

Abstract
Smoking behaviors, including amount smoked, smoking cessation, and tobacco-related diseases, are altered by the rate of nicotine clearance. Nicotine clearance can be estimated using the nicotine metabolite ratio (NMR) (ratio of 3’hydroxycotinine/cotinine), but only in current smokers. Advancing the genomics of this highly heritable biomarker of CYP2A6, the main metabolic enzyme for nicotine, will also enable investigation of never- and former- smokers. We performed the largest genome-wide association study (GWAS) to date of the NMR in European ancestry current smokers (n=5185), found 1255 genome-wide significant variants, and replicated the chromosome 19 locus. Fine-mapping of chromosome 19 revealed 13 putatively causal variants, with nine of these being highly putatively causal and mapping to CYP2A6, MAP3K10, ADCK4, and CYP2B6. We also identified a putatively causal variant on chromosome 4 mapping to TMPRSS11E and demonstrated an association between TMPRSS11E variation and a UGT2B17 activity phenotype. Together the 14 putatively causal SNPs explained ~38% of NMR variation, a substantial increase from the ~20–30% previously explained. Our additional GWASs of nicotine intake biomarkers showed that cotinine and smoking intensity (cotinine/cigarettes per day (CPD)) shared chromosome 19 and 4 loci with the NMR, and that cotinine and a more accurate biomarker, cotinine+3’hydroxycotinine, shared a chromosome 15 locus near CHRNA5 with CPD and Pack-Years (i.e. cumulative exposure). Understanding the genetic factors influencing smoking-related traits facilitates epidemiological studies of smoking and disease, as well as assists in optimizing smoking cessation support, which in turn will reduce the enormous personal and societal costs associated with smoking.
Keywords: genome-wide association study, cigarette smoking, nicotine metabolism, biomarkers, nicotine metabolite ratio
Introduction
Cigarette smoking persists in part due to the reinforcing properties of nicotine, the major psychoactive compound in cigarettes (1). The Nicotine Metabolite Ratio (NMR) (2), the ratio of major nicotine metabolites (3’hydroxycotinine (3HC)/cotinine (COT)), is heritable (h2~80% (3)) and is highly correlated (r=0.83) with the rate of nicotine metabolic clearance (2) and thus associates with numerous smoking behaviors. Higher NMR is associated with greater cigarette consumption and lower cessation (reviewed in (4)). Genetically variable CYP2A6 metabolically inactivates nicotine to COT; COT is then metabolized to 3HC exclusively by CYP2A6 (5, 6). A higher NMR indicates faster nicotine inactivation and CYP2A6 activity (2, 7–9). CYP2A6 also metabolically activates tobacco-specific nitrosamines (10); thus, the association between the NMR and lung cancer (11) may be influenced by both altered smoking quantity (i.e. carcinogen intake) and procarcinogen activation. Further, CYP2A6 can metabolize many other drugs (e.g. tegafur and letrozole) (12, 13).
The NMR prospectively predicts smoking cessation outcomes; smokers with higher NMR had higher quit rates on varenicline versus nicotine patch, whereas quit rates were similar in those with lower NMR (14). Additional tobacco products such as pipe, cigars, and smokeless tobacco contain nicotine, as do newer products (e.g. electronic cigarettes) (15). The NMR influences nicotine intake across many product types, including commercial smokeless tobacco and iqmik (16).
Virtually all of the genome-wide significant (GWS) SNPs in genome-wide association studies (GWASs) of the NMR (3, 17–19) reside in the chromosome 19 region that contains CYP2A6. Here we assessed five cohorts from Australia (20), Finland (3), and North America (14) to conduct the largest NMR meta-GWAS to date in 5185 European ancestry current smokers. We also ran GWASs of biomarkers of nicotine intake, COT+3HC and COT alone; COT+3HC is more accurate than COT due to the accumulation of COT in slow metabolizers (21). To directly investigate genetic similarities and differences between objective and self-reported nicotine intake phenotypes, we also performed GWASs for two self-reported measures, cigarettes smoked per day (CPD) and Pack-Years (i.e. cumulative exposure). Finally, we performed a GWAS of a biomarker of smoking intensity, COT/CPD, using COT given its use as a common nicotine intake biomarker. To our knowledge, these represent the first GWASs of COT+3HC and COT/CPD, and the first within-dataset comparison of the NMR and these numerous smoking biomarkers. This work will enhance NMR genomics for studies that include non-, former- and intermittent-smokers (as NMR can only be measured in current, regular smokers (7)), for example in tobacco-related disease risk assessments (22), as well as in precision medicine approaches for smoking cessation (14, 23) and for cancer due to the role of CYP2A6 in chemotherapeutic (e.g. letrozole, tegafur) drug metabolism (12, 13).
Materials and Methods
Study Samples and Phenotypes
European current smokers (n=5185) with cotinine levels ≥10 ng/ml were studied (see Supplementary information and Tables S1 and S2 for cohort details; cohort data is available from lead investigators). A power curve is shown in Figure S1. Individuals with cotinine <10 ng/ml, suggestive of non-daily smoking (24) and thus unstable NMR measurements, were excluded. The six phenotypes are shown in Figure S2. COT and 3HC were quantified from blood samples using LC-MS/MS or GC-MS (for FINRISK); these previously validated approaches yielded highly concordant results (25). Self-report variables were acquired from surveys. We set all CPD and Pack-Years values of zero to missing and replaced 3HC values below the limit of detection (LOD) of 1 ng/ml with (26). Phenotypes were rank-transformed using the ‘rntransform’ function in the R package ‘GenABEL’(27) to follow the standard normal distribution N(0,1).
GWAS Analyses
Contributing sites performed genotyping, post-genotyping quality control, imputation, and GWAS analyses (Table S2). Cohort-specific quality control of GWAS summary results was performed at the University of Helsinki. The variant inclusion criteria were: MAF>1%, imputation info score (Rsq)>0.7, call rate>0.9, and Hardy-Weinberg Equilibrium P>1×10-6. Base pair positions are reported according to GRCh37 (hg19).
GWAS analyses of the six phenotypes were performed using linear mixed models with Rvtests (http://zhanxw.github.io/rvtests/) or GEMMA (28). The continuous scale of the effect allele dose (0–2) was used to account for imputation uncertainties. A relatedness matrix, calculated from all variants with MAF >5% using the Balding-Nichols method in the vcf2kinship package in RVTESTS, accounted for the entire spectrum of genetic relatedness from familial relationships to more distant population structure. Additionally, population stratification was accounted for prior to genotype imputation by restricting the analyses to European ancestry participants (details by study cohort in Table S2). The genomic inflation factor (λ) was calculated with the function ‘estlambda’ from the R package ‘GenABEL’ (27) (Table S3); together with an LD score regression intercept test using LD Hub (29), we saw no evidence of significant inflation (Table S4). QQ-plots and Manhattan plots were acquired with the R package ‘qqman’ (30).
Covariates for the base analytic model included sex, age, and BMI (in kg/m2) (31), while the main analytic model further controlled for alcohol use (grams/week) (due to high co-use and associations with NMR (31–33)) and birth-year (due to trends in smoking attitudes, social acceptance, regulatory bans, and variation in recruitment years). Because the NMR influences CPD (31), and CPD may influence the NMR (via nicotine/tobacco inhibition of CYP2A6 (34)), we ran additional models for NMR controlling for CPD, and for CPD controlling for NMR. We also ran a model for COT and COT+3HC, controlling for CPD. Missing covariate values (all <10%) were recoded to the median value. Reported results reflect the main analytic model unless otherwise stated; alternative models are found in the supplementary data.
GWAS Meta-Analysis, Fine-mapping, and Annotation
Model- and phenotype-specific meta-analyses were performed using GWAMA (35), using fixed effects models, applying study-specific genomic control correction to adjust for remaining population stratification, and using a GWS association threshold of P<5×10-8. An association locus was defined as the region extending 2.5Mb in both directions (none were >2.5Mb) of the GWS SNP with the smallest P-value. Regional plots were created with LocusZoom (36); LD data were from 1000-Genomes. We additionally performed the largest COT GWAS (n=8885) by meta-analysing our base model results (as most similar) with those from Ware et al. (37) (n=4548) using non-overlapping samples.
To identify putatively causal SNPs in each GWS region, we performed a shotgun stochastic search with FINEMAP v1.2 (38) and a step-wise conditional regression with GCTA v1.91.3beta (39) (see Supplementary Information). For each locus, we calculated the heritability estimate of the causal variants using FINEMAP (see Benner et al. 2018; bioRxiv https://doi.org/10.1101/318618). We report the heritability estimate of the causal variants for each locus as calculated by FINEMAP (Benner et al. 2018; bioRxiv https://doi.org/10.1101/318618). Variant annotation included assessment of genomic location, functional consequence, and association with RNA expression levels and methylation pattern (see Supplementary Information).
Code availability
All scripts used for statistical analyses are available upon request.
Results
Altogether 1885 GWS (P<5×10−8) SNPs and six association loci were found on chromosomes 1, 4, 5, 9, 15, and 19 across the six phenotypes (Figure 1, Table 1, Table S5, Table S6). Using a leave-one-out approach, illustrated in cohort-specific plots, the results were largely consistent across the five cohorts (Figure S3 and S4). Correlations between the phenotypes are found in Table S7.
Figure 1. Chromosomal location and annotation for all genome-wide significant SNPs for all six phenotypes.
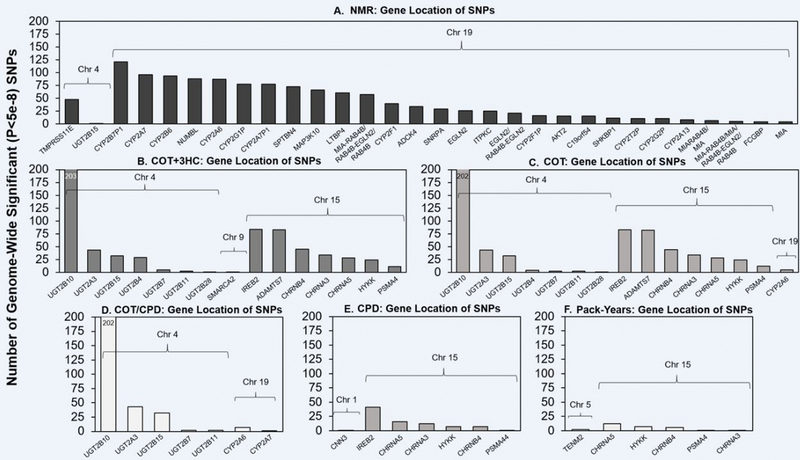
Annotations of genome-wide significant (GWS) SNPs in the main analytic models of the (A) NMR, (B) COT+3HC, (C) COT, (D) COT/CPD, (E) CPD, and (F) Pack-Years are shown. Annotations were performed using NCBI’s dbSNP (build 37). The GWS SNPs were found within or 5’ or 3’ (i.e. within 90kb of the gene) of the genes indicated. The only intergenic GWS SNP located >90kb from a gene was the chromosome 9 COT+3HC SNP (located 194kb 5’ of SMARCA1). For clarity, the NMR plot (A) excludes genes with <4 GWS SNPs: PSMC4, HNRNPUL1, LGALS14, ZNF780B, CNTD2, PLD3, CYP2S1, CEACAM5, RABAC1, LEUTX, ZNF546, TTC9B, PRX, SERTAD3, AXL, CCDC97, BCKDHA, CEACAM6, ARHGEF1, GRIK5, POU2F2, ERF, PSG3. Note: the number of genome-wide significant SNPs found is influenced by linkage disequilibrium patterns and the size of the gene.
Table 1.
Number of total and putatively causal GWS SNPs, top SNP, and heritability estimate for each association locus for each phenotype
| PHENOTYPE | CHR | # GWS SNPs | # PUTATIVELY CAUSAL SNPS 1 | REGIONAL HERITABILITY ESTIMATE (%)2 | TOP SNP | BP POSITION 3 | MAJOR/MINOR | MAF | LOCATION | eQTL4 | meQTL5 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NMR | 4 | 48 | 1 | 1.8 | rs34638591 rs36103218 rs34103191 |
69359223 69359253 69359280 |
C/T T/C G/A |
0.43 | TMPRSS11E (intron) | UGT2B15, UGT2B17, UGT2B29P,RP11–1267H10.2, RP11–1267H10.1 | None |
| NMR | 19 | 1207 | 13 | 36.4 | rs56113850 | 41353107 | C/T | 0.45 | CYP2A6 (intron) | EGLN2, CYP2A7, CYP2T2P | Cis, Trans |
|
| |||||||||||
| COT+3HC | 4 | 315 | 1 | 0.9 | rs10000284 | 69669183 | T/C | 0.08 | UGT2B10 (12.5kb 5’) | None | None |
| COT+3HC | 9 | 1 | 1 | 0.7 | rs12684930 | 1821338 | G/C | 0.08 | SMARCA2 (194kb 5’) | None | None |
| COT+3HC | 15 | 309 | 1 | 2.5 | rs2036527 | 78851615 | G/A | 0.35 | CHRNA5 (6.2kb 5’) | CHRNA3, CHRNA5, CHRNB4,IREB2, PSMA4, RP11–160C18.2,RP11–650L12.2 | Cis |
|
| |||||||||||
| COT | 4 | 286 | 1 | 0.8 | rs294775 | 69680933 | T/C | 0.08 | UGT2B10 (0.8kb 5’) | None | None |
| COT | 15 | 307 | 1 | 2.4 | rs2036527 | 78851615 | G/A | 0.35 | CHRNA5 (6.2kb 5’) | CHRNA3, CHRNA5, CHRNB4, IREB2, PSMA4, RP11–160C18.2, RP11–650L12.2 | Cis |
| COT | 19 | 5 | 1 | 0.8 | rs2316205 | 41346768 | C/T | 0.49 | CYP2A6 (2.7kb 3’) | EGLN2, CYP2A7 | Cis |
|
| |||||||||||
| COT/CPD | 4 | 281 | 1 | 0.2 | rs294778 | 69682555 | A/G | 0.08 | UGT2B10 (intron) | None | None |
| COT/CPD | 19 | 8 | 1 | 1.6 | rs56113850 | 41353107 | C/T | 0.45 | CYP2A6 (intron) | EGLN2, CYP2A7, CYP2T2P | Cis, Trans |
|
| |||||||||||
| CPD | 1 | 1 | 1 | 0.5 | rs860873 | 95387208 | G/A | 0.47 | CNN3 (intron) | RP11–86H7.7, RP4–639F20.1, RWDD3 | None |
| CPD | 15 | 84 | 1 | 0.6 | rs72740955 | 78849779 | C/T | 0.35 | CHRNA5 (8.1kb 5’) | CHRNA3, CHRNA5, IREB2, PSMA4,RP11–160C18.2, RP11–650L12.2 | Cis |
|
| |||||||||||
| PACK-YEARS | 5 | 2 | 1 | 0.7 | rs2337033 | 167392011 | A/G | 0.35 | TENM2 (intron) | None | None |
| PACK-YEARS | 15 | 27 | 1 | 0.5 | rs72740955 | 78849779 | C/T | 0.35 | CHRNA5 (8.1kb 5’) | CHRNA3, CHRNA5, IREB2, PSMA4,RP11–160C18.2, RP11–650L12.2 | Cis |
The results shown are for the main model that adjusted for population substructure, age, sex, BMI, alcohol use, and birth year. CHR, chromosome; GWS, genome-wide significant; SNP, single-nucleotide polymorphism; BP, base pair; MAJOR, the more common allele; MINOR, the less common allele; MAF, minor allele frequency; eQTL, expression quantitative loci; meQTL, methylation quantitative trait loci; NMR, the nicotine metabolite ratio; COT, cotinine; 3HC, 3-hydroxycotinine; CPD, cigarettes per day
Number of SNPs in the FINEMAP top configuration of putatively causal SNPs
Obtained with FINEMAP (full results in Table S5)
Base pair positions reported according to GRCh37 (hg19)
Full eQTL information is available in Table S5
Full meQTL information is available in Table S6
Associations for the NMR were found on chromosomes 4 and 19
Two association loci on chromosomes 4 and 19 (Figure 2) were found for the NMR, explaining 38.2% of variation (Table 1). We replicated the top SNP, rs56113850, located on chromosome 19 in an intron of CYP2A6 (Figure 2, Table 1) (3, 18, 19). The chromosome 19 locus explained 36.4% of NMR variation (Table 1) (vs. ~20–30% previously explained (3, 17, 19)) and consisted of several SNPs residing in previously unreported genes (Table S8). The chromosome 4 locus for the NMR was novel, with most of the GWS SNPs mapping to TMPRSS11E, a transmembrane serine protease (Figure 2), explaining 1.8% of NMR variation (Table 1). The smallest P-value was obtained for three SNPs in an intron of TMPRSS11E (Table 1, Table S5), in perfect linkage disequilibrium (LD) in Europeans from the 1000 Genomes Project. Controlling for CPD did not substantially alter the number of GWS SNPs (Table S5).
Figure 2. Associations in the meta-GWAS of the Nicotine Metabolite Ratio (NMR) were found in chromosome 4 and chromosome 19.

(A) Manhattan plot of the meta-GWAS of the NMR highlighting two association loci. The dashed line represents the genome-wide significance threshold of P<5×10-8. (B) The chromosome 4 association locus: the top signal comprised three intronic SNPs in TMPRSS11E (rs34638591 (C > T), rs36103218 (T > C), and rs34103191 (G > A)) with identical P-values. FINEMAP and GCTA analyses revealed one putatively causal and conditionally independent chromosome 4 signal for the NMR. (C) The chromosome 19 association locus: the top SNP was rs56113850 (C > T) found in intron 4 of CYP2A6. (D) FINEMAP and GCTA analyses revealed several putatively causal and conditionally independent chromosome 19 signals for the NMR. In plots (B) and (C), the light purple areas represent the genome-wide significant regions, and the linkage disequilibrium values are based on the 1000-Genomes reference panel. The top SNP has been indicated with a purple diamond, the red line represents the genome-wide significance threshold of P=5×10−8, and the blue line represents the suggestive threshold of P=1×10-5. Plots reflect the main analytic model. Base pair positions are provided according to GRCh37 (hg19).
FINEMAP analyses of chromosome 19 in cohorts 1–2 showed that nine out of 13 putatively causal SNPs were highly likely causal (probability>0.5) (Table 2, Table S9). Of these nine, two are known functional CYP2A6 variants (rs1801272/CYP2A6*2, rs28399433/CYP2A6*9; www.pharmvar.org), three were in/near to CYP2A6 (including rs113288603), two were in CYP2B6, one was in ADCK4, and one was near MAP3K10 (Table 2). Notably, only rs113288603 was among the conditionally independent SNPs in prior GWASs that used stepwise conditional analyses (3, 17–19). We also performed stepwise conditional analysis with GCTA using the meta-GWAS results, yielding six conditionally independent SNPs (Table S9); only one overlapped with the FINEMAP SNPs (Figure 2, Table S10). The GCTA-configuration includes by default the top SNP (i.e. rs56113850), which was not found with FINEMAP. Our further comparisons suggested that GCTA may not be appropriate (see Supplementary Information). For chromosome 4, FINEMAP and GCTA analyses highlighted only one SNP as being causal or conditionally independent (Table S9). Neither method could distinguish the causal or conditionally independent SNP among the highly correlated SNPs in the area.
Table 2.
FINEMAP top configuration of causal SNPs for the NMR on chromosome 19
| SNP | BP | MAJOR/MINOR | MAF | SNP PROB | BETA | SE | P-VALUE | LOCATION | NCBI dbSNP FUNCTIONAL ANNOTATION | eQTL | meQTL |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| RS11667982 | 41338761 | G/A | 0.14 | 1 | −0.42 | 0.05 | 1.48e-15 | CYP2A6 (10.7kb 3’) | NA | None | Cis |
| RS7250713 | 41355195 | C/G | 0.37 | 1 | −0.37 | 0.04 | 2.05e-25 | CYP2A6 | intron variant | EGLN2, CYP2A7, CYP2G1P | Cis |
| RS28399433 | 41356379 | A/C | 0.12 | 0.9995 | −0.81 | 0.04 | 9.46e-94 | CYP2A6 (0.02kb 5’) | NA (CYP2A6*9) | CYP2A7, CYP2G1P, C19orf54, ADCK4 | None |
| RS3181842 | 41523016 | T/C | 0.4 | 0.9621 | 0.42 | 0.07 | 1.28e-10 | CYP2B6 | downstream variant 500B,utr variant 3’ | None | Cis |
| RS1801272 | 41354533 | A/T | 0.02 | 0.9042 | −0.9 | 0.08 | 5.81e-29 | CYP2A6 | missense (CYP2A6*2) | None | None |
| RS148856862 | 41204624 | C/T | 0.01 | 0.8302 | −0.61 | 0.11 | 2.17e-08 | ADCK4 | intron variant | None | None |
| RS707265 | 41524087 | G/A | 0.4 | 0.8293 | −0.41 | 0.07 | 4.99e-10 | CYP2B6 | utr variant 3’ | None | Cis |
| RS113288603 | 41362293 | C/T | 0.15 | 0.5804 | 0.39 | 0.05 | 5.54e-15 | CYP2A6 (5.9kb 5’) | NA | None | None |
| RS12972927 | 40685483 | C/T | 0.01 | 0.5311 | −0.5 | 0.1 | 3.59e-07 | MAP3K10 (12.0kb 5’) | NA | None | Trans |
| RS11672345 | 41909719 | G/A | 0.02 | 0.3935 | −0.57 | 0.1 | 3.66e-09 | BCKDHA | intron variant | None | None |
| RS7248187 | 41437426 | C/G | 0.24 | 0.3430 | −0.25 | 0.03 | 2.05e-14 | CYP2B7P1 | intron variant | CYP2B7P1 | Cis |
| RS2604911 | 41296117 | C/G | 0.28 | 0.3296 | 0.19 | 0.03 | 7.05e-11 | MIA-RAB4B/RAB4B-EGLN2/RAB4B | intron variant | ADCK4 | Cis |
| RS11672809 | 41344101 | T/C | 0.03 | 0.2402 | 0.83 | 0.09 | 2.73e-20 | CYP2A6 (5.3kb 3’) | NA | CYP2A6 | Cis, Trans |
The results are from the main model. The most probable configuration consisted of 13 SNPs (depicted in the table) and their heritability estimate was 36.7% (95% CI: 32.9–41.0%). All betas listed are from the joint model including the 13 top configuration SNPs, and they are reported for the minor allele. SNP PROB indicates the posterior probability of being a causal SNP. FINEMAP gave a regional heritability estimate of 36.4% (95% CI: 32.6–40.4%) for the main model results and suggested that there are 9–14 causal SNPs within the region. SNP, single-nucleotide polymorphism; BP, base pair; MAJOR, the more common allele; MINOR, the less common allele; MAF, minor allele frequency; SE, standard error; eQTL, expression quantitative loci; meQTL, methylation quantitative trait loci; Cis, within 1Mb of the position of the SNP; Trans, > 1Mb away from the position of the SNP
Associations for COT+3HC and COT, biomarkers of nicotine intake, were found on chromosomes 4, 9, 15, and 19
For COT+3HC, three association loci on chromosomes 4, 9, and 15 (Figure S5, Figure S6) were found, explaining 4.1% of variation (Table 1). The chromosome 4 GWS SNPs mapped to seven UDP-glucuronosyltransferase (UGT) genes (Figure 1). The top chromosome 4 SNP was rs10000284, located ~13kb upstream of UGT2B10, encoding the UGT2B10 enzyme involved in nicotine and cotinine glucuronidation (40) (Table 1, Table S5, Figure S6). One GWS SNP was detected on chromosome 9 (Table 1, Table S5, Figure S6), located ~194kb upstream of SMARCA2 which putatively regulates transcription via chromatin remodelling (41). On chromosome 15, the GWS SNPs mapped to IREB2, ADAMTS7, CHRNB4, CHRNA3, CHRNA5, HYKK, and PSMA4 (Figure 1). The top SNP on chromosome 15 was rs2036527, located ~6kb upstream of CHRNA5 (Table S5, Figure S6) encoding the α5 nicotinic acetylcholine receptor (nAChR) subunit. FINEMAP and GCTA analyses revealed only the top SNP from chromosomes 4, 9, and 15 to be putatively causal and conditionally independent (Table S9).
The meta-GWAS of COT revealed association loci on chromosomes 4, 15 and 19 (Figure S5, Figure S6); the GWS SNPs explained 4.0% of variation (Table 1). The chromosome 4 and 15 genes were shared with COT+3HC; the chromosome 19 signal mapped to CYP2A6 (Figure 1). The chromosome 9 COT+3HC GWS SNP (rs12684930) was nearly GWS (P=8.1×10−8) for COT. FINEMAP and GCTA analyses of COT revealed the top SNP from chromosome 4, 15, and 19 to be putatively causal and conditionally independent (Table S9). After meta-analysing our COT results with those from Ware et al. (37) (total n= 8885), signals on chromosomes 4, 15, and 19 remained, whereas the chromosome 9 signal disappeared (Figure S7). The original Ware et al. COT GWAS detected variants on chromosomes 4 and 15, but not on 19 (37). Controlling for CPD reduced the number of GWS SNPs found on chromosome 15 in the COT+3HC and COT GWASs (Table S5).
Associations for COT/CPD, a measure of smoking intensity, were found on chromosomes 4 and 19
Two association loci on chromosome 4 and 19 (Figure S5, Figure S6) were identified for COT/CPD, explaining 1.7% of variation (Table 1). The chromosome 4 SNPs mapped to five UGTs (Figure 1). The top SNP on chromosome 4 was rs294778, located in an intron of UGT2B10 (Table 1, Table S5, Figure S6). The GWS SNPs on chromosome 19 mapped to CYP2A6 and CYP2A7; the top SNP was rs56113850 (Figure 1, Table 1, Table S5, Figure S6) as for the NMR. FINEMAP and GCTA analyses revealed the top SNP from chromosomes 4 and 19 to be putatively causal and conditionally independent (Table S9). As for the NMR, rs56113850 is unlikely to causally influence COT/CPD; rs56113850 did not remain significant for COT/CPD after conditioning on the 13 SNPs pinpointed by FINEMAP for the NMR (Table S10).
Associations for CPD and Pack-Years, self-reported measures of nicotine intake, were found on chromosomes 1, 5, and 15
For CPD, three association loci on chromosomes 1, 5, and 15 (Figure S5, Figure S6) explained 1.1% of variation (Table 1). There were single GWS SNP associations on chromosomes 1 (rs860873, located in an intron of CNN3) and 5 (rs2337033, located in an intron of TENM2), significant only in the base model (Table S5). The top SNP on chromosome 15 was rs72740955, located ~8.1kb upstream of CHRNA5 (Table S5); rs72740955 is in high LD (r2=0.9 and D’=0.97, 1000 Genomes) with the functional CHRNA5 variant rs16969968, which was associated with COT, COT+3HC and CPD (Table S6), replicating prior associations with smoking quantity (42). Our FINEMAP and GCTA analyses revealed only the top SNP from chromosomes 1, 5, and 15 to be putatively causal and conditionally independent (Table S9).
For Pack-Years, two association loci on chromosomes 5 and 15 (Figure S5, Figure S6) explained 1.2% of variation (Table 1). A single GWS SNP was detected on chromosome 5 (rs2337033, mapping to an intron of TENM2) (Table 1, Table S5, Figure S6) which was also GWS for CPD in the base model (Table S6). As for CPD, the top SNP on chromosome 15 was rs72740955, located ~8.1kb upstream of CHRNA5 (Table S5, Figure S6). FINEMAP and GCTA analyses revealed only the top SNP from chromosomes 5 and 15 to be putatively causal and conditionally independent (Table S9).
Many of the GWS SNPs were eQTL and/or meQTL
Many of the GWS SNPs were known expression quantitative trait loci (eQTL) (Table S11, Figure S8) and/or known methylation quantitative trait loci (meQTL) (Table S12, Figure S9). For instance, the T (vs. C) allele of the intronic TMPRSS11E rs36103218 SNP, associated with lower NMR, was associated with higher UGT2B17 expression in the liver in GTEx and higher UGT2B17 (glucuronidates 3HC (43)) activity in the PNAT2 cohort (Figure S10). The top chromosome 15 SNPs, rs2036527 (for COT+3HC and COT) and rs72740955 (for CPD and Pack-Years) were associated with altered expression of the α5 nAChR subunit in brain in GTEx (Figure S11).
Summary of genetic similarities and differences across the nicotine metabolism and smoking phenotypes
There was substantial overlap between the GWS SNPs and association loci found for the NMR and COT+3HC, COT, and COT/CPD (Figure 3, Table 1). Although none of the chromosome 4 SNPs were shared, they were found in the same region containing TMPRSS11E and UGT2 genes (Figure 3, Figure S6). In contrast to chromosome 4, all chromosome 19 SNPs that were GWS for COT and/or COT/CPD were shared with the NMR and mapped near to CYP2A6 (Figure 3, Table S6). Compared to the NMR, far fewer overall chromosome 19 SNPs (and within a narrower region) were observed for COT and COT/CPD (Table 1, Table S5). For COT+3HC, there were no GWS SNPs in chromosome 19. Comparing the NMR to the self-reported measures of nicotine intake, there was no overlap: neither of the two significant chromosomes for the NMR (4 and 19) were shared with CPD (1 and 15) or Pack-Years (5 and 15) (Table 1). We were unable to quantify genetic similarity using LD-score regression (LDSC) (44) across all phenotypes, because SNP-heritability estimates from LDSC were highly inaccurate (Table S4); LDSC likely requires a larger sample size and/or higher polygenicity for robust results.
Figure 3. Number and overlap of genome-wide significant SNP associations with the six phenotypes.

(A) Upset plot depicting exact numbers of overlapping SNP associations for all main model results (1 885 GWS SNPs across the six phenotypes). (B) The best fit proportional Venn diagram highlighting the proportions of GWS SNP associations and relationships between phenotypes for all main model results. The Venn diagram was drawn using the venneuler R package (59). It is approximative with the stress factor equaling 0.008 (<0.01 threshold advised), which indicates a good fit, but unlike the figure suggests, the NMR GWS SNPs do not overlap with the COT+3HC GWS SNPs. (C) Upset plots depicting overlaps of the main model results by chromosome. For chromosomes 1 and 9, one GWS SNP occurred for CPD and COT+3HC, respectively. For chromosome 5, two GWS SNPs occurred for Pack-Years.
Discussion
The associated loci captured 38% of NMR variation, 4% of variation in nicotine intake measured by objective biomarkers (COT+3HC, COT), 2% of variation in smoking intensity (COT/CPD), and 1% of variation in self-reported nicotine intake (CPD, Pack-Years).
We confirmed the chromosome 19 association locus found in prior NMR GWASs by us (3, 17) and others (18, 19). The number of NMR GWS SNPs was essentially unchanged after additionally controlling for CPD (Table S5), suggesting altered cigarette consumption does not impact the observed genomic influences on the NMR. The top SNP from the meta-GWAS, rs56113850, was not identified in the FINEMAP top configuration and our additional analyses suggested that rs56113850 is not a causal variant itself but tags multiple causal variants, consistent with its high correlation with four SNPs pinpointed by FINEMAP (Figure S12). We also identified an entirely novel chromosome 4 signal for the NMR mapping to TMPRSS11E, adjacent to UGT2B17. According to the GWAS Catalog (https://www.ebi.ac.uk/gwas/; accessed September 24, 2019), TMPRSS11E is associated with hematological and blood lipid traits (45–47). Altered UGT2B17 expression and activity (Figure S10) is a possible mechanism underpinning the association with NMR. Prior work in African American smokers, however, showed that the UGT2B17 gene deletion did not alter NMR (48, 49), suggesting there may be inter-ethnic variation in the relationship between UGT2B17 activity and the NMR.
Understanding NMR genomics permits extensions to epidemiological studies that include non-, former- and intermittent-users of tobacco products where the NMR itself cannot be reliably measured (7). The NMR is associated with tobacco-related disease (11), thus the assessment of NMR genomics in epidemiological studies could greatly improve our mechanistic understanding of disease risk pathways, and improve prevention efforts. A greater understanding of the contribution of CYP2A6 variation to NMR genomics could also advance precision medicine approaches in nicotine dependence; recently a CYP2A6 weighted genetic risk score was shown to replicate NMR-stratified cessation treatment findings (23). Moreover, NMR genomic information may be particularly useful in tailoring chemotherapy, as CYP2A6 metabolizes the chemotherapeutic agents letrozole and tegafur (12, 13), and other CYP2A6 substrate drug treatments (reviewed in 50, 51).
This study was the first GWAS of COT+3HC, an improved biomarker of nicotine intake compared to COT (21). While the chromosome 9 SNP (rs12684930, located ~194kb 5’ from SMARCA2) associated with COT+3HC was not previously associated with smoking-related traits, SMARCA2 has been linked in the GWAS Catalog to ADHD, bipolar disorder, MDD, and schizophrenia which are also associated with smoking (52–54). Variation in the chromosome 15 CHRNA5-CHRNA3-CHRNB4 cluster, which was significant in our analyses of COT+3HC, COT, CPD, and Pack-Years, has been robustly associated with nicotine phenotypes (37, 55, 56). The minor alleles of the top chromosome 15 SNPs for the objective nicotine biomarkers (i.e. rs2036527) and self-reported intake measures (i.e. rs72740955) were associated with higher intake and lower α5-nAChR expression, consistent with α5-nAChR knockout mice showing greater nicotine intake (57). We also found GWS SNPs in ADAMTS7 (for COT+3HC and COT) and IREB2 (for COT+3HC, COT, and CPD). A gene- and pathway-based analysis highlighted ADAMTS7 and IREB2 among the top genes associated with CPD (58), and the recent GWAS of CPD by the GSCAN consortium identified an intronic ADAMTS7 SNP as conditionally independent (42); additionally these genes are associated with tobacco-related diseases in the GWAS Catalog.
The lack of extensive genomic overlap between phenotypes may be due to poor capture of intake phenotype heritability (1–4%) versus NMR heritability (~38%) and low power for our CPD analysis. The GSCAN consortium’s CPD GWAS (>330 000 smokers) (42) identified rs56113850 among the conditionally independent SNPs, which was the top SNP for both the NMR and COT/CPD in our study: the rs56113850 T allele was associated with higher COT/CPD (beta=0.16) but lower NMR (beta=−0.69), likely due to slower removal of COT, the denominator in NMR (21). In support of this, a secondary analysis in PNAT2 showed no apparent impact of the rs56113850 T allele on increasing (COT+3HC)/CPD (Figure S13). Comparing between intake phenotypes, COT+3HC (vs. COT) was more genetically similar to CPD. In contrast, COT (vs. COT+3HC) was more genetically similar to the NMR, likely due to the impact of CYP2A6 activity (i.e. the NMR) on COT formation and metabolism (21).
Strengths of our study include the large sample size for biomarker analyses (e.g. NMR) and the use of biochemical intake measures. We were well-powered to detect variants that explain at least 0.76%, 1.0%, or 1.5% (power >80%, >96%, and >99%, respectively) of phenotypic variance. Fine-mapping of the association loci using a stochastic search allowed for any possible configuration of causal SNPs; this was valuable for the chromosome 19 NMR association locus which was not suited for stepwise conditional analyses. A limitation was that data from only two cohorts were available for the fine-mapping analysis. In addition, all study participants were of European descent, thus limiting the generalizability of our results to other populations. Studies in larger samples are needed to detect more variants with phenotypic impacts below 0.76%.
In conclusion, we identified over 1200 SNPs associated with the NMR, capturing ~38% of phenotype variation. Not surprisingly, the chromosome 19 locus, containing CYP2A6, comprised the majority of the GWS SNPs and explained ~36% of phenotype variation, a substantial increase from the ~20–30% variance previously accounted for (3, 17, 19). We also identified several novel loci influencing the NMR and nicotine exposure phenotypes, including a novel chromosome 4 region mapping to TMPRSS11E and several UGT2 genes. A greater understanding of the genomic influences on nicotine metabolism and tobacco exposure phenotypes may improve smoking cessation treatments and our understanding of tobacco-related disease risk.
Supplementary Material
Acknowledgements
This work is supported by the NIH (PGRN grant DA020830 and CA197461), CIHR (FDN-154294 and PJY-159710), a Canada Research Chair, Wellcome Trust Sanger Institute, Broad Institute, European Network for Genetic and Genomic Epidemiology, FP7-HEALTH-F4-2007, grant agreement number 201413, NIAAA (AA-12502/AA-00145/AA-09203/AA15416/K02AA018755), the Academy of Finland (100499/205585/118555/141054/264146/308248/312073/288509/312076/285380/312062/ 286284/134309/126925/121584/124282/129378/117787/41071/265240/263278), Finnish Foundation for Cardiovascular Research, Sigrid Juselius Foundation, University of Helsinki HiLIFE Fellow grant, Social Insurance Institution of Finland, Competitive State Research Financing of the Expert Responsibility area of Kuopio, Tampere and Turku University Hospitals (grant X51001), Juho Vainio Foundation, Paavo Nurmi Foundation, Finnish Cultural Foundation, Tampere Tuberculosis Foundation, Emil Aaltonen Foundation, Yrjö Jahnsson Foundation, Signe and Ane Gyllenberg Foundation, Diabetes Research Foundation of Finnish Diabetes Association, EU Horizon 2020 (755320), European Research Council (742927), Tampere University Hospital Supporting Foundation, Doctoral Program in Population Health (University of Helsinki), Finnish Research Foundation of the Pulmonary Diseases, Biomedicum Helsinki Foundation, and the Cancer Foundation Finland. We thank Dr. Aino Kankaanpää, Maria Novalen, Leanne McNeill, and Tabatha Goncalves for laboratory work.
Footnotes
Conflict of Interest
TK has consulted for Pfizer Finland. VS attended a conference trip sponsored by Novo Nordisk, received an advisory board meeting honorarium, and collaborates with Bayer ltd. RFT has consulted for Quinn Emmanual and Ethismos.
References
- 1.Benowitz NL. Nicotine addiction. N Engl J Med. 2010June17;362(24):2295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dempsey D, Tutka P, Jacob P, Allen F, Schoedel K, Tyndale RF, et al. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther. 2004July;76(1):64–72. [DOI] [PubMed] [Google Scholar]
- 3.Loukola A, Buchwald J, Gupta R, Palviainen T, Hallfors J, Tikkanen E, et al. A Genome-Wide Association Study of a Biomarker of Nicotine Metabolism. PLoS Genet. 2015;11(9):e1005498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chenoweth MJ, Tyndale RF. Pharmacogenetic Optimization of Smoking Cessation Treatment. Trends Pharmacol Sci. 2017January;38(1):55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakajima M, Yamamoto T, Nunoya K, Yokoi T, Nagashima K, Inoue K, et al. Role of human cytochrome P4502A6 in C-oxidation of nicotine. Drug Metab Dispos. 1996November;24(11):1212–7. [PubMed] [Google Scholar]
- 6.Nakajima M, Yamamoto T, Nunoya KI, Yokoi T, Nagashima K, Inoue K, et al. Characterization of CYP2A6 involved in 3’-hydroxylation of cotinine in human liver microsomes. J Pharmacol Exp Ther. 1996May;277(2):1010–5. [PubMed] [Google Scholar]
- 7.Lea RA, Dickson S, Benowitz NL. Within-subject variation of the salivary 3HC/COT ratio in regular daily smokers: prospects for estimating CYP2A6 enzyme activity in large-scale surveys of nicotine metabolic rate. J Anal Toxicol. 2006Jul-Aug;30(6):386–9. [DOI] [PubMed] [Google Scholar]
- 8.Mooney ME, Li ZZ, Murphy SE, Pentel PR, Le C, Hatsukami DK. Stability of the nicotine metabolite ratio in ad libitum and reducing smokers. Cancer Epidemiol Biomarkers Prev. 2008June;17(6):1396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.St Helen G, Jacob P 3rd, Benowitz NL Stability of the nicotine metabolite ratio in smokers of progressively reduced nicotine content cigarettes. Nicotine Tob Res. 2013November;15(11):1939–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossini A, de Almeida Simao T, Albano RM, Pinto LF. CYP2A6 polymorphisms and risk for tobacco-related cancers. Pharmacogenomics. 2008November;9(11):1737–52. [DOI] [PubMed] [Google Scholar]
- 11.Park SL, Murphy SE, Wilkens LR, Stram DO, Hecht SS, Le Marchand L. Association of CYP2A6 activity with lung cancer incidence in smokers: The multiethnic cohort study. Plos One. 2017May25;12(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamiya I, Yoshisue K, Ishii Y, Yamada H, Chiba M. Effect of CYP2A6 genetic polymorphism on the metabolic conversion of tegafur to 5-fluorouracil and its enantioselectivity. Drug Metab Dispos. 2014September;42(9):1485–92. [DOI] [PubMed] [Google Scholar]
- 13.Murai K, Yamazaki H, Nakagawa K, Kawai R, Kamataki T. Deactivation of anti-cancer drug letrozole to a carbinol metabolite by polymorphic cytochrome P450 2A6 in human liver microsomes. Xenobiotica. 2009November;39(11):795–802. [DOI] [PubMed] [Google Scholar]
- 14.Lerman C, Schnoll RA, Hawk LW, Cinciripini P, George TP, Wileyto EP, et al. Use of the nicotine metabolite ratio as a genetically informed biomarker of response to nicotine patch or varenicline for smoking cessation: a randomised, double-blind placebo-controlled trial. Lancet Resp Med. 2015February;3(2):131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vardavas CI, Filippidis FT, Agaku IT. Determinants and prevalence of e-cigarette use throughout the European Union: a secondary analysis of 26 566 youth and adults from 27 Countries. Tob Control. 2015September;24(5):442–8. [DOI] [PubMed] [Google Scholar]
- 16.Zhu AZX, Binnington MJ, Renner CC, Lanier AP, Hatsukami DK, Stepanov I, et al. Alaska Native smokers and smokeless tobacco users with slower CYP2A6 activity have lower tobacco consumption, lower tobacco-specific nitrosamine exposure and lower tobacco-specific nitrosamine bioactivation. Carcinogenesis. 2013January;34(1):93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chenoweth MJ, Ware JJ, Zhu AZX, Cole CB, Cox LS, Nollen N, et al. Genome-wide association study of a nicotine metabolism biomarker in African American smokers: impact of chromosome 19 genetic influences. Addiction. 2018March;113(3):509–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baurley JW, Edlund CK, Pardamean CI, Conti DV, Krasnow R, Javitz HS, et al. Genome-Wide Association of the Laboratory-Based Nicotine Metabolite Ratio in Three Ancestries. Nicotine & Tobacco Research. 2016September;18(9):1837–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel YM, Park SL, Han Y, Wilkens LR, Bickeboller H, Rosenberger A, et al. Novel Association of Genetic Markers Affecting CYP2A6 Activity and Lung Cancer Risk. Cancer Res. 2016October1;76(19):5768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pergadia ML, Glowinski AL, Wray NR, Agrawal A, Saccone SF, Loukola A, et al. A 3p26–3p25 genetic linkage finding for DSM-IV major depression in heavy smoking families. Am J Psychiatry. 2011August;168(8):848–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu AZ, Renner CC, Hatsukami DK, Swan GE, Lerman C, Benowitz NL, et al. The ability of plasma cotinine to predict nicotine and carcinogen exposure is altered by differences in CYP2A6: the influence of genetics, race, and sex. Cancer Epidemiol Biomarkers Prev. 2013April;22(4):708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SL, Murphy SE, Wilkens LR, Stram DO, Hecht SS, Le Marchand L. Association of CYP2A6 activity with lung cancer incidence in smokers: The multiethnic cohort study. Plos One. 2017;12(5):e0178435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Boraie A, Taghavi T, Chenoweth MJ, Fukunaga K, Mushiroda T, Kubo M, et al. Evaluation of a weighted genetic risk score for the prediction of biomarkers of CYP2A6 activity. Addict Biol. 2019February27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vartiainen E, Seppala T, Lillsunde P, Puska P. Validation of self reported smoking by serum cotinine measurement in a community-based study. J Epidemiol Community Health. 2002March;56(3):167–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanner JA, Novalen M, Jatlow P, Huestis MA, Murphy SE, Kaprio J, et al. Nicotine metabolite ratio (3-hydroxycotinine/cotinine) in plasma and urine by different analytical methods and laboratories: implications for clinical implementation. Cancer Epidemiol Biomarkers Prev. 2015August;24(8):1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornung RW, Reed LD. Estimation of Average Concentration in the Presence of Nondetectable Values. Applied Occupational and Environmental Hygiene. 1990 1990/01/01;5(1):46–51. [Google Scholar]
- 27.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007May15;23(10):1294–6. [DOI] [PubMed] [Google Scholar]
- 28.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012June17;44(7):821–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017January15;33(2):272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv. 2014. [Google Scholar]
- 31.Chenoweth MJ, Novalen M, Hawk LW Jr, Schnoll RA, George TP, Cinciripini PM, et al. Known and novel sources of variability in the nicotine metabolite ratio in a large sample of treatment-seeking smokers. Cancer Epidemiol Biomarkers Prev. 2014September;23(9):1773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKee SA, Weinberger AH. How Can We Use Our Knowledge of Alcohol-Tobacco Interactions to Reduce Alcohol Use? Annu Rev Clin Psycho. 2013;9:649–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gubner NR, Kozar-Konieczna A, Szoltysek-Boldys I, Slodczyk-Mankowska E, Goniewicz J, Sobczak A, et al. Cessation of alcohol consumption decreases rate of nicotine metabolism in male alcohol-dependent smokers. Drug Alcohol Depend. 2016June1;163:157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benowitz NL, Hukkanen J, Jacob P 3rd, Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 2009(192):29–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics. 2010May28;11:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010September;26(18):2336–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ware JJ, Chen XN, Vink J, Loukola A, Minica C, Pool R, et al. Genome-Wide Meta-Analysis of Cotinine Levels in Cigarette Smokers Identifies Locus at 4q13.2. Sci Rep-Uk. 2016February1;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benner C, Spencer CCA, Havulinna AS, Salomaa V, Ripatti S, Pirinen M. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016May15;32(10):1493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011January7;88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G, Blevins-Primeau AS, Dellinger RW, Muscat JE, Lazarus P. Glucuronidation of nicotine and cotinine by UGT2B10: loss of function by the UGT2B10 Codon 67 (Asp>Tyr) polymorphism. Cancer Res. 2007October1;67(19):9024–9. [DOI] [PubMed] [Google Scholar]
- 41.Koga M, Ishiguro H, Yazaki S, Horiuchi Y, Arai M, Niizato K, et al. Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum Mol Genet. 2009July1;18(13):2483–94. [DOI] [PubMed] [Google Scholar]
- 42.Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019January14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen G, Giambrone NE, Lazarus P. Glucuronidation of trans-3’-hydroxycotinine by UGT2B17 and UGT2B10. Pharmacogenet Genomics. 2012March;22(3):183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015March;47(3):291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell. 2016November17;167(5):1415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nature Genetics. 2018November;50(11):1514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffmann TJ, Theusch E, Haldar T, Ranatunga DK, Jorgenson E, Medina MW, et al. A large electronic-health-record-based genome-wide study of serum lipids. Nat Genet. 2018March;50(3):401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu AZ, Zhou Q, Cox LS, Ahluwalia JS, Benowitz NL, Tyndale RF. Variation in trans-3’-hydroxycotinine glucuronidation does not alter the nicotine metabolite ratio or nicotine intake. Plos One. 2013;8(8):e70938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taghavi T, St Helen G, Benowitz NL, Tyndale RF. Effect of UGT2B10, UGT2B17, FMO3, and OCT2 genetic variation on nicotine and cotinine pharmacokinetics and smoking in African Americans. Pharmacogenet Genomics. 2017April;27(4):143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonagh EM, Wassenaar C, David SP, Tyndale RF, Altman RB, Whirl-Carrillo M, et al. PharmGKB summary: very important pharmacogene information for cytochrome P-450, family 2, subfamily A, polypeptide 6. Pharmacogenet Genom. 2012September;22(9):695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanner JA, Tyndale RF. Variation in CYP2A6 Activity and Personalized Medicine. J Pers Med. 2017December;7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Checkoway H, Powers K, Smith-Weller T, Franklin GM, Longstreth WT, Swanson PD. Parkinson’s disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am J Epidemiol. 2002April15;155(8):732–8. [DOI] [PubMed] [Google Scholar]
- 53.Milberger S, Biederman J, Faraone SV, Chen L, Jones J. ADHD is associated with early initiation of cigarette smoking in children and adolescents. J Am Acad Child Psy. 1997January;36(1):37–44. [DOI] [PubMed] [Google Scholar]
- 54.Diaz FJ, James D, Botts S, Maw L, Susce MT, de Leon J. Tobacco smoking behaviors in bipolar disorder: a comparison of the general population, schizophrenia, and major depression. Bipolar Disord. 2009March;11(2):154–65. [DOI] [PubMed] [Google Scholar]
- 55.Wassenaar CA, Dong Q, Wei QY, Amos CI, Spitz MR, Tyndale RF. Relationship Between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 Variation and Smoking Behaviors and Lung Cancer Risk. J Natl Cancer I. 2011September;103(17):1342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010May;42(5):436–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011March31;471(7340):597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minica CC, Mbarek H, Pool R, Dolan CV, Boomsma DI, Vink JM. Pathways to smoking behaviours: biological insights from the Tobacco and Genetics Consortium meta-analysis. Mol Psychiatry. 2017January;22(1):82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilkinson L. Exact and approximate area-proportional circular Venn and Euler diagrams. IEEE Trans Vis Comput Graph. 2012February;18(2):321–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.