estimation (DRE). The method to compute E[M(b)] is described in the Supplementary Methods. The results reported in the main text use DRE due to its theoretically proved lower bias in the face of model misspecification.
Mediation Analysis
In our analysis, the patient covariates (C) and drug (propofol) exposure (D) are considered the exposures; burst suppression (B) is the mediator; and mortality (M) is the outcome The total effect of C->M or D->M can be decomposed into four components5.
Direct effect: The effect of the exposure (C/D) on mortality (M) in the absence of a mediator (B=0%);
Reference interaction: an effect on mortality (M) due to an interaction between the exposure (C/D) and the mediator (B), when the mediator takes the value it would have in the absence of the exposure;
Mediated interaction: an effect on mortality due to an interaction between the exposure (C/D) and the mediator (B), when the exposure has an effect on the mediator;
Pure indirect effect: The effect of the mediator (B) on mortality (M) without the exposure (C/D = 0) if the exposure (C/D) has an effect on the mediator (B); that is, a purely mediated effect.
Technical details are provided in the Supplementary Methods.
Sensitivity Analysis
The E-value was recently proposed as a way to assess the robustness of effect estimates to unmeasured confounding6. This value, calculated as , quantifies the minimum strength of unmeasured confounding on either the cause or effect that would be needed to explain away the estimated effect, expressed on the risk ratio (RR) scale.
Confidence Intervals
Confidence intervals were estimated nonparametrically using bootstrapping, which simulates the sampling process from an infinite population. Specifically, the effect of each arrow is calculated 200 times. In each calculation, a bootstrapped dataset the same size as the actual dataset was obtained by random sampling with replacement. The effects estimated across the bootstrap iterations formed a null distribution. The 95% confidence interval was estimated using the 2.5% and 97.5% percentiles of this null distribution. P-values are provided for univariate associations. P-values of the estimated effects are not stated, in accordance with recent literature7.
Results
Predicting Burst Suppression Burden and Mortality
In Table 3, we show the cross-validation performance of predicting burst suppression burden using patient covariates and propofol dose (B | C, D); and the cross-validation performance of predicting mortality using patient covariates, propofol dose, and burst suppression burden (M | B, C, D). Overall, the best performance is achieved by the random forest model, where the spearman’s correlation is 0.188 (0.097 – 0.277) for predicting burst suppression, and the area under ROC is 0.682 (0.631 – 0.732) for predicting mortality. The results suggest a nonlinear relationship between these variables. In the next subsection, we show the effect estimates based on random forest.
Table 3.
Prediction performance of each prediction model from external cross-validation
| Prediction Model | B | C, D Spearman’s rho correlation | M | B, C, D Area under ROC |
|---|---|---|
| Logistic regression | 0.158 (0.074 – 0.247) | 0.634 (0.581 – 0.689) |
| Gradient boosted trees | 0.174 (0.084 – 0.264) | 0.684 (0.632 – 0.737) |
| Random forest | 0.188 (0.097 – 0.277) | 0.682 (0.631 – 0.732) |
| Bayesian additive regression tree (BART) | 0.144 (0.054 – 0.230) | 0.680 (0.627 – 0.731) |
The ten most important variables for predicting burst suppression burden based on random forest are (descending order): Age, propofol, APACHE II, initial GCS, CCI, premorbid mRS, seizure / status epilepticus, structural brain injury, gender, and smoking. The ten most important variables for predicting mortality are (descending order): Age, CCI, propofol, APACHE II, septic shock, initial GCS, burst suppression burden, premorbid mRS, hemorrhagic stroke, and seizure / status epilepticus.
Estimated Direct an Mediated Effects
We assessed the effects of the burst suppression burden (B), exposure to propofol (D), and clinical condition (C) on mortality, by contrasting the expected mortality rate under two different levels for each of the explanatory variables B, D, and C. These effects represent the estimated difference we would expect to see in a series of counterfactual randomized clinical trials (cRCTs), each with two groups: one randomized to each of the two levels of the explanatory variables.
Effect of burst suppression on mortality (Figure 2A):
To quantify the effect of burst suppression on in-hospital mortality, we estimated the expected difference in outcome for a cRCT in which one group had their burst suppression burden set to 0%, and the other to 100%. The effect is a 12% [95% CI 7–17] increase in mortality, or a 1.45 times higher mortality risk ratio.
Effect of propofol exposure on burst suppression burden and on mortality (Figure 2B):
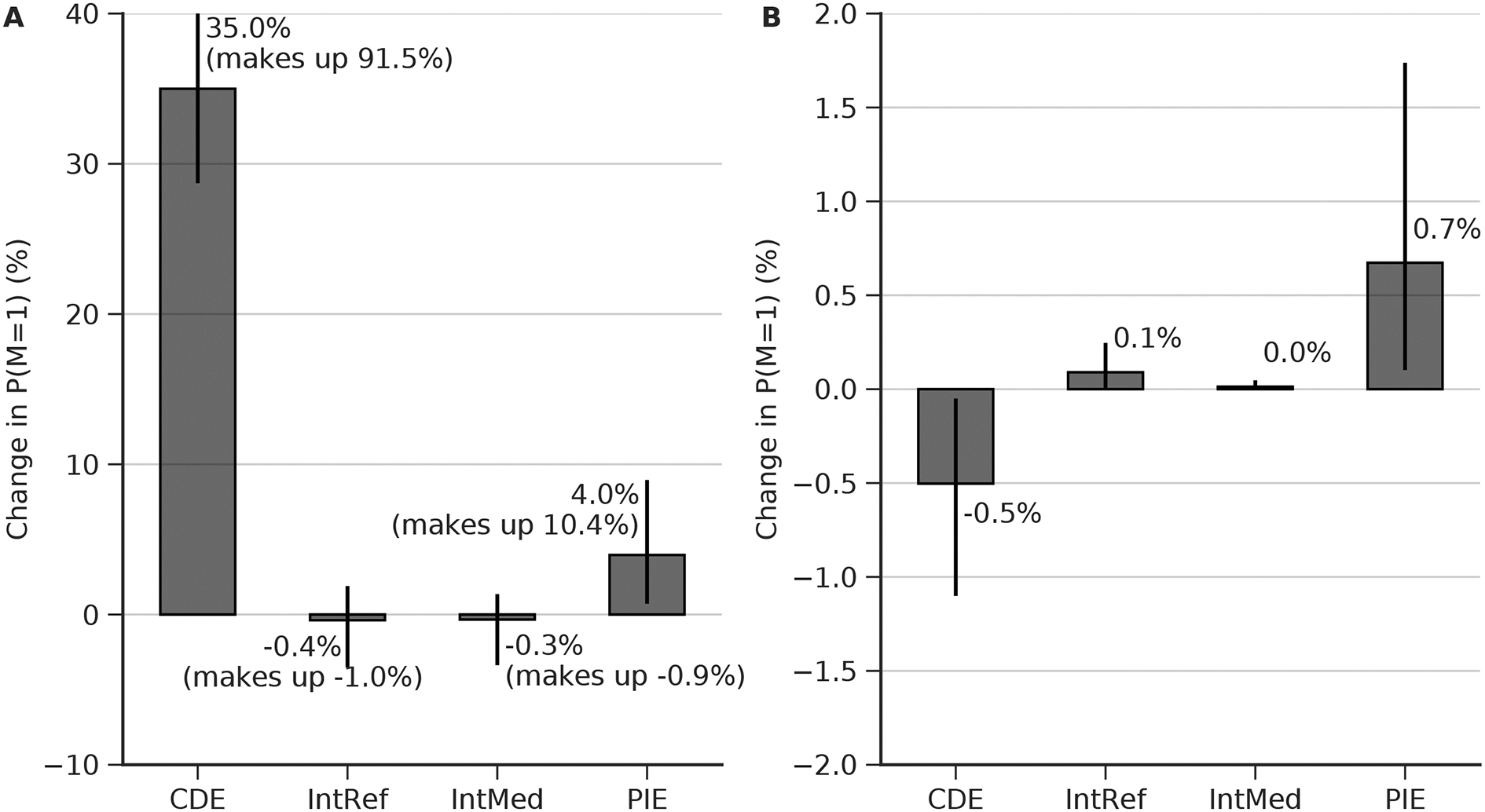
To measure the effect of propofol exposure on mortality, we estimated the expected difference in outcome for a cRCT in which one group received a -weight-normalized cumulative propofol dose over their ICU stay of 284.1 mg/kg (“low dose”), while the other group received 1315.5 mg/kg (“high dose”). In our analysis, propofol can affect mortality either directly, or indirectly by changing the burst suppression burden. The average direct effect is to decrease mortality slightly, by 0.5% [0.05–1]. However, this is offset by the indirect effects, which are to increase burst suppression burden 6% [1–12] (or by 1.14 [1.02 – 1.35] times on the risk ratio scale), which in turn slightly increases mortality by 0.7% [0.1 – 2.0] (pure indirect effect). In principle there are two other potential mediated effects (the reference interaction and mediated interaction effects); however, these turn out to be negligible. All four effects are shown in Figure 3B. The total effect (sum of all effects) of propofol exposure on in-hospital mortality is essentially negligible (an increase of 0.3% [−0.5 – 2.0]). The fact that the total effect is negligible is because the direct effect (slight benefit) and the mediated effect (slight harm) essentially cancel each other.
Figure 3.
(A) Four-way decomposition of the total effect from C to M. DE: direct effect. IntRef: reference interaction. IntMed: mediated interaction. PIE: pure indirect effect. The vertical line indicates 95% confidence interval, with the lower end 2.5% and upper end 97.5%. The percentage in the parenthesis indicates the percentage in the total effect. (B) the four-way decomposition of the total effect from D to M.
Effect of clinical condition on burst suppression burden and on mortality (Figure 2C):
To measure the effect of clinical condition on mortality, we estimated the expected difference in outcome for a cRCT in which one group of ICU patients is “assigned” to have mild critical illness, while the other group is assigned to be more severely ill. In our analysis, clinical condition can affect mortality either directly, or indirectly by changing the burst suppression burden. The average direct effect is to increase mortality by 35% [29–41] (8.42 [4.66–44.17] times increased risk ratio). In addition, we estimate a 39% [8–66] increase in burst suppression burden (1.88 [1.16 – 4.93] times increased risk ratio), which in turn leads to a 4% [0.7 – 9] (pure indirect effect) increase in mortality. In principle there are two other potential mediated effects (the reference interaction and mediated interaction effects); however, these turn out to be very small (not shown in Figure 2; all four effects are shown in Figure 3A). The total effect (sum of all effects) of propofol exposure on in-hospital mortality is 38% [32–44]. We note that the direct effect thus accounts for 91.5% of the total effect, while the effect on burst suppression mediates 10.4% of the total effect of critical illness on mortality.
The complete list of estimated effects is provided in the Supplementary Tables (Table S2).
Sensitivity Analysis
There are many potential unmeasured confounding variables in any cross-sectional study. In our case they are defined as any unmeasured variable that affects two or more variables among D, B, and M, which include (1) the differences between the seven ICUs such as room temperature and noise level; (2) other variables not included in the covariates C such as genetic factors.
The E-values for each of the estimated effects are shown in Figure 2C. For example, to explain away the estimated effect of burst suppression on in-hospital mortality as due to cofounding, there would need to be an unmeasured factor U that makes either the expected burst suppression burden or the expected in-hospital mortality rate 2.27 times more likely when U has a unit change (all other factors remaining constant). The same explanation applies to other pairs.
Is it likely that unmeasured confounding factors of this magnitude exist? It seems unlikely that variation in environmental factors such as room temperature or noise levels in different ICUs could have such a large effect on either burst suppression burden or in-hospital mortality. With regard to potential genetic factors, a rare genetic disorder (Ohtahara syndrome10) has been reported to have a high rate of patients with burst suppression. Although this syndrome is rare and was not present in our cohort, it may be instructive to (roughly) estimate the effect it might have had if present as an unmodeled confounder, to provide perspective on the calculated E values. In 33 patients diagnosed with Ohtahara syndrome, 28 had burst suppression, among whom 10 had a KCNQ2 gene variant; none of 5 patients without burst suppression had the KCNQ2 gene variant. Therefore in patients with KCNQ2 gene variant, 10 out of 10 had burst suppression (100%), and in patients without KCNQ2 gene variant, 18 (28-10) out of 23 (28-10+5-0) had burst suppression (78.3%), yielding a risk ratio of 100%/78.3% = 1.28, lower than the E-value 2.27 for B and M. These considerations lead us to conclude that, while it is likely that unmeasured confounding exists (as in any analysis of observational data; see discussion section), the estimated effects may nevertheless be relatively robust to the presence of unmeasured confounding.
Discussion
In this study, we investigated the mechanisms by which burst suppression and mortality occur in ICU patients. Our results suggest that critical illness itself, as opposed to exposure to high doses of anesthetic/sedative drugs, is the dominant driver of both burst suppression and mortality.
Prior studies have also shown that burst suppression is associated with increased mortality, which is consistent with our finding. A prior study on 125 mechanically ventilated adult ICU patients found that unintentional burst suppression (taken as a dichotomized variable) was associated with 6-month mortality, with hazard ratio of 2.04 (1.12 – 3.70) based on a time-dependent Cox proportional hazard regression model after adjusting for clinically import covariates, including CCI, baseline dementia, APACHE II, sequential organ failure assessment (SOFA), daily coma and delirium status1. In our case, we used the nonparametric model random forest, and doubly robust estimation which is robust such that it only requires either the burst suppression prediction or the mortality prediction model to be correctly specified3. Apart from differences in estimation methods, the difference between their results and ours suggests that the clinical setting and patient population, i.e., ICU vs. operating room, may be an effect modifier.
Our results argue that most unintentional burst suppression is probably not due to use of high propofol doses; rather, burst suppression is primarily attributable to critical illness itself. Two possible explanations exist. The first is that critical illness itself is primarily responsible for a large portion of burst suppression in the ICU. In support of this possibility, burst suppression in the EEG has been reported as one manifestation of severe sepsis11,12. Our analysis suggests that burst suppression is likely a manifestation of a wide range of severe medical illnesses. In fact, our mediation analysis suggests that burst suppression is an important mechanism (or readout for such mechanisms) by which critical illness leads to death, accounting for 10% in the increase of mortality. A second, compatible explanation is that severe illness alters the brain’s pharmacodynamic response to sedative drugs, such that low doses that would ordinarily induce only sedation instead suffice to induce burst suppression. Under either of these explanations, burst suppression serves a biomarker of neurological and medical “vulnerability”. This suggests that the problem is not primarily exposure to high doses of sedative drugs, and that avoidance of burst suppression by reducing sedative use would not likely have a large effect on mortality. Note that our results still do not rule out the possibility that closely monitoring a patient’s EEG in order to personalize propofol dosing might reduce mortality to a modest degree. That is, while merely avoiding “over-dosing” is unlikely to impact mortality, it is still possible that tailoring doses to the bare minimum needed for an individual (recognizing that some individuals require substantially lower-than-average doses to achieve the same pharmacodynamic effect) could be beneficial. A recent randomized clinical trial appears to provide some evidence along these lines: The trial was designed to test whether EEG-guided anesthesia could reduce the incidence of intra-operative burst suppression; while intra-operative burst suppression was reduced, this had no effect on delirium. Nevertheless, the reduction in burst suppression was associated with a small (2.4%) reduction in 30-day mortality13.
In terms of the validity of our effect estimates, the similarity in magnitude and direction of estimated effects (see Table S3 and Table S4 in the Supplementary Tables) among multiple methods suggests that these estimates are robust to model specification. The fact that two out of the three nonparametric models were more accurate than logistic regression in predicting burst suppression, and that all three nonparametric methods outperformed logistic regression in predicting mortality, suggests a nonlinear relationship exists between the variables. Therefore, nonparametric methods could be a more precise modeling method for predicting and effect estimation in a clinical setting, albeit at the price of reduced clinical interpretability.
Our study has several important limitations. (1) Our results may be influenced by selection bias. In current clinical practice, continuous EEG is not routinely performed in mechanically ventilated ICU patients; rather it is performed primarily in cases with suspected brain dysfunction such as subclinical seizure activity or encephalopathy, conditions themselves associated with increased mortality. The fact that more than 60% of patients in our dataset had structural brain injury reflects the fact that continuous EEG use is generally symptom-driven as opposed to randomly assigned. Thus, the effect sizes estimated in our study might not generalize to all ICU patients. (2) Our estimation of burst suppression burden (BSB) is coarse, being based on relatively long epochs (8–12 hours in duration, see Methods). Continuous quantitative evaluation of burst suppression would provide higher time resolution and might allow more granular insights into the effects. (3) The way we delineated mild illness from severe illness using 10% and 90% percentiles is arbitrary although reasonable. Due to its continuous nature, the effect size is dependent on the contrast we pick. The patient closest to 10th percentile is a 30 year old female with no comorbidities who presented with CNS infection and respiratory failure, representative of a typical young patient with acute illness in the ICU; while the patient closest to the 90th percentile is an 83 year old female with multiple comorbidities and severe cerebrovascular abnormalities, representative of a typical elderly ICU patient with chronic illness. (4) We do not have other post-illness morbidity scores. In-hospital mortality considered here is as an important and easy to obtain variable, but ultimately, neurocognitive morbidity is also important to investigate. (5) Our quantification of drug exposure using cumulative dose normalized by body weight ignores the finer temporal structure of drug administration history. Therefore, we do not know how prolonged low exposure vs. brief high doses might differentially affect burst suppression and mortality; knowing these details could provide more detailed guidance for drug dose management. (6) We represented many of the clinical variables as binary (e.g. respiratory failure, seizures or status epilepticus, toxic metabolic encephalopathy). This is a simplification, since in most cases these clinical problems can be given finer-grained characterizations according to etiology (e.g. cause of seizures) and severity (e.g. size of ischemic stroke). Nevertheless, given the variety of clinical problems in the cohort, we felt it prudent to binarize in order to avoid a profusion of small subsets of patients that would make the statistical analysis problematic. Future studies focused on more homogenous but more “deeply phenotyped” cohorts might yield further insights that were not accessible to our approach. (7) Along the same lines, patients came from 7 different ICUs, which have somewhat different patient populations. We did not explicitly adjust for ICU location, thus this is a potential source of unmeasured confounding in our graph. This limitation is mitigated to some extent by the broad range of medical condition variables that we did adjust for, which should to a large extent capture the reasons for patients being in the different ICUs. (8) Also in the same vein, seizures in ICU patients exist along a continuum, ranging from rare intermittent seizures, to frequent seizures, to continuous seizures. There is ongoing debate regarding whether seizures in critically ill patients contribute to poor outcomes vs. directly cause harm; and if they do directly worsen outcomes, there no consensus on the dose-response relationship between seizure burden and neurologic outcomes. Thus, seizure burden is a potentially important confounder omitted from our analysis, and deserves further investigation.
Conclusions
Unintentional burst suppression contributes substantially to in-hospital mortality. However, unintentional burst suppression appears to be caused more by critical illness itself, rather than by high exposure to propofol, and thus may be less modifiable than previously believed.
Supplementary Material
Acknowledgements
This work was supported by NIH-NINDS (1K23NS090900, 1R01NS102190, 1R01NS102574, 1R01NS107291) to MBW. This work was supported by SAGE Therapeutics to MBW, SZ, and ESR.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Declaration of Interests
The authors have disclosed that they do not have any competing interests.
Code Availability
Code for the algorithm development, evaluation, and statistical analysis is open source with no restrictions and is available from https://github.com/mghcdac/burst_suppression_causal_inference.
References
- 1.Watson PL, Shintani AK, Tyson R, Pandharipande PP, Pun BT, Ely EW. Presence of electroencephalogram burst suppression in sedated, critically ill patients is associated with increased mortality. Critical care medicine 2008;36:3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andresen JM, Girard TD, Pandharipande PP, Davidson MA, Ely EW, Watson PL. Burst suppression on processed electroencephalography as a predictor of post-coma delirium in mechanically ventilated ICU patients. Critical care medicine 2014;42:2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernán MARJ. Causal Inference: Boca Raton: Chapman & Hall/CRC; 2019.
- 4.Pearl J Causality: Cambridge University Press; 2009.
- 5.VanderWeele TJ. A unification of mediation and interaction: a four-way decomposition. Epidemiology (Cambridge, Mass) 2014;25:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.VanderWeele TJ, Ding P. Sensitivity analysis in observational research: introducing the E-value. Annals of internal medicine 2017. [DOI] [PubMed] [Google Scholar]
- 7.Lederer DJ, Bell SC, Branson RD, et al. Control of confounding and reporting of results in causal inference studies. Guidance for authors from editors of respiratory, sleep, and critical care journals. Annals of the American Thoracic Society 2019;16:22–8. [DOI] [PubMed] [Google Scholar]
- 8.Hirsch L, LaRoche S, Gaspard N, et al. American clinical neurophysiology society’s standardized critical care EEG terminology: 2012 version. Journal of clinical neurophysiology 2013;30:1–27. [DOI] [PubMed] [Google Scholar]
- 9.Chipman HA, George EI, McCulloch RE. BART: Bayesian additive regression trees. The Annals of Applied Statistics 2010;4:266–98. [Google Scholar]
- 10.Olson HE, Kelly M, LaCoursiere CM, et al. Genetics and genotype–phenotype correlations in early onset epileptic encephalopathy with burst suppression. Annals of neurology 2017;81:419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polito A, Eischwald F, Le Maho A-L, et al. Pattern of brain injury in the acute setting of human septic shock. Critical Care 2013;17:R204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosokawa K, Gaspard N, Su F, Oddo M, Vincent J-L, Taccone FS. Clinical neurophysiological assessment of sepsis-associated brain dysfunction: a systematic review. Critical care 2014;18:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wildes TS, Mickle AM, Abdallah AB, et al. Effect of Electroencephalography-Guided anesthetic administration on postoperative delirium among older adults undergoing major surgery: the engages randomized clinical trial. Jama 2019;321:473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.