

Abstract
The fungal indole alkaloids are a unique class of complex molecules that have a characteristic bicyclo[2.2.2]diazaoctane ring and frequently contain a spiro-oxindole moiety. While various strains produce these compounds, an intriguing case involves the formation of individual antipodes by two unique species of fungi in the generation of the potent anticancer agents (+)- and (−)-notoamide A. NotI and NotI′ have been characterized as flavin-dependent monooxygenases that catalyze epoxidation and semi-pinacol rearrangement to form the spiro-oxindole center within these molecules. This work elucidates a key step in the biosynthesis of the notoamides and provides an evolutionary hypothesis regarding a common ancestor for production of enantiopure notoamides.
Introduction
The fungal-derived prenylated indole alkaloids are a large class of natural products having a diverse range of biological activities relevant to many human and animal diseases.[1] They are assembled by fascinating biosynthetic mechanisms, and have been the subject of numerous unique and challenging bioinspired total syntheses.[2] This constantly expanding family of compounds includes the anthelmintic paraherquamides,[1e] calmodulin-inhibitory malbrancheamides,[1n] and anticancer notoamides,[3] stephacidins,[1m] and citrinadins,[4] among others. These alkaloids are typically composed of an initial prenylated dipeptide that is modified via a proposed intramolecular Diels-Alder (IMDA) reaction to form the characteristic bicyclo[2.2.2]diazaoctane core.[5],[6],[7],[2f, 8] A semi-pinacol rearrangement is predicted to generate the spiro-oxindole moiety found in the notoamides, paraherquamides, and spiromalbramide.[9],[10],[11] The respective enzymes involved in spirocycle formation are proposed to generate an initial indole-2,3-epoxide with facial selectivity, followed by controlled collapse of the epoxide giving rise to the observed spiro-oxindoles. Very few enzymes responsible for this type of reaction within the bicyclo-ring containing family have been characterized.[12],[13] NotB catalyzes 2,3-β-face epoxidation of notoamide E (1) to generate the non-spirocyclized terminal metabolites notoamides C (2) and D (3) that do not undergo IMDA cyclization. [12] Additionally, biochemical and structural analysis of PhqK provided the first insights into the mechanism of selective spirocyclization.[13] However, there has been little biochemical evidence to support the direct role of specific enzymes in the rearrangement reaction to form spirocyclized notoamide products, leaving an incomplete understanding of the formation of these molecules in secondary metabolism.[14]
The notoamides are a fascinating class of anticancer metabolites in the indole alkaloid family where two phylogenetically related fungal strains have evolved to generate terminal products based on catalytic processes with opposing enantioselectivity (Scheme 1). The natural products (−)-stephacidin A (4), (+)-notoamide B (5), (+)-6-epi-stephacidin A (6), (−)-6-epi-stephacidin A (7), and (+)-versicolamide B (8) are produced by the terrestrial strain Aspergillus amoenus (formerly Aspergillus versicolor NRRL 35600), while (+)-stephacidin A (9), (−)-notoamide B (10), (+)-6-epi-stephacidin A (6), and (+)-versicolamide B (8) are produced by the marine strain Aspergillus protuberus (formerly Aspergillus sp. MF297–2).[15],[16] Based on these isolation data, it was proposed that the two strains underwent an enantiodivergence with respect to the production of the notoamides.
Scheme 1.
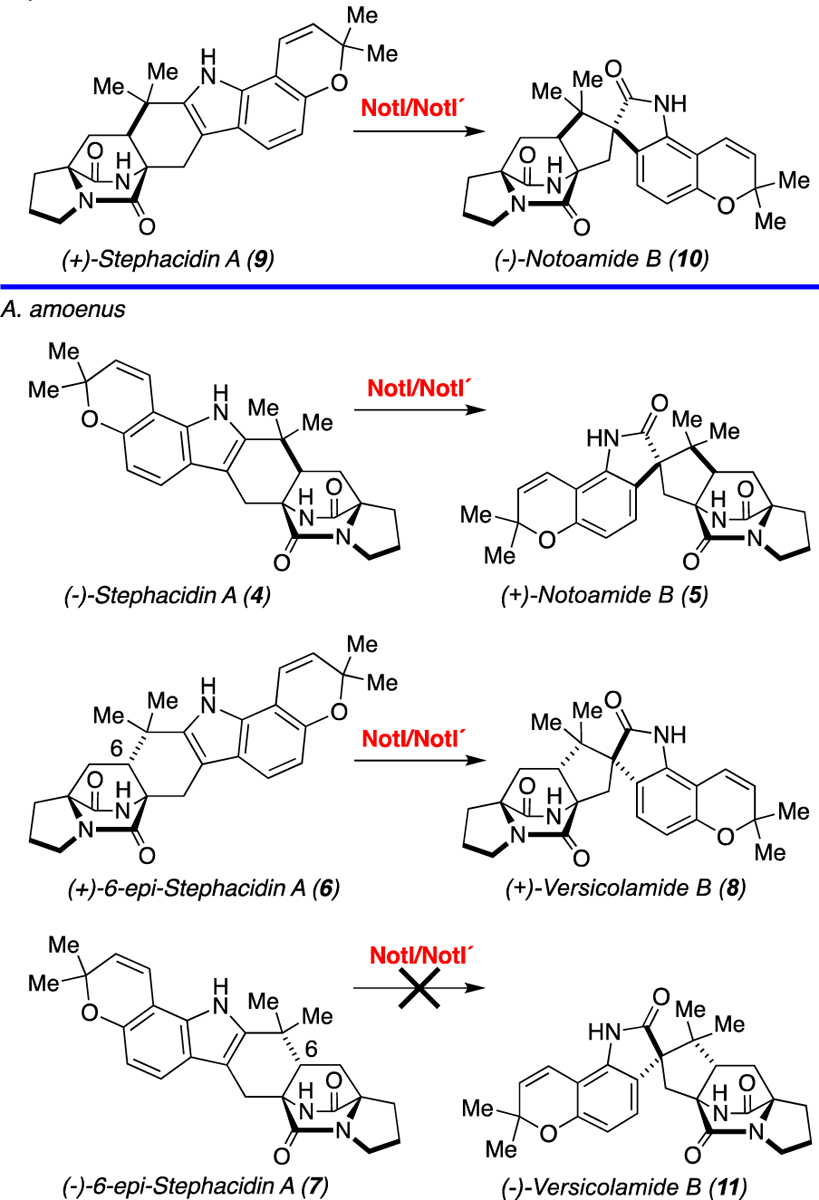
Reactions catalyzed by NotI/NotI′ in vitro and in the respective native fungal species.
The formation of enantiomeric natural products identified from one or more species is highly unusual,[17] and the existence of distinct metabolic systems that form exact antipodal pairs suggests that the strains have evolved one or more biosynthetic gene products that catalyze an identical enzymatic reaction to produce and further modify enantiomeric compounds.[18] Bioinspired synthetic schemes have been devised to generate stephacidin A and notoamide B, supporting the hypothesis that one or more biosynthetic enzymes determine the chirality of the molecule.[2d, 2h, 19] However, the molecular mechanisms that control this divergence within the respective organisms have remained a mystery.
As a starting point for these studies, we sequenced the genomes of A. amoenus (not′) and A. protuberus (not).[12] The not/not′ gene clusters were identified through in silico database mining followed by open reading frame (orf) and BLAST analysis to complete a more thorough annotation of the metabolic systems. These data, combined with previous isotopically enriched precursor incorporation studies[20] supported our proposed biosynthetic pathway for the notoamides.[18] Comparison of the not/not′ gene clusters showed an overall 71% DNA sequence identity (between notA-notJ/notA′-notJ′), indicating a closely related phylogeny.[21] However, bioinformatic analyses provided only limited understanding of the biosynthetic pathway and little mechanistic information about the sequence divergence observed between the two enantiomeric natural products 7 and 2. Consequently, we were motivated to investigate the biochemical transformations involved in the formation of the notoamides, and determine the basis for the structural branch-point that generates these antipodal indole alkaloid molecules.
Results and Discussion
Previous work from our laboratories revealed the role of early steps in notoamide assembly. NotF and NotC were characterized as reverse and normal prenyltransferases, respectively, while NotB was shown to be a FAD-dependent oxidase.[9],[12] Herein, we report the biochemical function of late-stage enzyme NotI as a flavin-dependent monooxygenase (FMO) (Figures S3, S4, and S5) that catalyzes the semi-pinacol rearrangement to generate the spiro-oxindole moiety present in many of the bicyclo[2.2.2]diazaoctane fungal indole alkaloids. We also report the biochemical function of NotI′ (85% sequence identity to NotI) as the first heterologously expressed and biochemically characterized gene product from A. amoenus.
The functions of NotI and NotI′ were investigated by separately incubating compounds (+)- and (−)- stephacidin A (9 and 4) with NotI or NotI′. The reactions were subjected to HPLC analysis and compared directly with synthetic standards or authentic natural products isolated from the respective fungal cultures (Scheme 1 and Figure 1). Both NotI and NotI′ catalyzed conversion of (+)- and (−)- stephacidin A (9 and 4) to (−)- and (+)- notoamide B (10 and 5), although a clear preference was observed for the conversion of (−)-stephacidin A (4) to (+)-notoamide B (5) (Table S4). Additionally, NotI and NotI′ converted (+)-6-epi-stephacidin A (6) to (+)-versicolamide B (8), but no reaction was observed with (−)-6-epi-stephacidin A (7) to produce (−)-versicolamide B (11). This is compatible with the conversion observed in A. amoenus where (+)-versicolamide B (8) was produced and (−)-6-epi-stephacidin A (7) was determined to be a shunt metabolite.[16] To further define this biocatalytic process, the reactions of NotI with (−)-stephacidin A (4) to generate (+)-notoamide B (5) were fit to Michaelis-Menten model kinetics (Figure S6). The Km and Vmax values were determined to be 37.4 ± 14.5 μM and 1.19 ± 0.13 μM/min, respectively. The relatively low conversion observed for this enzyme could be due to decoupling of the flavin redox chemistry from the epoxidation/semi-pinacol rearrangement. The measured rate of product formation (V0) was 0.06 μM/min at a standard substrate concentration of 200 μM, and the rate of NADH consumption was 1.43 μM/min, with an epoxidation efficiency of 4.2%, indicating decoupling (Figure S7). While a comparison of reactivity between the two enzymes would be intriguing, the necessity of using an MBP-tagged NotI′ precluded an accurate determination of the native reaction kinetics. Accordingly, we have confirmed that the enzymes may not be optimally folded because the flavin incorporation ratios for both enzymes are quite low (18% for NotI and 0.5% for NotI′).
Figure 1.
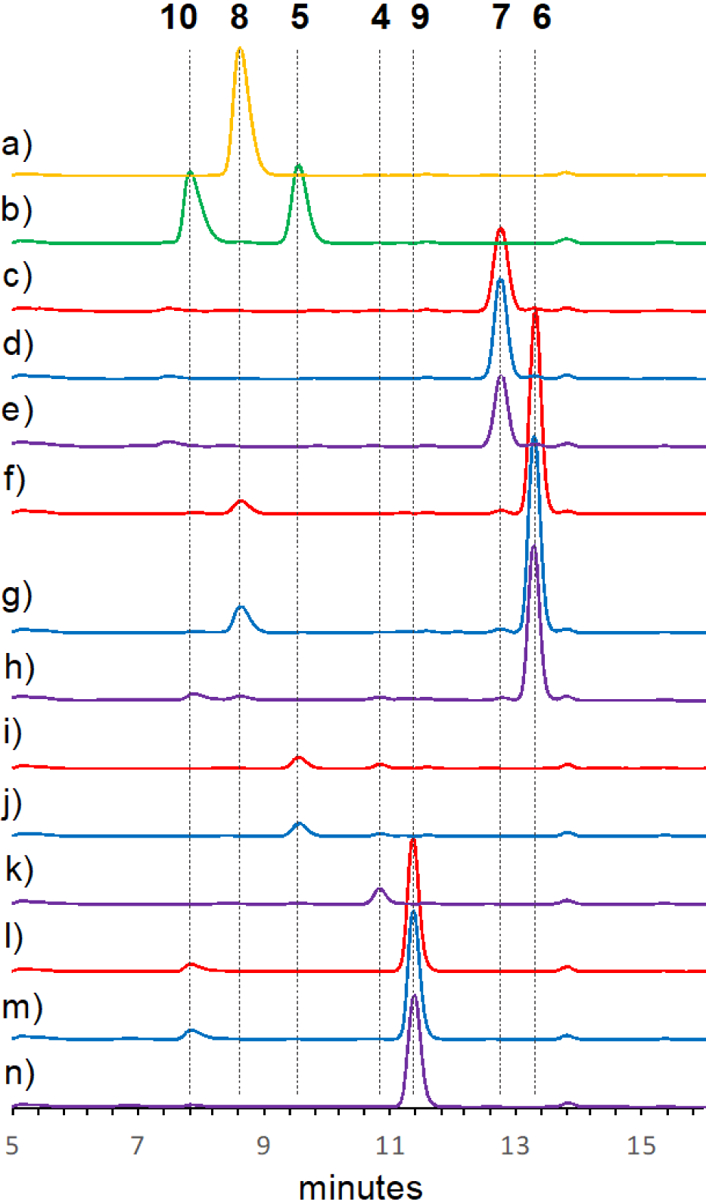
Biochemical activity assays with NotI and NotI′. (a) (+)-versicolamide B (8) standard. (b) (+)- and (−)-notoamide B (5 and 10) standards. (c) NotI′ reaction with (−)-6-epi-stephacidin A (7). (d) NotI reaction with 7. (e) No enzyme control with 7. (f) NotI′ reaction with (+)-6-epi-stephacidin A (6). (g) NotI reaction with 6. (h) No enzyme control with 6. (i) NotI′ reaction with (−)-stephacidin A (4). (j) NotI reaction with 4. (k) No enzyme control with 4. (l) NotI′ reaction with (+)-stephacidin A (9). (m) NotI reaction with 9. (n) No enzyme control with 9. No enzyme controls are shown in purple, reactions with NotI are shown in blue, and NotI′ reactions are shown in red. The HPLC data were collected at a wavelength of 240 nm.
Next, we sought to assess the timing of epoxidation and semi-pinacol rearrangement in notoamide biosynthesis. Reactions were conducted using NotI and NotI′ with pre-IMDA pathway intermediates including brevianamide F (12), deoxybrevianamide E (13), 6-OH-deoxybrevianamide E (14), notoamide S (15), notoamide E (1), and post-IMDA intermediates (+)-notoamide T (16), and (−)-notoamide T (17) and analyzed by QTOF LC-MS (Scheme 2 and Figures S12–S24). Both enzymes demonstrated a remarkable range of substrate tolerance, leading to new products with masses indicative of oxidation. In some cases, multiple products were generated, indicating either a loss of stereocontrol for collapse of the epoxide or the generation of an alternative oxidized product.
Scheme 2.
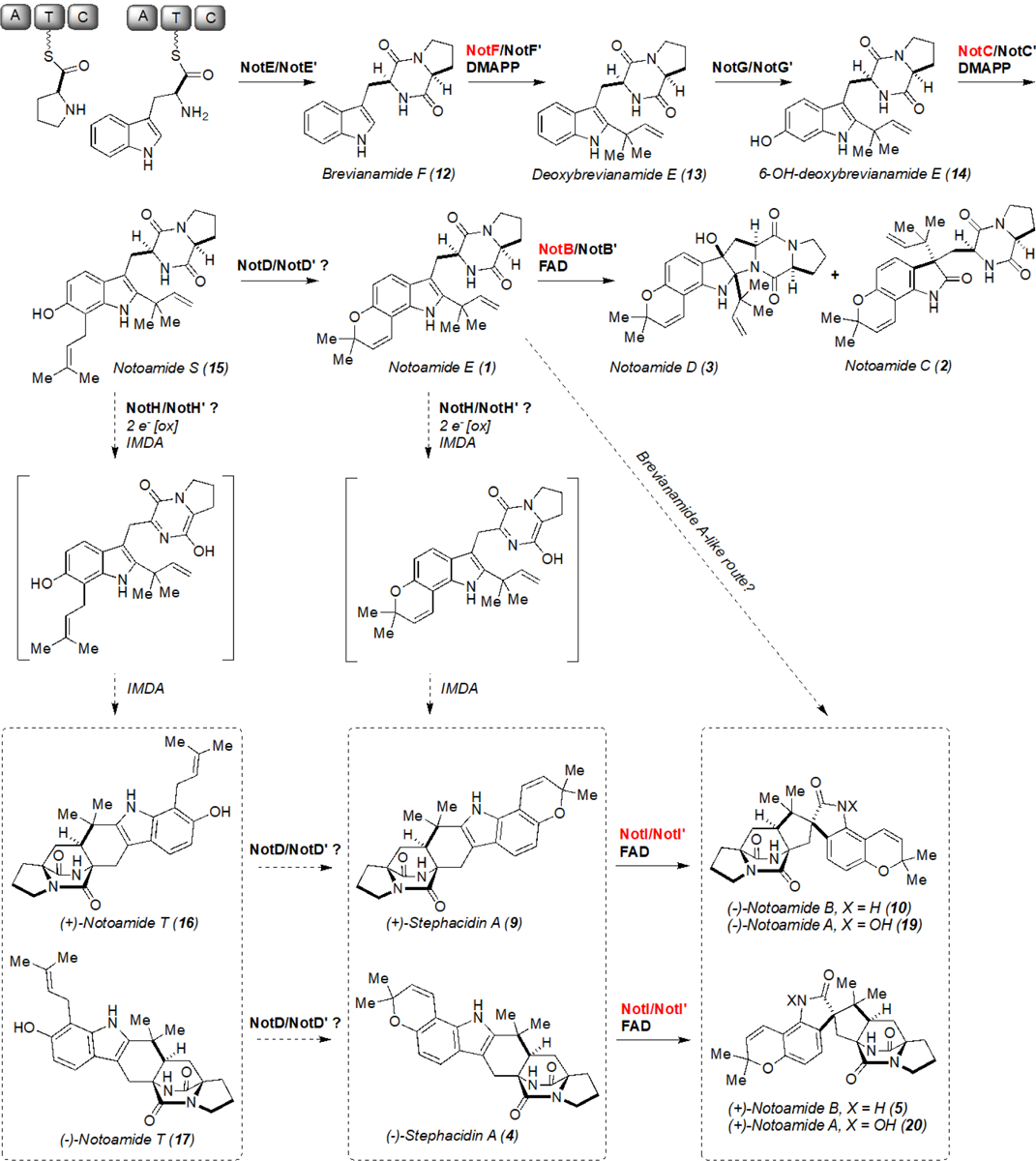
Proposed (−)/(+)-notoamide A (19 and 20) biosynthetic pathways. The functions of enzymes highlighted in red have been experimentally confirmed. The terminal N-hydroxylation to generate 19 and 20 is catalyzed by an additional enzyme subsequent to spirocyclization.
Based on the analysis above, we expected that the reactions between NotI/NotI′ and notoamide T (16 and 17) had formed a new metabolite, thus we determined the structure by NMR spectroscopy (Table S2 and Figures S8, S9, S25–S35). Racemic notoamide T was converted by NotI and purified by HPLC to yield approximately 2 mg of final product. The new compound notoamide TI (18) was obtained as a white amorphous solid and possessed a molecular formula of C26H31N3O4 as suggested by HRESIMS based on [M+H]+ ion peak at m/z 450.2414, representing thirteen degrees of unsaturation. Moreover, the UV spectrum in methanol with wavelength of maximum absorbance at 242, 309, and 335 (sh) nm was indicative of aromatic functionality.
Lastly, a comparison of NotI/NotI′ to the recently characterized PhqK,[13] provided insight into the potential reaction mechanism. The enzymes share 35% sequence identity and have highly similar native substrates; thus the Phyre2[22] homology models of NotI and NotI′ were aligned with the crystal structure of PhqK (RMSD = 2.28, PDB ID: 6pvi) to investigate the presence of possible catalytic amino acids (Figure 2). Arginine 195 in NotI/NotI′ aligns with the catalytic arginine in PhqK (Arg192), indicating that it may play a similar role in directing the collapse of the epoxide and spirocyclization. The C-terminus of the enzyme, which is important for binding the substrate and closing off the active site in PhqK, seems to be very different in NotI. This portion of the enzyme shared no homology with FMOs in the Protein Data Bank, including PhqK, indicating that the C-terminus may be a point of divergence in the evolution of these FMOs (Figures S10 and S11). While the substrate-binding regions of FMOs vary in structure and function, the architecture of the cofactor binding domain seems to be maintained over time.[23]
Figure 2.
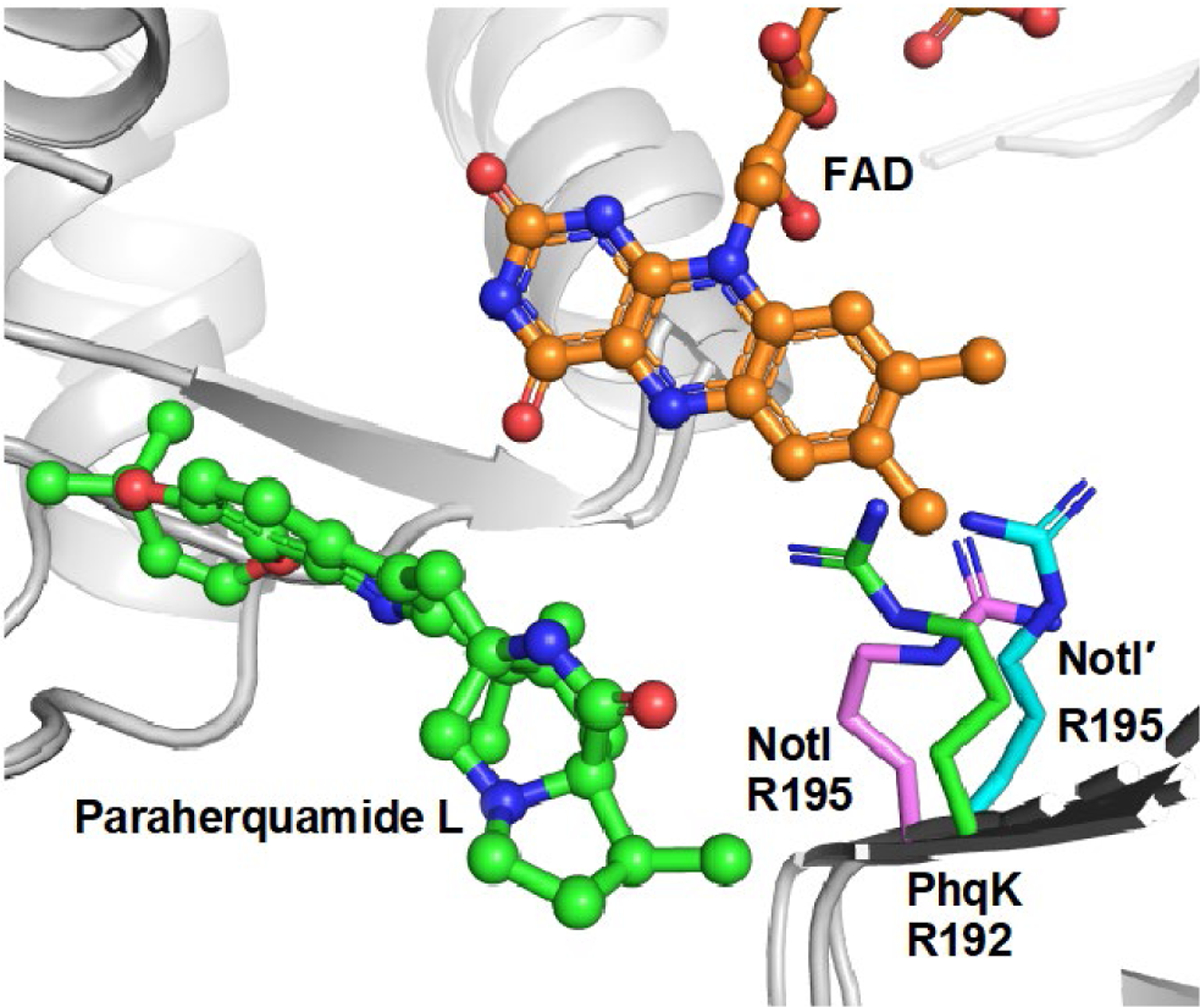
Alignment of PhqK (PDB ID: 6pvi) and Phyre2 models of NotI and NotI′ demonstrating the presence of the catalytic arginine in all three proteins. FAD conformation was modelled from urate oxidase HpxO (PDB ID: 3rp7).
Additionally, sequence comparison with homologous enzymes demonstrated that this catalytic arginine is conserved across a range of fungal FMOs, many of which are capable of catalyzing epoxidation and semi-pinacol rearrangement (Table S3 and Figure S11). In all cases, the catalytic arginine is bordered by a large N-terminal amino acid (Phe/Tyr/Trp) and a smaller C-terminal amino acid (Ala/Gly/Ser). The steric conservation around this pivotal amino acid indicates that the nearby residues may facilitate the range of motion required for the arginine to direct collapse of the indole epoxide species.
Conclusion
We initially hypothesized that NotI/NotI′ would be selective for their respective enantiomeric substrates. However, both NotI and NotI′ accepted either of the stephacidin A enantiomers, with a clear preference for the (−)-isomer (4). This indicates that the conversion of (+)-stephacidin A (9) may be an evolved trait from an ancestral organism previously only capable of converting (−)-stephacidin A. In contrast, the facial selectivity of the FMO-catalyzed oxidation appears to be highly diastereoselective, delivering the oxygen atom from the least-hindered face of the 2,3-disubstituted indole substrate. These findings support our proposed biosynthesis in which NotI and NotI′ are not candidates for the control of the enantio-divergence in compounds produced by the Aspergillus species, and that the formation of antipodal notoamides is instead due to an earlier step in the biosynthetic pathway.[15]
With our current knowledge of the biochemical transformations involved, we reason that the enzyme responsible for the IMDA is the likely candidate for the enantio-divergent step. Additionally, a novel indole alkaloid metabolite notoamide TI was generated through in vitro reactions, further expanding the chemical diversity of bicyclo[2.2.2]diazaoctane ring containing molecules. The production of notoamide TI suggests that there may be parallel pathways to the formation of notoamide B through either (+)/(−)-notoamide T (16/17) or (+)/(−)-stephacidin A (9/4) (Scheme 2). However, precursor feeding studies with isotopically labelled stephacidin A suggest that the proposed order involving initial pyran ring formation followed by semi-pinacol rearrangement is likely to be the preferred route.[20a] Moreover, we cannot eliminate the possibility of a brevianamide A-like route similar to what we elucidated recently.[24] This would involve the sequential FMO-mediated epoxidation, P450-catalyzed desaturation of the dioxopiperazine ring, and spontaneous IMDA to generate 5 or 10 without passing through 4 or 9 (Scheme 2). In this investigation, two group A FMOs were found to be involved in the formation of the spiro-oxindole center of various bicyclo[2.2.2]diazaoctane fungal indole alkaloids via semi-pinacol rearrangement.[25] The designation of NotI/NotI′ into this group is based on the presence of the DG fingerprint, which is involved in both FAD and NAD(P)H binding (Figure S11). The mechanism of this reaction has recently been investigated in a homologous paraherquamide-producing system and FMO PhqK.[13] It is hypothesized that the C2–C3 bond of the indole is epoxidized by the FMO on the less-hindered face, as has been reported in synthetic approaches,[2d, 15] and suggested for similar molecules such as the taichunamides and paraherquamides.[13, 24] Protonation of the reactive epoxide intermediate leads to ring opening to form a hydroxy cation and the subsequent deprotonation facilitates the 1,2 shift to provide notoamide B (Scheme 3). While the enzymes have evolved a mechanism for stereospecific collapse of the indole epoxide, unnatural substrates seem to evade this catalyst-controlled selectivity. As has been observed with other oxidative enzymes,[26] whether an epoxidation or hydroxylation reaction occurs can depend on positioning of the substrate in the active site. This indicates that, in some cases, the unnatural substrates may not be orientated properly for selective spirocycle formation.
Scheme 3.
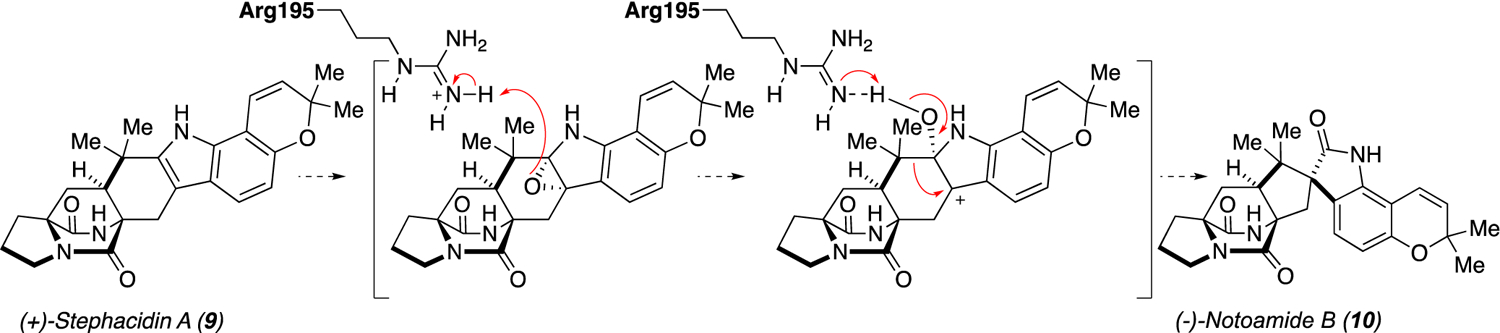
Proposed mechanism for epoxidation and spirocyclization catalysed by NotI and NotI′.
Through homology modelling of NotI and NotI′, and comparison to the high-resolution crystal structures of PhqK, we have determined that the catalytic arginine in PhqK may also be present in NotI/NotI′ (Figure 2). Arg195 in NotI/NotI′ is proposed to perform general acid catalysis to mediate the collapse of the epoxide with a stereoselective formation of the spiro-oxindole. These findings demonstrate that a catalyst-controlled semi-pinacol rearrangement reaction is involved in notoamide biosynthesis, and may have important implications for the inherently flexible FMOs in fungal indole alkaloid biosynthesis.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01 CA070375 to R.M.W. and D.H.S.), R35 GM118101, the Hans W. Vahlteich Professorship (to D.H.S.), and a Rackham Predoctoral Fellowship (to A.E.F.). The authors would also like to thank J.J. Schmidt and A.W. Robertson for helpful discussions regarding NMR data analysis.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of Interest
The authors declare no conflict of interest.
References:
- [1].a) Birch AJ, Wright JJ, Chem Commun 1969, 644–645; [Google Scholar]; b) Steyn PS, Tetrahedron 1973, 29, 107–120; [Google Scholar]; c) Polonsky J, Merrien MA, Prange T, Pascard C, Moreau S, J.C.S. Chem. Comm 1980, 601–602; [Google Scholar]; d) Prangé T, Billion MA, Vuilhorgne M, Pascard C, Polonsky J, Moreau S, Tetrahedron Lett 1981, 22, 1977–1980; [Google Scholar]; e) Yamazaki M, Okuyama E, Kobayashi M, Inoue H, Tetrahedron Lett 1981, 22, 135–136; [Google Scholar]; f) Ondeyka JG, Goegelman RT, Schaeffer JM, Kelemen L, Zitano L, J Antibiot 1990, 43, 1375–1379; [DOI] [PubMed] [Google Scholar]; g) Liesch JM, Wichmann CF, J Antibiot 1990, 43, 1380–1386; [DOI] [PubMed] [Google Scholar]; h) Blanchflower SE, Banks RM, Everett JR, Manger BR, Reading C, J Antibiot 1991, 44, 492–497; [DOI] [PubMed] [Google Scholar]; i) Blanchflower SE, Banks RM, Everett JR, Reading C, J Antibiot 1993, 46, 1355–1363; [DOI] [PubMed] [Google Scholar]; j) Whyte AC, Gloer JB, J Nat Prod 1996, 59, 1093–1095; [DOI] [PubMed] [Google Scholar]; k) Hayashi H, Nishimoto Y, Nozaki H, Tetrahedron Lett 1997, 38, 5655–5658; [Google Scholar]; l) Sugie Y, Dekker KA, Hirai H, Ichiba T, Ishiguro M, Shiomi Y, Sugiura A, Brennan L, Duignan J, Huang LH, Sutcliffe J, Kojima Y, J Antibiot 2001, 54, 1060–1065; [DOI] [PubMed] [Google Scholar]; m) Qian-Cutrone JF, Huang S, Shu YZ, Vyas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q, J Am Chem Soc 2002, 124, 14556–14557; [DOI] [PubMed] [Google Scholar]; n) Martínez-Luis S, Rodríguez R, Acevedo L, González MC, Lira-Rocha A, Mata R, Tetrahedron 2006, 62, 1817–1822. [Google Scholar]
- [2].a) Adams LA, Gray CR, Williams RM, Tetrahedron Lett 2004, 45, 4489–4493; [Google Scholar]; b) Adams LA, Valente MWN, Williams RM, Tetrahedron 2006, 62, 5195–5200; [Google Scholar]; c) Greshock TJ, Grubbs AW, Williams RM, Tetrahedron 2007, 63, 6124–6130; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Greshock TJ, Grubbs AW, Tsukamoto S, Williams RM, Angew Chem Int Edit 2007, 46, 2262–2265; [DOI] [PubMed] [Google Scholar]; e) Miller KA, Welch TR, Greshock TJ, Ding YS, Sherman DH, Williams RM, J Org Chem 2008, 73, 3116–3119; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Miller KA, Williams RM, Chem Soc Rev 2009, 38, 3160–3174; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Miller KA, Tsukamoto S, Williams RM, Nat Chem 2009, 1, 63–68; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Mukai K, de Sant’Ana DP, Hirooka Y, Mercado-Marin EV, Stephens DE, Kou KGM, Richter SC, Kelley N, Sarpong R, Nat Chem 2018, 10, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S, Angew Chem Int Edit 2007, 46, 2254–2256; [DOI] [PubMed] [Google Scholar]; b) Tsukamoto S, Kato H, Samizo M, Nojiri Y, Onuki H, Hirota H, Ohta T, J Nat Prod 2008, 71, 2064–2067; [DOI] [PubMed] [Google Scholar]; c) Tsukamoto S, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM, J Am Chem Soc 2009, 131, 3834–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tsuda M, Kasai Y, Komatsu K, Sone T, Tanaka M, Mikami Y, Kobayashi J, Org Lett 2004, 6, 3087–3089. [DOI] [PubMed] [Google Scholar]
- [5].Li SM, Nat Prod Rep 2010, 27, 57–78. [DOI] [PubMed] [Google Scholar]
- [6].Finefield JM, Frisvad JC, Sherman DH, Williams RM, J Nat Prod 2012, 75, 812–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Porter AEA, Sammes PG, J Chem Soc Chem Comm 1970, 1103. [Google Scholar]
- [8].Dan Q, Newmister SA, Klas KR, Fraley AE, McAfoos TJ., Somoza AD, Sunderhaus JD, Ye Y, Shende VV, Yu F, Sander JN, Brown WC, Zhao L, Paton RS, Houk KN, Smith JL, Sherman DH, Williams RM, Nat. Chem 2019, 11, 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li SY, Finefield JM, Sunderhaus JD, McAfoos TJ, Williams RM, Sherman DH, J Am Chem Soc 2012, 134, 788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Grubbs AW, Artman GD, Tsukamoto S, Williams RM, Angew Chem Int Edit 2007, 46, 2257–2261. [DOI] [PubMed] [Google Scholar]
- [11].Watts KR, Loveridge ST, Tenney K, Media J, Valeriote FA, Crews P, J Org Chem 2011, 76, 6201–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Cacho RA, Chooi YH, Zhou H, Tang Y, Acs Chem Biol 2013, 8, 2322–2330; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tsunematsu Y, Ishikawa N, Wakana D, Goda Y, Noguchi H, Moriya H, Hotta K, Watanabe K, Nat Chem Biol 2013, 9, 818–825. [DOI] [PubMed] [Google Scholar]
- [15].Greshock TJ, Grubbs AW, Jiao P, Wicklow DT, Gloer JB, Williams RM, Angew Chem Int Edit 2008, 47, 3573–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kato H, Nakahara T, Sugimoto K, Matsuo K, Kagiyama I, Frisvad JC, Sherman DH, Williams RM, Tsukamoto S, Org Lett 2015, 17, 700–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Finefield JM, Sherman DH, Kreitman M, Williams RM, Angew Chem Int Edit 2012, 51, 4802–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sunderhaus JD, Sherman DH, Williams RM, Isr J Chem 2011, 51, 442–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Artman GD, Grubbs AW, Williams RM, J Am Chem Soc 2007, 129, 6336–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ding YS, de Wet JR, Cavalcoli J, Li SY, Greshock TJ, Miller KA, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH, J Am Chem Soc 2010, 132, 12733–12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Finefield JM, Kato H, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM, Org Lett 2011, 13, 3802–3805; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Finefield JM, Sherman DH, Tsukamoto S, Williams RM, J Org Chem 2011, 76, 5954–5958; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Finefield JM, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM, Tetrahedron Lett 2011, 52, 1987–1989; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sunderhaus JD, McAfoos TJ, Finefield JM, Kato H, Li SY, Tsukamoto S, Sherman DH, Williams RM, Org Lett 2013, 15, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li S, Srinivasan K, Tran H, Yu FA, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH, Medchemcomm 2012, 3, 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fraley AE, Caddell Haatveit K, Ye Y, Kelly SP, Newmister SA, Yu F, Williams RM, Smith JL, Houk KN, Sherman DH, J Am Chem Soc 2020 142 (5), 2244–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ, Nat. Protoc 2015, 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mascotti ML, Ayub MJ, Furnham N, Thornton JM, Laskowski RA, J Mol Biol 2016, 428, 3131–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ye Y, Du L, Zhang X, Newmister SA, Zhang W, Mu S, Minami A, McCauley M, Alegre-Requena JV, Fraley AE, Androver-Castellano ML, Carney N, Shende VV, Oikawa H, Kato H, Tsukamoto S, Paton RS, Williams RM, Sherman DH, Li S, Nat. Catal 2020, DOI: 10.1038/s41929-020-0454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Huijbers MME, Montersino S, Westphal AH, Tischler D, van Berkel WJH, Arch. Biochem. Biophys 2014, 544, 2–17; [DOI] [PubMed] [Google Scholar]; b) Eppink MHM, Schreuder HA, van Berkel WJH, Protein Science 1997, 6, 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Newmister SA, Gober CM, Romminger S, Yu F, Tripathi A, Parra LLL, Williams RM, Berlinck RGS, Joullié MM, Sherman DH. J Am Chem Soc 2016, 138 (35), 11176–11184. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li S, Tietz DR, Rutaganira FU, Kells PM, Anzai Y, Kato F, Pochapsky TC, Sherman DH, Podust LM, J Biol Chem 2012, 287 (45), 37880–37890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.