

Abstract
Deubiquitinating enzymes (DUBs) appear to be a new class of regulators of cardiac homeostasis and disease. However, DUB-mediated signaling in the heart is not well understood. Herein we report a novel mechanism by which cylindromatosis (CYLD), a DUB mediates cardiac pathological remodeling and dysfunction. Cardiomyocyte-restricted (CR) overexpression of CYLD (CR-CYLD) did not cause gross health issues and hardly affected cardiac function up to age of one year in both female and male mice at physiological conditions. However, CR-CYLD overexpression exacerbated pressure overload (PO)-induced cardiac dysfunction associated with suppressed cardiac hypertrophy and increased myocardial apoptosis in mice independent of the gender. At the molecular level, CR-CYLD overexpression enhanced PO-induced increases in poly-ubiquitinated proteins marked by lysine (K)48 -linked ubiquitin chains and autophagic vacuoles containing undegraded contents while suppressing autophagic flux. Augmentation of cardiac autophagy via CR-ATG7 overexpression protected against PO-induced cardiac pathological remodeling and dysfunction in both female and male mice. Intriguingly, CR-CYLD overexpression switched the CR-ATG7 overexpression-dependent cardiac protection into myocardial damage and dysfunction associated with increased accumulation of autophagic vacuoles containing undegraded contents in the heart. Genetic manipulation of Cyld in combination with pharmacological modulation of autophagic functional status revealed that CYLD suppressed autolysosomal degradation and promoted cell death in cardiomyocytes. In addition, Cyld gene gain- and/or loss-of-function approaches in vitro and in vivo demonstrated that CYLD mediated cardiomyocyte death associated with impaired reactivation of mechanistic target of rapamycin complex 1 (mTORC1) and upregulated Ras genes from rat brain 7 (Rab7), two key components for autolysosomal degradation. These results demonstrate that CYLD serves as a novel mediator of cardiac pathological remodeling and dysfunction by suppressing autolysosome efflux in cardiomyocytes. Mechanistically, it is most likely that CYLD suppresses autolysosome efflux via impairing mTORC1 reactivation and interrupting Rab7 release from autolysosomes in cardiomyocytes.
Keywords: CYLD, Deubiquitinating enzymes, Autolysosome efflux, Pressure overload, Cardiomyocytes
Graphical Abstract
1. Introduction
Since ubiquitin was discovered in the earlier 1970’s, the ubiquitin proteasome system (UPS), which consists of ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), ubiquitin ligases (E3s), proteasomes, and deubiquitinating enzymes (DUBs), has been implicated in virtually all aspects of cell biology [1–5]. Protein ubiquitination, a process in which ubiquitin (Ub) is covalently conjugated by its terminal glycine (G76) onto a lysine (K) residue of a substrate protein by the sequential action of an E1, E2 and E3, is a reversible posttranslational modification. A single Ub molecule may be attached, which is defined as monoubiquitination. Several lysine residues can be tagged with single Ub molecules, giving rise to multiple monoubiquitination, also referred to as multiubiquitination. Since Ub has seven lysine residues (K36, K11, K27, K33, K48, K63) itself, Ub molecules can form different types of chains in an iterative process, known as polyubiquitination. In general, K48-linked poly-Ub chains represent a signal for proteasomal degradation of modified substrates, whereas mono-Ub and K63-linked poly-Ub chain modifications function as signaling devices for establishing protein-protein interactions and regulating cellular functions. The hydrolysis of ubiquitin linkage is conducted by DUBs. Eight E1s, a dozen different types of E2s, hundreds of E3s, and approximately 100 functional DUBs have been identified in humans.
During the past decades, an important role of Ub, E1s, several E2s and E3s, and proteasome in cardiac homeostasis and dysfunction has been well documented [6, 7]. However, the myocardial function of DUBs is less well understood. Studies have demonstrated that A20 (also known as TNFAIP3) is a negative regulator of cardiac maladaptive remodeling and dysfunction via its ability to suppress the activation of nuclear factor kappa B (NF-κB), mitogen-activated protein kinases (MAPKs) and transforming growth factor beta (TGFβ) signaling [8–10]. Subsequent data have shown that cardiomyocyte-restricted (CR) overexpression of ubiquitin-specific protease (USP)15 results in age-dependent cardiac hypertrophy, indicating a mediator role of USP15 in cardiomyopathy [11]. Notably, our data showed that cylindromatosis (CYLD) [12] which has DUB activity highly specific for K63-linked Ub chains and is capable of suppressing NF-κB, MAPK and TGF-β signaling [13], is likely a crucial mediator of pressure overload (PO)-induced cardiomyopathy via a mechanism independent of the NF-κB pathway [14]. These findings underscore the functional specificity of individual DUBs in the heart, thereby supporting the notion that the association of DUBs with substrate adaptors, scaffolds and inhibitors may result in regulatory interactions that drive specificity [15, 16]. Indeed, recent studies have revealed that USP4 and USP18 negatively regulate cardiac pathological remodeling [17, 18] whereas USP14 may act as a mediator of cardiac pathological hypertrophy [19]. Nevertheless, the functional significance of individual DUBs in cardiac homeostasis and disease remains poorly understood.
Autophagy is an evolutionarily conserved catabolic process that targets cytoplasmic components such as organelles, protein aggregates or individual proteins to the lysosome for degradation. Autophagy in mammals has been classed into three types: (i) macroautophagy, (ii) microautophagy and (iii) chaperone-mediated autophagy (CMA). The macroautophagy (thereafter referred to as autophagy) is the best characterized. It is a very dynamic process that leads to formation of a double-membrane bound vesicle termed the autophagosome, which sequesters the targeted cargos, followed by autophagosome fusion with lysosomes to form the autolysosome, resulting in autolysosomal degradation (autolysosome efflux) [20–22]. The role of autophagy in the heart remains controversial. Depending the nature of stresses as well as the timing of assessments, activation of cardiac autophagy has been shown to be either adaptive or maladaptive [23]. In PO-hearts, recent studies have revealed that autophagy activation is most likely adaptive [20, 23]. However, the definitive proof of autophagy-mediated cardiac protection in adult PO-hearts is still missing.
Recently, ubiquitination of substrates and protein components of autophagy machinery has emerged as a central regulatory mechanism of autophagy acting at various steps from autophagy induction to termination [24]. Therefore, it is not surprising that DUBs regulate autophagy [25]. Conceptually, it has been shown that otubain protease (OTUB)1, USP8, USP9X, USP10, USP13, USP19, USP20, and USP33 may act as positive regulators of autophagy, whereas A20, USP14, USP30, s-USP35, USP15, USP44, and ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) probably serve as negative regulators of autophagy [25]. However, it is still not fully elucidated how the Ub code regulates autophagy in terms of altering protein signaling and targeting regulatory proteins for degradation. In addition, the crosstalk between E3s which add K63- or K48-linked poly-Ub chains to the components of autophagic machinery and DUBs that counteract their actions in the control of autophagy remains unclear. To date, the pathophysiological relevance of DUB-mediated regulation of autophagy in the heart has not yet been studied. For example, it is unclear whether the USP14- or USP15-mediated autophagy inhibition is linked to their pro-hypertrophic effects on the heart [11, 18]. In addition, it remains unknown whether the A20-mediated autophagy inhibition documented in other pathways and processes such as self-directed immune responses [26] takes place in the heart as well.
In the present study, we demonstrated that CYLD suppresses autophagy at the stage of autolysosome efflux in cardiomyocytes, thereby exaggerating cardiac pathological remodeling and dysfunction in the setting of PO. These results uncovered a novel aspect of CYLD acting as an inhibitor of autophagy in the stressed hearts, thus providing the first evidence directly linking DUB-mediated autophagy regulation with cardiomyopathy.
2. Methods
Cardiomyocyte-restricted Cyld transgenic (CR-Cyld Tg) and CR-Atg7×tTA Tg mice were generated as we previously reported [27, 28]. Littermates of wild type (WT), CR-tTA Tg, CR-Atg7 Tg, CR-Cyld×tTA Tg, CR-Atg7×tTA Tg, and CR-Cyld×Atg7×tTA Tg mice were generated by crossbreeding between CR-Cyld Tg and CR-Atg7×tTA Tg mice. Global Cyld knockout mice were generated as we previously reported [14]. Transverse aortic arch constriction (TAC) model, echocardiography, histopathology, immunohistochemistry, and biochemical assays were performed as we previously described [14, 27, 29].
Detailed methods are provided in the Online-only Data Supplement.
3. Results
3.1. CR-CYLD overexpression exacerbates PO-induced cardiomyopathy
We have demonstrated that PO upregulates the expression of myocardial CYLD at both mRNA and protein levels and global KO of Cyld (CyldKO) protects against PO-induced cardiomyopathy, indicating a detrimental role of CYLD in the stressed hearts [14]. However, this notion remains to be verified by cardiac specific targeting of CYLD in vivo. Hence, we generated CR-Cyld Tg mice (Fig. S1A–E) and determined the impact of CR-CYLD overexpression on PO-induced cardiomyopathy in mice. At physiological conditions, CR-Cyld Tg mice did not develop any gross health issues with normal cardiac function up to age of one year (Fig. S1D), revealing that upregulation of cardiac CYLD alone under physiological conditions is not harmful. These results along with the previous findings [14] support a mediator role of CYLD in cardiac pathological remodeling and dysfunction. Accordingly, a cohort of mixed male and female littermates of adult non-transgenic wild type control (NG) and CR-Cyld Tg (TG) mice were subject to sham and TAC operations and TAC-induced cardiac dysfunction was monitored for 8 weeks. We found that TAC-induced onset and progression of cardiac dysfunction towards heart failure were dramatically worsened by CR-CYLD overexpression independent of gender differences (Fig. 1A and Table S3–5), demonstrating a mediator role of CYLD in PO-induced cardiomyopathy and heart failure. To gain pathological insights into the CYLD-mediated cardiomyopathy towards heart failure, a larger cohort of mixed male and female littermates of adult NG and TG mice were subject to sham and TAC operations for 2 weeks. We have demonstrated that a substantial number of wild type mice die of acute heart failure within 2 weeks after TAC and the mortality rate varies ranging from 20% to 40% depending on the genetic backgrounds [14, 29, 30]. Consistent with our report that TAC causes a mortality rate of ~20% in wild type ICR mice [29], we found that regardless of the gender, up to 23% of NG mice and 21% of TG mice died at 2 weeks after the severe TAC (Fig. S2). Kaplan Meier test showed that the survival rates of NG and TG mice are comparable independent of the gender (Fig. S2). These results suggest that CYLD may not mediate PO-induced acute heart failure. However, CR-CYLD overexpression exaggerated PO-induced cardiac dysfunction and myocardial apoptosis while suppressing PO-induced cardiomyocyte hypertrophy without affecting PO-induced cardiac fibrosis independent of the gender (Fig. 1B–E, Fig. S3A–C and Tables S6–11). Our previous studies demonstrated that knockdown of CYLD slightly enhances basal leucine uptake but diminishes angiotensin II-induced increases in leucine uptake in cultured neonatal rat ventricular myocytes [14]. These results indicate that CYLD facilitates pathological cardiomyocyte hypertrophy while compromising physiological adaptation of cardiomyocytes. Given that time period from 1-week to 2-week in the murine TAC model is a stage of transition of cardiac adaptation to maladaptive remodeling and dysfunction [29], it is conceivable that CYLD mediates cardiac pathological remodeling and dysfunction while suppressing adaptive cardiomyocyte hypertrophy.
Fig. 1.
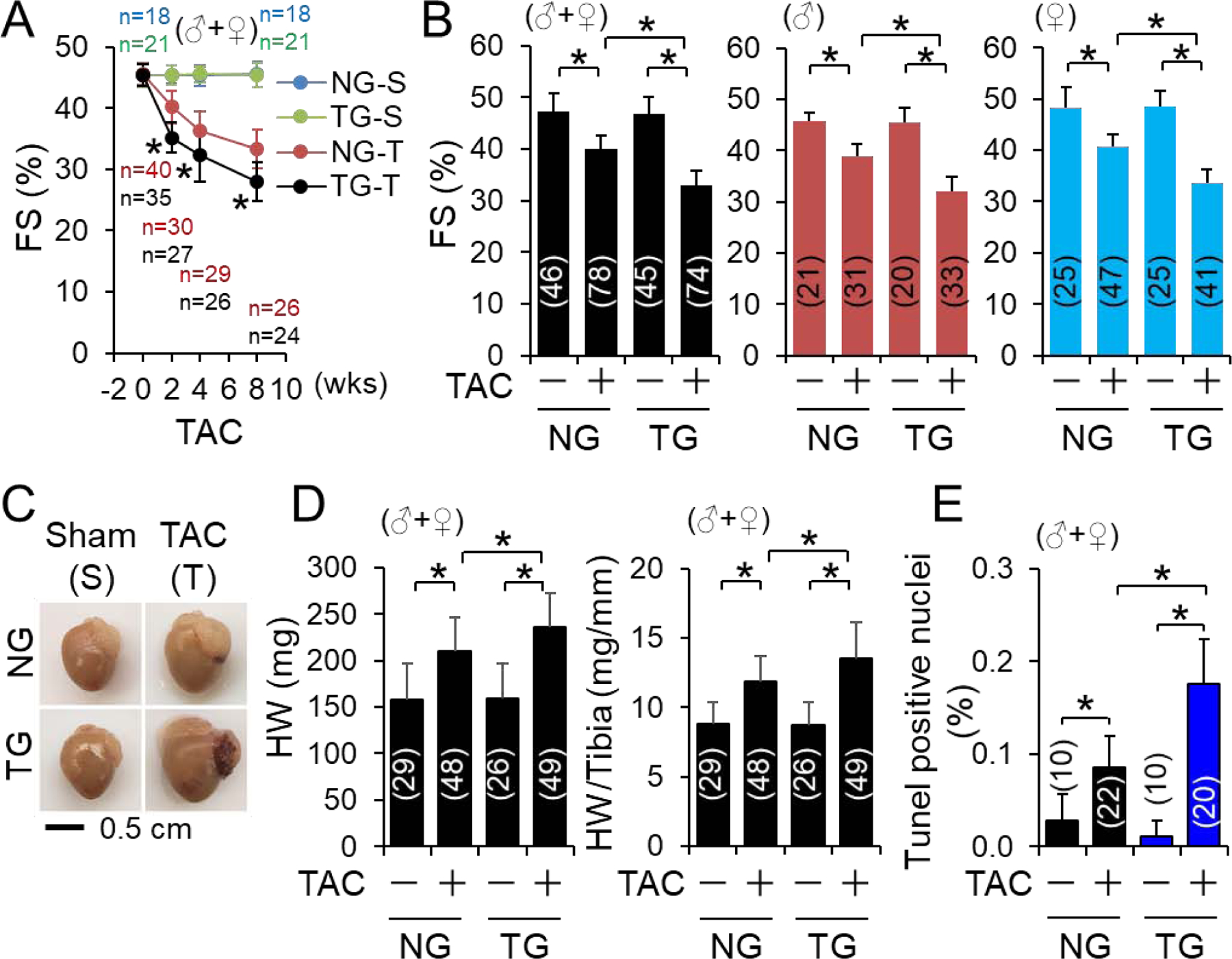
The impact of CR-Cyld overexpression on pressure overload-induced cardiomyopathy. (A) A time course study of the impact of CR-CYLD overexpression on TAC-induced cardiac dysfunction. Littermates of male (♂) and female (♀) non-transgenic wild type (NG) and CR-Cyld Tg (TG) mice at age of 10 weeks were subject to sham or TAC operations for 8 weeks. FS (%). was monitored weekly. (B-E) The impact of C R-CYLD overexpression on 2-week TAC-induced cardiac pathological remodeling and dysfunction: A large cohort study. (B) FS (%), (C) Representative heart images of NG and TG mice, (D) heart weight (HW) and HW to tibia ratio, and (E) myocardial apoptosis at 2 weeks after TAC. Animal numbers for each group are indicated in the parentheses. *, p<0.05 between indicated groups.
Although we did not find the gender differences in the role of CYLD in exacerbating PO-induced cardiomyopathy (Fig. 1, Figs. S1–2, and Table S3–11), we did notice that related to the male counterparts, such negative impact of CYLD was slightly weakened in the females, but not statistically significant (Fig. 1B and Tables S6–11). This feature may be attributed to long recognized sex-related differences in myocardial remodeling in which the remodeling process appears to be more favorable in females vs. males [31, 32]. To avoid subtle effects of the gender differences on CYLD-mediated biological actions, the mechanistic studies of CYLD-operated signaling with small cohorts of mice were then carried out in randomly reselected male or female mice.
3.2. CR-CYLD overexpression worsens PO-induced accumulation of K48-linked ubiquitinated proteins while deteriorating PO-induced autophagy insufficiency in the heart
Since there is a dramatic increase in myocardial protein aggregation in response to hemodynamic stress, PO-induced heart disease has been included in the category of proteinopathies [33]. In the heart, protein quality control (PQC) is tightly regulated by both the ubiquitin proteasomal degradation and autophagosome-lysosome pathways [7, 20]. Considering that ubiquitination is critical for both proteasomal and autophagic degradation [7, 20], we postulated that CYLD via its DUB activity interferes cardiac PQC, thereby contributing to PO-induced cardiomyopathy. Indeed, PO-induced accumulation of myocardial poly-ubiquitinated proteins (Ub-Ps) was increased in C R-Cyld Tg mice compared with NG mice (Fig. 2A). Of interest, CR-CYLD overexpression increased not only soluble but also insoluble Ub-Ps in PO-hearts (Fig. 2B). As expected, CR-CYLD overexpression decreased both soluble and insoluble Ub-Ps marked by K63-linked Ub chains with molecular weights larger than 75 kDa in both sham-operated and PO-hearts (Fig. S4), revealing that CYLD DUB activity is specific for a group of K63-linked ubiquitinated proteins with molecular weights larger than 75 kDa in the heart. However, CR-CYLD overexpression augmented both soluble and insoluble Ub-Ps marked by K48-linked Ub chains in PO-hearts alone (Fig. S4). These findings suggest that CYLD is capable of worsening PO-induced deficiency in proteasomal degradation of proteins marked by K48-linked Ub chains via a mechanism of K63-linked ubiquitination dependent PQC.
Fig. 2.
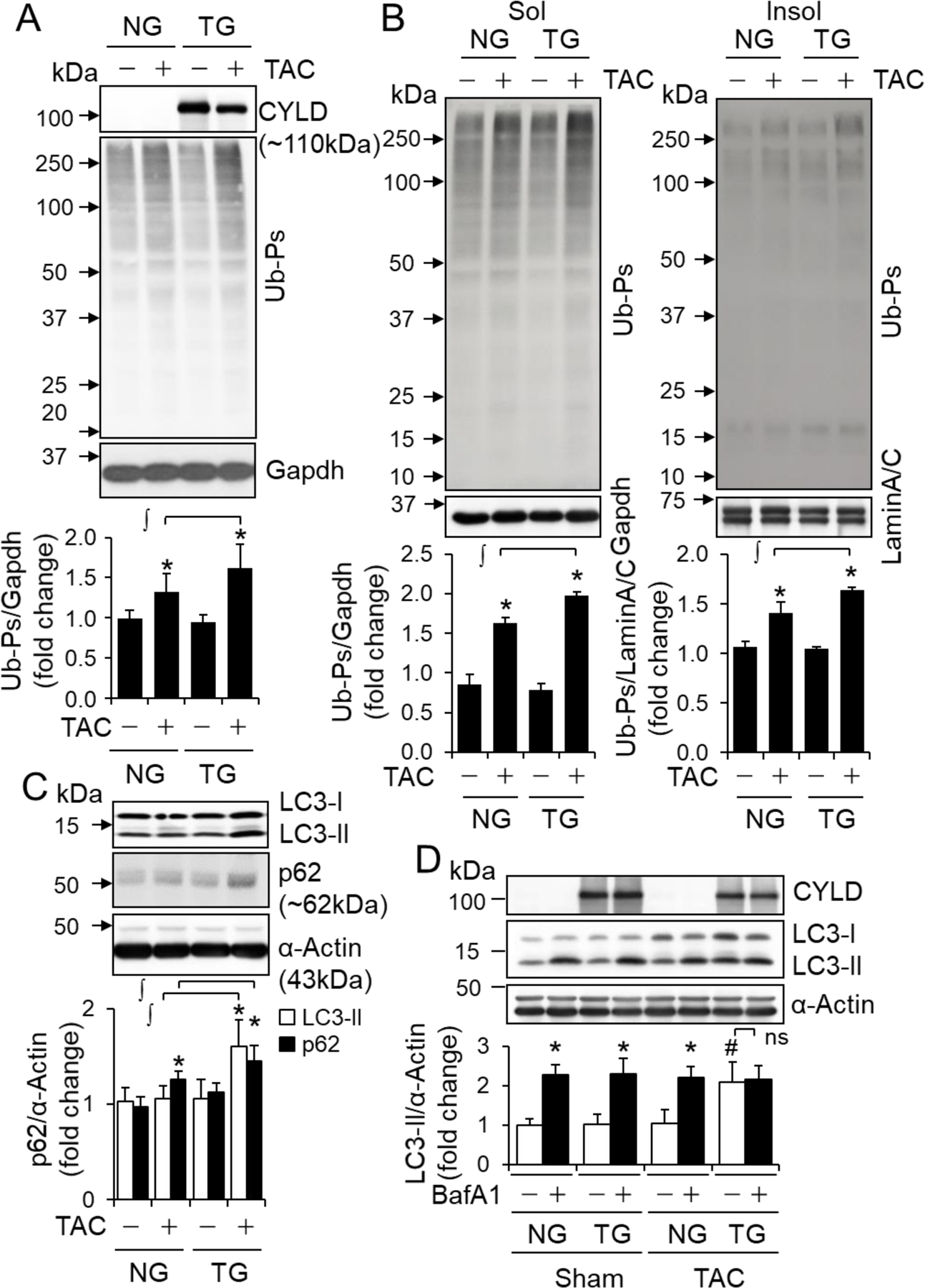
The impact of CR-Cyld overexpression on pressure overload-induced accumulation of poly-ubiquitinated proteins (Ub-Ps) and autophagic flux in the heart. Littermates of male non-transgenic wild type (NG) and CR-Cyld Tg (TG) mice at age of 10 weeks were subject to sham or TAC for 2 weeks and then randomly assigned for Western blot analyses: (A, B) Ub-Ps in left ventricles (LVs). Total LV lysates (A) as well as soluble (Sol) or insoluble (Insol) fractions (B) (n=4~6) were used for Ub-Ps assessments. ∫, p<0.05 between indicated groups; *, p<0.05 vs. TAC (−) in the same groups. (C) The protein levels of LC3-II and p62 in LVs. n=4–6, ∫, p<0.05 between indicated groups; *, p<0.05 vs. TAC (−) in the same groups. (D) Autophagic flux (n=3). *, p<0.05 vs. BafA1 (−) in the same groups. #, p<0.05 vs. NG TAC BafA1 (−) groups. ns, non-significant.
Usually, proteasomal degradation via ubiquitin-proteasome system (UPS) is efficient in clearing soluble damaged proteins; whereas autophagic degradation via the autophagosome-lysosome pathway functions in clearing organelles, and larger insoluble protein aggregates [7, 20]. We have demonstrated that severe TAC with the pressure gradient >60 mmHg leads to UPS functional deficiency in the heart within a week [34]. Thus, we believe that the observed accumulation of Ub-Ps in PO-hearts of NG mice at 2 weeks (Fig. 2A–B and Fig. S4) is most likely due to TAC-induced UPS deficiency because we used the same severe TAC model (Table S6–8) in this study. Previously, we have also demonstrated that autophagy inhibition compromises UPS performance in cardiomyocytes [35]. Importantly, we noticed that CR-CYLD overexpression-induced suppression of K63-linked ubiquitination is more dramatic in insoluble proteins in the heart (Fig. S4), that are presumably cleared by autophagic degradation aforementioned. Therefore, we questioned whether the observed phenotypes of increased accumulation of Ub-Ps in PO-hearts of TG mice may be attributed to CYLD-mediated autophagy inhibition compromising UPS performance in the heart. Indeed, CR-CYLD overexpression increased the steady protein level of LC3-II, an autophagosome marker in PO hearts and enhanced PO-induced accumulation of p62, an autophagy adaptor protein while dramatically suppressing autophagic flux, a more accurate measure of autophagy function [36] in PO hearts (Fig. 2C and D), indicating a negative impact of CYLD on cardiac autophagy. In addition, CR-CYLD overexpression led to increased accumulation of autophagic vacuoles with undegraded contents without affecting the total number of autophagic vacuoles (Fig. 3), suggesting that CYLD could suppress autophagic degradation without affecting autophagosome or autolysosome formation. To this end, our results indicate that CYLD affects PO-induced cardiomyopathy most likely via suppressing cardiac autophagy.
Fig. 3.
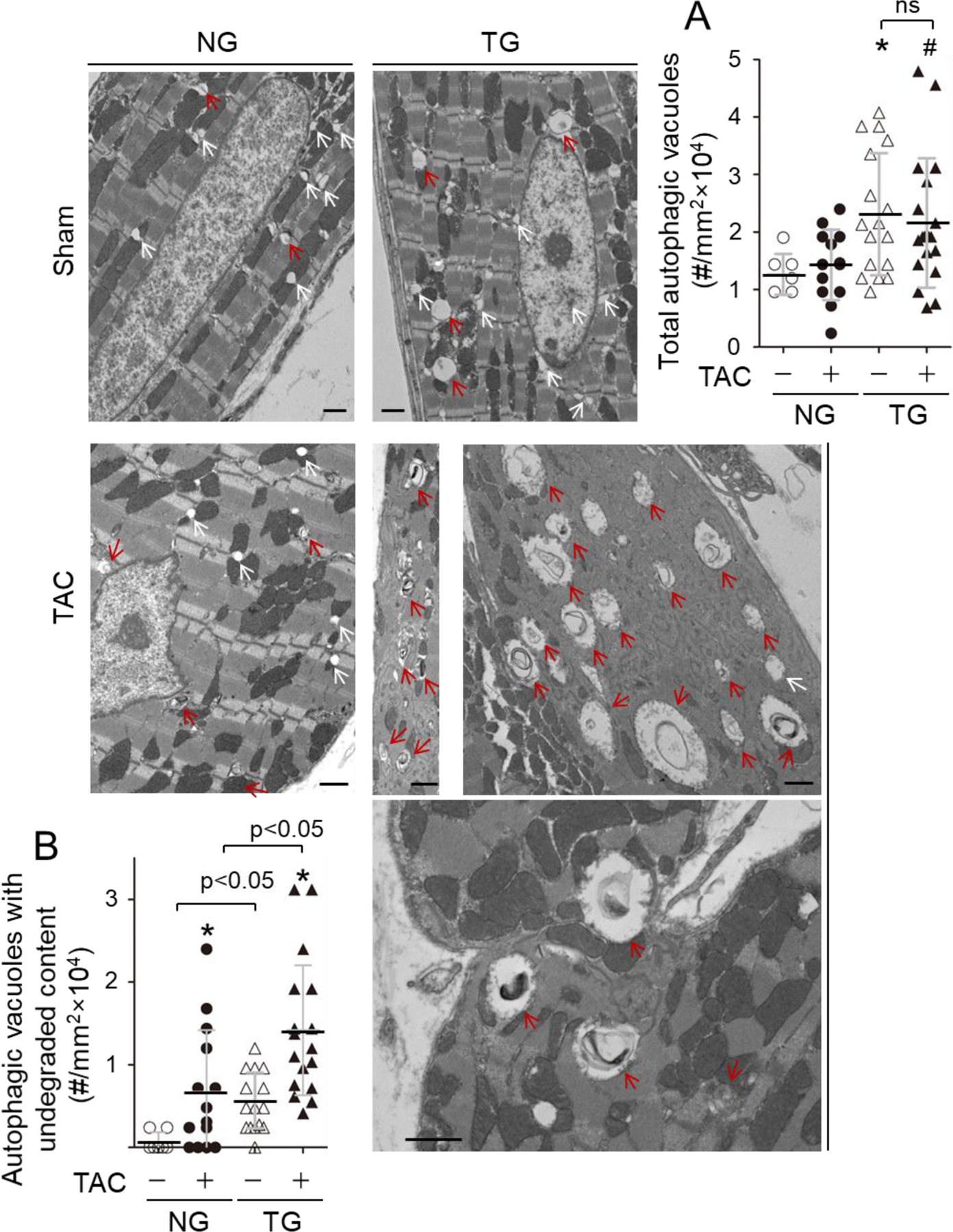
The impact of CR-Cyld overexpression on pressure overload-induced accumulation of autophagic vacuoles in the heart. Littermates of female non-transgenic wild type (NG) and CR-Cyld Tg (TG) mice at age of 10 weeks were subject to sham or TAC for 2 weeks and then to assessment of autophagic vacuoles via a transmission electron microscopy (TEM). (A) Total autophagic vacuoles in LVs. *, p<0.05 vs. TAC (−) groups; #, p<0.05 vs. NG TAC (+) group. ns, non-significant. (B) The autophagic vacuoles with undegraded contents in LVs. *, p<0.05 vs. TAC (−) in the same groups.
3.3. Activation of cardiac autophagy via CR-ATG7 overexpression protects against PO-induced cardiomyopathy
Recent studies have revealed that autophagy activation in PO-hearts is most likely adaptive [20]. However, the definitive proof of autophagy-mediated cardiac protection in adult PO-hearts is still missing. Hence, we determined the impact of CR-autophagy impairment or inhibition via CR-Atg5KO on PO-induced cardiomyopathy in adult mice. Of note, all these mice carried the MerCreMer transgene to minimize off-target effects of MerCreMer on the heart, and all received the same regime of tamoxifen treatment, which had minimal impact on the heart at baseline conditions. We found that CR-Atg5KO exacerbated PO-induced cardiomyopathy and heart failure in adult mice (data not shown), indicating the adaptive nature of autophagy activation in PO-hearts. To further verify the hypothesis, we tested if enhancement of myocardial autophagy via CR-Atg7 transgenic overexpression protects against PO-induced cardiomyopathy in mice. At physiological conditions, CR-ATG7 overexpression did not cause detectable pathology and had minimal impact on cardiac function up to age of 6 months (Fig. S5) as we previously observed [28]. However, PO-induced cardiac hypertrophy and dysfunction were dramatically ameliorated by CR-ATG7 overexpression (Fig. 4A and B, and Tables S12–14). Of note, cardiac function in all sham operated groups were similar, and tTA expression (CR-tTA Tg) and Atg7 insertion without expressing ATG7 (CR-Atg7 Tg) hardly affected PO-induced cardiac dysfunction (Tables S12–14). Therefore, we used nontransgenic WT littermates (NG) as the control groups thereafter in our studies. At the cellular level, CR-ATG7 overexpression suppressed PO-induced cardiomyocyte hypertrophy, cardiac fibrosis and apoptosis at 4 weeks (Fig. 4C). Of note, the beneficial effects of CR-ATG7 on PO-induced cardiomyopathy was independent of the gender (Fig. 4). Collectively, these results demonstrate that autophagy activation is an adaptive response in PO-hearts.
Fig. 4.
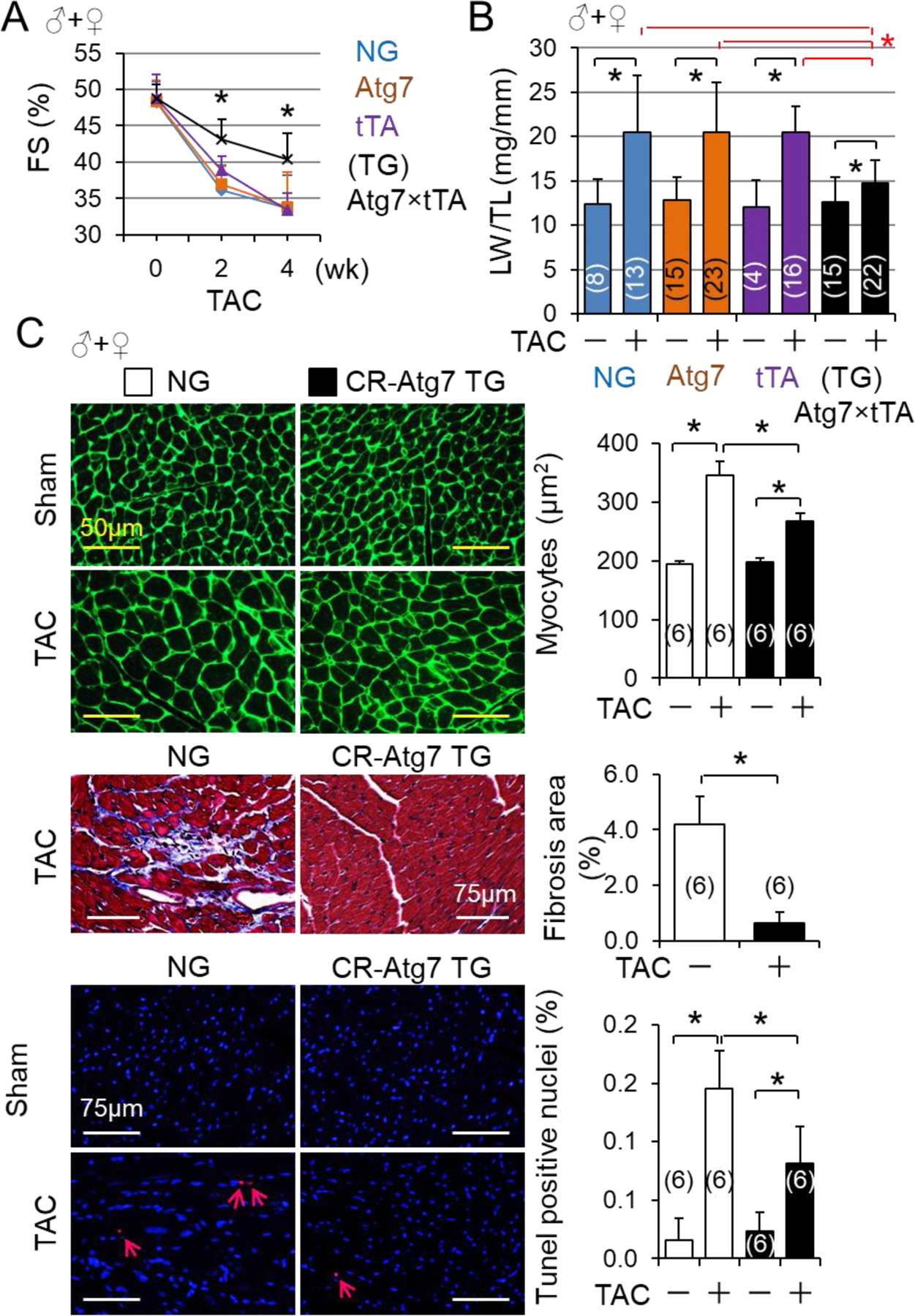
CR-Atg7 overexpression-mediated protection against pressure overload-induced cardiomyopathy. (A) Time course of TAC-induced cardiac dysfunction in NG and CR-Atg7xtTA Tg (TG) mice. Littermates of male (♂) and female (♀) mice with indicated genotypes at age of 10 weeks were subject to sham and TAC operations and then the cardiac function of these mice was monitored by echocardiography weekly. Animal numbers of each group were indicated in the parentheses. *, p<0.05 vs. NG, Atg7 or tTA groups. (B) Lung weight (LW)/tibia length (TL) ratios. LW/TL ratios of both male and female mice with indicated genotypes were measured at 4 weeks after TAC. Animal numbers of each group are indicated in the parentheses. *, p<0.05 between indicated groups. (C) Cardiac remodeling. The cardiac myocyte sizes, fibrosis and apoptosis in NG and CR-Atg7×tTA Tg mice were measured at 4 weeks after TAC. Animal numbers of each group (♂+♀, randomly selected 3 of each gender mice) are indicated in the parentheses. *, p<0.05 between indicated groups.
3.4. Upregulation of myocardial CYLD turns on ATG7-mediated cardiomyopathy while switching off ATG7-dependent cardiac protection in the setting of PO
ATG7 plays a critical role in autophagosome biosynthesis but not in the fusion of autophagosomes with lysosomes [20]. We questioned whether CR-ATG7 overexpression-induced cardiac protective autophagy could reverse CR-CYLD overexpression-mediated cardiac autophagy inhibition and dysfunction. We generated littermates of non-transgenic WT (Non), CR-Atg7xtTA Tg (Atg7), CR-CyldxtTA Tg (Cyld), and CR-Atg7xtTA Cyld Tg (Duo) mice and performed TAC operations in these mice with mixed genders for 2 weeks. TAC-induced cardiac dysfunction was ameliorated by CR-ATG7 overexpression whereas worsened by CR-CYLD overexpression (Fig. 5A and Tables S15–16) as observed in the experiments aforementioned (Figs. 3 and 4). Unexpectedly, the adverse cardiac hypertrophy and dysfunction due to CR-CYLD overexpression were not attenuated by additional CR-ATG7 overexpression; instead, they became even worse due to increased ATG7 expression (Fig. 5A and B, and Tables S15–16). These effects were independent of gender (data not shown). Transmission electron microscopy revealed that TAC-induced accumulation of autophagic vacuoles with undegraded contents was decreased by CR-ATG7 overexpression but increased by CR-CYLD overexpression; and surprisingly, the increases in autophagic vacuoles with undegraded contents due to CR-CYLD overexpression were exaggerated by additional CR-ATG7 overexpression in the hearts (Fig. 5C). These results indicate that in PO-hearts, CR-ATG7 overexpression enhances autophagosome formation, fusion with lysosomes, and autolysosomal degradation (autolysosome efflux); however, CR-CYLD overexpression does not affect autophagosome formation and fusion with lysosomes but suppresses autolysosome efflux. Accordingly, CYLD does not interfere with ATG7-mediated autophagosome synthesis while suppressing ATG7-dependent autolysosome efflux, thereby leading to accumulation of toxic autophagic vacuoles in PO-hearts. Thus, CYLD can turn on ATG7-dependent accumulation of toxic autophagic vacuoles in cardiomyocytes, which exaggerates PO-induced cardiomyopathy, while switching off ATG7-mediated execution of autophagy, which is cardiac protective.
Fig. 5.
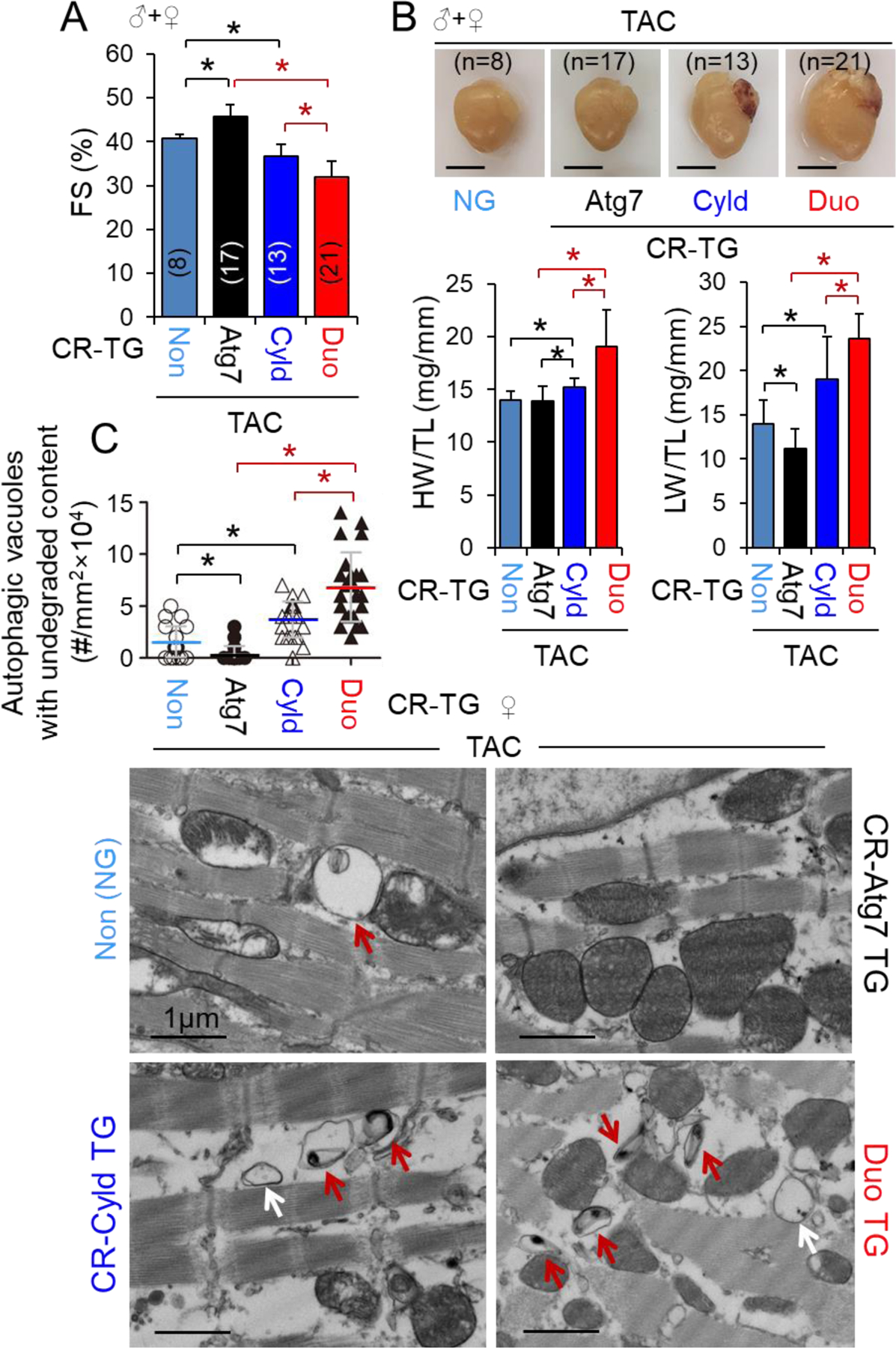
The adverse impact of CR-Cyld overexpression on CR-Atg7 overexpression-mediated cardiac protection in pressure overloaded hearts. Littermates of male and female mice with indicated genotypes at age of 10 weeks were subject to TAC for 2 weeks. (A) FS (%). *, p<0.05 between indicated groups. (B) Representative heart images (top panels), heart weight (HW)/tibia length (TL) and lung weight (LW)/TL ratios (lower panels). *, p<0.05 between indicated groups. (C) Autophagic vacuoles in LVs. Two female mice with indicated genotypes were randomly assigned for the assessment. At least 6 randomly chosen fields of LV TEM images for each group were measured. *, p<0.05 between indicated groups.
3.5. CYLD suppresses autophagy at a stage of autolysosome efflux in cardiomyocytes
It has been well-established that lysosomotropic reagents such as bafilomycin A1 (BafA1) and chloroquine (CQ) can disturbs lysosomal stability by increasing lysosomal pH, thereby disrupt autophagosomes fusion with lysosomes [36]. Since microtubule-associated protein 1 light chain 3 (LC3)-II usually exists on autophagosomes and the LC3-II associated with the inner membrane of autophagosomes is degraded by lysosomal enzyme after the fusion of autophagosomes with lysosomes, the difference in LC3-II levels in the presence and absence of lysosome inhibitors such as BafA1 or CQ represents the amount of autophagosomes that is fused with lysosomes for autophagic degradation, i.e., autophagic flux in the cell [36]. Of note, a short-term treatment (~6 h) of BafA1 or CQ mainly inhibits the fusion of autophagosomes with lysosomes; however, a long-term treatment (~22 h) of BafA1 or CQ can also activates adaptive autophagosome formation in cultured cells [37]. Thus, the accumulated LC3-II by a long-term treatment (>22 h) of BafA1 or CQ is likely a net result of autophagosome induction (autophagosome formation and fusion with lysosomes) versus autophagosome clearance via lysosomes, reflecting a status of lysosomal degradation of fused autophagosomes, i.e, autolysosome efflux. Utilizing these features, an elegant study established an experimental system which measures the capacities of autophagosome induction and fusion with the lysosome as well as autolysosome efflux in cultured neonatal rat ventricular myocytes (NRVMs) with lentiviral expression of mCherry-green fluorescent protein (GFP) tandem-tagged LC3 [38]. The fluorescent signal of GFP is quenched by the low pH inside the lysosome, whereas red fluorescent protein (RFP) such as mCherry exhibits more stable fluorescence in the acidic compartment [36]. In the cells expressing mRFP-GFP tandem-tagged LC3, autophagosomes and autolysosomes are labeled with yellow (punctate dots that fluoresce green and red, i.e., yellow) and red (punctate dots that fluoresce only red) signals, respectively [36]. Therefore, the relative abundance of cellular autophagosomes and autolysosomes were morphologically traced by exploiting the differences in these two fluorescent proteins (i.e., lysosomal quenching of GFP fluorescence versus lysosomal stability of RFP fluorescence) in these cardiomyocytes [38]. In the cardiomyocytes cultured in full growth medium, constitutive (basal) autophagy is characterized by a preponderance of autolysosomes (punctate dots red only) and a few autophagosomes (yellow punctate dots). Treatment of rapamycin at a final dose of 5 µM for 24 hours markedly increases abundance of autolysosomes without a discernible accumulation of autophagosomes, indicating enhanced autophagic flux. The rapamycin treatment also causes a decline in cumulative LC3-II abundance, suggesting consumption of autophagosomes. Thus, this setting activates the whole process of autophagy ranging from autophagosome induction and fusion with the lysosome to completion of autophagic degradation, reaching a steady state in which most of the autophagosomes fused with lysosomes are degraded by lysosomal enzymes, i.e., complete autolysosome efflux. Co-treatment of rapamycin with CQ at a dose of 10 µM for 24 hours results in accumulation of autophagosomes and near the absence of autolysosomes. Therefore, under the optimized experimental conditions, in the presence of rapamycin, the CQ-induced accumulation of autophagosomes reflects the capacity of autophagosome formation or the amount of autophagosomes fused with lysosomes, whereas the CQ-induced depletion of autolysosomes indicates at least in part the capability of autolysosome efflux, in cardiomyocytes.
Taken with these advantages, we carried out two sets of experiments to verify the role of CYLD in regulating autophagic flux and autolysosome efflux in vitro. In mouse embryonic fibroblasts (MEF), stable overexpression of Cyld shRNAs did not affect LC3-II accumulation induced by a short-term (4 hours) treatment of BafA1 or CQ, but enhanced LC3-II accumulation induced by a long-term (24 hours) treatment of BafA1 or CQ (Fig. 6A–D). These results suggest that CYLD does not affect autophagosome formation or the fusion of autophagosomes with lysosomes, but may suppress autophagosome clearance after the fusion, i.e., autolysosome efflux. Because not the basal but BafA1-induced cell death in MEF was inhibited by knockdown of CYLD (Fig. 6E), it is likely that CYLD mediates cell death in stressed cardiomyocytes via suppressing autophagy at the stage of autolysosome efflux. With this in mind, we determined autophagic flux and autolysosome efflux in cardiomyocyte-like H9C2 cells with transient expression of mCherry-GFP-LC3 reporter and HA-Flag-tagged Cyld plasmids (Fig. 7A–C). We recapitulated the rapamycin-induced autophagic flux and autolysosome efflux in H9C2 cells. We also demonstrated that CYLD overexpression did not affect basal autophagy but selectively inhibited rapamycin-induced autolysosome efflux, rather than rapamycin-induced autophagosomes fusion with lysosomes (Fig. 7A–C). In addition, stable overexpression of CYLD did not affect cell death at basal condition but enhanced BafA1- or MG132-induced cell death in H9C2 cells (Fig. 7D). Importantly, MG132-induced adaptive activation was wiped out by CYLD overexpression (Fig. 7D), underscoring a critical role of CYLD in suppressing induced autophagy. Taken together, these results indicate that CYLD can promote autophagy inhibition-induced cell death most likely via suppressing autolysosome efflux in cardiomyocytes.
Fig. 6.
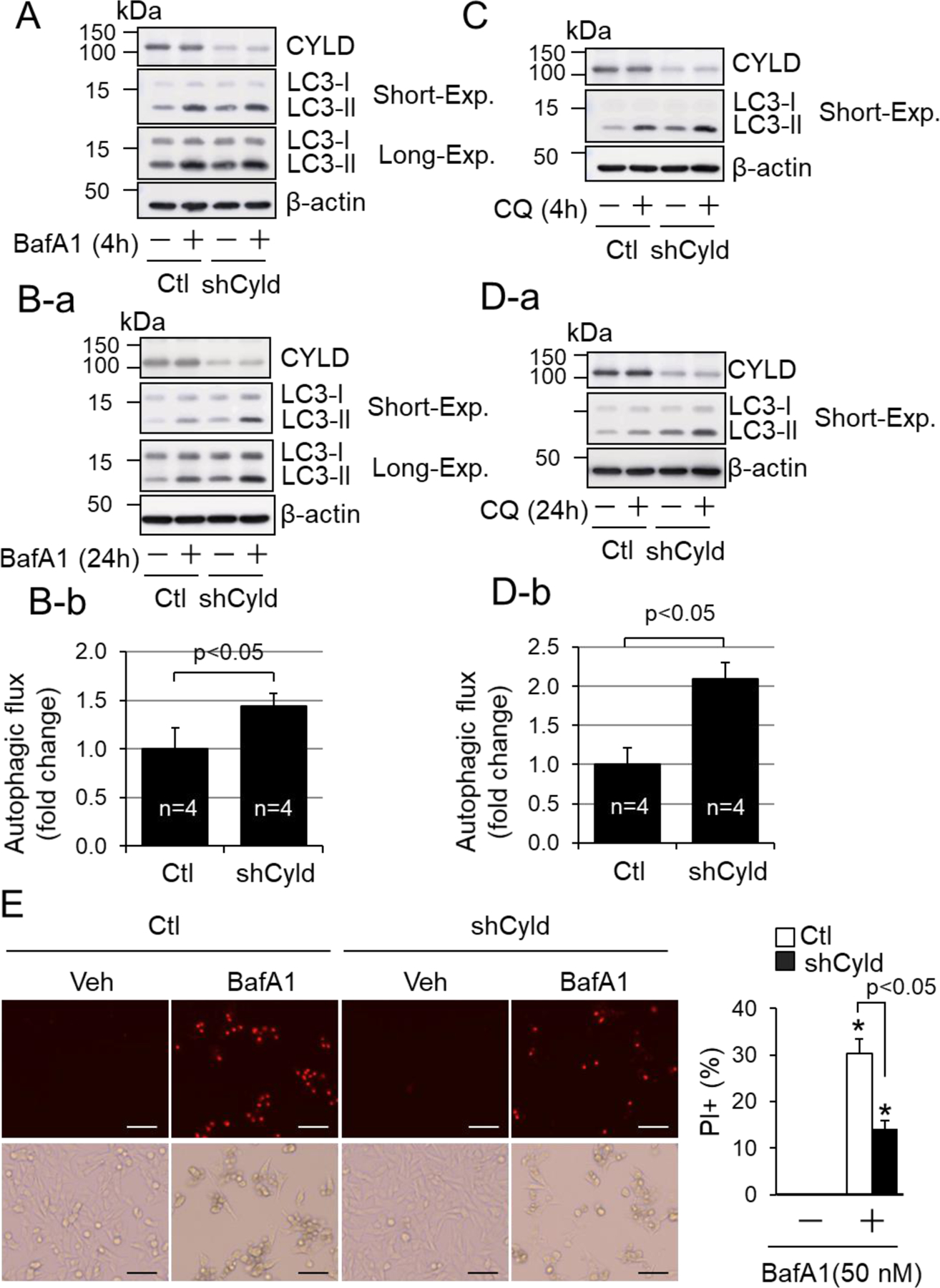
CYLD-mediated suppression of autophagic flux and cell death in MEF. (A, B, C, D) The impact of Cyld loss-of-function on autophagic flux in MEF. MEF with stably infected lentivirus of control (Ctl) or shCyld (shCyld) were treated with or without BafA1 (100 nM) (A) or CQ (50 µM) (C) for 4 hours or BafA1 (10 nM) (B) or CQ (10 µM) (D) for 24 hours in full growth culture medium. Results are from 4 separated experiments. Short-Exp, exposure with a short time period. (B-b, D-b) Semi-quantified results of autophagic flux induced by 24-hour treatment of BafA1 (10 nM) and CQ (10 µM) in Ctl and shCyld groups (n=4). (E) The impact of Cyld loss-of-function on BafA1 induced-cell death in MEF. MEF with stably infected lentivirus of control (Ctl) or shCyld (shCyld) were treated with or without BafA1 (50 nM) for 24 hours i n full growth culture medium (n=6). More than 10,000 cells were counted for each group. *, p<0.05 vs. vehicle control, Veh (−) in same group.
Fig. 7.
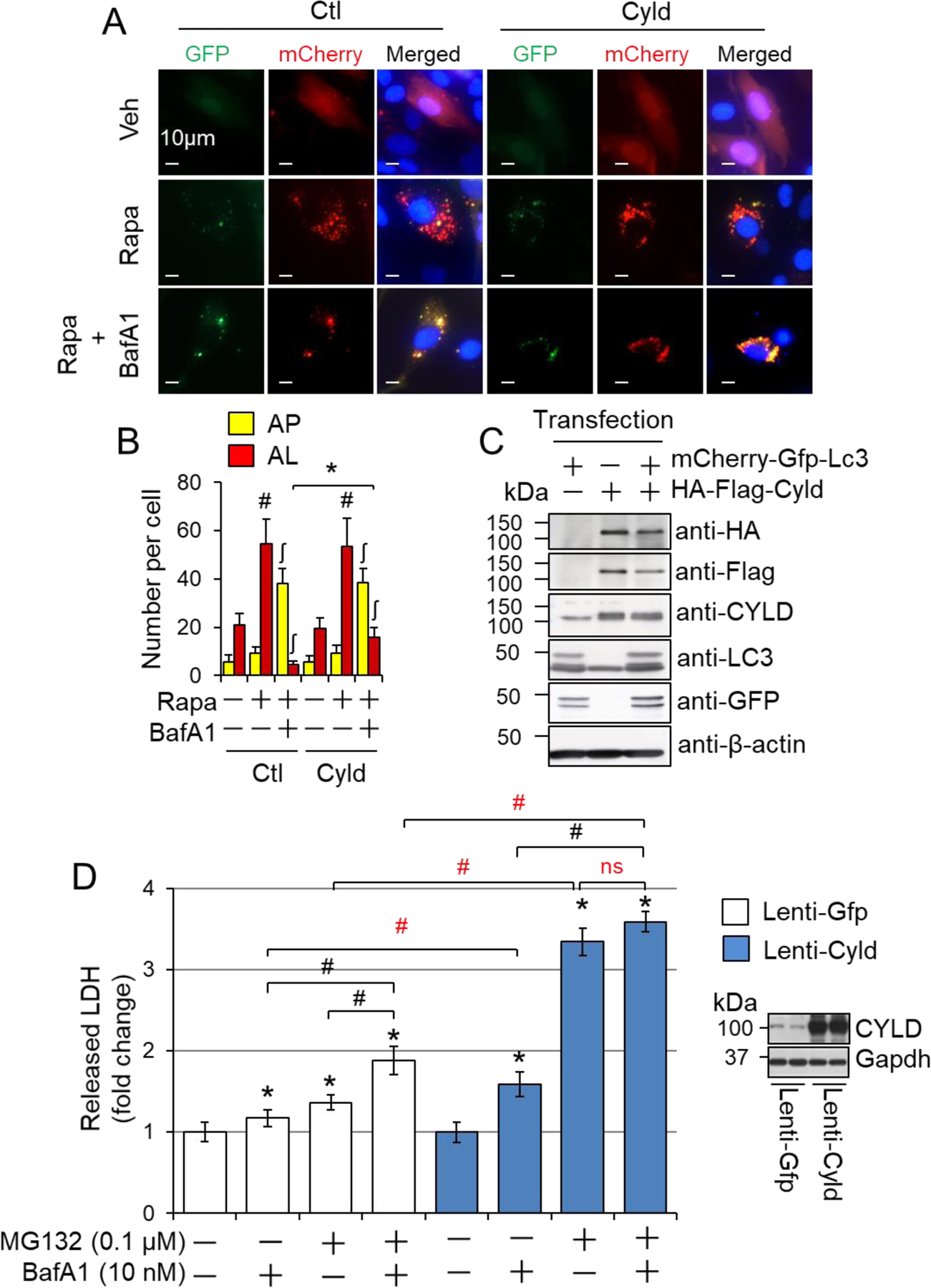
CYLD-mediated suppression of autolysosome clearance and cell death in cardiomyocytes. (A) The representative images of rapamycin (Rapa)-induced autolysosome efflux in H9C2 cells. (B) Quantified numbers of autophagosomes (AP) and autolysosomes (AL). H9C2 cells transfected with mCherry-GFP-LC3 and HA-flag-Cyld plasmids were treated with 1 µM Rapa and 10 nM BafA1 in full growth medium for 24 hours. 50 cells were counted for each group. # or ʃ, p<0.05 vs. vehicle treated controls in the same groups; *, p<0.05 between indicated groups. (C) Verification of transfection efficacy via Western blot analysis for (A) and (B). (D) CYLD overexpression-mediated cell death in H9C2 cells. H9C2 cells stably infected with lentivirus of Gfp (Lenti-Gfp) and lentivirus of Cyld (Lenti-Cyld) were treated with or without MG132 (0.1 µM) and/or BafA1 (10 nM) in serum-free DMEM for 24 hours. The amount of LDH released into supernatants was measured (n=6). The right panel shows the representative immunoblots of CYLD expression in these cells. *, p<0.05 vs. the control (−) in the same group; #, p<0.05 between indicated groups.
3.6. CYLD inactivates mechanistic target of rapamycin complex 1 (mTORC1) reactivation, upregulates Ras genes from rat brain 7 (Rab7) and enhances cardiomyocyte death in pressure overloaded hearts.
Unlike the early stages of autophagy including autophagosome formation and maturation or autophagosome fusion with lysosomes to form the autolysosome, the late stages of autophagy such as autolysosomal degradation after autolysosome formation have not been well investigated [20–22]. While Spinster (Spin) has been identified as an inhibitor of autolysosomal degradation or autolysosome efflux in normal rat kidney cells [39], C-type lectin domain family 16 A (Clec16a) and nuclear protein 1 (Nupr1) have been demonstrated to suppress autolysosome efflux in cancer cells [22, 40]. Strikingly, the impaired autolysosome efflux is characterized by an accumulation of autolysosomes containing undigested contents, which can be recapitulated by the treatment of CQ or BafA1. At molecular level, it is most likely that reactivation of mTORC1 and release of Rab7 from autolysosome membrane are critical for autolysosome efflux [39, 41]. These phenotypes of the cells with impaired autolysosome efflux were reminiscent of those in PO-hearts of CR-Cyld Tg mice. Therefore, we questioned whether mTORC1 and Rab7 are also involved in CYLD-mediated suppression of autolysosome efflux as well as escalation of cell death in cardiomyocytes which we uncovered in previous experiments [14], thereby exaggerating PO-induced cardiomyopathy. A time course study showed that after PO, myocardial mTORC1 activity evidenced by phosphorylation status of its downstream p70 ribosomal protein S6 kinase (p70S6K) [42] was increased at 3 days and then declined almost to the basal level at 7 days, thereafter reactivated at 14 days and maintained by 56 days in WT FVB/N mice (Fig. S6). TAC-induced cardiac dysfunction appeared at 14 days and then progressively deteriorated by 56 days in these mice (data not shown). In consistent with our previous observation in WT C57BL/6J mice [14], the expression of myocardial CYLD proteins was upregulated at 3 days, peaked at 14 days, and the declined by 56 days in WT FVB/N mice after PO (Fig. S6). These results suggest that upregulation of CYLD is potentially linked to mTORC1 reactivation in PO-hearts. Indeed, more detailed assessments of mTORC1 signaling revealed that 2-week TAC-induced phosphorylation of p70S6K was selectively blocked by CR-CYLD overexpression associated with upregulated protein levels of lysosomal-associated membrane protein 1 (Lamp1), Lamp2 and Rab7 in the heart (Fig. 8A and B). These results suggest that CYLD may hijack a mechanism which suppresses mTORC1 reactivation while interrupting Rab7 release from autolysosomes, thereby downregulating autolysosomal degradation. CYLD may also control the substrate access to mTORC1 without affecting mTORC1 activation because CR-CYLD overexpression had minimal impact on TAC-induced phosphorylation of Unc 51-like autophagy activating kinase 1 (ULK1) at Serine (S) 757 (Fig. 8A), which is phosphorylated by mTORC1 thus suppressing autophagy induction [43]. In addition, CR-CYLD overexpression did not affect TAC-induced phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) (Fig. 8A), another typical substrate of mTORC1 [42]. CR-CYLD overexpression did not affect 2-week TAC-induced decreases in phosphorylation of myocardial Akt or increases in protein expression of myocardial p53, two substrates of CYLD in the other tissues [44, 45] (Fig. S7), suggesting a tissue specific role of CYLD. However, like the effect of CYLD knockdown in cultured NRVMs [14], CR-CYLD overexpression also slightly increased extracellular signal-regulated kinases (ERK) activity in 2-week PO-hearts (Fig. S7). Although not conclusive at this point, the CYLD-mediated increases in ERK activity is likely contributing to the inhibition of mTORC1 reactivation as previously reported [46]. On the other hand, TAC-induced myocardial death was dramatically suppressed by Cyld KO (Fig. 8C). Taken together, these results indicate that upregulation of CYLD suppresses mTORC1 reactivation and autolysosome clearance, thereby contributing to cardiomyocyte death in PO-hearts (Fig. 8D).
Fig. 8.
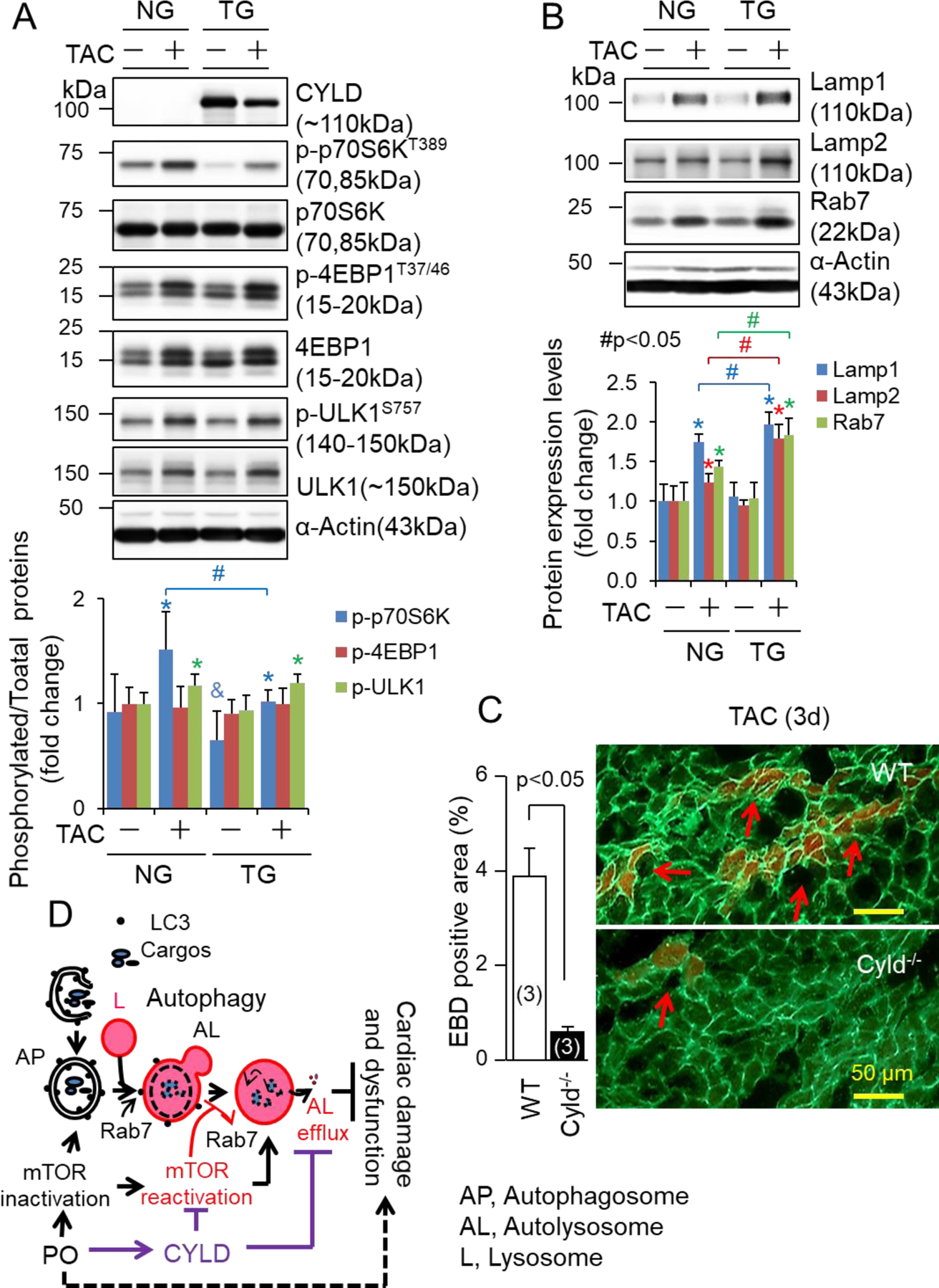
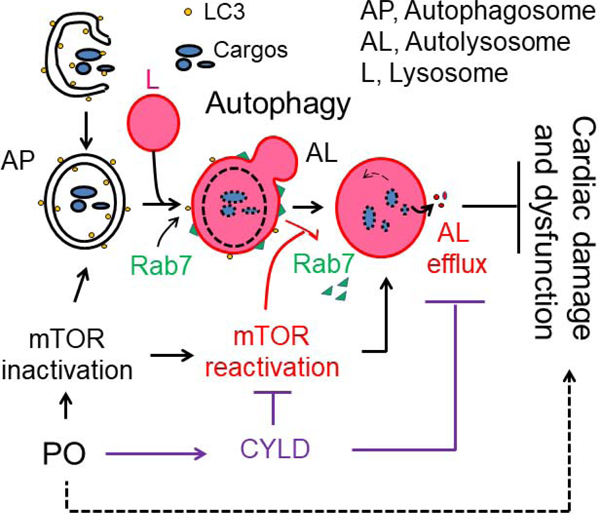
The impact of CR-Cyld overexpression on mTORC1 signaling, the expression of Lamp1, Lapm2 and Rab7 and myocardial necrosis in PO-hearts. (A, B) Littermates of male nontransgenic wild type (NG) and CR-Cyld Tg (TG) mice at age of 10 weeks were subject to sham or TAC operations for 2 weeks. LV lysates of these mice were subject to Western blot analysis of (A) CYLD and mTORC1 signaling and (B) the expression of lysosomal membrane proteins of Lamp1 and Lamp2, as well as Rab7. n=4, *, p<0.05 vs. TAC (−) in the same groups, &, p<0.05 vs. NG TAC (−); #, p<0.05 between indicated groups. (C) The impact of Cyld KO on TAC-induced myocardial death. Littermates of wild type (WT) and Cyld KO mice at age of 8 weeks were subject to TAC for 3 days and then received a single intraperitoneal injection of Evans blue dye (EBD) 18 hours prior to heart harvest. Arrows indicate BED positive necrotic myocardial cells. Animal numbers are indicated in the parentheses. (D) A scheme of our working hypothesis. AP, autophagosomes; L, lysosomes; AL, autolysosomes; PO, pressure overload.
4. Discussion
In the present study, we demonstrate that CYLD is a mediator of PO-induced cardiomyopathy independent of gender differences. Mechanistically, CYLD suppresses autophagy by selectively interrupting autolysosome efflux in cardiomyocytes, thereby switching autophagic adaptation into autophagic damage in PO-hearts. At the molecular level, it is likely that CYLD inhibits mTORC1 reactivation and prevents Rab7 release from autolysosomes, both of which are required for autolysosome efflux in cardiomyocytes. These data support the hypothesis that DUBs serve as a unique class of cardiac autophagy regulators, able to affect cardiac homeostasis and disease. In addition, our findings provide novel insight into autophagy-mediated regulation of cardiac pathological remodeling and dysfunction and help to explain the sometimes contradictory data for the roles of cardiac autophagy.
CYLD DUB activity is highly specific for K63-linked Ub chains but has been shown to act on K48-linked Ub chains as well [47]. We noticed that TAC-induced increases in myocardial poly-ubiquitinated proteins is not attenuated but enhanced by CR-CYLD overexpression. These results indicate that CYLD deteriorates proteotoxicity via impairing the protein degradation pathways for protein quality control in PO-hearts. Indeed, our results demonstrate that upregulation of CYLD in cardiomyocytes impairs autophagy at the stage of autolysosome efflux and enhances myocardial death in PO-hearts. The adverse phenotypes observed in 2-week TAC-CR-CYLD mice were reminiscent of those in CR-mTOR KO mice, including inhibited p-p70S6K, accumulated autophagic vacuoles with undegraded contents, cardiomyocyte death, and cardiac dysfunction [48], supporting the axis of CYLD–mTORC1 inactivation–autolysosome efflux inhibition–cardiomyocyte death in PO-hearts. In addition, a downstream effector role of Rab7 in CYLD-mediated autophagy inhibition is reinforced by a previous study showing that upregulated Rab7 protein expression is associated with accumulated autophagic vacuoles with undegraded content and cell death in cardiomyocytes in UM-X7.1 hamster model of human dilated cardiomyopathy [49]. On the other hand, previous studies demonstrated that ubiquitination of mTOR through K63-linked poly-Ub chains are required for amino acid-mediated activation of mTORC1 in human embryonic kidney (HEK) 293T cells [50], and ubiquitination of Rab7 is critical for both its stability and signaling although the type of Ub modification is unclear [51]. These results indicate that CYLD suppresses mTORC1 reactivation potentially via deubiquitination of K63-linked poly-Ub chains from mTOR while trapping Rab7 on the membrane of autolysosomes presumably via removing K63-linked and/or K48-linked poly-Ub chains from Rab7 in stressed cardiomyocytes.
It is worthy to note that the contradictory roles of autophagy in the heart proposed by previous studies [20, 23] are at least partly due to the interpretations of autophagy per se. Conceptually, if autophagy is considered as a dynamic process for targeted degradation of cytoplasmic components, most of the studies investigating the role of cardiac autophagy have demonstrated its adaptive nature of autophagy in PO-hearts [20]. In this regard, our CR-Atg5KO and CR-Atg7 overexpression studies provide definitive proof of autophagy-mediated cardiac protection in adult PO-hearts. On the other hand, the inhibition of some particular components of autophagic machinery such as genetic disruption of Beclin-1 and Atg16 does appear to be beneficial to the stressed hearts including PO-hearts, thereby supporting the concept that hyper-, over- or massive activation of cardiac autophagy is detrimental [20, 23]. This concept has been further promoted by the observations that loss of Atg5, Atg7 or Becline-1 reduced non-apoptotic cell death, so called autophagic cell death, in MEF and other types of cells in vitro [52, 53]. However, these components of autophagic machinery may act in nonautophagic functions [52]; the precise mechanism by which autophagy regulates cardiomyocyte death is poorly understood and whether autophagic cell death occurs in pathophysiologically relevant conditions in the heart remains to be determined [53]. In addition, emerging evidence has demonstrated that Beclin-1 or Atg16 are capable of suppressing autophagy in the heart [20, 23]. A recent report showed that brozopine ameliorates PO-induced upregulation of myocardial Beclin-1, LC3-II and autophagic vacuoles, and cardiomyopathy in C57BL/6J mice at 10 weeks after TAC [54], when the myocardial autophagy is impaired due to the sustained PO [30]. Therefore, not autophagy, but a dysregulated form of autophagic responses such as accumulation of autophagosomes without increases in autophagic flux, or augmentation of autolysosome formation without enhancing autolysosome efflux has been likely misinterpreted into ‘activated autophagy’ that contributes to cardiac pathological remodeling and dysfunction. Importantly, our findings support this hypothesis and provide alternative explanations to previous observations regarding the contradictory roles of autophagy, not only in cardiomyocytes but potentially in the other cell types as well. For example, in cultured endothelial cells, Cyld was upregulated; autophagosomes accumulated; and knockdown of Cyld or Atg7 similarly rescued cell death in a setting of autophagic flux by palmitic acid overloading [55]. These results were interpreted as autophagy-mediated cell death in endothelial cells [55]. We propose that palmitic acid-induced upregulation of Cyld leads to inhibition of autolysosome efflux, thereby facilitating cell death as we observed in cultured H9C2 cells, i.e., an autophagy inhibition-mediated cell death, rather than autophagy-mediated cell death. We therefore hypothesize that the rescue effect of Atg7 knockdown is due to the downregulation of cytotoxic autophagosome accumulation in this specific experimental setting.
The major limitation of the present study is that the molecular links between CYLD, inhibition of mTORC1 reactivation, Rab7 upregulation, and autolysosome efflux suppression have not been fully dissected. A reasonable expectation is that there are yet unidentified substrates that are autophagy machinery components, helping to mediate cardiac autolysosome efflux regulation. Indeed, a previous proteomics study of the human endogenous coregulatory complexome defined several CYLD interacting proteins, including tripartite motif containing 28 (TRIM28), TRIM37, and ULK1 as candidates [56]. TRIM28 and TRIM37 may act as upstream signaling molecules of mTORC1 in autophagy suppression [57, 58] while ULK1 could serve as a downstream effector of mTORC1 in autophagy induction and/or execution in the cell [59, 60]. However, our findings showed that CYLD is not likely regulating the axis of mTORC1 -ULK1 (S757 phosphorylation) in autophagy inhibition. Thus, except aforementioned K63 deubiquitination of mTOR and Rab7, the potential of TRIM28 or TRIMP37 and CYLD interaction in mTORC1 reactivation and suppressing autolysosome efflux in the heart deserves further investigation. In addition, the substrate cargoes of autophagy that CYLD regulates are not well characterized. Previous studies have shown that K63-linkded Ub-chains decorate protein aggregates and damaged mitochondria for aggrephagy and mitophagy, respectively [61]. Whilst the identities of protein aggregates for aggrephagy in the heart are poorly established, the elimination of damaged mitochondria via mitophagy has been demonstrated to play a critical role in cardiac diseases [20, 23]. Since it has been demonstrated that mitophagy protects against PO-induced myocardial mitochondrial dysfunction and pathological remodeling, and heart failure [62, 63], it arises an interesting question whether CYLD regulates mitophagy in heart. Indeed, our pilot studies showed that CYLD is redistributed and accumulated in mitochondria in stressed cardiomyocytes and enhances cell death probably via interrupting adaptive mitophagy in cardiomyocytes (data not shown). Moreover, pathological settings which could activate the CYLD-mediated autophagy inhibition remain to be further determined. Given that autophagy inhibition is often associated with metabolic cardiac diseases such as diabetic cardiomyopathy [64, 65], it is intriguing whether CYLD may play a critical role in dysregulation of cardiac autophagy contributing to diabetic cardiomyopathy. Further investigation of these subjects will lead to a better understanding of CYLD-mediated autophagy inhibition in cardiac pathological remodeling and dysfunction.
5. Perspectives
Pressure overload initially enhances cardiac autophagy to protect against cardiac pathological remodeling and dysfunction. However, pressure overload over time increases the protein level of CYLD, leading to cardiac autophagy inhibition and contributing to cardiac maladaptive remodeling and dysfunction. At the molecular level, CYLD does not affect autophagosome biosynthesis and fusion with the lysosome but suppresses autolysosomal degradation after the autolysosome formation in cardiomyocytes. As a result, CYLD-mediated autophagy inhibition is characterized by the accumulation of autophagic vacuoles with nondegraded contents, as well as the conversion of adaptive responses of early autophagic processes into pathological damage (Fig. 8D). Our findings highlight a unique role of CYLD in suppressing autophagy at the step of autolysosome efflux in pressure overloaded hearts and suggest that targeting CYLD may be a novel therapeutic approach for the treatment of adverse cardiac remodeling and dysfunction associated with hypertensive heart disease.
Supplementary Material
Highlights.
Cardiac CYLD is a mediator of pressure overload-induced cardiomyopathy.
Activation of cardiac autophagy is adaptive in pressure overloaded hearts.
ATG7 mainly promotes autophagic responses prior to the step of autolysosome efflux.
CYLD inhibits mTORC1 reactivation and autolysosome efflux in cardiomyocytes.
CYLD enables early autophagic adaptive responses to become detrimental to the heart.
Sources of Funding
This research was supported by grants from the National Institute of Health (P01 AT003961, R01 HL131667) and American Diabetes Association (1-16-IBS-059), and a scholarship of China Scholarship Council (No. 201506220204).
Nonstandard Abbreviations and Acronyms
- CYLD
Cylindromatosis
- DUB
Deubiquitinating enzyme
- CR
Cardiomyocyte-restricted
- Tg
Transgenic
- PO
Pressure overload
- TAC
Transverse aortic arch constriction
- mTORC1
Mechanistic target of rapamycin complex 1
- Rab7
Ras genes from rat brain 7
- E1
Ubiquitin-activating enzymes
- E2
Ubiquitin-conjugating enzymes
- E3
Ubiquitin ligases
- Ub
Ubiquitin
- USP
Ubiquitin-specific protease
- TNFAIP3
Tumor necrosis factor alpha-induced protein 3
- PQC
Protein quality control
- LC3
Microtubule-associated protein 1 light chain 3
- p70S6K
p70 ribosomal protein S6 kinase
- 4EBP1
Eukaryotic translation initiation factor 4E-binding protein 1
- Lamp1
Lysosomal-associated membrane protein 1
- ERK
Extracellular signal-regulated kinases
- ANF
Atrial natriuretic factor
- BNP
Brain natriuretic peptide
- αMHC
alpha-myosin heavy chain
- βMHC
beta-myosin heavy chain
- SERCA
Sarcoplasmic reticulum calcium ATPase2a
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
References
- [1].Haglund K, Dikic I, Ubiquitylation and cell signaling, Embo J 24(19) (2005) 3353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R, A genomic and functional inventory of deubiquitinating enzymes, Cell 123(5) (2005) 773–86. [DOI] [PubMed] [Google Scholar]
- [3].Mukhopadhyay D, Riezman H, Proteasome-independent functions of ubiquitin in endocytosis and signaling, Science 315(5809) (2007) 201–5. [DOI] [PubMed] [Google Scholar]
- [4].Demartino GN, Gillette TG, Proteasomes: machines for all reasons, Cell 129(4) (2007) 659–62. [DOI] [PubMed] [Google Scholar]
- [5].Schulman BA, Harper JW, Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways, Nat Rev Mol Cell Biol 10(5) (2009) 319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Willis MS, Patterson C, Into the heart: the emerging role of the ubiquitin-proteasome system, J Mol Cell Cardiol 41(4) (2006) 567–79. [DOI] [PubMed] [Google Scholar]
- [7].Wang X, Robbins J, Heart failure and protein quality control, Circ Res 99(12) (2006) 1315–28. [DOI] [PubMed] [Google Scholar]
- [8].Cook SA, Novikov MS, Ahn Y, Matsui T, Rosenzweig A, A20 is dynamically regulated in the heart and inhibits the hypertrophic response, Circulation 108(6) (2003) 664–7. [DOI] [PubMed] [Google Scholar]
- [9].Huang H, Tang QZ, Wang AB, Chen M, Yan L, Liu C, Jiang H, Yang Q, Bian ZY, Bai X, Zhu LH, Wang L, Li H, Tumor suppressor A20 protects against cardiac hypertrophy and fibrosis by blocking transforming growth factor-beta-activated kinase 1-dependent signaling, Hypertension 56(2) (2010) 232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, Yang Q, Huang Y, Wei YS, Liu PP, Liu DP, Liang CC, Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction, Circulation 115(14) (2007) 1885–94. [DOI] [PubMed] [Google Scholar]
- [11].Isumi Y, Hirata T, Saitoh H, Miyakawa T, Murakami K, Kudoh G, Doi H, Ishibashi K, Nakajima H, Transgenic overexpression of USP15 in the heart induces cardiac remodeling in mice, Biochem Biophys Res Commun 405(2) (2011) 216–21. [DOI] [PubMed] [Google Scholar]
- [12].Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, Jones C, Hansen J, Blair E, Hofmann B, Siebert R, Turner G, Evans DG, Schrander-Stumpel C, Beemer FA, van Den Ouweland A, Halley D, Delpech B, Cleveland MG, Leigh I, Leisti J, Rasmussen S, Identification of the familial cylindromatosis tumour-suppressor gene, Nat Genet 25(2) (2000) 160–5. [DOI] [PubMed] [Google Scholar]
- [13].Mathis BJ, Lai Y, Qu C, Janicki JS, Cui T, CYLD-mediated signaling and diseases, Current drug targets 16(4) (2015) 284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang H, Lai Y, Mathis BJ, Wang W, Li S, Qu C, Li B, Shao L, Song H, Janicki JS, Sun SC, Wang XL, Tang D, Cui T, Deubiquitinating enzyme CYLD mediates pressure overload-induced cardiac maladaptive remodeling and dysfunction via downregulating Nrf2, J Mol Cell Cardiol 84 (2015) 143–53. [DOI] [PubMed] [Google Scholar]
- [15].Amerik AY, Hochstrasser M, Mechanism and function of deubiquitinating enzymes, Biochim Biophys Acta 1695(1–3) (2004) 189–207. [DOI] [PubMed] [Google Scholar]
- [16].Reyes-Turcu FE, Ventii KH, Wilkinson KD, Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes, Annu Rev Biochem 78 (2009) 363–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].He B, Zhao YC, Gao LC, Ying XY, Xu LW, Su YY, Ji QQ, Lin N, Pu J, Ubiquitin-Specific Protease 4 Is an Endogenous Negative Regulator of Pathological Cardiac Hypertrophy, Hypertension 67(6) (2016) 1237–48. [DOI] [PubMed] [Google Scholar]
- [18].Ying X, Zhao Y, Yao T, Yuan A, Xu L, Gao L, Ding S, Ding H, Pu J, He B, Novel Protective Role for Ubiquitin-Specific Protease 18 in Pathological Cardiac Remodeling, Hypertension 68(5) (2016) 1160–1170. [DOI] [PubMed] [Google Scholar]
- [19].Liu N, Chai R, Liu B, Zhang Z, Zhang S, Zhang J, Liao Y, Cai J, Xia X, Li A, Liu J, Huang H, Liu S, Ubiquitin-specific protease 14 regulates cardiac hypertrophy progression by increasing GSK-3beta phosphorylation, Biochem Biophys Res Commun 478(3) (2016) 1236–41. [DOI] [PubMed] [Google Scholar]
- [20].Wang X, Cui T, Autophagy modulation: a potential therapeutic approach in cardiac hypertrophy, Am J Physiol Heart Circ Physiol 313(2) (2017) H304–H319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhao YG, Zhang H, Autophagosome maturation: An epic journey from the ER to lysosomes, The Journal of cell biology 218(3) (2019) 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mu Y, Yan X, Li D, Zhao D, Wang L, Wang X, Gao D, Yang J, Zhang H, Li Y, Sun Y, Wei Y, Zhang Z, Chang X, Yao Z, Tian S, Zhang K, Terada LS, Ma Z, Liu Z, NUPR1 maintains autolysosomal efflux by activating SNAP25 transcription in cancer cells, Autophagy 14(4) (2018) 654–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sciarretta S, Maejima Y, Zablocki D, Sadoshima J, The Role of Autophagy in the Heart, Annual review of physiology 80 (2018) 1–26. [DOI] [PubMed] [Google Scholar]
- [24].Grumati P, Dikic I, Ubiquitin signaling and autophagy, J Biol Chem 293(15) (2018) 5404–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jacomin AC, Taillebourg E, Fauvarque MO, Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities?, Cells 7(8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Abbasi A, Forsberg K, Bischof F, The role of the ubiquitin-editing enzyme A20 in diseases of the central nervous system and other pathological processes, Frontiers in molecular neuroscience 8 (2015) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang W, Li S, Wang H, Li B, Shao L, Lai Y, Horvath G, Wang Q, Yamamoto M, Janicki JS, Wang XL, Tang D, Cui T, Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins, J Mol Cell Cardiol 72 (2014) 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J, Enhanced autophagy ameliorates cardiac proteinopathy, J Clin Invest 123(12) (2013) 5284–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li J, Ichikawa T, Villacorta L, Janicki JS, Brower GL, Yamamoto M, Cui T, Nrf2 protects against maladaptive cardiac responses to hemodynamic stress, Arterioscler Thromb Vasc Biol 29(11) (2009) 1843–50. [DOI] [PubMed] [Google Scholar]
- [30].Qin Q, Qu C, Niu T, Zang H, Qi L, Lyu L, Wang X, Nagarkatti M, Nagarkatti P, Janicki JS, Wang XL, Cui T, Nrf2-Mediated Cardiac Maladaptive Remodeling and Dysfunction in a Setting of Autophagy Insufficiency, Hypertension 67(1) (2016) 107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Du XJ, Gender modulates cardiac phenotype development in genetically modified mice, Cardiovasc Res 63(3) (2004) 510–9. [DOI] [PubMed] [Google Scholar]
- [32].Piro M, Della Bona R, Abbate A, Biasucci LM, Crea F, Sex-related differences in myocardial remodeling, Journal of the American College of Cardiology 55(11) (2010) 1057–65. [DOI] [PubMed] [Google Scholar]
- [33].Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ Jr., Rothermel BA, Hill JA, Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy, Circulation 117(24) (2008) 3070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ranek MJ, Zheng H, Huang W, Kumarapeli AR, Li J, Liu J, Wang X, Genetically induced moderate inhibition of 20S proteasomes in cardiomyocytes facilitates heart failure in mice during systolic overload, J Mol Cell Cardiol 85 (2015) 273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tian Z, Wang C, Hu C, Tian Y, Liu J, Wang X, Autophagic-lysosomal inhibition compromises ubiquitin-proteasome system performance in a p62 dependent manner in cardiomyocytes, PLoS One 9(6) (2014) e100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mizushima N, Yoshimori T, Levine B, Methods in mammalian autophagy research, Cell 140(3) (2010) 313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Li M, Khambu B, Zhang H, Kang JH, Chen X, Chen D, Vollmer L, Liu PQ, Vogt A, Yin XM, Suppression of lysosome function induces autophagy via a feedback downregulation of MTOR complex 1 (MTORC1) activity, J Biol Chem 288(50) (2013) 35769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A, Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury, Circulation 125(25) (2012) 3170–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rong Y, McPhee CK, Deng S, Huang L, Chen L, Liu M, Tracy K, Baehrecke EH, Yu L, Lenardo MJ, Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation, Proc Natl Acad Sci U S A 108(19) (2011) 7826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Redmann V, Lamb CA, Hwang S, Orchard RC, Kim S, Razi M, Milam A, Park S, Yokoyama CC, Kambal A, Kreamalmeyer D, Bosch MK, Xiao M, Green K, Kim J, Pruett-Miller SM, Ornitz DM, Allen PM, Beatty WL, Schmidt RE, DiAntonio A, Tooze SA, Virgin HW, Clec16a is Critical for Autolysosome Function and Purkinje Cell Survival, Sci Rep 6 (2016) 23326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ, Termination of autophagy and reformation of lysosomes regulated by mTOR, Nature 465(7300) (2010) 942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Laplante M, Sabatini DM, mTOR signaling in growth control and disease, Cell 149(2) (2012) 274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim J, Kundu M, Viollet B, Guan KL, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1, Nature cell biology 13(2) (2011) 132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lim JH, Jono H, Komatsu K, Woo CH, Lee J, Miyata M, Matsuno T, Xu X, Huang Y, Zhang W, Park SH, Kim YI, Choi YD, Shen H, Heo KS, Xu H, Bourne P, Koga T, Xu H, Yan C, Wang B, Chen LF, Feng XH, Li JD, CYLD negatively regulates transforming growth factor-beta-signalling via deubiquitinating Akt, Nature communications 3 (2012) 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fernandez-Majada V, Welz PS, Ermolaeva MA, Schell M, Adam A, Dietlein F, Komander D, Buttner R, Thomas RK, Schumacher B, Pasparakis M, The tumour suppressor CYLD regulates the p53 DNA damage response, Nature communications 7 (2016) 12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T, A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1, J Biol Chem 284(32) (2009) 21412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Blake PW, Toro JR, Update of cylindromatosis gene (CYLD) mutations in Brooke-Spiegler syndrome: novel insights into the role of deubiquitination in cell signaling, Hum Mutat 30(7) (2009) 1025–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N, Condorelli G, MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice, J Clin Invest 120(8) (2010) 2805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Miyata S, Takemura G, Kawase Y, Li Y, Okada H, Maruyama R, Ushikoshi H, Esaki M, Kanamori H, Li L, Misao Y, Tezuka A, Toyo-Oka T, Minatoguchi S, Fujiwara T, Fujiwara H, Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony-stimulating factor, Am J Pathol 168(2) (2006) 386–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT, K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells, Molecular cell 51(3) (2013) 283–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Modica G, Lefrancois S, Post-translational modifications: How to modulate Rab7 functions, Small GTPases (2017) 1–7. [DOI] [PMC free article] [PubMed]
- [52].Kroemer G, Levine B, Autophagic cell death: the story of a misnomer, Nat Rev Mol Cell Biol 9(12) (2008) 1004–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nah J, Fernandez AF, Kitsis RN, Levine B, Sadoshima J, Does Autophagy Mediate Cardiac Myocyte Death During Stress?, Circ Res 119(8) (2016) 893–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang B, Shen D, Tang J, Li J, Xiao Y, Chen X, Cao C, Han D, Gao E, Zhao W, Zhang J, Chang J, Sodium (+/−)-5-bromo-2-(alpha-hydroxypentyl) benzoate ameliorates pressure overload-induced cardiac hypertrophy and dysfunction through inhibiting autophagy, J Cell Mol Med 23(9) (2019) 6048–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Khan MJ, Rizwan Alam M, Waldeck-Weiermair M, Karsten F, Groschner L, Riederer M, Hallstrom S, Rockenfeller P, Konya V, Heinemann A, Madeo F, Graier WF, Malli R, Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells, J Biol Chem 287(25) (2012) 21110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, Ding C, Shi Y, Yucer N, Krenciute G, Kim BJ, Li C, Chen R, Li W, Wang Y, O’Malley BW, Qin J, Analysis of the human endogenous coregulator complexome, Cell 145(5) (2011) 787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pineda CT, Potts PR, Oncogenic MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by ubiquitinating and degrading AMPK in cancer, Autophagy 11(5) (2015) 844–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang W, Xia Z, Farre JC, Subramani S, TRIM37 deficiency induces autophagy through deregulating the MTORC1-TFEB axis, Autophagy 14(9) (2018) 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang C, Wang H, Zhang D, Luo W, Liu R, Xu D, Diao L, Liao L, Liu Z, Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy, Nature communications 9(1) (2018) 3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jung CH, Ro SH, Cao J, Otto NM, Kim DH, mTOR regulation of autophagy, FEBS Lett 584(7) (2010) 1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Erpapazoglou Z, Walker O, Haguenauer-Tsapis R, Versatile roles of k63-linked ubiquitin chains in trafficking, Cells 3(4) (2014) 1027–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xu X, Hua Y, Nair S, Bucala R, Ren J, Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy, Hypertension 63(3) (2014) 490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J, Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure, Circulation 133(13) (2016) 1249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ouyang C, You J, Xie Z, The interplay between autophagy and apoptosis in the diabetic heart, J Mol Cell Cardiol 71 (2014) 71–80. [DOI] [PubMed] [Google Scholar]
- [65].Kobayashi S, Liang Q, Autophagy and mitophagy in diabetic cardiomyopathy, Biochim Biophys Acta 1852(2) (2015) 252–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.