

Abstract
Nicotine use disorder has been associated with glutamatergic alterations within the basal ganglia that might contribute to relapse. Specifically, initiation of cue-induced nicotine seeking produces rapid, transient synaptic potentiation (t-SP) in nucleus accumbens core (NAcore) medium spiny neurons (MSNs), defined as increases in spine head diameter and AMPA to NMDA current ratios (A/N). Ifenprodil, which inhibits nicotine reinstatement when administered systemically, antagonizes GluN2B-containing NMDA receptors, has affinity for serotonin receptors, and blocks serotonin transporters (SERT). The mechanisms underlying its therapeutic efficacy, however, remain unknown. Using pharmacological and genetic approaches, the current study examined the role of NAcore GluN2B receptors as well as SERT in mediating cue-induced nicotine seeking and associated MSN structure and physiology. Prior to reinstatement, rats received intra-NAcore injections of either ifenprodil, citalopram or artificial cerebral spinal fluid (15 min prior), or GluN2B or control siRNAs (3 consecutive days prior). Rats were sacrificed after a 15-minute cue-induced reinstatement session for dendritic spine analysis, western blotting or whole-cell electrophysiology. Intra-NAcore ifenprodil blocked nicotine-seeking behavior and promoted a higher frequency of shorter spines on MSN dendrites. However, a decrease in membrane-bound GluN2B receptor expression did not prevent cue-induced nicotine seeking or associated MSN cell physiology. Interestingly, intra-NAcore citalopram, an SSRI, prevented cue-induced nicotine seeking. Together, these results indicate that the therapeutic effects of ifenprodil on cue-induced nicotine seeking may, in part, be due to its actions at SERT rather than GluN2B, which may be specific to nicotine-seeking as opposed to other drugs of abuse.
Keywords: Nicotine, Ifenprodil, Citalopram, GluN2B, NMDA, SERT, Nucleus Accumbens, Medium Spiny Neurons
Introduction
Nicotine is the primary component in combustible cigarettes that maintains smoking behavior in humans (Stolerman and Jarvis 1995), and is the primary addictive component of e-cigarettes (Electronic Nicotine Delivery Systems, or ENDS; (St Helen et al. 2016)). Recent decreased use of combustible cigarettes has been paralleled with a drastic rise in the use of e-cigarettes, especially among adolescents (Wang et al. 2018). Nicotine vaping prevalence among adolescents rapidly increased from 2017–2018, with rates translating to roughly an additional 1.3 million new ENDS users compared to the previous year (Miech et al. 2019). Given the current regulatory effort from the Food and Drug Administration to decrease nicotine and tobacco use, understanding neural mechanisms that drive nicotine addiction and relapse is essential for developing therapeutics that promote long-term cessation.
Cue-induced nicotine seeking is associated with dysregulation in glutamate homeostasis within corticostriatal circuitry (Knackstedt et al. 2009; Powell et al. 2019, Gipson et al. 2013). Glutamatergic alterations within this system following withdrawal from nicotine self-administration render synapses of medium spiny neurons (MSNs) in the nucleus accumbens core (NAcore) in a potentiated state. Upon re-exposure to nicotine-conditioned cues, glutamatergic release from prefrontal glutamatergic afferents targeting the NAcore is enhanced, resulting in post-synaptic alterations within the NAcore (Stefanik et al. 2016; Gipson et al. 2013; Gipson et al. 2013). Further, drug exposure (both contingent and non-contingent administration) induces an increase in extrasynaptic GluN2B-containing NMDA receptors (Shen et al. 2011; Papouin et al. 2012). The rapid rise in glutamate released by afferents targeting the NAcore following drug-paired cue exposure likely activates these extrasynaptic GluN2B-containing receptors, triggering mechanisms of synaptic plasticity that promote drug seeking and taking (Papouin et al. 2012; Petralia 2012). Specifically, silent synapses, which are commonly observed following drug exposure (i.e. contingent and non-contingent cocaine), appear to contain mainly GluN2B receptors (Huang et al. 2009; Russo et al. 2010) and are present in dendritic spines with small head diameters (Russo et al. 2010), consistent with a developmental morphology. Thus, drug use may promote a switch in NMDA receptor composition, leading to enhanced expression of GluN2B-containing receptors (Sheng et al. 1994; Elias et al. 2008). This modification may render reward neural circuitry more prone to synaptic modifications, thus remodeling synaptic contacts in a drug- and activity-dependent manner (Dong and Nestler 2014).
Synaptic alterations have been reported within NAcore MSNs following cue-induced nicotine reinstatement. For example, changes in cellular and dendritic morphology as well as synaptic physiology, measured by alterations in the ratio of AMPA to NMDA mediated postsynaptic currents (A/N), occur within 15 minutes of exposure to contingent nicotine-paired cues. Following cue-initiated nicotine seeking, rapid and transient synaptic plasticity occurs, measured as increased dendritic spine head diameter and A/N ratios (Gipson et al. 2013). Given that changes in MSN dendritic morphology and physiology can be correlated with nicotine-seeking behavior (Gipson et al. 2014; Gipson et al. 2013; Russo et al. 2010), it is thought that this may be a maladaptive neural event that drives relapse (Mulholland et al. 2016).
Previous studies have shown that blockade of GluN2B-containing NMDA receptors with ifenprodil inhibits cue-induced reinstatement of nicotine, cocaine, and heroin seeking (Shen et al. 2011; Schumann et al. 2009; Gipson et al. 2013). Previously, we have shown that NAcore GluN2B-containing receptors are upregulated following withdrawal from nicotine self-administration, and importantly, systemic administration of ifenprodil, an antagonist of GluN2B-containing NMDA receptors, decreases nicotine-seeking behavior without reducing locomotion (Gipson et al. 2013). Given the role of NAcore GluN2B-containing NMDA receptors in mediating drug seeking behavior (Shen et al. 2011; Gipson et al. 2013), it is possible that these receptors within this key brain region associated with addiction critically regulate nicotine reinstatement-associated synaptic changes (Scofield et al. 2016; Scofield et al. 2016). While ifenprodil is primarily an antagonist of GluN2B-containing NMDA receptors, this compound also heavily interacts with serotonergic signaling through antagonism of the ligand-gated cation channel 5-HT3 (McCool and Lovinger 1995) as well as 5-HT1a and 5-HT2 (Chenard et al. 1991). Interestingly, activation of 5-HT3 increases accumbens dopamine release which is reversed by co-administration of a 5-HT3 antagonist (Chen et al. 1991). Further, accumbens dopamine release is regulated by serotonin neurotransmission from the dorsal raphe nucleus (Yoshimoto and McBride 1992). Thus, ifenprodil may reduce nicotine seeking behavior through 5-HT3-mediated blockade of NAcore dopamine release. In addition, analogues of ifenprodil inhibit the serotonin transporter (SERT; (Manepalli et al. 2012; Talbot et al. 2016), and ifenprodil potentiates the antidepressant-like effects of selective serotonin reuptake inhibitors (SSRIs) such as imipramine in vivo (Poleszak et al. 2014). These findings suggest that ifenprodil interacts with serotonin uptake, and studies exploring these additional therapeutic avenues of ifenprodil are warranted.
Due to the high comorbidity of depression and nicotine dependence (Mineur and Picciotto, 2009), it is not surprising that, although complex, a link between the two has been established (Shoaib and Buhidma, 2018). Further, antidepressants are frequently prescribed as long-term smoking cessation aids with clinical success similar to nicotine replacement (Hughes et al. 2014). While most antidepressants, including citalopram, target SERT, and given that ifenprodil has binding affinity for the serotonin system, here we examined whether intra-NAcore citalopram, a selective SERT inhibitor (Bezchlibnyk-Butler et al. 2000; Schrantee et al. 2019), mediates cue-induced nicotine seeking.
In the present study, we examined the role of GluN2B-containing NMDA receptors in regulating cue-induced nicotine reinstatement and associated MSN morphology and physiology. Specifically, we examined (1) if intra-NAcore administration of ifenprodil inhibits cue-induced nicotine reinstatement and associated structural plasticity, measured via MSN dendritic spine head diameter and (2) the role of GluN2B receptors within the NAcore in nicotine seeking and MSN physiology (as measured by A/N ratios) using small interfering RNAs (siRNAs) to knockdown GluN2B expression. Lastly, in an effort to explore additional therapeutic mechanisms of action of ifenprodil, we examined whether citalopram, an SSRI, was effective in decreasing cue-induced reinstatement. We hypothesized that intra-NAcore ifenprodil treatment would decrease cue-induced nicotine reinstatement and alter MSN spine morphology through reducing dendritic spine head diameter. Additionally, we hypothesized that due to the upregulation of GluN2B receptors during nicotine withdrawal as well as the established role of GluN2B receptors in mediating drug seeking, downregulation of these receptors using GluN2B-specific siRNAs would inhibit cue-induced nicotine reinstatement and prevent cue-evoked changes in MSN A/N ratios. Additionally, we hypothesized that due to its action on the serotonin system, citalopram would also inhibit cue-induced nicotine reinstatement, which may lend insight into additional therapeutic actions of ifenprodil.
Materials and Methods
Subjects
Two to three month old male Sprague Dawley rats (n=78; 250–300 g; Charles River, Riverside, CA) were housed on a 12-hour reverse-light cycle and had ad libitum access to food and water prior to surgical and self-administration procedures. All animals were kept in a temperature- and humidity-controlled animal facility. All animals were handled daily, and all animal use practices were approved by the Institutional Animal Care and Use Committee (IACUC) of Arizona State University.
Surgical Procedures
Rats were anesthetized with ketamine HCL (80–100 mg/kg, i.m.) and xylazine (8 mg/kg, i.m.) and underwent surgical implantation of intravenous jugular catheters. Silastic catheters (Dow Corning, Midland, MI, USA) were inserted 2.5–3 cm into the right jugular vein and tunneled subcutaneously to exit the skin on the dorsum between the scapulae. The distal end of the catheter was attached to a stainless steel cannula (Plastics1, Roanoke, VA, USA) with a harness assembly (Instech Laboratories, Plymouth Meeting, PA, USA). Following jugular vein insertion, rats were transferred to a stereotaxic apparatus where guide cannulae were bilaterally inserted into the NAcore (+1.5 mm anterior/posterior (A/P), +2.0 mm medial/lateral (M/L), and −5.5 mm dorsal/ventral (D/V), according to the stereotaxic atlas (Paxinos et al. 2013)). Rats were given cefazolin (100 mg/kg, i.v.), heparin (10 usp, i.v.), and meloxicam (1 mg/kg, s.c.) on the day of surgery and daily during a 7-day post-surgical recovery period. Heparin was administered into the catheter daily throughout the duration of post-operative recovery as well as throughout self-administration of nicotine to maintain patency.
Operant chambers
All behavioral testing (food training, nicotine self-administration, extinction and reinstatement) was conducted in 28 modular test chambers (13 ENV-008, 15 ENV-007; Med Associates, St. Albans, VT). Sound-attenuating cubicles, each equipped with ventilation fans, housed all chambers. Each chamber was composed of clear Plexiglas, which constituted the front and back walls as well as the ceiling, and two aluminum sidewalls. Thin metal bars positioned above a catch pan, filled with bedding, made up the floor of each chamber. Each chamber was equipped with an intelligence panel that included two levers (active and inactive), a food hopper, two stimulus lights as well as a house light. The levers were located 4.3 cm from the chamber base, and were separated by a flanked pellet receptacle, used for food training. Each chamber contained two jewel lights, one above each lever, as well as a house light, which was located on the opposite wall. The house light was only used for food-training purposes. The right lever was designated as the “active” lever, which yielded one nicotine intravenous infusion and initiation of the nicotine-paired cues (i.e. tone + light) when pressed. The left “inactive” lever did not yield a nicotine intravenous infusion or initiation of nicotine-paired cues when pressed, and resulted in no programmed consequences. The drug delivery tether was connected to a nicotine-containing syringe located in a single-speed automated drug infusion pump (PHM-100), was threaded through a 3-cm diameter circular hole at the top of each chamber and attached to a single-channel liquid swivel mounted to the top of the chamber enclosure. These chambers have also been described in further detail in our previous study (Overby et al. 2018).
Food Training Procedures
Following 6 days of post-operative care, rats were food restricted (20 g of chow/day) a minimum of 2 hours before food training. Rats underwent overnight food training (15 hrs) in which one lever press (fixed-ratio-1, or FR-1, schedule of reinforcement) resulted in the delivery of one food pellet (Bio Serv, 45 mg/pellet). Pellet delivery was not paired with any discrete cues other than the sound of the pellet dispenser. Rats were required to achieve a 2:1 ratio of active to inactive lever presses throughout food training and were required to accumulate over 200 active lever presses throughout the session. If criteria were not met, animals underwent additional training two days following the first training session. Additional food training was necessary for 8 out of 78 animals (10%). Following food training, animals were food restricted to 20g of chow/day for the remainder of the experiment.
Drugs
(−)Nicotine tartrate (MP Biomedicals, Solon, OH) was dissolved in 0.9% sterile saline and adjusted to pH 7.2–7.4 with 1 M NaOH. The final concentration was 0.2 mg/mL free base. Ifenprodil hemitartrate (Tocris Bioscience, cat. #: 0545/10) and citalopram (Sigma-Aldrich, cat. # C7861) were dissolved in sterile artificial cerebrospinal fluid (aCSF; Tocris, cat. #3525) to a concentration of 10 pmol per 2μl and 2μg/mL, respectively. Concentrations were based on previous literature using intra-cranial microinjections (Sarro et al. 1993; Inoue et al. 2004). GluN2B and scrambled negative control siRNAs were purchased from Ambion, and reconstituted to a stock solution of 50 μM in nuclease free H2O. Stock solution was diluted to 2.2μM in nuclease free H2O, which was further diluted to 200 nM using X-tremeGENE siRNA transfection reagent (Sigma-Aldrich, cat. #: 04 476 093 001) before microinjections according to the manufacturer’s directions. GluN2B (cat. #: ab65783; Lot #: GR242023–1) and Goat Anti-rabbit (HRP) (cat #: ab97080; Lot #: GR258796–3) antibodies were purchased from Abcam and the GAPDH antibody was purchased from Cell-Signaling Technology (cat. # D16H11; Lot #: 6).
Self-administration, Extinction, and Reinstatement Procedures
One week after jugular vein catheterization surgery, rats were trained to self-administer intravenous nicotine (0.02 mg/kg/infusion; see Figure 1) on a fixed ratio-1 (FR-1) schedule of reinforcement Nicotine was delivered during a 5.9-s infusion after a lever press on a designated active lever. Concurrent with each infusion, cue lights located above the levers were illuminated and a tone (2900 Hz) was presented, followed by a 20-s timeout period during which additional lever presses were recorded by had no programmed consequences (Goenaga et al. 2019; Powell et al. 2019; Gipson et al. 2013). An inactive lever was present at all times and presses on this lever were recorded but produced no programmed consequences at any time during the experiment. All sessions were 2 hr in duration, and during the first session the maximum number of infusions that could be obtained was capped at 25 infusions to prevent aversion to nicotine. All animals were required to complete at least 10 sessions where >10 infusions per session were obtained and at least a 2:1 active/inactive lever press ratio was observed. Rats that achieved acquisition criteria were then moved into extinction training. Only animals meeting self-administration criteria were included in behavioral data analysis. Extinction training consisted of daily 2-hour sessions with active and inactive levers present, but neither lever produced any programmed consequences. All animals received 14 days of extinction training, where a criterion of less than 25 active lever presses over the last 2 days of extinction was required prior to reinstatement sessions.
Figure 1. Ifenprodil inhibits cue-induced nicotine reinstatement following nicotine self-administration and extinction training.
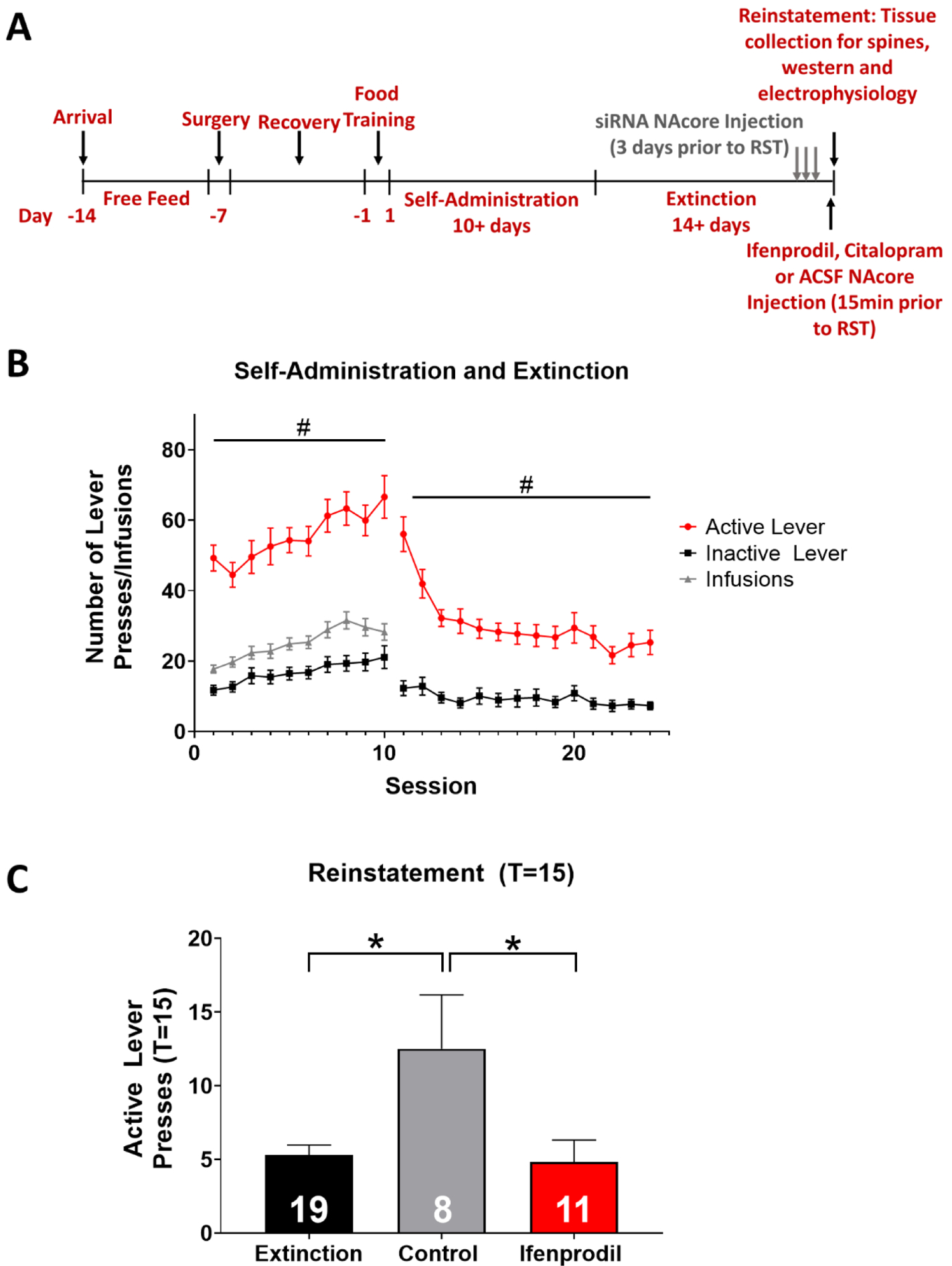
(A) Timeline of experimental procedures. (B) Rats acquired self-administration, distinguishing between the active (red) and inactive (gray) lever to receive intravenous nicotine infusions (black). Active lever pressing significantly increased throughout self-administration training (#p<0.05, significant main effect of session on active lever presses). During extinction training, active lever pressing decreased, and the slope of active versus inactive lever presses significantly differed, where active lever pressing was reduced across session (#p<0.05). (C) Ifenprodil prevented cue-induced nicotine reinstatement, as measured by number of active lever pressing during the reinstatement session compared to the last 2 days of extinction training (*p<0.05, significant main effect of treatment). Numbers within bars indicate the number of animals per group.
Intra-NAcore Microinjections
Following extinction, animals underwent a 15-minute cue-induced reinstatement session (T(time)=15) where active lever pressing resulted in re-exposure to previously paired nicotine cues (light + tone), but no nicotine was infused. NAcore microinjections were conducted in all experiments, however, the time at which animals were microinjected varied. Ifenprodil, citalopram and aCSF control animals were microinjected directly into the NAcore (2 uL/hemisphere; ifenprodil: 10 pmol (Sarro et al. 1993); citalopram: 2μg/mL) 15 minutes prior to reinstatement. siRNAs specific for GluN2B-containing NMDA receptors or control sequences were infused once daily for 3 consecutive days prior to reinstatement at a rate of 2 uL/hemisphere [200 nM (Shen et al. 2011)] 15 min prior to extinction sessions. A scrambled negative control siRNA, which has no significant sequence similarity to mouse, rat or human GluN2B gene sequences, was used as a control. For siRNA-treated animals, reinstatement was initiated 24 hrs following the last siRNA microinjection. Immediately following the reinstatement session, rats were either anesthetized with a ketamine/xylazine mixture, CO2, or rapidly decapitated for dendritic spine morphology assessment, electrophysiology or western blotting, respectively.
Dendritic Spine Morphology
For dendritic spine morphological measurements, rats were anesthetized using a ketamine/xylazine overdose and brains were harvested following transcardial perfusion. 200 μm thick NAcore containing slices were made using vibratome (Leica) and a gene gun was used to impregnate fixed sections of tissue with DiI (1,1-dioctadecyl-3,3,3’,3’,-tetramethyl-indocarbocyanine perchlorate) using previously published methods (Foster Olive, Del Franco, and Gipson 2018). NAcore MSNs were imaged using a confocal microscope (Leica SP5). DiI was excited using a helium/neon laser line (543 nm) and spines were observed using a 63x oil immersion objective (HCX PL APO CS 63x/1.4–0.6NA). Image resolution was set to 512 × 512 pixels with a pixel scale of 0.15 × 0.15 μm, and 0.21 μm scanning intervals were used to image each dendrite (see complete methods in Powell et al. (2019)). Only dendrites >50 μm from the soma qualified for further examination, and each dendritic length was between 45 and 55 μm. Additionally, only cells with processes that did not overlap with other cells were chosen for analysis. Images were deconvolved by Autoquant (Media Cybernetics) and were then processed into a 3-D reconstruction using Bitplane Imaris software Spines were then traced using the Filament module of the Imaris software package. The minimum and maximum spine terminal point spine head diameter (dh) parameters were set at 0.143 μm and 0.900 μm, respectively. For each spine, dh and spine neck length were quantified using the Filament module of Imaris software. Spines that were not within the minimum and maximum parameters as well as areas that had saturated concentrations of DiI were omitted from analysis.
Western Blotting
Following reinstatement, animals were rapidly decapitated and NAcore tissue was crude dissected over ice. To examine membrane-enriched subfractions (i.e. cell surface receptors), tissue was homogenized in 200μL of sucrose buffer (containing 0.48g HEPES and 21.91g sucrose per 200mL water), supplemented with 1:100 protease and phosphatase inhibitors directly prior to homogenization. The sample was then centrifuged at 1,000 × g for 10 min at 4°C. The supernatant was collected in an additional tube and the pellet was then re-suspended in an additional 200μL of sucrose buffer. The pellet/sucrose mixture was centrifuged at 1,000 × g for 10 min at 4°C, and the supernatant was collected and combined with the first supernatant. The supernatant was then centrifuged at 12,000 × g for 20 min. The resulting supernatant was collected and discarded. The resulting pellet was re-suspended in 100uL of ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer containing 1:100 protease and phosphatase inhibitors. Homogenates were centrifuged at 10,000 × g for 5 min at 4°C and the supernatant was collected and stored at −80°C. Using a BCA kit, protein concentrations of each sample were assessed. Equal protein quantities were loaded onto a 4–12% Bis-Tris gel (Invitrogen) and dry-transferred to a nitrocellulose membrane (iBlot, Invitrogen). Membranes were blocked for 2 hours in either tris-buffered saline plus 0.1% Tween-20 (TBST) containing 5% nonfat milk or bovine serum albumin, made in TBST for GAPDH and GluN2B, respectively. Following blocking, membranes were washed 3x for 5 min each with TBST, and incubated overnight at 4°C in primary antibody (GAPDH: 1:1,000; GluN2B 1:1,000). Membranes were washed 6 × 5 min in blocking buffer and then incubated in secondary antibody for 2 hr at room temperature. Secondary antibodies were used at a dilution of 1:10,000 and 1:5,000 for GluN2B and GAPDH, respectively. Membranes were then washed an additional 6 X for 5 min in TBST. An enhanced chemiluminescence substrate (Thermo Scientific) was used to activate membranes and protein expression was detected using x-ray film. Band density was quantified using NIH ImageJ software and protein expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). For antibody concentrations and source information, see Table 1.
Electrophysiology
Following reinstatement, animals were anesthetized with CO2 and rapidly decapitated. Brains were rapidly removed and submerged in ice-cold carbogen (95% O2 / 5% CO2) saturated cutting solution (cutting aCSF) containing (in mmol/L): NaCl, 120; NaHCO3, 25; Dextrose, 10; KCl, 3.3; NaH2PO4, 1.23; CaCl2, 1.8; MgCl2, 2.4. Solution osmolarity was adjusted to 295 ± 5 mOsm and pH adjusted to 7.40 ± 0.03. Brains were then transferred to a cutting chamber of a vibrating tissue slicer (Leica, VT1000S) and 300μm thick coronal slices of the NAcore were prepared in ice-cold cutting aCSF. Slices were then placed in a holding chamber filled with recording aCSF solution containing (in mmol/L): NaCl, 120; NaHCO3, 25; KCl, 3.3; NaH2PO4, 1.23; CaCl2, 0.9; MgCl2, 2.0; dextrose, 10, osmolarity adjusted to 295 ± 5 mOsm and pH adjusted to 7.40 ± 0.03. The holding chamber aCSF was continuously bubbled with carbogen (95% O2 / 5% CO2) and incubated at 34°C for 45 minutes and then allowed to cool to room temperature before slice recording. Prior to experiments, slices were transferred to a recording chamber where they were perfused continuously at a flow rate of 1–2 mls/min with filtered, carbogen-saturated recording aCSF solution. MSNs were visually identified using infrared DIC microscopy with an Olympus BX51WI microscope. Whole-cell recordings were made from the soma of MSNs neurons after establishing a giga-ohm seal (Resistance range: 1–10 GΩ). Recording pipettes (7–15 mΩ), made from thin-walled capillary tubes were filled with an intracellular solution containing (in mmol/L): K-gluconate, 135; NaCl, 12; K-EGTA, 1; HEPES, 10; Mg-ATP, 2 and tris-GTP, 0.38. Osmolarity was adjusted to 285±5 mOsm and pH adjusted to 7.30 ± 0.01. Upon establishing a giga-seal, the cell membrane was ruptured and held at −80mV. Resting membrane potential, cellular capacitance, membrane resistance and pipet resistance were monitored throughout the duration of the recording. Only cells that exhibited thin, over shooting action potentials, normal resting membrane potential, and changes in uncompensated access resistance less than 20 mΩ were included in analysis. Recordings were initiated 10 minutes after cell membrane rupture to allow for diffusion of the internal solution into the cell. A stimulating electrode was placed in the dorsal region of the NAcore to activate prelimbic excitatory fibers targeting the NAcore. AMPA currents, evoked by electrical stimulation, were first measured at −80 mV. The membrane potential was then gradually increased to +40 mV. The cell was left to stabilize at +40 mV for 5 minutes. Excitatory post-synaptic currents (EPSCs) composed of both AMPA and NMDA receptor mediated currents were then elicited at +40mV. DNQX (20 μM) was then bath applied for 5 minutes and NMDA receptor mediated currents were obtained. AMPA currents were then obtained by subtracting the isolated NMDA receptor-mediated current from the whole EPSC. A/N ratios were calculated by measuring the peak amplitude of each current and taking a ratio. NMDA decay time was also calculated from the NMDA receptor-mediated current by obtaining the time for the cell to reach 37% of its initial amplitude. All recordings were conducted using the recording software Axograph. Responses were digitized at 10 kHz and saved on a disk using digidata interface (Axon Instruments) and analyzed offline using Axograph.
Data Analysis
Behavioral and spine data were analyzed using analysis of variance (ANOVAs) with post-hoc Bonferroni-corrected t-tests where appropriate. Behavioral analysis during self-administration, extinction, and reinstatement was performed using a two-way mixed measures ANOVA where treatment was considered a main factor and session (extinction vs. reinstatement where applicable) was a repeated-measure factor. Linear regression analyses were used to examine changes in lever pressing across sessions. All behavioral analyses were conducted as described previously (Powell et al. 2019). Electrophysiological data was analyzed using unpaired t-tests to compare overall differences between the two treatment groups. Statistical analyses were performed in GraphPad Prism 7.0, and p<0.05 was considered statistically significant. Values presented are represented as mean ± standard error of the mean (SEM). Specific analyses used are presented within each figure legend.
Results
Nicotine self-administration and extinction
Only rats that readily distinguished between active and inactive levers were used for analysis in the current study. Thirteen out of ninety-one (14%) animals were dropped from the study for not reaching self-administration criteria. A linear regression analysis revealed a significant difference in the slope of active versus inactive lever pressing, indicating active lever pressing increased across sessions whereas inactive lever pressing was unchanged (F1,1262 = 4.7, p<0.05; Figure 1B). For extinction training, a two-way ANOVA revealed that active lever pressing remained significantly higher than inactive lever pressing (F13,1733 = 3.4, p<0.05); However, active lever pressing significantly decreased across sessions (F13,1733 = 6.7, p<0.05; Figure 1B), and a linear regression analysis revealed a significant difference in the slope of active and inactive lever pressing across session (F1,1757 = 23.5, p<0.05; Figure 1B). Extinction criteria were the same as those used in prior studies (Powell et al. 2019; Goenaga et al. 2019), where active lever pressing was below 30 presses and was significantly reduced compared to session one of extinction training.
Intra-NAcore ifenprodil treatment decreases nicotine cue-induced reinstatement
There were no significant differences in total nicotine infusions between treatment groups randomized to receive either control (aCSF) or ifenprodil treatment (p>0.05). An ANOVA with Bonferroni corrections revealed a significant main effect of treatment on reinstatement of nicotine seeking (F2,34 = 5.2, p<0.05; Figure 1C), where aCSF-treated rats had significantly more active lever presses during the reinstatement session (at T=15) than observed during the first 15 min of extinction. Cue-induced reinstatement was not observed in animals treated with intra-NAcore ifenprodil (p>0.05). Further, control rats had significantly higher active lever pressing during reinstatement compared to ifenprodil treated animals. These results indicate that ifenprodil treatment inhibited cue-induced nicotine seeking relative to control treated rats when administered directly into the NAcore.
Intra-NAcore ifenprodil promotes a higher frequency of shorter dendritic spines in MSNs
Spine morphological measures were analyzed at T=15 following intra-core aCSF and ifenprodil treatment. A representative DiI labeled MSN within the NAcore and representative micrographs of dendritic spines are shown in Figure 2A. Comparison of mean morphological parameters revealed no significant differences in spine density (t12 = 0.3, p>0.05), spine head diameter (t12 = 0.5, p>0.05), spine neck length (t12 = 0.1, p>0.05), or the ratio for spine head diameter/neck length (t12 = 0.7, p>0.05). These results are shown in figure 2C, D, G, and H, respectively. A two-way ANOVA revealed that ifenprodil increased the frequency of shorter spines, relative to control (main effect of treatment: F10,132 = 2.8, p<0.01; Figure 2E), yet there was no effect on the cumulative frequency of spine head diameter (F10,132 = 0.8, p>0.05; Figure 2F).
Figure 2. Intra-NAcore ifenprodil treatment increases the frequency of shorter spine necks present on MSNs within the NAcore.
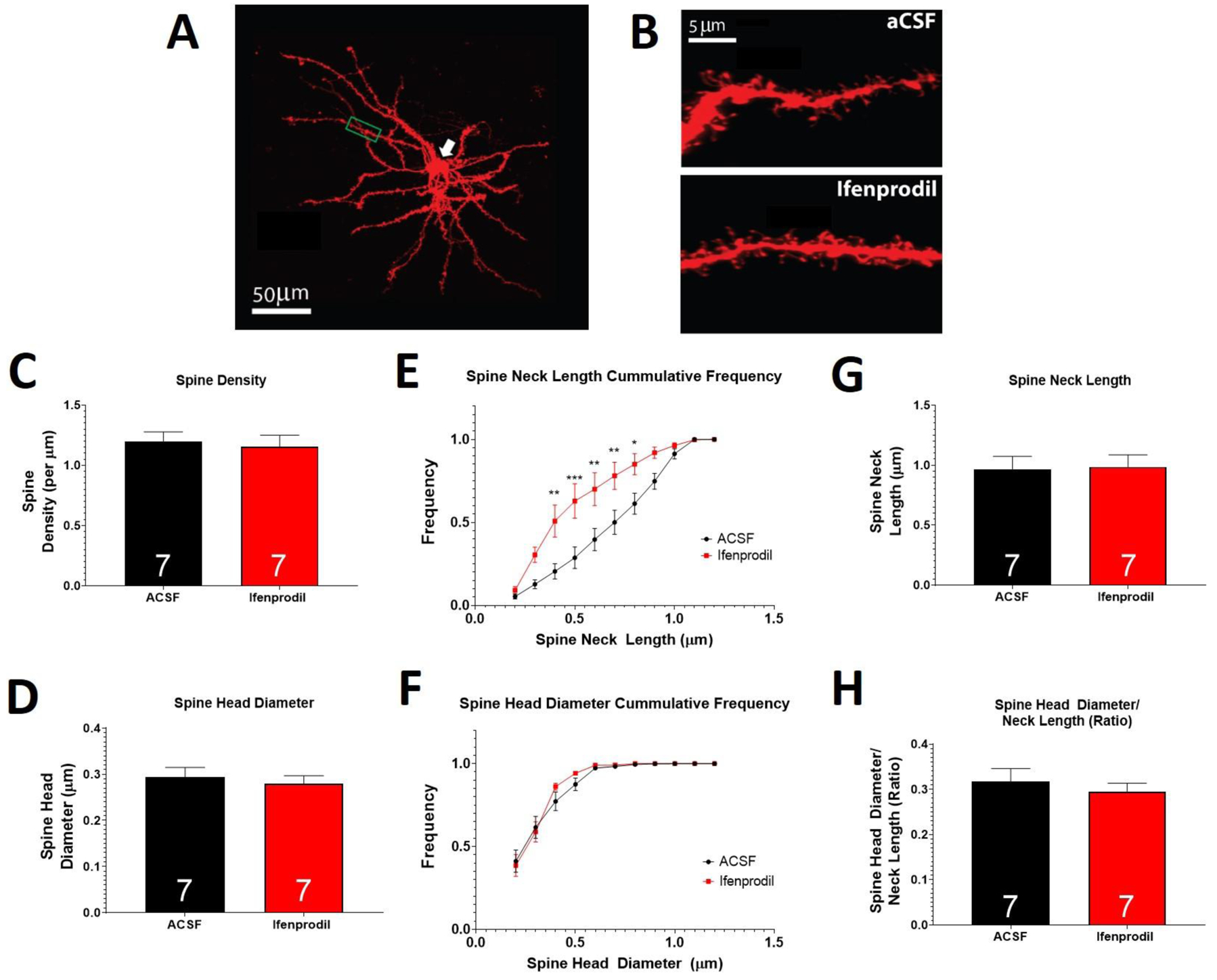
(A) Representative DiI labeled MSN and (B) representative dendritic segments used for spine analysis. Ifenprodil did not alter spine density (C), spine head diameter (D), spine neck length (G) or the ratio of spine head diameter/spine neck length (H). Ifenprodil induced a greater number of spines with shorter necks, relative to control (E), yet had no effect on spine head diameter cumulative frequency (F). A main effect of treatment is indicated by * p<0.05; ** p<0.01; *** p<0.001. Numbers within bars indicate the number of animals per group.
To further explore the role of GluN2B containing NMDA receptors in cue-induced nicotine seeking, siRNAs were used to reduce GluN2B receptor expression (online resources 1 and 2). While ifenprodil was successful in reducing nicotine cue-induced reinstatement, reducing the expression of GluN2B containing NMDA receptors was insufficient for preventing cue-induced reinstatement (see online resource 2). Furthermore, knockdown of GluN2B-containing NMDA receptors did not alter MSN synaptic physiology, including resting membrane potential, cellular capacitance, input/output responses, AMPA/NMDA ratio or NMDA decay time (please see online resources 1 and 2 for statistical analyses and figures).
SERT blockade with citalopram inhibits nicotine-cue-induced reinstatement.
There were no significant differences in total nicotine infusions between treatment groups randomized to receive either control (aCSF) or citalopram treatment (p>0.05). An ANOVA with Bonferroni corrections revealed a significant main effect of treatment on reinstatement of nicotine seeking (F2,58 = 5.7, p<0.05; Figure 3), where aCSF-treated rats had significantly more active lever presses during the reinstatement session (at T=15) than observed during the first 15 min of extinction. Cue-induced reinstatement was not observed in animals treated with intra-NAcore citalopram (p>0.05). Further, bonferroni multiple comparison analyses revealed that control rats had significantly higher active lever pressing during reinstatement compared to citalopram-treated animals (t29 = 3.11; p>0.05; Figure 3). No differences were found in inactive lever pressing. These results indicate that citalopram treatment inhibits cue-induced nicotine seeking relative to control treated rats when administered directly into the NAcore.
Figure 3. Intra-NAcore citalopram prevents cue-induced nicotine reinstatement.
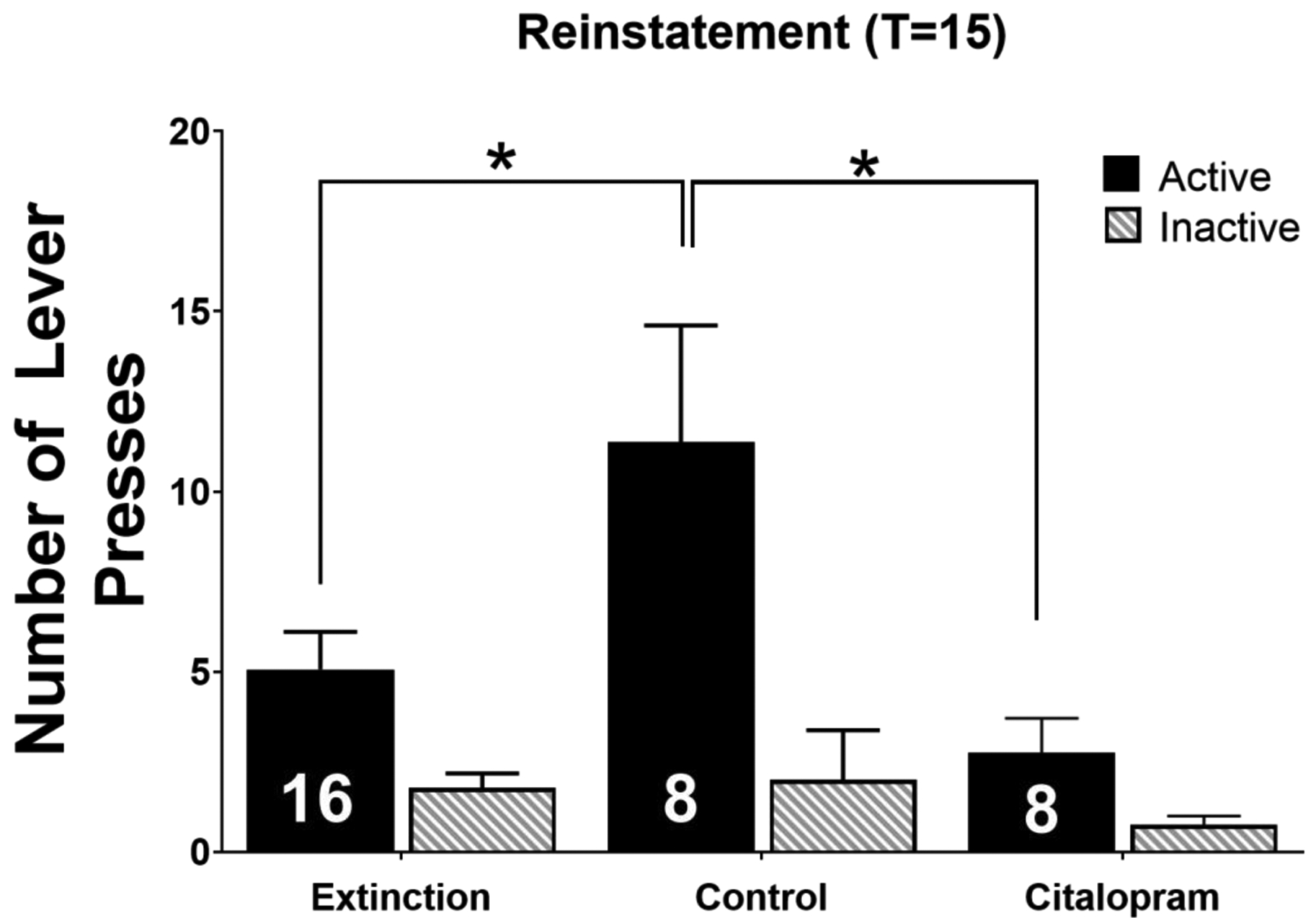
In control treated animals, active lever pressing was significantly increased during cue-induced reinstatement compared to extinction. Intra-NAcore citalopram inhibited active lever pressing during cue-induced reinstatement and did not significantly differ from extinction. No differences in inactive lever pressing were observed. * depicts p<0.05. Inset numbers represent the number of animals.
Discussion
The present set of studies explored the role of GluN2B-containing NMDA receptors and SERT in nicotine cue-induced reinstatement and associated synaptic plasticity. Intra-NAcore ifenprodil administration inhibited cue-induced nicotine seeking and increased the frequency of occurrence of shorter spines on MSN dendrites. Dendritic spine head diameter, however, was not altered following NAcore ifenprodil, contrary to our hypothesis. Further, GluN2B-specific siRNA treatment significantly reduced membrane-bound GluN2B-containing NMDA receptors within the NAcore but did not decrease cue-induced nicotine seeking and GluN2B-specific siRNA treatment did not alter MSN neuronal physiology, including baseline neuronal characteristics, A/N ratio, or NMDA current decay time (see Online Resource 1). However, citalopram prevented cue-induced nicotine seeking when administered directly into the NAcore. Together, these results show that while ifenprodil successfully prevented cue-induced nicotine seeking following self-administration and extinction training, partial knockdown of GluN2B-containing NMDA receptor expression was insufficient to reduce cue-induced nicotine seeking or underlying MSN neuronal physiology. Lastly, blockade of SERT using citalopram was found to prevent cue-induced nicotine seeking, suggesting that ifenprodil may exhibit its therapeutic efficacy, at least in part, by acting on the serotonin system.
GluN2B-containing NMDA receptors have been associated with drug seeking behavior (Gipson et al. 2013; Shen et al. 2011; Ma et al. 2007; Schumann et al. 2009), where increased expression is thought to promote drug seeking. Cue-induced nicotine seeking is associated with dysregulations in glutamate homeostasis within the corticostriatal circuitry, and some characteristics of these changes overlap with heroin while others overlap with cocaine seeking behavior. Specifically, re-exposure to nicotine-associated cues induces glutamatergic overflow within the NAcore, and GluN2B-containing NMDA (similar to heroin, see (Shen et al. 2011)) and GluA1-containing AMPA (similar to cocaine (Conrad et al. 2008)) receptors are upregulated in this brain region. Interestingly, GluN2B-containing NMDA receptors that are expressed extrasynaptically likely require glutamate overflow to become activated (Misra et al. 2000; Papouin et al. 2012; Shen et al. 2011). Thus, it is hypothesized that glutamatergic overflow from prefrontal glutamatergic afferents promotes activation of GluN2B-containing NMDA receptors located extrasynaptically, and this enhances drug seeking motivation. Interestingly, both GluA1-containing AMPA and GluN2B-containing NMDA receptors are prominent early in development (van Zundert et al. 2004), most undergo a developmental switch and are subsequently replaced by GluA2 and GluN2A subunits, respectively, which reflects the maturation of synaptic contacts. Following exposure to drugs of abuse, it is thought that a subunit switch back to developmental conditions occurs as a consequence of activity-dependent synaptic strengthening (Dong and Nestler 2014; Halt et al. 2012). Taken together, these findings indicate GluN2B-containing NMDA receptors may play a role in restructuring neural circuitry during chronic drug use, and thus NMDA receptor antagonism may be a pharmacotherapeutic avenue for individuals with substance use disorders (Ma et al. 2007; Gipson et al. 2013; Tomek et al. 2013; Bisaga et al. 2000). Further, NMDA receptor antagonists have been clinically utilized to treat various disorders including depression (Sanacora et al. 2017) and chronic pain (Bell and Kalso 2018; Bell et al. 2006; Li and Chen 2019). It should be noted, however, that clinical utility of NMDA receptor antagonists for SUDs may be limited due to poor receptor specificity, negative side effects and abuse potential (Herman et al. 1995).
Akin to other studies, here we have shown efficacy of ifenprodil to prevent cue-induced drug seeking. Previous studies have shown that systemically administered ifenprodil (via intraperitoneal injections) reduces cue-induced nicotine seeking (Gipson et al. 2013) and morphine-primed reinstatement (Ma et al. 2007) in a dose-dependent manner. As well, systemic ifenprodil administration has also been shown to prevent cue-induced and heroin-primed reinstatement (Shen et al. 2011). Like results reported here, intra-NAshell and intra-NAcore microinjections of ifenprodil have been shown to prevent morphine-primed (Ma et al. 2007) and heroin-primed (Shen et al. 2011) reinstatement, respectively. The current data lend additional support to the therapeutic efficacy of ifenprodil in treating tobacco use disorder.
To our knowledge, the current study is one of the first to show reduced cue-induced nicotine reinstatement following intra-NAcore ifenprodil treatment. Additionally, we are not aware of other studies reporting that intra-NAcore ifenprodil administration induces a greater frequency of shorter spines, which may affect cell excitability. In line with our results, GluN2B knockout mice show a decrease in filamentous actin, rendering a decrease in spine density (Akashi et al. 2009) and cell excitability. Although changes in spine neck length were not evaluated in this study, the loss of filamentous actin would render spines incapable of forming long protrusions (Hotulainen and Hoogenraad 2010). As well, ifenprodil has been shown to reduce Ras activation, a signaling molecule associated with LTP induction and associated spine growth (Patterson and Yasuda 2011). Taken together, ifenprodil may promote dendritic spine destabilization by reducing filamentous actin which would prevent actin recruitment and assembly as well as Ras activation. In combination, this may lead to the increase in the frequency of shorter spines as observed in the current study. In this way, ifenprodil may have inhibited spine functionality relevant to nicotine seeking, blocking cue-induced nicotine reinstatement.
Contrary to other studies showing that decreased GluN2B expression was sufficient to reduce cue- and heroin-primed reinstatement, the current results show that a reduction in membrane-bound GluN2B expression was insufficient to inhibit cue-induced nicotine reinstatement. Additionally, downregulation of GluN2B receptor expression did not result in changes in MSN physiology, including various cellular properties (i.e. cellular capacitance, resting membrane potential or input/output curves), A/N ratio, or NMDA current decay time. While the reduction observed in the current study is similar to that reported by Shen and colleagues, it is possible that this slight reduction in membrane bound GluN2B protein expression is sufficient for reducing heroin seeking, but is not sufficient in reducing cue-induced nicotine seeking. Given that the primary mechanisms of action of heroin and nicotine are different (e.g., agonism of μ opioid receptors (Sim-Selley et al. 2000) versus nicotinic acetylcholine receptors (nAChRs) (Dajas-Bailador and Wonnacott 2004)), it is possible that neural mechanisms driving cue-induced heroin versus nicotine seeking differ, and thus a greater reduction in accumbens GluN2B receptor expression is needed for robust decreases in nicotine seeking. Taken together, the negative results from the current siRNA study are informative for the field, as they demonstrate a lack of utility of this technique in preclinical models of nicotine seeking.
As mentioned previously, in addition to its actions at GluN2B-containing NMDA receptors, ifenprodil also has non-specific binding at serotonin receptors (McCool and Lovinger 1995; Maksay et al. 2005), G protein-activated inwardly rectifying potassium (GIRK) channels (Kobayashi et al. 2006) and is known to interact with alpha-1 adrenergic receptors (Chenard et al. 1991). In the current study, we found that SERT blockade, using citalopram, prevented cue-induced nicotine reinstatement. While it has been shown that ifenprodil acts on the serotonin system and that analogues of ifenprodil inhibit the SERT (Manepalli et al. 2012; Talbot et al. 2016), it is likely that ifenprodil can elicit therapeutic effects through mechanisms other than GluN2B receptor antagonism. In line with this hypothesis, ifenprodil reduces depressive-like behaviors through activation of mTOR (Yao et al. 2020) and has been shown to potentiate the antidepressant-like effects of SSRIs such as imipramine and fluoxetine (Poleszak et al. 2014), which are both known to act on SERT. Given that antidepressants have also been linked to smoking cessation (Hughes et al. 2014; Hitsman et al. 2001), it is possible that ifenprodil induces its therapeutic efficacy for nicotine seeking via SERT.
In conclusion, we report that local administration of the GluN2B-containing receptor antagonist and ifenprodil directly into the NAcore prevents cue-induced nicotine seeking behavior and promotes a higher frequency of shorter dendritic spines on MSNs in this region. However, modest reductions in GluN2B expression are not sufficient to prevent cue-induced nicotine reinstatement or associated MSN plasticity, suggesting that ifenprodil does not act through GluN2B receptor for therapeutic efficacy in reducing nicotine seeking behavior. Importantly, we found that SERT blockade via citalopram was sufficient to prevent cue-induced nicotine seeking. Taken together, ifenprodil may prevent cue-induced nicotine seeking through actions on the serotonin system. While we do not rule out the possibility that potent antagonism of GluN2B receptors is necessary to prevent cue-induced nicotine reinstatement, the data presented here suggest that ifenprodil may in part act on SERT to reduce nicotine seeking. It is also notable that NMDA receptor antagonism may not be a viable therapeutic avenue to treat nicotine use disorder, thus a more thorough understanding of the mechanisms of action of ifenprodil may yield more translationally beneficial outcomes. Thus, further exploration of ifenprodil’s action on the serotonin system are warranted to understand its therapeutic efficacy.
Supplementary Material
Acknowledgements:
The authors thank Hanaa Ulangkaya, Vincent Carfagno, Amanda Bull, Mark Namba, and Ngoc Van Do for their technical assistance. We also thank the laboratories of Dr. M. Foster Olive and Dr. Heather A. Bimonte-Nelson for providing equipment necessary for western blot experiments.
Funding: This work was supported by the Arizona Alzheimer’s Consortium, DA036569, DA044479, DA046526, and DA045881 (to CDG), and AA027962 (to JMLJ).
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest
Ethical Approval: All procedures performed in the studies involving animals were in accordance with the ethical standards of the Arizona State University Institutional Animal Care and Use Committee. Protocol number: 18–1642R RFC 18
References:
- Akashi K, Kakizaki T, Kamiya H, Fukaya M, Yamasaki M, Abe M, Natsume R, Watanabe M, and Sakimura K. 2009. “NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses.” J Neurosci 29 (35):10869–82. doi: 10.1523/JNEUROSCI.5531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RF, Dahl JB, Moore RA, and Kalso E. 2006. “Perioperative ketamine for acute postoperative pain.” Cochrane Database Syst Rev (1):CD004603. doi: 10.1002/14651858.CD004603.pub2. [DOI] [PubMed] [Google Scholar]
- Bell RF, and Kalso EA. 2018. “Ketamine for pain management.” Pain Rep 3 (5):e674. doi: 10.1097/PR9.0000000000000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezchlibnyk-Butler K, Aleksic I, and Kennedy SH. 2000. “Citalopram--a review of pharmacological and clinical effects.” J Psychiatry Neurosci 25 (3):241–54. [PMC free article] [PubMed] [Google Scholar]
- Bisaga A, Popik P, Bespalov AY, and Danysz W. 2000. “Therapeutic potential of NMDA receptor antagonists in the treatment of alcohol and substance use disorders.” Expert Opin Investig Drugs 9 (10):2233–48. doi: 10.1517/13543784.9.10.2233. [DOI] [PubMed] [Google Scholar]
- Chen JP, van Praag HM, and Gardner EL. 1991. “Activation of 5-HT3 receptor by 1-phenylbiguanide increases dopamine release in the rat nucleus accumbens.” Brain Res 543 (2):354–7. [DOI] [PubMed] [Google Scholar]
- Chenard BL, Shalaby IA, Koe BK, Ronau RT, Butler TW, Prochniak MA, Schmidt AW, and Fox CB. 1991. “Separation of alpha 1 adrenergic and N-methyl-D-aspartate antagonist activity in a series of ifenprodil compounds.” J Med Chem 34 (10):3085–90. doi: 10.1021/jm00114a018. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, and Wolf ME. 2008. “Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving.” Nature 454 (7200):118–21. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador F, and Wonnacott S. 2004. “Nicotinic acetylcholine receptors and the regulation of neuronal signalling.” Trends Pharmacol Sci 25 (6):317–24. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dong Yan, and Nestler Eric J.. 2014. “The neural rejuvenation hypothesis of cocaine addiction.” Trends in Pharmacological Sciences 35 (8):374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, and Nicoll RA. 2008. “Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development.” Proc Natl Acad Sci U S A 105 (52):20953–8. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster Olive M, Del Franco AP, and Gipson CD. 2018. “Diolistic Labeling and Analysis of Dendritic Spines.” Methods Mol Biol 1727:179–200. doi: 10.1007/978-1-4939-7571-6_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, and Kalivas PW. 2014. “Rapid, transient synaptic plasticity in addiction.” Neuropharmacology 76 Pt B:276–86. doi: 10.1016/j.neuropharm.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Reissner KJ, Kupchik YM, Smith AC, Stankeviciute N, Hensley-Simon ME, and Kalivas PW. 2013. “Reinstatement of nicotine seeking is mediated by glutamatergic plasticity.” Proc Natl Acad Sci U S A 110 (22):9124–9. doi: 10.1073/pnas.1220591110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, and Kalivas PW. 2013. “Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation.” Neuron. doi: 10.1016/j.neuron.2013.01.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenaga J, Powell GL, Leyrer-Jackson JM, Pina J, Phan S, Prakapenka AV, Koebele SV, Namba MD, McClure EA, Bimonte-Nelson HA, and Gipson CD. 2019. “N-acetylcysteine yields sex-specific efficacy for cue-induced reinstatement of nicotine seeking.” Addict Biol. doi: 10.1111/adb.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, and Hell JW. 2012. “CaMKII binding to GluN2B is critical during memory consolidation.” EMBO J 31 (5):1203–16. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman BH, Vocci F, and Bridge P. 1995. “The effects of NMDA receptor antagonists and nitric oxide synthase inhibitors on opioid tolerance and withdrawal. Medication development issues for opiate addiction.” Neuropsychopharmacology 13 (4):269–93. doi: 10.1016/0893-133X(95)00140-9. [DOI] [PubMed] [Google Scholar]
- Hitsman B, Spring B, Borrelli B, Niaura R, and Papandonatos GD. 2001. “Influence of antidepressant pharmacotherapy on behavioral treatment adherence and smoking cessation outcome in a combined treatment involving fluoxetine.” Exp Clin Psychopharmacol 9 (4):355–62. doi: 10.1037//1064-1297.9.4.355. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, and Hoogenraad CC. 2010. “Actin in dendritic spines: connecting dynamics to function.” J Cell Biol 189 (4):619–29. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schlüter OM, Zukin RS, and Dong Y. 2009. “In vivo cocaine experience generates silent synapses.” Neuron 63 (1):40–7. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR, Stead LF, Hartmann-Boyce J, Cahill K, and Lancaster T. 2014. “Antidepressants for smoking cessation.” Cochrane Database Syst Rev (1):CD000031. doi: 10.1002/14651858.CD000031.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, Markou A, and Kalivas PW. 2009. “The role of cystine-glutamate exchange in nicotine dependence in rats and humans.” Biol Psychiatry 65 (10):841–5. doi: 10.1016/j.biopsych.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Washiyama K, and Ikeda K. 2006. “Inhibition of G protein-activated inwardly rectifying K+ channels by ifenprodil.” Neuropsychopharmacology 31 (3):516–24. doi: 10.1038/sj.npp.1300844. [DOI] [PubMed] [Google Scholar]
- Li Z, and Chen Y. 2019. “Ketamine reduces pain and opioid consumption after total knee arthroplasty: A meta-analysis of randomized controlled studies.” Int J Surg 70:70–83. doi: 10.1016/j.ijsu.2019.08.026. [DOI] [PubMed] [Google Scholar]
- Ma YY, Chu NN, Guo CY, Han JS, and Cui CL. 2007. “NR2B-containing NMDA receptor is required for morphine-but not stress-induced reinstatement.” Exp Neurol 203 (2):309–19. doi: 10.1016/j.expneurol.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Maksay G, Bíró T, and Bugovics G. 2005. “Allosteric modulation of 5-HT3 serotonin receptors.” Eur J Pharmacol 514 (1):17–24. doi: 10.1016/j.ejphar.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Manepalli S, Surratt CK, Madura JD, and Nolan TL. 2012. “Monoamine transporter structure, function, dynamics, and drug discovery: a computational perspective.” AAPS J 14 (4):820–31. doi: 10.1208/s12248-012-9391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, and Lovinger DM. 1995. “Ifenprodil inhibition of the 5-hydroxytryptamine3 receptor.” Neuropharmacology 34 (6):621–9. [DOI] [PubMed] [Google Scholar]
- Miech R, Johnston L, O’Malley PM, Bachman JG, and Patrick ME. 2019. “Adolescent Vaping and Nicotine Use in 2017–2018 - U.S. National Estimates.” N Engl J Med 380 (2):192–193. doi: 10.1056/NEJMc1814130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra C, Brickley SG, Farrant M, and Cull-Candy SG. 2000. “Identification of subunits contributing to synaptic and extrasynaptic NMDA receptors in Golgi cells of the rat cerebellum.” J Physiol 524 Pt 1:147–62. doi: 10.1111/j.1469-7793.2000.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Chandler LJ, and Kalivas PW. 2016. “Signals from the Fourth Dimension Regulate Drug Relapse.” Trends Neurosci. doi: 10.1016/j.tins.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overby PF, Daniels CW, Del Franco A, Goenaga J, Powell GL, Gipson CD, and Sanabria F. 2018. “Effects of nicotine self-administration on incentive salience in male Sprague Dawley rats.” Psychopharmacology (Berl) 235 (4):1121–1130. doi: 10.1007/s00213-018-4829-4. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, and Oliet SH. 2012. “Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists.” Cell 150 (3):633–46. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Patterson M, and Yasuda R. 2011. “Signalling pathways underlying structural plasticity of dendritic spines.” Br J Pharmacol 163 (8):1626–38. doi: 10.1111/j.1476-5381.2011.01328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS 2012. “Distribution of extrasynaptic NMDA receptors on neurons.” ScientificWorldJournal 2012:267120. doi: 10.1100/2012/267120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleszak E, Wosko S, Serefko A, Wlaz A, Kasperek R, Dudka J, Wrobel A, Nowak G, and Wlaz P. 2014. “The effects of ifenprodil on the activity of antidepressant drugs in the forced swim test in mice.” Pharmacol Rep 66 (6):1031–6. doi: 10.1016/j.pharep.2014.06.016. [DOI] [PubMed] [Google Scholar]
- Powell GL, Leyrer-Jackson JM, Goenaga J, Namba MD, Piña J, Spencer S, Stankeviciute N, Schwartz D, Allen NP, Del Franco AP, McClure EA, Olive MF, and Gipson CD. 2019. “Chronic treatment with N-acetylcysteine decreases extinction responding and reduces cue-induced nicotine-seeking.” Physiol Rep 7 (1):e13958. doi: 10.14814/phy2.13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, and Nestler EJ. 2010. “The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens.” Trends Neurosci 33 (6):267–76. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Frye MA, McDonald W, Mathew SJ, Turner MS, Schatzberg AF, Summergrad P, Nemeroff CB, and American Psychiatric Association (APA) Council of Research Task Force on Novel Biomarkers and Treatments. 2017. “A Consensus Statement on the Use of Ketamine in the Treatment of Mood Disorders.” JAMA Psychiatry 74 (4):399–405. doi: 10.1001/jamapsychiatry.2017.0080. [DOI] [PubMed] [Google Scholar]
- Schrantee A, Solleveld MM, Schwantje H, Bruin WB, Mutsaerts HM, Adriaanse SM, Lucassen P, Booij J, and Reneman L. 2019. “Dose-dependent effects of the selective serotonin reuptake inhibitor citalopram: A combined SPECT and phMRI study.” J Psychopharmacol:269881119836229. doi: 10.1177/0269881119836229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann J, Matzner H, Michaeli A, and Yaka R. 2009. “NR2A/B-containing NMDA receptors mediate cocaine-induced synaptic plasticity in the VTA and cocaine psychomotor sensitization.” Neurosci Lett 461 (2):159–62. doi: 10.1016/j.neulet.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Scofield MD, Heinsbroek JA, Gipson CD, Kupchik YM, Spencer S, Smith AC, Roberts-Wolfe D, and Kalivas PW. 2016. “The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis.” Pharmacol Rev 68 (3):816–71. doi: 10.1124/pr.116.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD, Li H, Siemsen BM, Healey KL, Tran PK, Woronoff N, Boger HA, Kalivas PW, and Reissner KJ. 2016. “Cocaine Self-Administration and Extinction Leads to Reduced Glial Fibrillary Acidic Protein Expression and Morphometric Features of Astrocytes in the Nucleus Accumbens Core.” Biol Psychiatry 80 (3):207–15. doi: 10.1016/j.biopsych.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Moussawi K, Zhou W, Toda S, and Kalivas PW. 2011. “Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors.” Proc Natl Acad Sci U S A 108 (48):19407–12. doi: 10.1073/pnas.1112052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, and Jan LY. 1994. “Changing subunit composition of heteromeric NMDA receptors during development of rat cortex.” Nature 368 (6467):144–7. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Selley DE, Vogt LJ, Childers SR, and Martin TJ. 2000. “Chronic heroin self-administration desensitizes mu opioid receptor-activated G-proteins in specific regions of rat brain.” J Neurosci 20 (12):4555–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Helen G, Havel C, Dempsey DA, Jacob P 3rd, and Benowitz NL. 2016. “Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes.” Addiction 111 (3):535–44. doi: 10.1111/add.13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanik MT, Kupchik YM, and Kalivas PW. 2016. “Optogenetic inhibition of cortical afferents in the nucleus accumbens simultaneously prevents cue-induced transient synaptic potentiation and cocaine-seeking behavior.” Brain Struct Funct 221 (3):1681–9. doi: 10.1007/s00429-015-0997-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolerman IP, and Jarvis MJ. 1995. “The scientific case that nicotine is addictive.” Psychopharmacology (Berl) 117 (1):2–10; discussion 14–20. [DOI] [PubMed] [Google Scholar]
- Talbot JN, Geffert LM, Jorvig JE, Goldstein RI, Nielsen CL, Wolters NE, Amos ME, Munro CA, Dallman E, Mereu M, Tanda G, Katz JL, Indarte M, Madura JD, Choi H, Leak RK, and Surratt CK. 2016. “Rapid and sustained antidepressant properties of an NMDA antagonist/monoamine reuptake inhibitor identified via transporter-based virtual screening.” Pharmacol Biochem Behav 150-151:22–30. doi: 10.1016/j.pbb.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomek SE, Lacrosse AL, Nemirovsky NE, and Olive MF. 2013. “NMDA Receptor Modulators in the Treatment of Drug Addiction.” Pharmaceuticals (Basel) 6 (2):251–68. doi: 10.3390/ph6020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, and Constantine-Paton M. 2004. “Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal.” Trends Neurosci 27 (7):428–37. doi: 10.1016/j.tins.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Wang TW, Asman K, Gentzke AS, Cullen KA, Holder-Hayes E, Reyes-Guzman C, Jamal A, Neff L, and King BA. 2018. “Tobacco Product Use Among Adults - United States, 2017.” MMWR Morb Mortal Wkly Rep 67 (44):1225–1232. doi: 10.15585/mmwr.mm6744a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Ju P, Liu H, Wu X, Niu Z, Zhu Y, Zhang C, and Fang Y. 2020. “Ifenprodil rapidly ameliorates depressive-like behaviors, activates mTOR signaling and modulates proinflammatory cytokines in the hippocampus of CUMS rats.” Psychopharmacology (Berl). doi: 10.1007/s00213-020-05469-0. [DOI] [PubMed] [Google Scholar]
- Yoshimoto K, and McBride WJ. 1992. “Regulation of nucleus accumbens dopamine release by the dorsal raphe nucleus in the rat.” Neurochem Res 17 (5):401–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.