

Abstract
Neuroblastoma, the most common extracranial solid tumor in children, accounts for nearly 8% of childhood cancers in the United States. It is a disease with pronounced clinical and biological heterogeneities. The amplification of MYCN, whose key tumorigenic functions include the promotion of proliferation, facilitation of the cell’s entry into the S phase and prevention of cells from leaving the cell cycle, correlates with poor prognosis. Patients with a high proliferation index disease have low survival rates. Neuroblastoma is one of the most radioresponsive of all human tumors. To exploit this radiosensitivity, radioactive guanidine (R)-(–)-5-[125I]iodo-3’-O-[2-(ε-guanidinohexanoyl)-2-phenylacetyl]-2’-deoxyuridine (9, GPAID) was designed. This compound enters neuroblastoma cells much like meta-iodobenzylguanidine (MIBG). Additionally, it co-targets DNA of proliferating cells, an attribute especially advantageous in the treatment of MYCN-amplified tumors. GPAID was synthesized from the trimethylstannyl precursor with an average yield of >90% at the no-carrier-added specific activities. The norepinephrine transporter-aided delivery of GPAID to neuroblastoma cells was established in the competitive uptake studies with nonradioactive MIBG. The intracellular processing and DNA targeting properties were confirmed in the subcellular distribution experiments. Studies in a mouse model of neuroblastoma demonstrated the therapeutic potential of GPAID. The tin precursor of GPAID can be used to prepare compounds radiolabeled with SPECT- and PET-compatible radionuclides. Accordingly, these reagents can function as theranostics useful in the individualized and comprehensive treatment strategies comprising treatment planning and the assessment of tumor responses as well as the targeted molecular radiotherapy employing treatment doses derived from the imaging data.
Keywords: radioactive guanidines, theranostics, neuroblastoma, norepinephrine transporter, DNA-targeted, MIBG, Auger electron emitters
Graphical Abstract
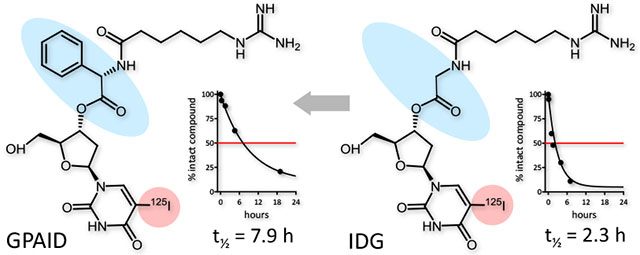
GPAID was designed to treat high risk neuroblastoma, including relapsed or refractory disease. GPAID targets neuroblastoma cells in the same way as MIBG. It also co-targets DNA of proliferating cells, an attribute especially advantageous in the treatment of MYCN-amplified tumors.
1. INTRODUCTION
Childhood cancers represent less than 2% of all human cancers and yet in children one year or older, these malignancies are responsible for more deaths than all other diseases combined.1 Every day, 43 children are diagnosed with cancer. In the U.S., one in five children with cancer will not survive even after intensive multi-modality treatments. Moreover, majority of childhood cancer survivors experience late, chronic side effects that adversely affect their adult lives.2 New treatments are needed to eliminate, or at least appreciably diminish, acute toxicities, minimize long-term morbidities of current therapies and improve cure rates for children who do not respond to the existing treatments. The purpose of this study was to develop novel radioactive drugs for children with neuroblastoma (NB) that might afford them not only cures but also lives free of the lifelong morbidities of many existing treatments.
Targeted molecular radiotherapy with 131I-metaiodobenzylguanidine (131IMIBG), a treatment option for patients with relapsed or refractory NB, takes advantage of the intrinsic radiosensitivity of this malignancy.3,4 131IMIBG was introduced nearly 30 years ago.5 At present, it is used only in clinical trials because its place in the management of NB remains uncertain6,7 and it still needs the FDA approval for high-risk NB. Published data indicate that this treatment is relatively safe but complete tumor responses are rare.7 Side effects include hematologic toxicities8, secondary malignancies,9–11 damage to ovaries12 and thyroid disorders affecting >80% of the long-term survivors treated with 131IMIBG.13 Recently, we reported a series of norepinephrine transporter-targeted (NET) and DNA co-targeted theranostics developed to ameliorate these deficiencies of MIBG.14 Employing Auger electron emitters such as 123I, 124I or 125I yields agents that are virtually harmless when located in the cytoplasm or extracellular spaces but are extraordinarily radiotoxic when either within the structure of DNA or in its immediate vicinity. Nearly all of the electron energy associated with the 125I decay is deposited within a sphere smaller than cell nucleus.15,16 Our Auger electron-emitting drugs enter NB cells by the same mechanism as MIBG and are subsequently catabolized to a product that can participate in the DNA synthesis, scheduled on unscheduled.14 With this dual target approach, we expect to deliver therapeutic radiation doses to NB cells while sparing normal tissues. A potential clinical candidate 5-[125I]iodo-3′-O-(ε-guanidinohexanoyl)-2′-deoxyuridine (IDG) emerged from this study. However, its half-life in human serum was relatively short, ~2.3 h. The lengthening of biological half-lives is thought to produce more effective theranostics.17,18 In the present work, with the intention to increase the biological stability, (R)-(–)-5-iodo-3’-O-[2-(ε-guanidinohexanoyl)-2-phenylacetyl]-2’-deoxyuridine (GPAID) was prepared and evaluated. The addition of bulky phenylacetyl group provides steric hindrance of the 3’-ester. Moreover, D-enantiomers of natural amino acids are not recognized by most mammalian enzymes and thereby exhibit increased enzymatic stability. For instance, D-amino acid esters are hydrolyzed >70 times slower than the corresponding esters of L-amino acids.19–22 Properties of the new derivative were evaluated in vitro using neuroblastoma cells lines and in vivo in NB allografts in mice.
2. EXPERIMENTAL
2.1. General
Chemicals and reagents purchased from commercial suppliers were used without further purification, unless indicated. Sodium [125I]iodide in 1×10−5 NaOH (pH 8-11), with specific activities of >78,000 GBq/mmol was from PerkinElmer (Billerica, MA). All target nonradioactive compounds were ≥98% pure by the rigorous HPLC analysis (Supporting Information, SI). Radioiodinated products were identified and evaluated through the independently prepared non-radioactive reference standards whose HPLC retention times (tR) of UV signals were compared with tR of the radioactivity HPLC signals of the corresponding radioactive products. Additional detailed information is included in SI (page S3).
2.2. Chemistry
2.2.1. (R)-(–)-5-Iodo-5’-O-dimethoxytrityl-3’-O-[2-(ε-(N,N′-bis(tert-butyloxycarbonyl)guanidino)hexanoyl)-2-phenylacetyl]-2’-deoxyuridine (3)
To a solution of 5-iodo-5’-O-DMTr-2’-deoxyuridine (2.16 g, 3.29 mmol), (R)-(–)-2-[6-N-Boc-amino)hexanamido]-2-phenylacetic acid (2) (1.28 g, 3.51 mmol) and DMAP (0.087 g, 0.71 mmol) in 25 mL of DCM, dicyclohexylcarbodiimide (0.72 g, 3.49 mmol) were added at room temperature. The reaction mixture was stirred for 4 h, filtered and the solvent removed under reduced pressure. The residue was purified by column chromatography on a silica gel, using a gradient of MeOH in DCM (0.3-0.5:10), to give 2.22 g (67% yield) of 3 as a pale yellow solid; Rf = 0.71 (DCM/MeOH, 10:0.5). HPLC analysis: tR = 34.1 min (≥97.1% pure at 254 nm and 280 nm) on ACE C18 100 Å column, (5μm, 4.6×250 mm) at the elution rate of 0.8 mL/min with a linear gradient of MeCN in water from 50% to 95% over 90 min, then 95% MeCN kept for 10 min. 1HNMR (CDCl3, 600 MHz) δ 10.34 (s, 1H, H3- uridine), 8.39 (s, 1H, H6-uridine), 7.88-7.79 (m, 2H, HN-Boc, HN3), 7.43-7.27 (m, 10H, 2×5H, aryl; 4H, 2×H2-aryl-OMe, 2×H6-aryl-OMe), 6.84-6.79 (m, 4H, 2×H3-aryl-OMe, 2×H5-aryl-OMe), 6.28 (dd, H1’, 3J1’,2’=6.45 Hz, 3J1’,2”=4.40 Hz), 5.35-5.28 (m, 1H, H2), 5.35-5.28 (m, 1H, H3’), 4.66-3.86 (m, 3H, H4’, H5’), 3.78 (s, 6H, 2×OCH3-DMTr), 3.21-3.17 (2H, H9), 2.34-2.27 (m, 2H, H2’), 2.20-2.12 (m, 2H, H5), 1.85-1.64 (m, 2H, H8), 1.43 (s, 9H, 3×CH3 - t-butyl), 1.43-1.28 (m, 4H, H6, H7) ppm. HR-MS (ESI), (m/e): [M+H]+ calcd for [C49H55IN4O11 + 1H]+, 1003.2985; found 1003.2977.
2.2.2. (R)-(–)-5-Iodo-3’-O-[2-(ε-(N,N′-bis(tert-butyloxycarbonyl)guanidino)hexanoyl)-2-phenylacetyl]-2’-deoxyuridine (4)
Anhydrous 3 (1.81 g, 1.81 mmol) was dissolved in MeCN (10 mL) and ZrCl4 (0.51 g, 2.19 mmol) was added. The mixture was stirred at room temperature for ~1 h. The solvent was evaporated in a vacuum. Fifty mL of EtOAc/water mixture (1/1, v/v) was added to the residue and briefly sonicated. The organic layer was separated, washed with brine and dried over MgSO4. After evaporation of the solvent, a crude product was purified on a silica gel column to give 1.05 g, (83% yield) of the title compound; Rf = 0.43 (DCM/MeOH, 10:0.5). HPLC analysis: tR = 32.5 min (≥96.6% pure at 254 nm and 280nm) on Jupiter C18 100 Å column (5μm, 4.6×250 mm) at the elution rate of 0.8 mL/min with a linear gradient of MeCN in water from 10% to 95% over 45 min, then 95% MeCN kept for 15 min. 1HNMR (CDCl3, 600 MHz) δ 10.56 (s, 1H, H3-uridine), 8.54 (s, 1H, H6-uridine), 8.02-7.87 (m, 2H, HN-Boc, HN3), 7.36-7.25 (m, 5H, aryl), 6.18 (dd, 1H, H1’, 3J1’,2’=6.45 Hz, 3J1’,2”=4.40 Hz), 5.76-5.69 (m, 1H, H2), 5.35-5.18 (m, 2H, H3’- uridine, OH-C5’- uridine), 3.86-3.74 (m, 3H, H4’- uridine, H5’-uridine), 3.19-3.12 (2H, H9), 2.34-2.27 (m, 2H, H2’- uridine), 2.18-2.12 (2H, H5), 1.57-1.51 (2H, H8), 1.41 (s, 9H, 3×CH3-t-butyl), 1.33-1.28 (4H, H6, H7) ppm. 13CNMR (CDCl3, 100 MHz) δ: 172.3 (O=C4), 170.6 (O=C1), 163.5 (O=C4-uridine), 159.3 (O=C-O-t-butyl), 153.2 (O=C2 - uridine), 145.3 (C6- uridine), 135.1 (C1- aryl), 129.3 (C2- aryl), 129.3 (C6- aryl), 128.5 (C3- aryl), 128.4 (C5-aryl), 127.3 (C4- aryl), 86.7 (C1’- uridine), 85.6 (C4’- uridine), 79.8 (C1- t-butyl), 76.2 (C3’- uridine), 68.8 (C5- uridine), 62.5 (C5’- uridine), 57.4 (C2), 40.8 (C9), 37.79 (C2’- uridine), 35.8 (C5), 28.4 (C8), 28.3 (C2- t-butyl), 26.2 (C7), 25.2 (C6). HR-MS (ESI), (m/e): [M+H]+ calcd for [C28H37IN4O9 + 1H]+, 701.1606; found 701.1611.
2.2.3. (R)-(–)-5-Iodo-3’-O-[2-(ε-(N,N′-bis(tert-butyloxycarbonyl)guanidino)hexanoyl)-2-phenylacetyl]-2’-deoxyuridine (5)
The Boc group of 4 (1.33 g, 1.89 mmol) was cleaved with ~25% TFA in DCM. When reaction was completed (monitoring by TLC), the mixture was evaporated under a vacuum at room temperature. The oily residue was treated with 15 mL of EtOAc/hexane (1:1, v/v) and sonicated briefly a few times. The solvent was carefully decanted from solids, a crude product washed with diethyl ether and a solvent was drawn off again. This washing procedure was repeated and then the remaining TFA salt was placed under a high vacuum to dry. The reaction vial containing a dried TFA salt suspended in DCM (15 mL) was placed on an ice bath and TEA (278 μL, 2.0 mmol) was added to a stirred mixture immediately followed by N,N’-bis(tert-butoxycarbonyl)-N”-trifluoromethanesulfonyl guanidine (0.83 g, 2.10 mmol). The mixture was stirred for 5 min and a second portion of TEA (278 μL, 2.0 mmol) added to a resulted clear solution. The stirring continued for 6 h (TLC monitoring) at room temperature. Upon the completion, an excess of amines and triflic amide were removed by aqueous workup with 5% citric acid and saturated brine. Organic phase was dried over MgSO4, filtered and evaporated. A crude product was purified on a silica gel column using a gradient of MeOH in DCM (0.8-2:10) to give 1.11 g of 5 in 69% yield. Rf = 0.38 (DCM/MeOH, 10:0.8). HPLC analysis: tR = 36.9 min (≥95.7% pure at 254 nm and 280nm) on ACE C18 100 Å column (5μm, 4.6×250 mm) at the elution rate of 0.8 mL/min with a linear gradient of MeCN in water from 10% to 95% over 45 min, then 95% MeCN kept for 25 min. 1HNMR (CDCl3, 600 MHz) δ: 11.29 (s, 1H, HN3- uridine), 8.32-8.28 (m, 2H, Boc-NH- guanidine, HN3), 8.24 (s, 1H, H6-uridine), 7.40-7.36 (m, 5H, aryl), 6.36 (dd, 1H, H1’-uridine, 3J1’,2’=6.45 Hz, 3J1’,2”=4.20 Hz), 6.22-6.05 (m, 1H, H2), 5.54-5.38 (m, 2H, H3’- uridine, H4’-uridine), 4.22-3.82 (m, 3H, H5’-uridine, OH-C5’-uridine), 3.44-3.34 (m, 2H, H9), 2.60-2.56 (m, 1H, HN10 - guanidine), 2.54-2.37 (m, 2H, H2’- uridine), 2.27-2.21 (2H, H5), 1.78-1.55 (m, 2H, H8), 1.46 (s, 9H, 3×CH3-t-butyl), 1.41 (s, 9H, 3×CH3-t-butyl), 1.40-1.35 (m, 4H, H6, H7) ppm. 13CNMR (CDCl3, 100 MHz) δ: 172.8 (O=C4), 170.63 (O=C1), 163.6 (O=C4-uridine), 159.6 (N=C-NH, guanidine), 156.2 (NC(O)-O-t-butyl, guanidine), 156.1 (HN-C(O)-O-t-butyl, guanidine), 153.3 (O=C2- uridine), 149.8 (C6- uridine), 135.1 (C1- aryl), 129.4 (C2- aryl), 129.2 (C6 - aryl), 127.4 (C3- aryl), 127.4 (C5 - aryl), 127.3 (C4 - aryl), 86.3 (C1’- uridine), 85.2 (C4’- uridine), 83.2 (C1- t-butyl), 79.3 (C1- t-butyl), 76.0 (C3’- uridine), 68.6 (C5 - uridine), 62.4 (C5’- uridine), 57.1 (C2), 40.8 (C9), 37.8 (C2’- uridine), 35.8 (C5), 28.3 (C8), 28.2 (d, C2- t-butyl), 28.1 (d, C2- t-butyl), 26.3 (C7), 25.2 (C6). HR-MS (ESI), (m/e): [M+H]+ calcd for [C34H47IN6O11 + 1H]+, 843.2420; found 843.2411.
2.2.4. (R)-(–)-5-Iodo-3’-O-[2-(ε-guanidinohexanoyl)-2-phenylacetyl]-2’-deoxyuridine (6)
A sample of this compound was prepared for use as the nonradioactive reference standard by the treatment of 5 (25 mg, 30 μmol) with TFA (0.1 mL) at room temperature for ~15 min. TFA was evaporated with a stream of nitrogen, the residue was triturated with ethyl acetate, briefly sonicated then evaporated again and the residue was triturated with diethyl ether. The solvent was evaporated using a rotary evaporator and the remaining TFA salt was kept under a high vacuum overnight to give 6 as white rigid foam. HPLC analyses: tR = 11.4 min (≥98.5% pure at 254 nm and 280 nm) on ACE C18 100 Å column, (5μm, 4.6×250 mm) eluted at 0.8 mL/min with a linear gradient of MeCN in water; both solvents contained 0.07% TFA (v/v), first from 20% to 50% over 10 min, then to 95% MeCN during additional 10 min and eluting for further 15 min. 1HNMR (CD3OD/D2O, 600 MHz) δ: 8.45 (s, 1H, H6-uridine), 8.24 (bs, 1H, HN3), 7.38-7.32 (m, 5H, aryl), 6.31 (dd, 1H, H1’-uridine, 3J1’,2’=6.40 Hz, 3J1’,2”=4.14 Hz), 6.12-6.03 (m, 1H, H2), 5.49-5.34 (m, 2H, H3’- uridine, H4’-uridine), 4.12-3.74 (m, 2H, H5’-uridine), 3.35-3.28 (m, 2H, H9, 2.54-2.37 (m, 2H, H2’- uridine), 2.27-2.21 (2H, H5), 1.68-1.47 (m, 2H, H8), 1.32-1.24 (m, 4H, H6, H7) ppm. HR-MS (ESI), (m/e): [M+H]+ calcd for [C24H31IN6O7 + 1H]+, 643.1336; found 643.2103.
2.2.5. (R)-(–)-5-Trimethylstannyl-3’-O-[2-(ε-(N,N′-bis(tert-butyloxycarbonyl)guanidino)hexanoyl)-2-phenylacetyl]-2’-deoxyuridine (7)
Stannylation of 5 was conducted in 30 mL MeCN twice: 244 mg (0.290 mmol) of 5 and repeated with 605 mg (0.718 mmol) of 5, using 165 mg (0.50 mmol) and 400 mg (1.22 mmol) hexamethylditin, respectively, with the palladium catalyst (~50 mg, 0.07 mmol) and TEA (200 μL, ~1.43 mmol) in both reactions. Each reaction mixture was gently refluxed under nitrogen for as long as the starting 5 was detectable (~3 h). The reaction progress was checked by TLC. After cooling to ambient temperature, the mixture was filtrated through a thin pad of silica prewashed with EtOAc/hexanes (2:1; v/v) to separate the remaining catalyst. The filtrate was evaporated to dryness. Two major products were present: the desired trimethylstannane (high TLC mobility) and the protodestannylated form of starting iodide (slower on TLC). Crude products were separated and purified by repeating silica gel flash column chromatography with mixtures of EtOAc/hexanes (2:1-2:2, v/v) and/or various gradients of MeOH in DCM (0.3-0.7:10), to give 6 (94 mg and 278 mg) with 37% and 41% yields for the two reaction, Rf = 0.55 (DCM/MeOH, 10:0.8). To reach ≥ 98% purity of stannylated product, the successive preparative HPLC purifications were required. Purified, anhydrous samples of stannane 7 in 100-μg aliquots were stored with the exclusion of light under nitrogen at −20°C. HPLC analyses: tR = 31.3 min (>98% pure at 254 nm and 280nm) on ACE C18 100 Å column (5 μm, 4.6×250 mm) eluted at 0.8 mL/min with a linear gradient of MeCN in water, first from 50% to 60% over 20 min, then to 70% MeCN during 10 min and increased to 95% MeCN in 15 min eluting for further 15 min. 1HNMR (CDCl3, 600 MHz) δ: 11.48 (s,1H, HN3-uridine), 8.31 (bs, 1H, Boc-NH-guanidine), 8.03 (d, 1H, HN3, J = 5.20 Hz), 7.85 (s, 1H, H6-uridine,3JSn,H=8.36Hz), 7.38-7.26 (m, 5H, aryl), 6.32 (dd, 1H, H1’, 3J1’,2’=6.45 Hz, 3J1’,2”=4.20 Hz), 5.81-5.77 (m, 1H, H2), 4.23-4.18 (m, 1H, OH-C5’-uridine), 4.08-3.76 (m, 2H, H3’-uridine, H4’-uridine), 3.34-3.30 (m, 2H, H5’-uridine), 2.77-2.71 (m, 1H, HN10-guanidine), 2.66-2.59 (m, 2H, H9), 2.56-2.42 (m, 4H, H2’-uridine, H5), 2.32-2.29 (m, 2H, H8), 1.52 (s, 9H, 3×CH3-t-butyl), 1.49 (s, 9H, 3×CH3-t-butyl), 1.47-1.34 (m, 4H, H6, H7), 0.29 (s, 9H, 3xSnCH3, 2JSn,H=29.5Hz) ppm. 13CNMR (CDCl3, 100 MHz) δ: 172.6 (O=C4), 170.7 (O=C1), 163. 6 (C4-uridine), 160.3 (N=C-NH, guanidine), 158. 2 (=NC(O)-O-t-butyl, guanidine), 157.1 (-HN-C(O)-O-t-butyl, guanidine), 150.7 (C2- uridine), 144.8 (C6- uridine), 135.4 (C1- aryl), 129.3 (C2- aryl), 129.3 (C6 - aryl), 127.4 (C3 - aryl), 127.3 (C5 - aryl), 127.2 (C4 - aryl), 113.3 (C5- uridine), 87.3 (C1’- uridine), 85.2 (C4’- uridine), 83.1 (C1- t-butyl), 79.3 (C1- t-butyl), 76.2 (C3’- uridine), 62.7 (C5’- uridine), 56.9 (C2), 40.7 (C9), 36.9 (C2’- uridine), 35.8 (C5), 28.7 (C8), 28.3 (d, C2- t-butyl), 28.1 (d, C2- t-butyl), 26.2 (C7), 25.10 (C6), −9.3 ((CH3)3-Sn). HR-MS (ESI), (m/e): [M+H]+ calcd for [C37H56SnN6O11 + 1H]+, 873.3128; found at 873.3122 using the 112Sn isotope.
2.2.6. (R)-(–)-5-[125I]Iodo-3’-O-[2-(ε-(N,N′-bis(tert-butyloxycarbonyl)guanidino)hexanoyl)-2-phenylacetyl]-2’-deoxyuridine (8)
Into a glass tube containing the tin precursor 7 (100 μg, 0.1 μmol) in MeCN (50 μL), a solution of Na125I/NaOH (10 μL-50 μL, 44.4-373.7 MBq) was added followed by a solution of 30% H2O2 in water (5 μL) and 2 min later, a solution of TFA (50 μL, 0.1% in MeCN). The resulting mixture, briefly vortexed and/or sonicated, was left for 15 min at room temperature and then quenched with a solution of Na2S2O3 (90 μg-100 μg) in water (60 μL). The reaction mixture was taken up into a syringe and the reaction tube washed twice with a solution of MeCN/H2O (50 μL, 1:1, v/v). The reaction mixture and washes were combined, injected onto the HPLC system and separated on the C18 reverse phase column with a linear gradient of MeCN in water. Eluted fractions with radioiodinated product 8 were pooled, evaporated with a stream of dried nitrogen or were dissolved in dry MeCN (~18.5 MBq/mL) for further analysis or storage. This standard radioiodination of stannane 7 was carried out several times with amounts of Na125I ranging from 44.4 MBq to 377.4 MBq giving overall 482 MBq of 8. The average yield of the isolated radioactive product was 94%. A crude reaction mixture was separated and purified by HPLC on Jupiter C18 100 Å column (5 μm, 4.6×250 mm) eluted at 0.8 mL/min, using a linear gradient of MeCN in water from 50% to 60% over 20 min, then from 60% to 95% in 25 min and 95% MeCN was held for further 15 min. The product was collected within 19.5 min-22 min after the injection and an excess of the unreacted tin precursor 7, eluting ~11 min later, was fully separated,. HPLC analysis: tR = 19.6 min, (≥98% radiochemical purity, Bioscan NI(T)/UV 254 nm).
2.2.7. (R)-(–)-5-[125I]Iodo-3’-O-[2-(ε-guanidinohexanoyl)-2-phenylacetyl]-2’-deoxyuridine (9)
The deprotection (HPLC monitoring) of 8 to attain 9 in a solution of 30% TFA/MeCN at room temperature required approximately 70 min. The treatment of 8 (dried residue, 21.8 MBq-185 MBq) with neat TFA (30 μL) at ~65 °C accelerated the elimination of the guanidine Boc-protection and was completed in ~15 min. To remove the guanidine Boc-protective groups, neat TFA (100 μL) was added to the dried residue of 8 and the resulted mixture was vortexed and then heated in a sealed vial at 55°C-65°C for 20 min-35 min. After cooling, the mixture diluted with CH3CN (200 μL) was evaporated repetitively with a stream of nitrogen, each time leaving ~20 μL of the liquid in the reaction vial to prevent the adhesion of the product to the vial’s walls. This process was repeated at least three times, to ensure that the excess of TFA was eliminated. The residue was then dissolved in a solution of 50% MeCN in water, injected onto the HPLC system and was separated on ACE C18 100 Å column (5 μm, 4.6×250 mm) eluted at 0.8 mL/min with a linear gradient of MeCN from 20% to 50% over 10 min, then to 95% MeCN over 10 min and eluting for further 15 min. Both solvents contained 0.07% TFA (v/v); tR = 11.6 min (≥ 98% radiochemical purity, Bioscan/UV 280 nm).The product eluting within 10.5 min-12.0 min after the injection was collected in three fractions, which were combined and evaporated. An average yield of the purified 9 from four preparations was 89%. Alternatively, if the initial separation of Boc-protected radioiodinated 8 was not essential, a crude radioiodination mixture of 7 was evaporated with a stream of nitrogen, and/or kept under a high vacuum, then was directly treated with neat TFA at elevated temperature and separated using HPLC as described above. To fully eliminate the presence of TFA and CH3CN, the combined HPLC fractions containing 9 were evaporated with a stream of nitrogen and to the residue ethanol (80 μL) was added followed by potassium phosphate buffer (20 μL, 10 mM PB, pH ~6.1). The resulted solution was injected again onto the HPLC system equipped with Luna CN column (5μ, 4.5×250 mm) and eluted at 0.8 mL/min with a linear gradient of 50% to 80% EtOH in 10 mM PB (pH 6.1) over a 30-min period. The product was collected within 23.0 min-24.5 min after the injection (average tR = 23.4 min, ≥98% radiochemical purity, Bioscan). In every HPLC separation or analysis of radiolabeled products, the eluate from a column was monitored with the radioactivity detector connected to an outlet of the UV detector (detection at 220 nm and 280 nm). Solutions containing the product were reconstituted in a required solvent and medium at the desired concentrations and then filtered through a sterile 0.22-μm filter (MilliporeSigma, Burlington, MA) into a sterile evacuated vial. The identity of the radiolabeled product was confirmed by the evaluation of UV signals of nonradioactive iodo-analog 6 with UV and radioactivity signals of 9, tR from the radio-HPLC analyses, and by comparing Rf obtained from the radio-TLC.
2.3. In Vitro evaluation
2.3.1. Stability of (R)-(–)-5-[125I]iodo-3’-O-[2-(ε-guanidinohexanoyl)-2-phenylacetyl]-2’-deoxyuridine
Aliquots of GPAID (1.4 MBq-2.3 MBq) in 120 μL of 25% EtOH/PB (10 mM, pH 6.1) were added to 6.88 mL PBS, cell culture media or serum (mouse, pig or human). Mixtures were briefly vortexed and incubated at 37°C, 5% CO2. At selected times, 1 mL of GPAID incubation mixture was withdrawn, 1 mL MeCN was added, the mixture was vortexed and centrifuged at 2,000 rpm for 15 min at 4°C. The supernatant was removed and the radioactivity measured. Supernatant (250 μL-500 μL) were acidified to pH ~6 with 0.05 N TFA (2 μL-8 μL). The excess of MeCN was evaporated with a stream of nitrogen and 100 μL of water added. This mixture was passed through a 0.22-μm filter and 100 μL (111 kBq-185 kBq) injected onto the HPLC system. HPLC analysis of extracts proceeded on the ACE C18 100 Å column (5 μm, 4.6×250 mm) eluted at 0.8 mL/min with a linear gradient of MeCN from 10% to 95% over 45 min and kept at 95% MeCN for 15 min the radioactivity detection (Bioscan). Both solvents contained 0.07% TFA (v/v).
2.3.2. Cells
Two human NB cell lines SK-N-SH (SK) and BE(2)-C (BE) and one murine NB cell line N1E-115 (N1E) were purchased from the American Type Culture Collection (ATCC; Manassas, VA). Cells were maintained according to the vendor’s instructions. For animal studies, cells were harvested when ~70% confluent and cryopreserved until ready to use.
2.3.3. Time- and concentration-dependent cellular uptake and subcellular distribution of GPAID
Previously established protocols were used in these experiment.14,28–29 Briefly, NB cells plated in TPP T25 flasks (Midwest Scientific, Valley Park, MO) at 5×105 cells per flask were maintained in culture for 48 h-72 h, depending on the doubling time of the cell line, until 60%-70% confluent. For time-dependent uptake studies, GPAID was added to the growth medium to produce the average concentrations of 28.2±5.5 kBq/mL, 27.6±2.8 kBq/mL and 30.7±1.0 kBq/mL in SK, BE and N1E cells, respectively. Radioactive medium was removed after 1 h, 5 h, 16 h, 24 h and 48 h in culture (n=4 per time). Cells were harvested by scraping and enumerated using Cellometer® (Nexcelom Bioscience, Lawrence, MA). Cell suspensions were centrifuged for 10 min at 1,500 rpm, cell pellets resuspended in ice-cold PBS, transferred into microfuge tubes and centrifuged at 500×g for 10 min. The radioactive content of cell pellets was measured in a gamma counter. The cellular uptake was calculated in terms of mBq per cell. For subcellular fractionation experiments, NB cells grown as monolayers were treated with GPAID and processed as described previously.14 In the concentration-dependent uptake experiments, GPAID in the appropriate medium at several concentrations was added to the cell monolayers. Cells were processed after 1 h incubation. Radioactive medium was removed, monolayers washed once with full nonradioactive medium and PBS. Cells were harvested, enumerated and their radioactive content measured.
2.3.4. Competition with MIBG
Cells were plated into T25 flasks and allowed to grow until ~60%-70% confluent. Fresh medium (3 mL) was added to one set of flasks (n=4 per time). To the second set of flasks (n=4 per time), fresh medium (3 mL) containing 100 μM nonradioactive MIBG was added. All cells were returned to the incubator for 30 min after which time 3 mL of medium containing GPAID (51.5±1.4 kBq/mL) was added to produce the final concentration of 50 μM MIBG and 25.8±0.7 kBq/mL GPAID. Cells were returned to the incubator for additional 1 h, 24 h and 48 h. At designated times, media were removed, monolayer washed 1× with full medium, 2× with PBS and SK and BE cells were harvested with the non-enzymatic cell dissociation solution (Sigma-Aldrich). N1E cells were harvested by flushing the monolayer with spent media. Cell numbers were determined and their radioactive content measured in a gamma counter. The whole cell uptake was calculated on terms of mBq/cells. The clonogenic survival was calculated as the ratio of the number of colonies produced by cells treated with nonradioactive MIBG and GPAID to the number of colonies produced by cells treated with GPAID only. The cell survival at 24 h was expressed as the ratio of viable cells in treatment groups to cells in untreated controls.
2.4. In Vivo evaluation
2.4.1. Mice
Research involving animals was performed in accordance with the UNMC institutional guidelines and protocols as defined by the Institutional Animal Care and Use Committee for U.S. institutions. Athymic NCr nude mice (spontaneous mutant) were purchased from Taconic (Rensselaer, NY). The intraperitoneal route (IP) was used for the tumor implant and dose administration. All experiments were conducted in mice of both genders four to eight weeks-old.
2.4.2. Therapy
Four-week old NCr nu/nu mice, male (n = 10) and female (n = 10), received IP implant of 5×106 N1E cells. One week later, mice were randomly assigned via a lottery to either control group or treatment group. Control mice were given IP injection of the vehicle. Mice in the therapy group received a bolus IP injection of 3.64±0.10 MBq GPAID per mouse (~145 MBq/kg bw). Triplicate aliquots of the injected dose (ID) were counted in a gamma counter. Syringes containing the dose and syringes after the injection were weighed on the analytical balance to determine ID. Whole body radioactivity was also measured immediately after each injection to confirm ID. Necropsy was performed at the termination of the experiment. Blood was collected via cardiac puncture. Aliquots were reserved for determination of hemoglobin (Hb; HemoCue®, Ängelholm, Sweden) and hematocrit (Hct).
2.5. Statistical analyses
All variables are expressed as average ± standard deviation or ± standard error. The cellular uptake, cell survival and other biological properties were analyzed using two-sided Student’s t-test. Tumor weights at necropsy were used as the final measure of response. These data were analyzed using the Mann-Whitney test. P values <0.05 were considered statistically significant. SigmaPlot/SigmaStat (Systat Software, Inc. Point Richmond, CA) and GraphPad InStat ( La Jolla, CA) were used for these analyses.
3. RESULTS AND DISCUSSION
3.1. Synthesis of GPAID (9)
In an effort to enhance the biological half-life of IDG, a potential clinical candidate selected from a large series of 5-[125I]iodo-5’-O- and -3’-O- alkanoylguanidino-2’-deoxyuridine derivatives,14 we decided to increase the stability of its 3’-ester linkage by producing sterically hindered (R)-(–)-5-[125I]iodo-3’-O-[2-(ε-guanidinohexanoyl)-2-phenylacetyl]-2’- deoxyuridine (9, GPAID). GPAID was prepared by inserting the (R)-(–)-2-amino-2-phenylacetyl segment at the 3’-O-position of 5-iodo-2’-deoxyuridine (IUdR). Nonradioactive (R)-(–)-5-iodo-3’-O-[2-(6-N,N’-bis(tert-butoxycarbonylguanidino)-hexanamido)-2-phenylacetyl]-2’-deoxyuridine 5 was attained as shown in Scheme 1 starting with the synthesis of (R)-methyl 2-[6-(Boc-amino)hexanamido)]-2-phenylacetate 1 through the coupling of 6-Boc-aminocaproic acid with (R)-(–)-2-phenylglycine methyl ester hydrochloride with activation mediated by 2-chloro-1,3-dimethyl-2-hexafluorophosphate (CIP)/DMAP in the presence of N,N-diisopropylethylamine (DIPEA)23,24 to give 1 in 88% yield without detectable racemization (details in SI p. S3-S4). In subsequent steps, (R)-2-[6-(tert-butoxycarbonylamino)hexanamido]-2-phenylacetic acid 2, prepared in 91% yield by hydrolysis of the methyl ester group of 1, was reacted with the 5’-O-DMTr-2’-deoxyuridine generating (R)-(–)-5-iodo-5’-O-DMTr-3’-O-[2-(6-(tert-butoxycarbonylamino)-hexanamido)-2-phenylacetyl]-2’-deoxyuridine 3 in 67% yield. The 5’-O-DMTr protecting group in 3 was cleaved with ZrCl4.25 The 5’-O-deprotected 4 was isolated in 83% yield. In the final step, the N-Boc-amino protection of 4 was removed with ~50% TFA/MeCN and the guanidinylation of the free 6-aminohexanoyl group was achieved with N,N’-bis(tert-butoxycarbonyl)-N”-trifluoromethanesulfonyl guanidine,26 to provide 5 in the overall yield of 69%. Stannane 7 was obtained with an average yield of ~39% through the stannylation of iodide 5 with hexamethylditin in the reaction catalyzed by bis-(triphenylphosphine)-palladium(II) dichloride based on the modified Stille coupling,27 previously utilized by us to efficiently produce 125IUdR28 and various radioiodinated compounds.14,29–32 Successive HPLC purifications were necessary to achieve ≥98% purity of the stannane 7. The identity of nonradioactive compounds were validated by means of 1HNMR and 13CNMR, high-resolution mass spectrometry and meticulous HPLC analyses (documented in SI to this paper). Once purified, anhydrous 7 stored with the exclusion of light under nitrogen at −20°C was stable for several months (≤7% decomposition by HPLC). Higher hydrophobicity of stannane 7 allowed for its complete separation from the [125I]iodinated product 8 in a single HPLC purification even if a large volume (~1 mL) of the crude reaction mixture was injected onto the column. The guanidine Boc-protective groups of 8 were removed with neat TFA at 55°C-65°C. GPAID 9 was isolated on HPLC (SI p. 10). Alternatively, if the separation of Boc-protected 8 was not required, a dried crude radioiodination mixture was directly treated with neat TFA at elevated temperatures. To eliminate TFA and CH3CN from GPAID samples intended for biological studies, combined HPLC fractions containing GPAID were re-injected on HPLC and eluted with a linear gradient of EtOH in potassium phosphate buffer (SI pp. S11-S12). Identities of all radiolabeled products were confirmed by comparing their UV signals and tR from the radio-HPLC analysis with the UV signals of the independently prepared nonradioactive reference iodo-analogs as well as Rf obtained from radio-TLC. Non-carrier-added [125I]iodinations conducted with 22.2 MBq up to 148 MBq 125I always produced GPAID in high radiochemical yields of >80%-92% and radiochemical purity of ≥95%. Two% to 6% of the protodestannylated side product was detectable in some crude reaction mixtures. This by-product originated primarily from samples of stannane 7 stored at −20°C for prolonged periods. If the radiolabeled product was stored for >18 h, it was repurified just before use even though HPLC analyses of such samples rarely indicated the radiochemical purity <95%.
Scheme 1.
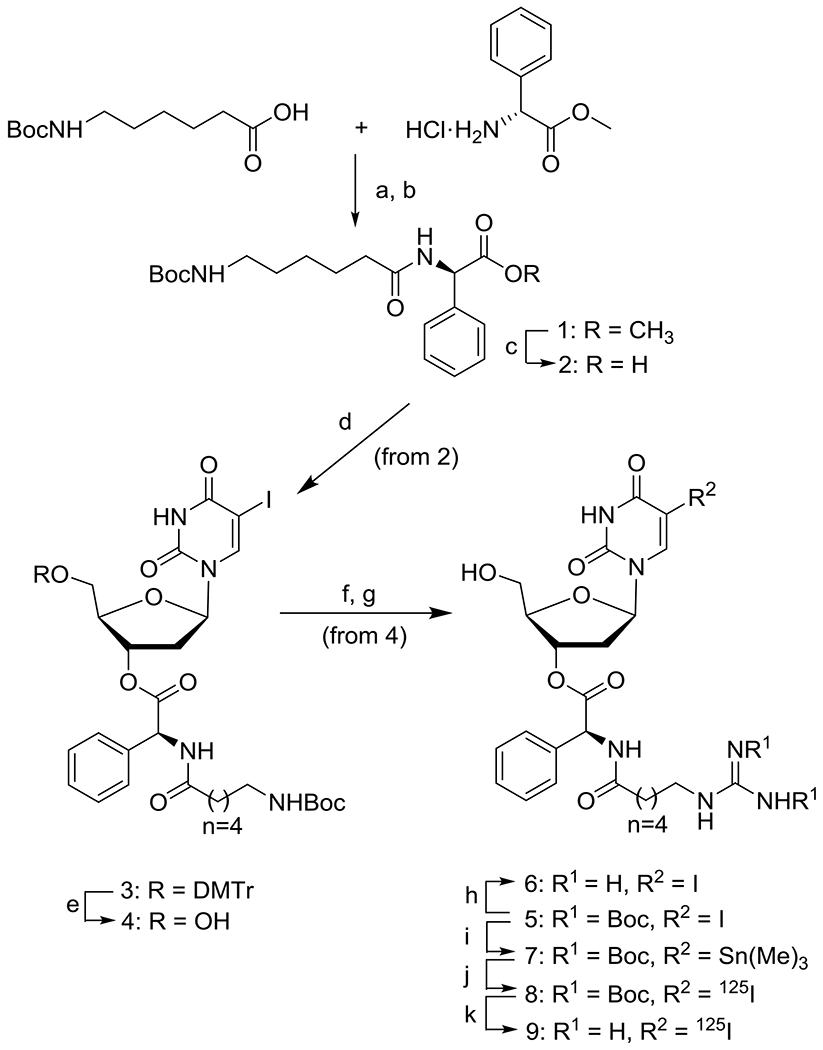
Reagents and conditions: (a) 0°C → 25°C, activated by CIP/DMAP in the presence of DIPEA; 88% yield after 2 h; (b) silica gel column chromatography; (c) (i) MeOH/H2O (3:1, v/v), KOH (1.6 equiv), 3-5 h at rt, (ii) 10% HCl, pH ~2; (d) (i) 5’-O-DMTr-2’-IUdR, (R)-(–)-2-[6-N-Boc-amino)hexanamido]-2-phenylacetic acid 2, DCC/DMPA in DCM, 4 h at rt; (ii) silica gel column chromatography; (e,f) ZrCl4 (1 equiv), CH3CN, 15 min, rt; 83% yield of 4; (g) (i) 4 in ~25% TFA/DCM, rt; (ii) evaporation under a vacuum at rt, treated with 15 mL of EtOAc/hexane (1:1, v/v) and sonicated; (iii) washed with Et2O, TFA salt dried under a high vacuum; (iv) N,N’-bis(tert-butoxycarbonyl)-N”-trifluoromethanesulfonyl guanidine (1.1 equiv), TEA (2.2 equiv), 0°C → rt, 6 h; (v) 5% citric acid and saturated brine, dried over MgSO4; (vi) silica gel column; 69% of 5; (h) (i) TFA (0.1 mL) at rt ~15 min; (ii) evaporation with a stream of nitrogen, residue triturated with EtOAc and Et2O; (iv) TFA salt of 6 under a high vacuum to give 6 as white rigid foam; (i) Sn2(CH3)6, (1.25-1.70 equiv) in CH3CN, TEA (2-4 equiv), (Ph3P)Pd(II)Cl2 (0.06-0.10 equiv), refluxed under nitrogen (1-3 h); (j) Na125I in NaOH (22.2-148 MBq), 30% H2O2 (5 μL), TFA/CH3CN (0,1% v/v), 1 min sonication, 6-15 min reaction time, HPLC purification; (k) TFA (neat, 100 μL), sealed vial at 55-65°C, 20-35 min, average radiochemical yield >80%.
3.2. In vitro evaluation
3.2.1. Stability studies
Ex vivo stability of GPAID was assessed in PBS, cell culture medium without NB cells and in the presence of NB cells, and in human, mouse and porcine sera after various times of incubation. The analytical details are provided in SI (pp. S14-S16). Half-lives in sera were calculated from the area under the peak of the intact GPAID recovered from the incubation mixtures plotted as a function of the incubation time. The degradation curves followed the first order kinetics. Data were fitted into the monoexponential equations with the intercept set at 100% (Figure 1A). In a typical experiment, a final concentration of GPAID in serum approximated radioactivity levels of 131IMIBG in blood of children receiving therapeutic doses.33,34 Similarly to IDG and other published derivatives,14 GPAID is catabolized to 125IUdR ensuring that the DNA co-targeting can be realized. Half-lives of GPAID in pig and human sera are 5.8±2.1 h and 7.9±0.7 h (avg±std err), respectively. For comparison, the corresponding IDG half-lives were 3.6 h±0.1 h and 2.3 h±0.1 h.14 The addition of the D-amino acid and a bulky phenyl group substantially increased GPAID stability in human and pig sera as compared to IDG. GPAID is not stable in mouse serum (Figure 1D), which contains several esterases including carboxylesterase. Human and porcine sera do not contain any carboxylesterase activity.35,36
Figure 1.
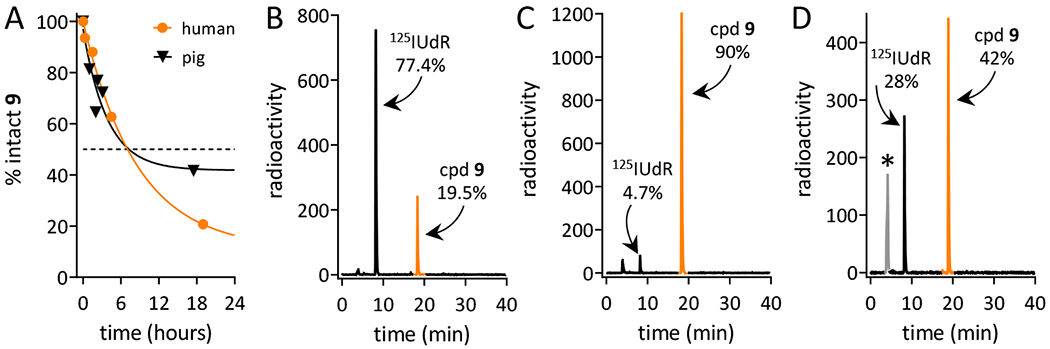
Stability of (R)-(–)-5-[125I]iodo-3’-O-[2-(ε-guanidinohexanoyl)-2-phenylacetyl]-2’-deoxyuridine (cpd 9, GPAID). HPLC profiles of radioactive species recovered from various incubation mixtures: A. Stability curves derived from HPLC analyses of GPAID incubated in human serum (orange circles) and pig serum (black triangles). Solid lines are the monoexponential fit of the percent intact GPAID as a function of various times of incubation. B. Composition of the mixture recovered after 17-min incubation of GPAID in mouse serum. C. GPAID recovered from culture medium without cells incubated for 24 h at 37°C, 5% CO2. D. Radioactivity profile of medium collected from SK-N-SH cells cultured with GPAID for 24 h at 37°C, 5% CO2; * this peak contains ~30% of total radioactivity in the form of either free 125I and 125I-uracil.
Prior to the in vitro studies, we also measured GPAID’s stability in PBS and in cell culture media to confirm that the data acquired in these experiments represent properties of the intact compound. GPAID is stable in PBS (SI p. S14) and in cell culture in the absence of NB cells. Figure 1C shows the radioactive HPLC profile of GPAID incubated in cell culture medium for 24 h at 37°C in 5% CO2 atmosphere. After 24 h at these conditions, >90% of GPAID remain intact. When GPAID is added to the exponentially growing monolayers of NB cell (Figures 1D) and incubated with cells for 24 h, the recovered media contain ~42% of the intact GPAID and ~28% of 125IUdR. Since GPAID is stable in media without cells under the identical set of conditions, the considerable loss of the intact compound and appearance of 125IUdR in the extracellular spaces are indicative of the uptake and intracellular processing. Nucleosides cross the plasma membrane of mammalian cells by a facilitated diffusion process that is bidirectional.37,38 It is therefore evident that 125IUdR is produced intracellularly. If it is not taken up into DNA, 125IUdR diffuses freely through the cellular membrane and emerges in the cell culture media.
3.2.2. Cellular uptake and subcellular distribution of GPAID
GPAID is taken up by NB cells in a time- and concentration-dependent manner (Figures 2A, 2B). The uptake is directly proportional to the incubation time up to 24 h and it also dependents on the cell population doubling time (TD). When cells are incubated with GPAID for 1 h, washed and processed immediately, cellular uptake is nearly identical in BE cells (TD=19 h) as it is in SK cells (TD=40 h). However, a 5-h incubation results in the radioactivity levels of 5.9±0.5 mBq per BE cell and 0.9±0.1 mBq per SK cell indicating greater levels of 125I incorporation into the DNA of rapidly proliferating BE cells. At later times, the uptake per cell levels off because the number of cells more than doubles. Similarly to the MIBG uptake, the cellular radioactivity is directly proportional to the extracellular concentration of GPAID within the entire tested range up to 200 kBq/mL (2.45×10−9 M). MIBG uptake is linear up to ~1×10−7 M and begins to saturate at concentrations >5×10−7 M.39 Cells incubated with different radioactive concentrations of GPAID end up with different amounts of intracellular 125IUdR. However, in this study all cells were washed after the incubation with GPAID to remove unbound radioactivity. With this approach, as validated in the subcellular fractionation experiments, within 24 h of GPAID treatment virtually all 125I is incorporated into DNA. Thus, in calculating disintegrations accumulated in 24 h (Figure 2C), we assumed that extranuclear 125I is low, if any, and therefore will not contribute to the radiotoxicity. NB cells accumulate >20 disintegrations per cell after a brief 1-h exposure to GPAID at all tested concentrations >0.015 MBq/mL. D37 for 125I in DNA of human cells is ~8.7 disintegrations/cell per 24 h (1.3 mBq/cell per 18 h40,41). Extracellular concentrations of GPAID required to achieve this level of 125I incorporation into DNA are expected to be easily attainable in a clinical setting. It is also noteworthy that all tested extracellular concentrations of GPAID are well below the clinical concentration of 131IMIBG estimated on the assumption of a uniform distribution throughout the body (denoted with an asterisk in Figure 2C).
Figure 2.
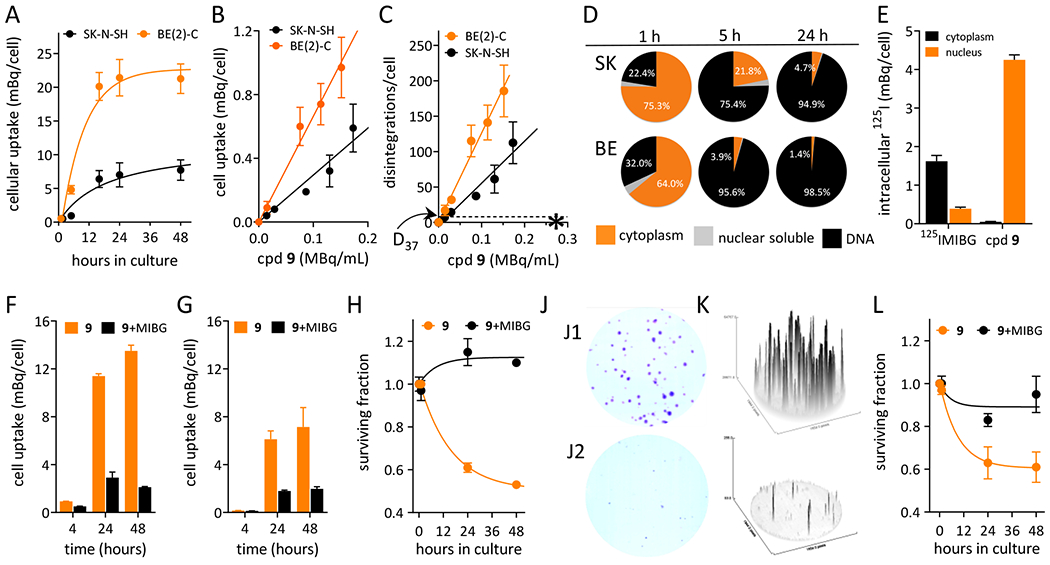
In vitro evaluation of GPAID (cpd 9) in neuroblastoma cells. A. Effects of the treatment time on the uptake of GPAID by BE(2)-C and SK-N-SH cells. B. Concentration-dependent uptake of GPAID by BE(2)-C and SK-N-SH cells after 1 h treatment. C. Total 125I intracellular disintegrations accumulated over 24 h after incubation of BE(2)-C and SK-N-SH cells with various concentrations of GPAID for 1 h. * indicates an estimated systemic concentration of 131IMIBG in a 20-kg child at the lowest clinically used dose of 5 GBq. Arrow points to D37 of 8.7 disintegrations per cell reported for 125I incorporated into DNA of human cells. D. Subcellular distribution of GPAID in neuroblastoma cells after various treatment times at the average GPAID concentration of 28±5 kBq/mL. E. Comparison of the cytoplasmic and nuclear distribution of GPAID and 125IMIBG in N1E-115 cells after a 24-h treatment at an average concentration of 29±3 kBq/mL. Nearly 100% of 125I activity in the nuclei of cells treated with GPAID is bound to DNA. F. Cellular uptake of GPAID by BE(2)-C cells in the absence (orange bars) and the presence (black bars) of 0.05 mM nonradioactive MIBG. G. Cellular uptake of GPAID by SK-N-SH cells in the absence (orange bars) and the presence (black bars) of 0.05 mM nonradioactive MIBG. H. Survival of BE(2)-C cells exposed to GPAID without (orange symbols) and with (black symbols) 0.05 mM nonradioactive MIBG. J. Colonies of BE(2)C cells three weeks after treatment. J1. Control cells treated with MIBG alone. J2. Cells treated with 25.8±0.7 kBq/mL GPAID for 24 h. K. ImageJ analyses of colonies shown in J1 and J2. L. Survival of SK-N-SH cells exposed to GPAID without (orange symbols) and with (black symbols) 0.05 mM nonradioactive MIBG.
Subcellular distribution studies confirmed the intracellular processing of GPAID and production of 125IUdR as measured by the DNA-associated radioactivity (Figure 2D). After 1 h with GPAID, SK cells retain ~22% of the total recovered 125I in their DNA compared to 32% in DNA of BE cells. When cells are incubated for 1 h with GPAID, washed, returned into the incubator with fresh nonradioactive medium for additional 4 h and then processed, nearly all cellular 125I, >95% of the total radioactivity, is associated with DNA of rapidly dividing BE cells while only ~75% 125I is associated with DNA of slower growing SK cells. At 24 h, practically all 125I is associated with DNA in both cells lines. The dissimilarities in the subcellular distribution of 125IMIBG and GPAID (TD=27 h) is illustrated in Figure 2E using N1E cells. The majority of 125IMIBG after 24 h incubation was recovered in the cytoplasmic fraction. Only ~19% of 125I was retrieved in the soluble nuclear fraction and 0% in DNA. In contrast, in cells exposed to GPAID, practically all 125I was recovered in the DNA pellet (>99%).
GPAID has pKa ~12 and like MIBG exists at physiological pH as a mono-protonated cation, which results in a poor cell membrane permeability and hinders its cellular uptake by passive diffusion.14,42 To determine if the NET-directed uptake of GPAID is competed by MIBG, a competitive uptake assay was conducted in SK and BE cells. These cells represent a well-established robust model for the NET-specific MIBG uptake.39,43 The cell monolayer method14 was employed. Experiments in both cell lines confirmed that the active uptake of GPAID is competitively inhibited by nonradioactive MIBG indicating the same transport mechanism (Figures 2F, 2G). Fifty μM MIBG inhibited the GPAID uptake by 70% and 85% in exponentially growing monolayers of SK and BE cells, respectively. The survival of NB cells was assessed at each time post treatment using the trypan blue exclusion method (Figures 2H, 2L) followed by the clonogenic assay (Figures 2J, 2K) to determine the effect of GPAID alone and when its uptake was competed with 50 μM MIBG. Both assays show significant reduction in survival when NB cells are treated with GPAID alone. As expected, the survival also depended on the duration of exposure (Figures 2H, 2L). Cells exposed to nonradioactive MIBG alongside GPAID produced on average 3× more colonies (Figure 2J1) compared to cells treated with GPAID alone. After the treatment with GPAID at the extracellular concentration of 25.8±0.7 kBq/mL, on average only 32% of BE cells survived, retained their reproductive integrity and formed colonies (Figure 2J2). Colonies were considerably smaller and less intensely stained with crystal violet compared to cells treated with GPAID and MIBG. The intensity of staining is proportional to the number of cells in each colony (Figure 2K).44
3.3. In vivo evaluation
The therapeutic potential of GPAID was evaluated in mice bearing IP N1E allografts. Two factors contributed to the choice of this tumor model. First, to avoid rapid degradation of GPAID by carboxylesterases present in mouse blood (Figure 1B), N1E cells were implanted IP and IP injections were used as the route of GPAID administration. Second, we previously determined that a mouse NB allograft represents a more realistic model of NB.45 Mice of both genders were treated with a single IP dose of 3.64±0.10 MBq/mouse GPAID one week after the tumor implant. The experiment was terminated 14 days later when control mice presented with significant abdominal distensions indicative of large tumor nodules. At necropsy, the average tumor weights were 2.61±0.57 g and 0.88±0.54 g in NT and GPAID-treated mice, respectively (Figure 3A). The difference between treatment and control groups is statistically significant (P = 0.03). Hb and HCt levels in mice treated with GPAID were within the normal range values (Figure 6F) and support the postulated low toxicity of 125I-labeled compounds. Biodistribution conducted at necropsy 14 days after the dose of GPAID shows only marginal radioactivity retention in all tissues. Elevated levels of GPAID are still present in the adrenal glands 0.034±0.016 % ID/g.
Figure 3.
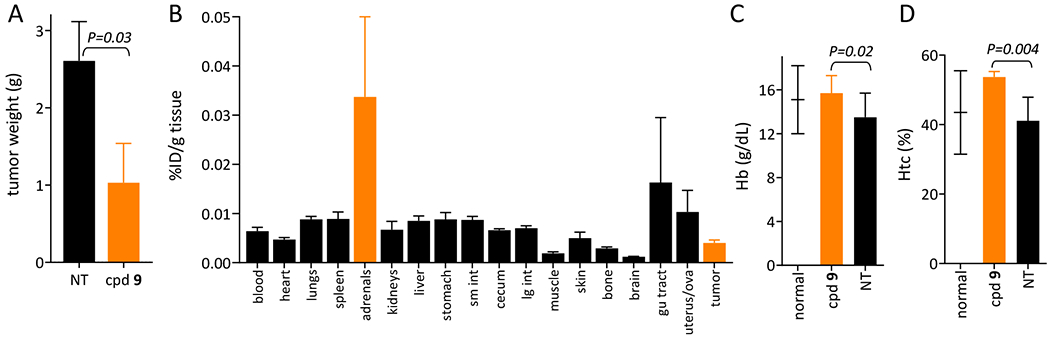
Tumor sizes, biodistribution of GPAID (cpd 9) and hemoglobin and hematocrit values in mice from the therapy and control (NT) groups. A. Weights of intraperitoneal N1E-115 tumors extirpated from control mice (black bar) and from mice treated with an average dose of 3.64±0.10 MBq/mouse GPAID (orange bar). B. Residual levels of 125I in various tissues and tumors extirpated from mice treated with GPAID at the termination necropsy 14 days after treatment. C. Hemoglobin values in tumor bearing controls NT (black bar) and mice treated with GPAID (orange bar). D. Hematocrit values in tumor bearing controls NT (black bar) and mice treated with GPAID (orange bar). Values for normal healthy mice are indicated by a short horizontal line with the standard deviation.
4. Conclusions
131IMIBG, the first theranostic5 introduced to the management of NB nearly 30 years ago became an essential component of treatment strategies for relapsed and refractory disease. However, the role of 131IMIBG in NB is still debated.6,7,46 Response rates, even with high-dose 131IMIBG therapy, are suboptimal. Tumor responses are only transient. High-dose therapies are associated with acute and chronic adverse effects related to the radiation exposure of normal tissues prohibiting further dose escalations.33 New therapies must attain a balance between improved survival rates and the morbidity of side effects. Molecular radiotherapeutics labeled with Auger‐electron emitting radionuclides are expected to have more favorable toxicity profiles permitting the administration of curative doses. We report 125I-labeled guanidine GPAID that was designed to deliver radiotoxic doses of radiation to NB cell’s DNA while sparing normal tissues. GPAID is a more stable analogue of IDG14. It can be efficiently synthesized from 5-iodo-2’-deoxyuridine, a molecular radiotherapy platform with clinically proven minimal toxicities and DNA targeting properties. Incorporation of an unnatural amino acid residue into the structure of radioactive guanidine increases stability. GPAID undergoes intracellular processing and its catabolite 125IUdR is incorporated into DNA of NB cells with high proliferation activities, i.e., cells in the high‐risk and aggressive disease. Tumor responses to clinically manageable doses of GPAID are significant in mouse allografts of NB. The chemical structure accommodates therapeutic as well as diagnostic radionuclides. Biological properties of GPAID suggest its significant potential as a novel theranostic for the management of NB.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Ed Ezell of the Fred & Pamela Buffett Cancer Center Structural Biology Facility Shared Resource (National Cancer Institute award P30 CA036727) for his help with the NMR analyses. Mass spectrometry analyses were conducted by the NIH/NIGMS Mass Spectrometry Resource, Washington University in St. Louis School of Medicine, St. Louis, MO.
Sponsors:
National Cancer Institute Grant R21CA187548
State of Nebraska LB 905
Contributor Information
Zbigniew P. Kortylewicz, University of Nebraska Medical Center, Department of Radiation Oncology, J. Bruce Henriksen Cancer Research Laboratories
Don W. Coulter, University of Nebraska Medical Center, Department of Pediatrics, Division of Hematology and Oncology
Guang Han, Huazhong University of Science and Technology, Hubei Cancer Hospital, Tongji Medical College, Department of Radiation Oncology.
Janina Baranowska-Kortylewicz, University of Nebraska Medical Center, College of Pharmacy, Department of Pharmaceutical Sciences.
REFERENCES
- 1.GBD 2017 Childhood Cancer Collaborators. The global burden of childhood and adolescent cancer in 2017: an analysis of the Global Burden of Disease Study 2017. Lancet Oncol. 2019(9); doi: 10.1016/S1470-2045(19)30339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips SM, Padgett LS, Leisenring WM, Stratton KK, Bishop K, Krull KR, Alfano CM, Gibson TM, de Moor JS, Hartigan DB, Armstrong GT, Robison LL, Rowland JH, Oeffinger KC, Mariotto AB. Survivors of childhood cancer in the United States: prevalence and burden of morbidity. Cancer Epidemiol Biomarkers Prev. 2015;24(4):653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deacon JM, Wilson PA, Peckham MJ. The radiobiology of human neuroblastoma. Radiother Oncol. 1985;3(3):201–209. [DOI] [PubMed] [Google Scholar]
- 4.Wheldon TE, Livingstone A, Wilson L, O’Donoghue J, Gregor A. The radiosensitivity of human neuroblastoma cells estimated from regrowth curves of multicellular tumour spheroids. Br J Radiol. 1985;58(691):661–664. [DOI] [PubMed] [Google Scholar]
- 5.Kimmig B, Brandeis WE, Eisenhut M, Bubeck B, Hermann HJ, zum Winkel K. Scintigraphy of a neuroblastoma with I-131 meta-iodobenzylguanidine. J Nucl Med. 1984;25(7):773–775. [PubMed] [Google Scholar]
- 6.Gaze MN, Gains JE, Walker C, Bomanji JB. Optimization of molecular radiotherapy with [131I]-meta Iodobenzylguanidine for high-risk neuroblastoma. Q J Nucl Med Mol Imaging. 2013;57(1):66–78. [PubMed] [Google Scholar]
- 7.Wilson JS, Gains JE, Moroz V, Wheatley K, Gaze MN. A systematic review of 131I-meta iodobenzylguanidine molecular radiotherapy for neuroblastoma. Eur J Cancer. 2014;50(4):801–815. [DOI] [PubMed] [Google Scholar]
- 8.Bleeker G, Schoot RA, Caron HN, de Kraker J, Hoefnagel CA, van Eck BL, Tytgat GA. Toxicity of upfront 131I-metaiodo-benzyl-guanidine (131I-MIBG) therapy in newly diagnosed neuroblastoma patients: a retrospective analysis. Eur J Nucl Med Mol Imaging. 2013;40(11):1711–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polishchuk AL, Dubois SG, Haas-Kogan D, Hawkins R, Matthay KK. Response, survival, and toxicity after iodine-131-metaiodobenzylguanidine therapy for neuroblastoma in preadolescents, adolescents, and adults. Cancer. 2011;117(18):4286–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Incesoy-Ozdemir S, Bozkurt C, Yüksek N, Oren AC, Sahin G, Bozkurt S, Ertem U. Secondary childhood acute myeloid leukemia with complex karyotypic anomalies including monosomy 7, monosomy 5 and translocation (1;10) after 131I-metaiodobenzylguanidine therapy for relapsed neuroblastoma. Turk J Pediatr. 2011;53(1):83–86. [PubMed] [Google Scholar]
- 11.Garaventa A, Gambini C, Villavecchia G, Di Cataldo A, Bertolazzi L, Pizzitola MR, De Bernardi B, Haupt R. Second malignancies in children with neuroblastoma after combined treatment with 131I-metaiodobenzylguanidine. Cancer. 2003;97(5):1332–1338. [DOI] [PubMed] [Google Scholar]
- 12.Clement SC, Kraal KC, van Eck-Smit BL, van den Bos C, Kremer LC, Tytgat GA, van Santen HM. Primary ovarian insufficiency in children after treatment with 131I-metaiodobenzylguanidine for neuroblastoma: report of the first two cases. J Clin Endocrinol Metab. 2014;99(1):E112–116. [DOI] [PubMed] [Google Scholar]
- 13.Clement SC, van Eck-Smit BL, van Trotsenburg AS, Kremer LC, Tytgat GA, van Santen HM. Long-term follow-up of the thyroid gland after treatment with 131I-Metaiodobenzylguanidine in children with neuroblastoma: importance of continuous surveillance. Pediatr Blood Cancer. 2013;60(11):1833–1838. [DOI] [PubMed] [Google Scholar]
- 14.Kortylewicz ZP, Coulter DW, Han G, Baranowska-Kortylewicz J. Norepinephrine-transporter-targeted and DNA-co-targeted theranostic guanidines. J Med Chem. 2019. doi: 10.1021/acs.jmedchem.9b00437. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.Booz J, Paretzke HG, Pomplun E, Olko P. Auger-electron cascades, charge potential and microdosimetry of iodine-125. Radiat Environ Biophys. 1987;26(2):151–162. [DOI] [PubMed] [Google Scholar]
- 16.Adelstein SJ, Kassis AI. Radiobiologic implications of the microscopic distribution of energy from radionuclides. Int J Rad Appl Instrum B. 1987;14(3):165–169. [DOI] [PubMed] [Google Scholar]
- 17.Chen H, Jacobson O, Niu G, Weiss ID, Kiesewetter DO, Liu Y, Ma Y, Wu H, Chen X. Novel “Add-On” Molecule based on Evans blue confers superior pharmacokinetics and transforms drugs to theranostic agents. J Nucl Med. 2017;58(4):590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bandara N, Jacobson O, Mpoy C, Chen X, Rogers BE. Novel structural modification based on Evans blue dye to improve pharmacokinetics of a somastostatin-receptor-based theranostic agent. Bioconjug Chem. 2018;29(7):2448–2454. [DOI] [PubMed] [Google Scholar]
- 19.Guichard G, Benkirane N, Zeder-Lutz G, van Regenmortel MH, Briand JP, Muller S. Antigenic mimicry of natural L-peptides with retro-inversopeptidomimetics. Proc. Natl. Acad. Sci. U.S.A 1994;91(21):9765–9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Lee CP, Smith PL, Borchardt RT. Metabolism, uptake, and transepithelial transport of the stereoisomers of Val-Val-Val in the human intestinal cell line, Caco-2. Pharm Res. 1996;13(11):1663–7. [DOI] [PubMed] [Google Scholar]
- 21.Vig BS, Lorenzi PJ, Mittal S, Landowski CP, Shin HC, Mosberg HI, Hilfinger JM, Amidon GL. Amino acid ester prodrugs of floxuridine: synthesis and effects of structure, stereochemistry, and site of esterification on the rate of hydrolysis. Pharm Res. 2003;20(9):1381–8. [DOI] [PubMed] [Google Scholar]
- 22.Tsume Y, Incecayir T, Song X, Hilfinger JM, Amidon GL. The development of orally administrable gemcitabine prodrugs with D-enantiomer amino acids: enhanced membrane permeability and enzymatic stability. Eur J Pharm Biopharm. 2014;86(3):514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamiński ZJ. 2-Chloro-4,6-disubstituted -1,3,5-triazines a novel group of condensing reagents. Tetrahedron Lett. 1985;26(24),2901–2904. [Google Scholar]
- 24.Akaji K, Kuriyama N, Kiso Y. Efficient coupling of α,α-dimethyl amino acid using a new imidazolidium reagent, CIP. Tetrahedron Lett. 1994,35(20),3315–3318. [Google Scholar]
- 25.Sharma GVM, Reddy ChG, Krishna PR. Zirconium(IV) chloride catalyzed new and efficient protocol for the selective cleavage of p-methoxybenzyl ethers. J. Org. Chem 2003;68(11),4574–4575. [DOI] [PubMed] [Google Scholar]
- 26.Baker TJ, Tomioka M, Goodman M. Preparation and use of N,N’-di-Boc-N”-triflylguanidine. Org Synth. 2002,78,91–98. [Google Scholar]
- 27.The Stille Reaction recent reviews:; (a) Williams R Org Synth. 2011;88,197–201, [PMC free article] [PubMed] [Google Scholar]; (b) Selig R, Schollmeyer D, Albrecht W, Laufer S. Tetrahedron 2011;67(47),9204–9213. [Google Scholar]
- 28.Baranowska-Kortylewicz J, Helseth LD, Lai J, Schneiderman MH, Schneiderman GS, Dalrymple GV. Radiolabeling kit/generator for 5-radiohalogenated uridines. J Label Compd Radiopharm. 1994;34(6):513–521. [Google Scholar]
- 29.Kortylewicz ZP, Nearman J, Baranowska-Kortylewicz J. Radiolabeled 5-iodo-3’-O-(17beta-succinyl-5alpha-androstan-3-one)-2’-deoxyuridine and its 5’-monophosphate for imaging and therapy of androgen receptor-positive cancers: synthesis and biological evaluation. J Med Chem. 2009;52(16):5124–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kortylewicz ZP, Kimura Y, Inoue K, Mack E, Baranowska-Kortylewicz J. Radiolabeled cyclosaligenyl monophosphates of 5-iodo-2’-deoxyuridine, 5-iodo-3’-fluoro-2’,3’-dideoxyuridine, and 3’-fluorothymidine for molecular radiotherapy of cancer: synthesis and biological evaluation. J Med Chem. 2012;55(6):2649–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han G, Kortylewicz ZP, Enke T, Baranowska-Kortylewicz J. Co-targeting androgen receptor and DNA for imaging and molecular radiotherapy of prostate cancer: in vitro studies. Prostate. 2014;74(16):1634–1646. [DOI] [PubMed] [Google Scholar]
- 32.Kortylewicz ZP, Mack E, Enke CA, Estes KA, Mosley RL, Baranowska-Kortylewicz J. Preclinical evaluation of investigational radiopharmaceutical RISAD-P intended for use as a diagnostic and molecular radiotherapy agent for prostate cancer. Prostate. 2015;75(1):8–22. [DOI] [PubMed] [Google Scholar]
- 33.Pandit-Taskar N, Zanzonico P, Hilden P, Ostrovnaya I, Carrasquillo JA, Modak S. Assessment of organ dosimetry for planning repeat treatments of high-dose 131I-MIBG therapy: 123I-MIBG versus posttherapy 131I-MIBG imaging. Clin Nucl Med. 2017;42(10):741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matthay KK, DeSantes K, Hasegawa B, Huberty J, Hattner RS, Ablin A, Reynolds CP, Seeger RC, Weinberg VK, Price D. Phase I dose escalation of 131I-metaiodobenzylguanidine with autologous bone marrow support in refractory neuroblastoma. J Clin Oncol. 1998;16(1):229–236. [DOI] [PubMed] [Google Scholar]
- 35.Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, Lockridge O. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol 2005;70(11):1673–1684. [DOI] [PubMed] [Google Scholar]
- 36.Bahar FG, Ohura K, Ogihara T, Imai T. Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci. 2012;101(10):3979–88. [DOI] [PubMed] [Google Scholar]
- 37.Plagemann PG, Richey DP. Transport of nucleosides, nucleic acid bases, choline and glucose by animal cells in culture. Biochim Biophys Acta 1974;344(3-4):263–305. [DOI] [PubMed] [Google Scholar]
- 38.Paterson AR, Kolassa N, Cass CE. Transport of nucleoside drugs in animal cells. Pharmacol Ther. 1981;12(3):515–536. [DOI] [PubMed] [Google Scholar]
- 39.Smets LA, Loesberg C, Janssen M, Metwally EA, Huiskamp R. Active uptake and extravesicular storage of m-iodobenzylguanidine in human neuroblastoma SK-N-SH cells. Cancer Res. 1989;49(11):2941–2944. [PubMed] [Google Scholar]
- 40.Kassis AI, Fayad F, Kinsey BM, Sastry KS, Taube RA, Adelstein SJ. Radiotoxicity of 125I in mammalian cells. Radiat Res. 1987;111(2):305–18. [PubMed] [Google Scholar]
- 41.Makrigiorgos GM, Kassis AI, Baranowska-Kortylewicz J, McElvany KD, Welch MJ, Sastry KS, Adelstein SJ. Radiotoxicity of 5-[123I]iodo-2’-deoxyuridine in V79 cells: a comparison with 5-[125I]iodo-2’-deoxyuridine. Radiat Res. 1989;118(3):532–44. [PubMed] [Google Scholar]
- 42.Wieland DM, Brown LE, Rogers WL, Worthington KC, Wu JL, Clinthorne NH, Otto CA, Swanson DP, Beierwaltes WH. Myocardial imaging with a radioiodinated norepinephrine storage analog. J Nucl Med. 1981;22(1):22–31. [PubMed] [Google Scholar]
- 43.Montaldo PG, Lanciotti M, Casalaro A, Cornaglia-Ferraris P, Ponzoni M. Accumulation of m-iodobenzylguanidine by neuroblastoma cells results from independent uptake and storage mechanisms. Cancer Res. 1991;51(4):4342–4346. [PubMed] [Google Scholar]
- 44.Guzmán C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9(3):e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coulter DW, Boettner AD, Kortylewicz ZP, Enke SP, Luther JA, Verma V, Baranowska-Kortylewicz J. Butyrylcholinesterase as a Blood Biomarker in Neuroblastoma. J Pediatr Hematol Oncol. 2017;39(4):272–281. [DOI] [PubMed] [Google Scholar]
- 46.Streby KA, Shah N, Ranalli MA, Kunkler A, Cripe TP. Nothing but NET: a review of norepinephrine transporter expression and efficacy of 131I-mIBG therapy. Pediatr Blood Cancer. 2015;62(1):5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.