

Abstract
To characterize the influence of APOE genotype on cerebral Aβ load and longitudinal Aβ trajectories, [11C]PiB PET imaging studies were performed in a cohort of 428 participants with known APOE genotype and a range of clinical diagnoses from cognitively normal elderly to Alzheimer’s disease (AD). [11C]PiB PET imaging was used to assess amyloid load in a clinically heterogeneous cohort of 428 elderly participants. Serial [11C]PiB data and a repeated measures model were used to model amyloid trajectories in a subset of 235 participants classified on the basis of APOE genotype. We found that APOE-ε4 was associated with increased Aβ burden and an earlier age of onset of Aβ positivity, whereas APOE-ε2 appeared to have modest protective effects against Aβ. APOE class did not predict rates of Aβ accumulation. The present study suggests that APOE modifies AD risk through a direct influence on amyloidogenic processes, which manifests as an earlier age-of-onset of Aβ positivity, although it is likely that other genetic, environmental, and lifestyle factors are important.
Keywords: Alzheimer’s disease, apolipoprotein-E, positron emission tomography (PET), amyloid, genetics
1.0. INTRODUCTION
An evolving view of late-onset Alzheimer’s disease (LOAD) is that it is a complex and multifactorial disease in which genetic, environmental, and lifestyle factors modulate risk, age of onset, and disease progression (Bertram et al., 2007; Borenstein et al., 2006; Gatz et al., 2006). Evidence suggests that LOAD is driven by impairments in Aβ clearance rather than overproduction (Mawuenyega et al., 2010), although it is not well understood how LOAD risk factors conspire to modify disease pathophysiology and/or lower the threshold for detection of disease symptoms.
Three major polymorphic alleles of the APOE gene (ε2, ε3, ε4) give rise to three apolipoprotein-E (apoE) isoforms (Rall et al., 1982; Roda et al., 2019). Much evidence suggests functional roles for apoE in regulating Aβ aggregation and clearance, and that these functions are affected by the apoE genotype/isoform. The presence of one or more ε4 alleles of the APOE gene is associated with an increased risk of LOAD and an earlier age-of-onset in a gene-dose dependent manner (Bekris et al., 2011; Bird, 2008; Corder et al., 1993; Farrer et al., 1997; Saunders et al., 1993; Tang et al., 1996), whereas the APOE-ε2 allele may have protective effects (Morris et al., 1995; Nagy et al., 1995; Wilson et al., 2002). Previous studies have examined the relationship between APOE genotype and the AD-related phenotype of cerebral Aβ deposition in positron emission tomography (PET) imaging studies. In general, these studies show that APOE-ε4 is associated with increased cerebral Aβ load (Jansen et al., 2015; Mecca et al., 2018; Morris et al., 2010; Resnick et al., 2015; Wirth et al., 2014; Yan et al., 2018) and an accelerated rate of Aβ deposition (Grimmer et al., 2010; Villemagne et al., 2011), whereas APOE-ε2 appears to protect against Aβ accumulation (Kim et al., 2017; Morris et al., 2010).
The present study seeks to provide further support for the concept of genetic modulation of amyloidogenic processes by replicating observed associations between APOE genotype and brain Aβ burden in a cross-sectional cohort (n=428) and to model the influence of APOE genotype on amyloid trajectories in a subcohort (n=235) with serial [11C]PiB assessments.
2.0. METHODS
2.1. Study Participants and Study Design
432 participants were identified retrospectively from a multi-study cohort with [11C]PiB PET image data, cognitive assessments, and APOE genotyping. Subjects comprising this multi-study cohort were drawn from 14 different studies, although several ongoing studies of normal aging were the largest contributors of longitudinal data (Aizenstein et al., 2008; Butters et al., 2008; Lopez et al., 2018; Mathis et al., 2013; Nadkarni et al., 2019; Tudorascu et al., 2019). [11C]PiB data were acquired over a 14-year period beginning in 2003.
Comprehensive multi-domain neuropsychological assessment was completed with all participants as previously described (Mathis et al., 2013; Snitz et al., 2015). Only subjects with consensus diagnoses of cognitively normal (CN), mild cognitive impairment (MCI) (Albert et al., 2011), probable AD (McKhann et al., 2011), and dementia of unknown origin (DUO) were included. A DUO diagnosis applies to subjects with atypical dementia who fell into any of the following categories (Wolk et al., 2012) : (1) possible Alzheimer’s disease (pAD) who received this diagnosis due to an atypical presentation, usually reflecting non-memory cognitive symptoms or behavioral issues more prominent than typical AD; (2) patients with posterior cortical atrophy (PCA) syndrome; (3) patients with primary progressive aphasia (PPA); and (4) patients in whom no diagnosis could be determined. Like MCI, DUO subjects are heterogeneous in terms of [11C]PiB retention characteristics with SUVR values that range from control-like to AD-like (Wolk et al., 2012). It is believed that DUO subjects as a group are characterized by a spectrum of brain pathologies, often mixed or overlapping, where Aβ is a co-pathology in approximately one-third to one-half of cases
Demographic information, clinical diagnoses, and baseline global [11C]PiB retention indices for the full cohort are presented in Table 1. Serial [11C]PiB data was available in a subcohort of 235 participants (Table 2). For cross-sectional analyses, only baseline [11C]PiB scans were used, whereas all available [11C]PiB scans were used to model longitudinal trajectories. To examine the potential effects of selection bias, where MCI and AD subjects carry elevated genetic risk for APOE-ε4 positivity, we performed parallel analyses in subcohorts of cognitively normal (control) subjects. Demographic information is presented in Supplemental Table S1 for cross-sectional (n=246) and Supplemental Table S2 (n=173) for longitudinal control subjects.
Table 1.
Demographics and Clinical Characteristics for Cross-Sectional Cohort (n=428) by APOE Genotype Class
| Characteristic | APOE-ε3ε3 | APOE-ε2+ | APOE-ε4+ | Test Statistic (df), p-value |
|---|---|---|---|---|
| n | 241 | 43 | 144 | |
| Age at Baseline, y, mean (SD) | 75.60 (9.81) | 75.23 (10.28) | 71.19 (9.87) | F: 9.31 (2), p=0.0001 |
| Education, y, mean (SD) | 14.99 (2.71) | 15.33 (2.82) | 15.72 (3.13) | F: 2.88 (2), p=0.058 |
| MMSE, score, median (IQR) | 28 (27–29) | 28 (27–29) | 27 (23–29) | KW: 16.9 (2), p=0.0002 |
| Female sex, No.(%) | 131 (54.13) | 22 (52.38) | 75 (52.08) | χ2 : 0.27 (2), p=0.873 |
| Nonwhite race/ethnicity, No. (%) | 14 (5.81) | 4 (9.3) | 13 (9.03) | Fisher’s Exact: p=0.371 |
| Clinical Dx, No. (%) | χ2 : 57.83 (6), p<0.0001 | |||
| Control | 166 (67.5) | 27 (11.0) | 53 (21.5) | |
| MCI | 47 (47.0) | 13 (13.0) | 40 (40.0) | |
| AD | 17 (27.9) | 1 (1.6) | 43 (70.5) | |
| DUO | 11 (52.4) | 2 (9.5) | 8 (38.1) | |
| Positive PiB status, No. (%) | 80 (33.2) | 8 (18.6) | 93 (64.58) | χ2 : 47.4 (2), p<0.0001 |
| Baseline [11C]PiB SUVR, mean (SD) | 1.54 (0.44) | 1.36 (0.34) | 1.84 (0.5) | F: 28.49 (2), p<0.0001 |
Abbreviations:
Alzheimer’s disease (AD), dementia of unknown origin (DUO), interquartile range (IQR), Kruskall-Wallis (KW) test, standard deviation (SD), mild cognitive impairment (MCI), mini-mental state examination (MMSE), number (No.), Pittsburgh Compound-B (PiB), standard deviation (SD), standardized uptake value ratio (SUVR), years (y).
Table 2.
Subject Demographics and Clinical Characteristics for Longitudinal Subcohort (n=235) by APOE Genotype Class
| Characteristic | APOE-ε3ε3 | APOE-ε2+ | APOE-ε4+ | Test Statistic (df), p-value |
|---|---|---|---|---|
| n | 142 | 28 | 65 | |
| Age at Baseline, y, mean (SD) | 78.9 (8.92) | 75.6 (10.2) | 73.5 (9.23) | F: 7.98 (2), p=0.00045 |
| Education, y, mean (SD) | 15.1 (2.66) | 15.8 (2.80) | 16.1 (3.19) | F: 2.84 (2), p=0.0602 |
| MMSE score, median (IQR) | 29 (27–29) | 28 (27–29) | 28 (26–29) | KW: 4.00 (2), p=0.14 |
| Female sex, No.(%) | 74 (52.11) | 11 (39.29) | 28 (43.08) | χ2 : 2.44 (2), p=0.29 |
| Nonwhite race/ethnicity, No.(%) | 10 (7.04) | 3 (10.71) | 6 (9.23) | Fisher’s Exact: p=0.68 |
| Follow-up period, y, mean (SD) | 4.48 (2.34) | 4.03 (2.40) | 3.39 (1.80) | F: 5.49 (2), p=0.00468 |
| Number of Visits, mean (SD) | 3.23 (0.97) | 3.25 (1.4) | 2.91 (0.84) | F: 2.45 (2), p=0.0889 |
| Clinical Dx., No. (%) | Fisher’s Exact: p<<0.001 | |||
| Control | 119 (68.8) | 19 (11.0) | 35 (20.2) | |
| MCI | 21 (44.7) | 9 (19.1) | 17 (36.2) | |
| AD | 2 (14.3) | 0 (0) | 12 (85.7) | |
| DUO | 0 (0.0) | 0 (0.0) | 1 (100.0) | |
| Globally PiB Positive, No. (%) | 52 (36.62) | 4 (14.29) | 44 (67.69) | χ2 : 28.00 (2), p<<0.001 |
| Baseline [11C]PiB SUVR, mean(SD) | 1.56 (0.40) | 1.33 (0.35) | 1.86 (0.47) | F: 19.03 (2), p<<0.001 |
Abbreviations:
Alzheimer’s disease (AD), dementia of unknown origin (DUO), interquartile range (IQR), Kruskall-Wallis (KW) test, standard deviation (SD), mild cognitive impairment (MCI), mini-mental state examination (MMSE), number (No.), Pittsburgh Compound-B (PiB), standard deviation (SD), standardized uptake value ratio (SUVR), years (y).
APOE genotyping was performed on DNA extracts from blood as previously described (Kamboh et al., 1995). Participants were divided into three APOE classes: APOE-ε2 carriers (APOE-ε2+), APOE-ε3ε3, and APOE-ε4 carriers (APOE-ε4+). Four participants having an APOE-ε2ε4 genotype were excluded as they could not be unambiguously assigned to one APOE genotype class.
2.2. Standard Protocol Approvals, Registrations, and Patient Consents
All participants or their proxies provided written consent, and all studies were performed with approval of the Institutional Review Board of the University of Pittsburgh.
2.3. Data Acquisition
2.3.1. Magnetic Resonance (MR) Imaging
T1-weighted MR images were acquired on one of two MR imaging systems: MR images acquired in 2009 and earlier (49%) were acquired on a 1.5T GE Signa scanner whereas later scans (51%) were acquired on a 3.0T Siemens Magnetom Trio. Participants scanned on the 1.5T GE Signa scanner were positioned in a standard head coil and a brief scout T1-weighted image was obtained. A volumetric spoiled gradient recalled (SPGR) sequence with parameters optimized for contrast among gray matter, white matter, and CSF were acquired in the coronal plane (TE/TR = 5/25, flip angle = 40°, NEX = 1, slice thickness = 1.5 mm/0 mm interslice). MR images acquired on the 3T Siemens Magnetom Trio used a magnetization prepared rapid acquisition gradient echo (MPRAGE) sequence (TI/TE/TR = 900/2.98/2300 ms, flip angle = 9°, slice thickness = 1.2 mm, matrix size = 160 × 240 × 256).
2.3.2. Positron Emission Tomography (PET) imaging
PET emission data were acquired on one of two Siemens ECAT HR+ PET scanners as previously described (Mathis et al., 2013). Briefly, participants were positioned in the scanner approximately 35 min after [11C]PiB injection. A 10 min transmission scan was acquired using rotating 68Ge/68Ga rod sources to correct for photon attenuation, followed by a 20 minute emission scan (4 × 5-minute frames) beginning 50 minutes after [11C]PiB injection (15 mCi). PET emission data were reconstructed using filtered back-projection (Direct Inverse Fourier Transform) into a 128 × 128 × 63 matrix with voxel sizes of 2.06 × 2.06 × 2.43 mm3. Images were filtered with a 3 mm Hann window.
2.4. Data Processing
MRI and [11C]PiB PET images were processed as previously described (Rosario et al., 2011). Briefly, dynamic PET images were corrected for interframe motion and summed over 50–70 minutes (McNamee et al., 2009). MRI images were manually skull-stripped and reoriented with axial image planes parallel to the anterior-posterior commissure line. [11C]PiB images were registered to skull-stripped MRIs using rigid body registration in AIR v3.0, and MRIs were resliced to PET resolution.
Manual region-of-interest (ROI) tracings were performed using ROITool software (Siemens Medical Systems, Knoxville, TN, USA) on skull-stripped MRIs. ROIs were defined bilaterally on 3 to 5 contiguous transverse image planes for anterior cingulate gyrus (ANC), anteroventral striatum (AVS), frontal cortex (FRC), parietal cortex (PAR), lateral temporal cortex (LTC), posterior cingulate/precuneus (PRC), and cerebellar gray matter (CER) and subsequently used to sample co-registered PET images as previously described (Cohen et al., 2009; Price et al., 2005). Standardized uptake value ratios (SUVR) were computed for all regions normalized to CER. A six-region composite global [11C]PiB SUVR index (GBL6) was also computed as previously described (Aizenstein et al., 2008).
To investigate the dilutional effects of atrophy on [11C]PiB retention indices and Aβ trajectories, an MR-based two-compartment (brain and CSF) partial volume correction (Meltzer et al., 1990) was applied to SUVR outcomes as previously described (Rosario et al., 2011). CSF-corrected [11C]PiB SUVR values were compared to uncorrected SUVR in all analyses.
[11C]PiB positivity thresholds were determined using a sparse k-means clustering algorithm as previously described (Cohen et al., 2013). Subjects were adjudicated to be PiB-positive (PiB+) or negative (PiB−) at baseline and follow-up examinations using the GBL6 composite index with a positivity threshold of SUVR = 1.51 (Cohen et al., 2013).
2.5. Statistical Methods
Mean (standard deviation, SD) is presented for normally distributed variables such as age, education, follow-up period, number of visits, and baseline [11C]PiB SUVR. Median (interquartile range, IQR) is provided for MMSE. Categorical characteristics such as sex, ethnicity, diagnosis, and frequency of global [11C]PiB positivity are summarized with frequencies and percentages (Tables 1, 2). A one-way ANOVA was used for normally distributed variables having similar variances (age, education). A chi-square test for categorical variables was used to test for differences in gender composition and frequency of [11C]PiB positivity. Fisher’s exact test was used to compare some categorical variables with few subjects in one or more cells (e.g. race, diagnostic classification). The Kruskall-Wallis rank test was used to test continuous variables such as MMSE. The overall tests were followed up with post-hoc pairwise comparisons using t-tests or the Wilcoxon signed rank test as appropriate. All pairwise comparisons were Bonferroni corrected.
After examining the relationship between [11C]PiB SUVR and age by APOE class using basic data plots to characterize the type of association (e.g. linear, quadratic), our initial approach was to apply a repeated measures model with only the main effects of age and APOE genotype. Additional terms, quadratic terms, and their interactions with APOE were included to expand the basic model, which included age2, age2 × APOE interaction, diagnosis, and diagnosi × APOE interaction. We evaluated the Akaike Information Criterion (AIC), Bayesian Information Criterion (BIC), and t-value for each added term. Terms were added or dropped from the model based on this evaluation. The Kenward-Rogers method (Kenward and Roger, 1997) was used for computing the degrees of freedom.
The final model included fixed factors of APOE genotype, age (time-dependent predictor), age2, and their interactions to test the associations between APOE genotype and age over time, and to determine the age at which Aβ trajectories crossed the PiB positivity threshold (GBL6 SUVR > 1.51). The model included a random intercept and linear term to account for within-subject correlation. The following repeated measures model was fit for global [11C]PiB SUVR (y) for subject i and observation j :
where:
β0 represents the average [11C]PIB GBL6 SUVR value for the reference group (ε3ε3) at baseline (intercept)
APOEi is a dummy variable for the APOE group factor for subject i, with 2 levels (APOE2 for the ε2 allele and APOE4 for the ε4 allele, while the ε3 allele is the reference category)
Ageij represents age at serial observation j for subject i, fitted as continuous;
-
v0i is the subject-specific variation from average intercept effect, and v1i is the subject specific variation from the average slope with the following variance/covariance matrix:
and is the random error term at the jth observation for subject i.
To determine the Aβ trajectories for the three APOE genotype classes, estimates of fit parameters (β0 – β5) were substituted into the model equation above and solved over an age range of 45–95 years as previously described (Tudorascu et al., 2019). The age-of-onset was determined by solving this equation for the age at which each trajectory crossed the global [11C]PiB positivity threshold of SUVR=1.51.
We also computed annualized changes in PiB retention (SUVR/year) for all longitudinal subjects over the longest follow-up interval. Incident [11C]PiB positivity was defined as a change in [11C]PiB status from negative to positive between baseline and terminal examinations.
SAS 9.4 (SAS institute, Cary, NC) and R 3.4.2 (GNU General Public License), were used for statistical analyses.
3.0. RESULTS
3.1. Cross-Sectional Analyses
Of the 428 participating subjects, 43 (10.0%) were classified as APOE-ε2+, all of whom expressed the APOE-ε2ε3 genotype. The APOE-ε3ε3 genotype was the most abundant (241/428 subjects, 56.3%), whereas APOE-ε4+ subjects (144/428, 33.6%) were a mixture of ε3ε4 (111/428, 25.9%) and ε4ε4 (33/428, 7.7%) genotypes. APOE-ε2, -ε3, and -ε4 allele frequencies in the full cohort were 0.050, 0.743, and 0.207, respectively, which are comparable to those reported in American populations (Eichner et al., 1990; Singh et al., 2006). A notable difference in our sample is an increased APOE-ε4 allele frequency compared to the local population frequency (0.207 vs 0.122) (Eichner et al., 1990), although this finding is consistent with memory clinic settings (van der Flier et al., 2008). All APOE classes had similar (p=0.873) and nearly equal proportions of males and females (Table 1). APOE-ε4+ subjects were younger at baseline than either APOE-ε2+ (p=0.0085) or ε3ε3 (p=0.000022) subjects. APOE-ε4+ subjects showed greater impairment on the MMSE (median [IQR]: 27 [23–29] than either APOE-ε2+ (28 [27–29]; p=0.025) or APOE-ε3ε3 (28 [27–29]; p=0.000059) subjects despite being significantly younger (Table 1, Figure 1A). We also found statistically significant differences (p<0.001) in the distribution of diagnostic groups between APOE classes for both cross-sectional (Table 1) and longitudinal cohorts (Table 2). There were no differences in cognition between APOE-ε2+ and APOE-ε3ε3 subjects (p=0.54).
FIGURE 1: Distribution of Age, MMSE, and Baseline [11C]PiB Retention.
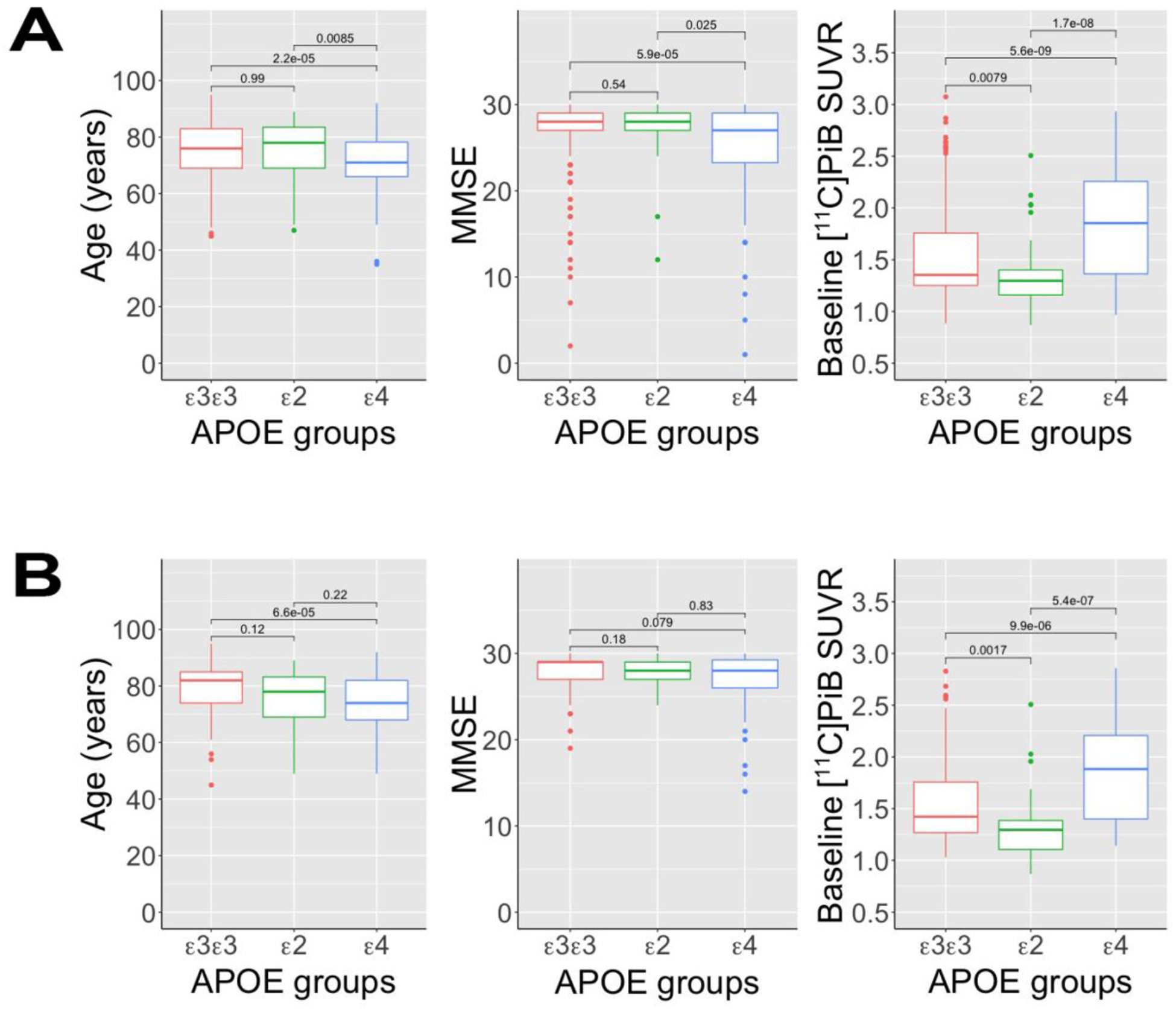
Box and whisker plots comparing the distribution of age, MMSE, and baseline [11C]PiB retention between three APOE genotype classes for: (A) the full (n=428) cohort; and (B) and the longitudinal sub-cohort (n=235). The box represents the interquartile range (IQR), where the median is marked by a horizontal line. The upper and lower whiskers extend up to 1.5 IQR from either the first or third quartile. Outliers are defined as points that fall beyond of the range of the whiskers (>1.5 IQR) and are plotted individually. Multiple comparisons were used to test for significant differences between pairs of APOE groups.
Non-CSF corrected GBL6 [11C]PiB SUVR was increased in APOE-ε4+ subjects (1.84±0.5) compared to either APOE-ε2+ (1.36±0.34; p ≪ 0.0001) or APOE-ε3ε3 (1.54±0.44); p≪0.0001) subjects, whereas [11C]PiB SUVR values in APOE-ε2+ subjects was reduced compared to APOE-ε3ε3 subjects (p=0.0079) (Figure 1A). These findings persisted after CSF correction (Supplemental Figure E1).
In the control-only subcohort (n=246), we observed significantly higher global [11C]PiB retention in APOE-ε4+ control subjects relative to both APOE-ε3ε3 (p=0.026) and APOE-ε2+ (p=0.005) controls (Table S1). There were no differences in education (p>0.673) or MMSE (p>0.378) between any APOE groups of control subjects (p > 0.37), although APOE-ε4+ control subjects were significantly younger (~4.2 y) than APOE-ε3ε3 control subjects (Table S1, p=0.013).
3.2. Longitudinal Analyses
The full longitudinal subcohort (n=235) averaged approximately three visits over a span of ~4 years. The distribution of APOE genotypes in the longitudinal subcohort was similar to that in the full cohort (Table 2). Of the APOE-ε4+ subjects, 11/65 (16.9%) were APOE-ε4 homozygotes. In general, the longitudinal subcohort mirrored the full cohort in terms of the distribution of age, cognition, gender balance, ethnic composition, and baseline [11C]PiB retention indices (Table 2), although not all significant findings in the full cohort reached statistical significance in the longitudinal subcohort (Figure 1). Demographic information for the group of 173 control subjects with longitudinal is shown in Supplemental Table S2. Statistically significant differences in the age distribution, frequency of [11C]PiB positivity and global [11C]PiB retention indices between APOE classes were maintained in the controls-only group.
Individual trajectories of [11C]PiB SUVR values are shown in Figure 2. Among the longitudinal subjects 95 (40.4%) were globally PiB-negative at baseline and remained globally PiB-negative throughout follow-up. A similar proportion of subjects (97/235, 41.3%) were globally PiB-positive at baseline and remained PiB-positive. Of the remaining 43 subjects, 40 (17.0%) converted from PiB-negative to PiB-positive, whereas 3 subjects (1.3%) who were PiB-positive at baseline reverted to PiB-negative (all were near the PiB-positivity cutoff). The proportion of PiB-positive subjects at baseline was highest for APOE-ε4+ subjects (44/65; 67.7%) and lowest for APOE-ε2+ subjects (4/28; 14.3%), whereas APOE-ε3ε3 subjects were intermediate (52/142, 36.6%). For subjects that were PiB-negative at baseline, conversion rates to PiB-positive (positive at latest examination) were identical for APOE-ε4+ (7/21; 33.3%) and APOE-ε3ε3 (30/90; 33.3%) subjects, whereas less than one-half as many APOE-ε2+ subjects (3/24; 12.5%) converted.
FIGURE 2: Individual Aβ Trajectories.
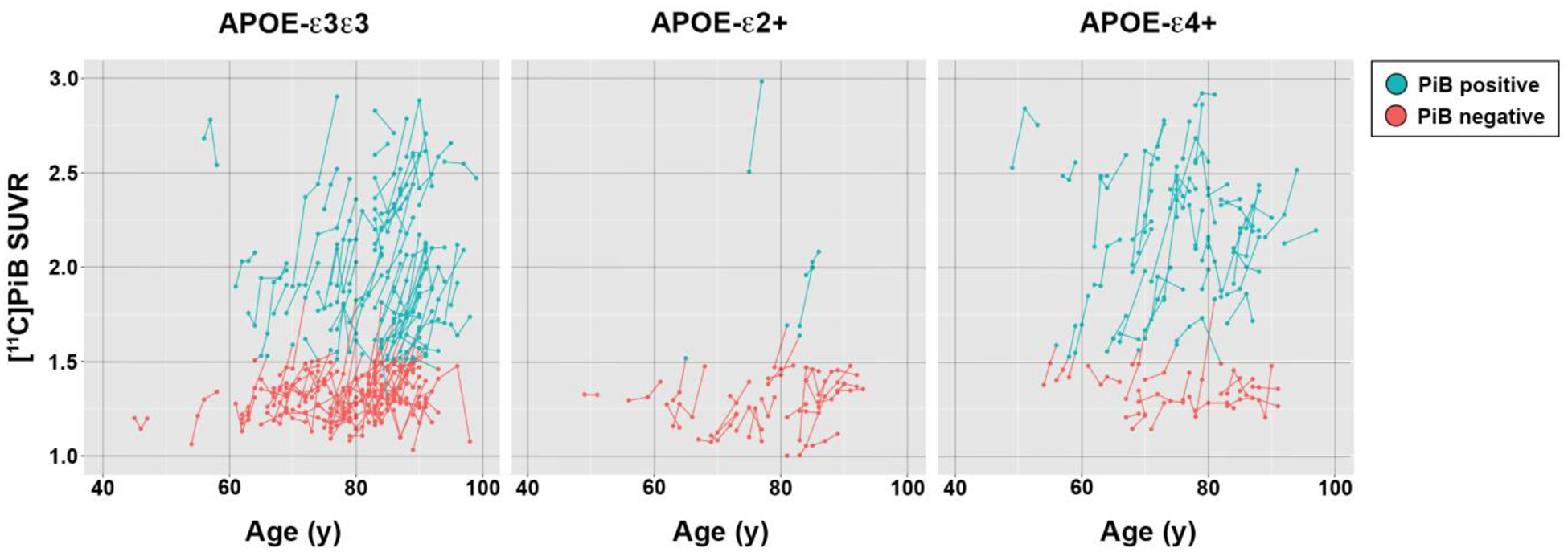
Individual trajectories showing the change in global [11C]PiB SUVR values by chronologic age are shown for longitudinal participants (n=235) for three APOE genotype classes. For each participant, serial [11C]PiB assessments are plotted as individual points where blue indicates Aβ positive values and red to indicate Aβ negative values. The points are connected by line segments to match the positivity status of the earlier vertex.
Table 3 represents the estimates and their standard errors of the intercepts and slopes for each main effect and interaction term in the model. Also shown in Table 3 are t-statistics and degrees of freedom, p-values, and the 95% CI for each estimate. The repeated measures model revealed significant interactions between age and APOE-ε4+ genotype (p=0.0116) as well as between age2 and APOE-ε4+ genotype (p=0.0097) (Table 3). The estimate for the age2 x APOE-ε4+ interaction is β7 = −0.0009 with a 95% confidence interval of (−0.0016, −0.00023). The estimate β7 = −0.0009 represents the mean difference in the slopes of [11C]PIB SUVR and age2 between APOE-ε4+ and APOE-ε3ε3 classes with all other model parameters fixed. Similarly, the estimate β8 = 0.0001 represents the mean difference in the slopes of [11C]PIB SUVR and age2 between APOE-ε2+ and APOE-ε3ε3 classes.
Table 3.
Repeated measures analysis estimates
| Effect | β (SE) | t (DF) | p-value | 95% CI for β |
|---|---|---|---|---|
| Intercept | β0 : 1.7924 (1.2353) | 1.45 (157) | 0.1488 | (−0.6476, 4.2324) |
| APOE-ε4+ | β1 : −4.7669 (2.1597) | −2.21 (198) | 0.0284 | (−9.0258, −0.5081) |
| APOE-ε2+ | β2 : 0.3025 (2.7657) | 0.11 (149) | 0.9131 | (−5.1624, 5.7674) |
| APOE-ε3ε3 | 0 | - | - | - |
| Age | β3 : −0.0366 (0.0304) | −1.27 (174) | 0.2299 | (−0.09666, 0.02338) |
| Age × APOE-ε4+ | β4 : 0.1410 (0.0554) | 2.54 (225) | 0.0116 | (0.03177, 0.2502) |
| Age × APOE-ε2+ | β5 : −0.0127 (0.0704) | −0.18 (167) | 0.8575 | (−0.1515, 0.1262) |
| Age × APOE-ε3ε3 | 0 | - | - | - |
| Age2 | β6 : 0.0004 (0.0002) | 2.26 (187) | 0.0250 | (0.000054, 0.000796) |
| Age2 × APOE-ε4+ | β7 : −0.0009 (0.0004) | −2.61 (244) | 0.0097 | (−0.00164, −0.00023) |
| Age2 × APOE-ε2+ | β8 : 0.0001 (0.0005) | −0.2 (180) | 0.8433 | (−0.00080, 0.000978) |
| Age2 × APOE-ε3ε3 | 0 | - | - | - |
Aβ trajectories showed distinct differences in the age of onset of global [11C]PiB positivity for the full longitudinal cohort (Figure 3). The trajectory for APOE-ε3ε3 subjects had an average age of onset of global [11C]PiB positivity of 77.9 yrs and a slope of 0.029 SUVR/year (95% CI: 0.023–0.036 SUVR/year) at the GBL6 positivity threshold (SUVR=1.51). The APOE-ε4+ trajectory showed an earlier age of onset of [11C]PiB positivity of 61.1 yrs and a rate of accumulation of 0.043 SUVR/yr (95% CI: 0.022–0.063 SUVR/year). In contrast, APOE-ε2+ showed a delayed age of onset of [11C]PiB positivity of 82.0 yrs and a rate of accumulation of 0.035 SUVR/yr (95% CI: 0.018–0.052 SUVR/year). Similar longitudinal trajectories were observed in the control-only longitudinal subcohort (n=173), although the age-of-onset of [11C]PiB positivity in APOE-ε4+ control subjects was found to be ~8 years later (69.3 y) than that observed for the full longitudinal cohort, whereas for both APOE-ε2+ and APOE-ε3ε3 control subjects it was delayed only by ~2 years (Figure 4). Compared to the full longitudinal cohort, rates of Aβ accumulation predicted by the model were unchanged for APOE-ε3ε3 control subjects (0.029 SUVR/yr), whereas rates for APOE-ε4+ control subjects were slightly less (0.033 SUVR/yr) and slightly greater (0.044 SUVR/yr) for APOE-ε2+.
FIGURE 3: Modeling of Aβ Trajectories.
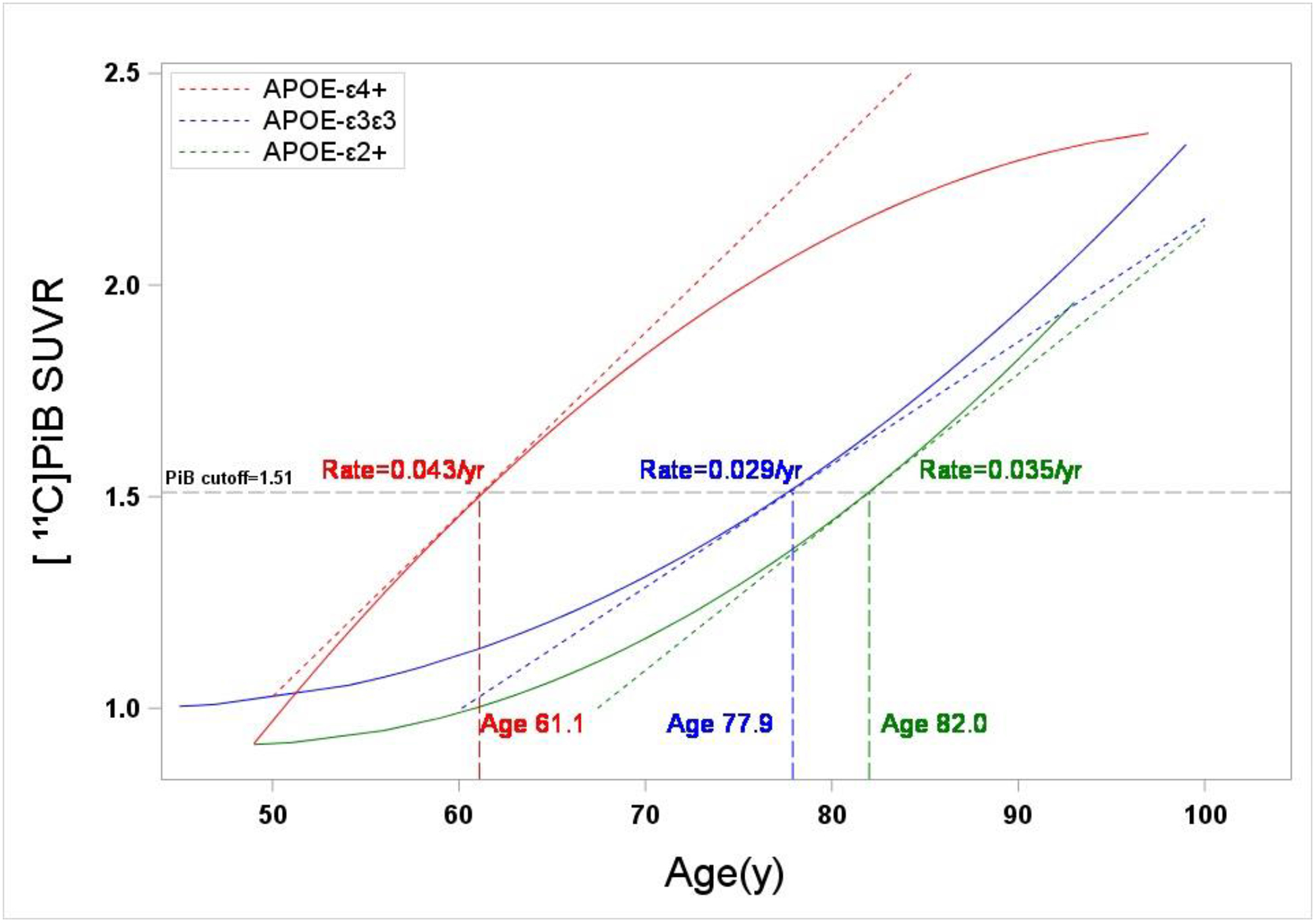
Global [11C]PiB SUVR trajectories for the three APOE genotype classes based on a repeated measures model of serial data from a longitudinal subcohort (n=235). The GBL6 positivity threshold of 1.51 SUVR is indicated by a black hatched line. Indicated also is the slope of a line tangent to each trajectory at the [11C]PiB positivity threshold, which corresponds to the rate of Aβ accumulation at the threshold. Also shown is the age at which each trajectory crosses the positivity threshold. APOE-ε4+ participants showed the earliest age of onset of Aβ positivity (60.9 y), while trajectories for APOE-ε3ε3 and APOE-ε2+ participants indicated a delay in the age of onset of Aβ positivity of 16.6 and 20.8 years, respectively.
FIGURE 4: Modeling of Aβ Trajectories in Control Subjects.
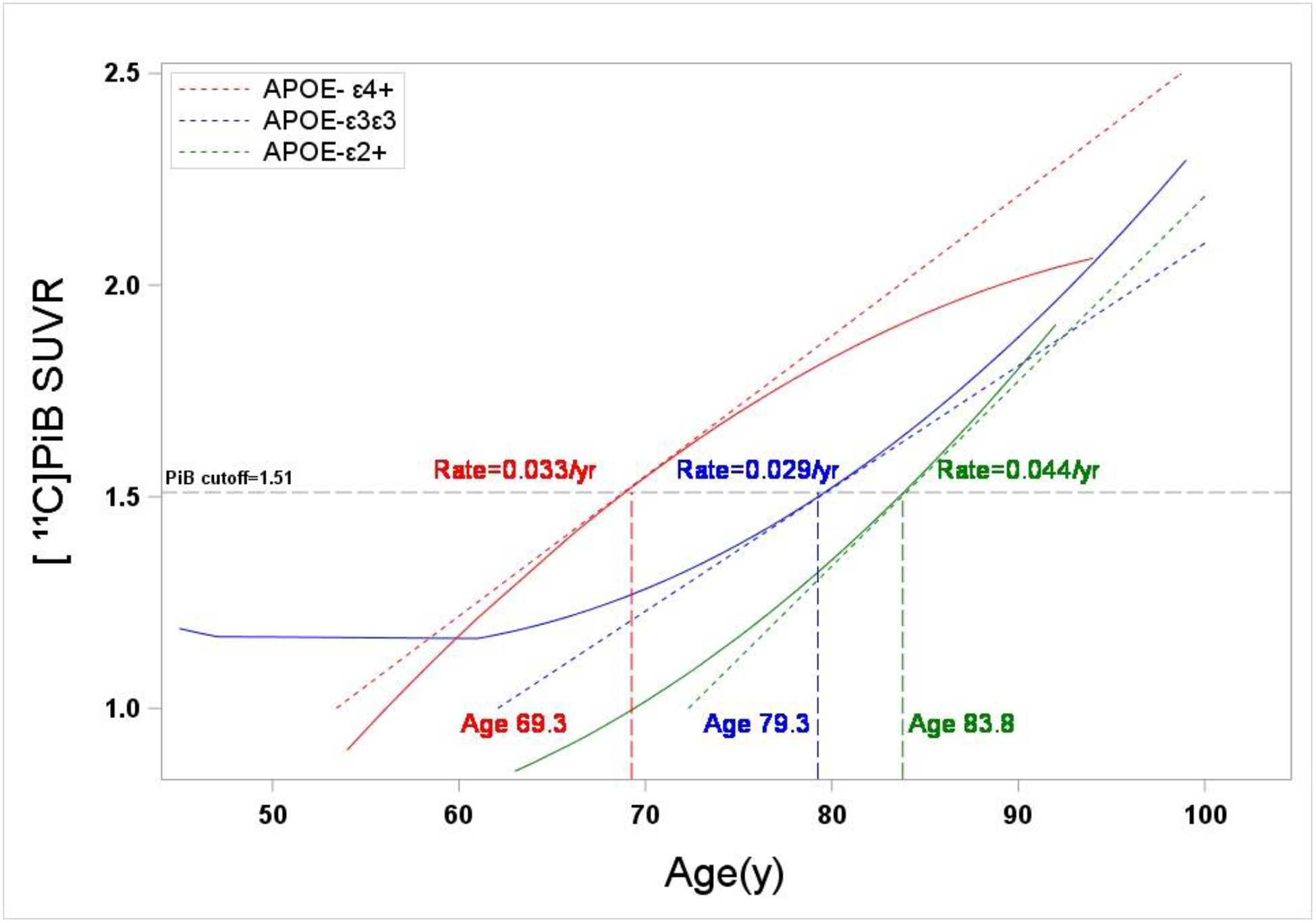
Global [11C]PiB SUVR trajectories for the three APOE genotype classes based on a repeated measures model of serial data from a subgroup of control longitudinal subjects (n=173). The GBL6 positivity threshold of 1.51 SUVR is indicated by a black hatched line. Indicated also is the slope of a line tangent to each trajectory at the [11C]PiB positivity threshold, which corresponds to the rate of Aβ accumulation at the threshold. Also shown is the age at which each trajectory crosses the positivity threshold. APOE-ε4+ control subjects showed the earliest age of onset of Aβ positivity (69.3 y), while trajectories for APOE-ε3ε3 and APOE-ε2+ control subjects indicated a delay in the age of onset of Aβ positivity of 10.0 and 14.8 years, respectively.
Individual rates of Aβ accumulation, as indexed by annualized changes in [11C]PiB SUVR, showed no significant effect of APOE genotype (Table 4) when compared for the full longitudinal subcohort (p=0.178; n=235), subjects PiB-positive at baseline (p=0.551; n=100), or subjects PiB-negative at baseline (p=0.340; n=125). Pairwise comparisons between APOE genotype classes also showed no significant differences in rates of amyloid accumulation, although small subsample sizes often limited comparisons to those between APOE-ε3ε3 and APOE-ε4+. Subjects PiB-negative at baseline (n=125) included a subgroup that converted to PiB-positive (n=40) and represent cases of incident amyloid positivity. In this group, we found APOE-ε4+ subjects to exhibit a faster rate of Aβ accumulation compared to APOE-ε3ε3 (0.09±0.03 SUVR/year vs 0.06±0.03 SUVR/year) which was significant (p=0.029).
Table 4.
Annualized changes in [11C]PiB retention measures by APOE genotype class
| Comparison | APOE-ε3ε3 | APOE-ε2+ | APOE-ε4+ | Test Statistic (df), p-value |
|---|---|---|---|---|
| All Participants | ||||
| n | 142 | 28 | 65 | |
| Change (SUVR/yr), mean (SD) | 0.03(0.05) | 0.04 (0.06) | 0.05 (0.07) | F: 1.74(2), p=0.178a |
| ε3ε3 vs ε2+ | 0.03(0.05) | 0.04 (0.06) | p=0.481b | |
| ε3ε3 vs ε4+ | 0.03(0.05) | 0.05 (0.07) | p=0.103b | |
| ε2+ vs ε4+ | 0.04 (0.06) | 0.05 (0.07) | p=0.574b | |
| Participants PiB+ at Baseline | ||||
| n | 52 | 4 | 44 | |
| Change (SUVR/yr), mean (SD) | 0.05 (0.07) | 0.11 (0.09) | 0.05 (0.08) | |
| Change (SUVR/yr), median (IQR) | 0.06 (0.01–0.10) | 0.08 (0.04–0.18) | 0.06 (0.0–0.10) | KW: 1.19 (2), p=0.551c |
| ε3ε3 vs ε4+ | 0.06 (0.01–0.10) | 0.06 (0.0–0.10) | p=0.936d | |
| Participants PiB- at Baseline | ||||
| n | 90 | 24 | 21 | |
| Change (SUVR/yr), mean (SD) | 0.02 (0.04) | 0.03 (0.04) | 0.04 (0.06) | F: 1.0 (2), 0.369a |
| Change (SUVR/yr), median (IQR) | 0.02 (0.0–0.05) | 0.02 (0.0–0.06) | 0.04 (0.0–0.08) | |
| ε3ε3 vs ε4+ | 0.02 (0.04) | 0.04 (0.06) | p=0.340e | |
| Incident PiB+ participants | ||||
| n | 30 | 3 | 7 | |
| Change (SUVR/yr), mean (SD) | 0.06 (0.03) | 0.09 (0.03) | 0.09 (0.03) | |
| Change (SUVR/yr), median (IQR) | 0.06 (0.04–0.08) | 0.10 (0.05–0.11) | 0.09 (0.06–0.12) | KW: 6.24(2), 0.044c |
| ε3ε3 vs ε4+ | 0.06 (0.04–0.08) | 0.09 (0.06–0.12) | p=0.029d | |
p-value from one-way ANOVA
p-value from paired t-test
p-value from Kruskall-Wallis test
p-value from non-parametric Wilcoxon test
p-value from independent two sample t-test
CSF correction of [11C]PiB SUVR values had a modest impact on the shapes of Aβ trajectories. Using CSF corrected data, the repeated measures model identified the same main effects as uncorrected data (APOE-ε4, age2) and interactions (age×APOE-ε4 and age2×APOE-ε4) with similar levels of significance (data not shown). The CSF-corrected data identified age to be a significant main effect (p=0.02), whereas uncorrected data did not (p=0.23). Differences between CSF-corrected and uncorrected Aβ trajectories became more prominent with increasing age, although the shape and the relative differences between the three trajectories was preserved with CSF correction applied (Supplemental Figure e-1).
4.0. DISCUSSION
A main objective of this work was to demonstrate that APOE genotype is predictive of cerebral Aβ burden, the age of onset of Aβ positivity, and also the rate of Aβ accumulation. In our cohort of 428 subjects, we found the prevalence of Aβ positivity among APOE-ε4+ to be approximately 65%, whereas the prevalence was two-fold lower in APOE-ε3ε3 subjects (33.2%), and nearly four-fold lower in APOE-ε2+ (18.6%) (Table 1). These prevalence estimates are consistent with previous [11C]PiB imaging studies (Bilgel et al., 2016; Jansen et al., 2015; Morris et al., 2010), including a recent meta-analysis (Jansen et al., 2015). We found that [11C]PiB SUVR as a continuous measure was higher in APOE-ε4+ than either APOE-ε3ε3 or APOE-ε2+ groups (p ≪ 0.001) and APOE-ε2+ was lower (p = 0.0079) than APOE-ε3ε3 (Figure 1A). These findings are consistent with earlier studies in smaller cohorts, which in general show increased Aβ burden in subjects with one or more APOE-ε4 alleles (Mecca et al., 2018; Wirth et al., 2014; Zwan et al., 2016), and also with post-mortem neuropathological studies relating plaque burden and APOE genotype (Schmechel et al., 1993).
Acknowledging that APOE-ε4 is associated with both an increased prevalence of Aβ positivity and increased Aβ burden for elderly subjects of a given age, a logical question to investigate is whether the increased Aβ burden is due to an earlier age of onset of amyloidogenic processes, an accelerated rate of Aβ accumulation, or both. The clinical onset of symptoms in LOAD is variable, even in subjects who are homozygous for APOE-ε4 (Blacker et al., 1997; Durmaz et al., 2019; Jack et al., 2015; Sando et al., 2008). It is not known to what extent variability in the clinical age of onset of dementia parallels the age of onset of Aβ positivity, although evidence suggests that the clinical age of onset is influenced by a complex interaction of factors including comorbidities (i.e. cerebrovascular disease, diabetes), cognitive resilience factors, and likely other genetic and lifestyle factors.
Although there are few reports of longitudinal [11C]PiB imaging studies designed to explore the association between APOE genotype and age of onset of Aβ positivity, the available data consistently show an earlier age-of-onset in APOE-ε4+ subjects (Bilgel et al., 2016; Fleisher et al., 2013; Jansen et al., 2015; Mishra et al., 2018). Our study is consistent with these observations, where we found the age-of-onset of global [11C]PiB positivity to be 61.1 years in APOE-ε4+ subjects, which precedes APOE-ε3ε3 subjects by ~16 years (Figure 3). We found this association to be evident, albeit attenuated, in a sub-cohort of subjects with normal cognition with an age-of-onset of 69.3 years (Figure 4), suggesting that the finding is not solely driven by those with impaired cognition at study entry. The trajectory model based on the full study cohort (Figure 3) suggests that the inclusion of cognitively impaired subjects may bias the results towards an earlier age-of-onset of Aβ positivity. However, the model that includes only cognitively normal subjects (Figure 4) is likely biased in the direction of a later age-of-onset as certainly some degree of cognitive impairment is expected in a group of subjects aged 75 years and older, of whom 20% are APOE-ε4+ (Table S2). Indeed, recent population studies suggest that at least 30% of APOE-ε4+ subjects age 75 and older meet the criteria for MCI or dementia, which can be higher or lower based on the cohort or APOE-ε4 gene dose (Bonham et al., 2016; Qian et al., 2017). It should be noted that, while some studies have suggested [11C]PiB positivity cutoffs such as those used in this study are too high (Villeneuve et al., 2015), Figure 3 suggests that similar or perhaps exaggerated findings would have resulted from the application of lower cutoff values.
The question of if and to what extent APOE-ε4 positivity influences the rate of Aβ accumulation is less clear. Some longitudinal Aβ imaging studies have suggested that APOE-ε4 positivity is associated with an accelerated rate of Aβ accumulation (Grimmer et al., 2010; Mishra et al., 2018; Villemagne et al., 2011), whereas others have failed to show such an association (Jack et al., 2013; Resnick et al., 2015). Still other studies suggest a more complex association where age and/or disease state potentially modify the relationship between APOE-ε4 and the rate of Aβ accumulation (Lim et al., 2017; Mishra et al., 2018). Indeed, for a sigmoidal biomarker trajectory like those suggested by Jack and Holtzman for Aβ (Jack and Holtzman, 2013), the transition from normal to abnormal biomarker status must be accompanied by an increase in the rate of change in the biomarker from zero during the antecedent normal period to some non-zero abnormal biomarker state. The observation of increased rates of Aβ accumulation during early disease phases would therefore be consistent with a sigmoidal biomarker trajectory. Once the biomarker reaches an abnormal value, the rate of biomarker change may remain constant or, as some studies suggest (Jack et al., 2013), reach a plateau at a more advanced age.
In the present study, longitudinal [11C]PiB SUVR change measures (SUVR/year) determined for individual subjects (Table 4) did not show any effects of APOE genotype on the rate of Aβ accumulation that reached the level of significance, except for the subgroup of incident [11C]PiB positive cases (n=40) where APOE-ε4 positivity was associated with a higher rate of Aβ accumulation than non-carriers (p = 0.029, Table 4). While significant, this result must be interpreted with caution due to the small number of APOE-ε4+ incident PiB-positive subjects (n=7).
Our Aβ trajectory models were also used to assess rates of Aβ accumulation by calculating the slope of the trajectory at the global [11C]PiB positivity threshold. Trajectory models showed strong agreement with individual change measures of [11C]PiB retention (Table 4), predicting rates of Aβ accumulation in APOE-ε4 carriers of 0.043 SUVR/year and 0.029 SUVR/year in APOE-ε3ε3 subjects. However, the 95% confidence intervals for the slope values were largely overlapping between APOE genotype classes and therefore the model does not support an effect of APOE genotype on the rate of Aβ accumulation. Individual measures of [11C]PiB retention show mean annualized changes of 0.05±0.07 and 0.03±0.05 SUVR/year for APOE-ε4 and APOE-ε3ε3 subjects, respectively, which also were not significantly different (p=0.155). Villemagne and colleagues (Villemagne et al., 2011) report a mean rate of Aβ accumulation of APOE-ε4 carriers (0.041 SUVR/year) that was similar to that observed in our study (0.05 SUVR/year) over a comparable follow-up period (38 months), although they observed a significantly slower rate of Aβ accumulation in APOE-ε4 non-carriers (0.016 SUVR/year) than we observed in either APOE-ε3ε3 (0.03 SUVR/year) or APOE-ε2+ (0.04 SUVR/year) subject groups. It is possible that this discrepancy is explained by differences in the age distribution of subjects within APOE subgroups (our APOE-ε3ε3 subjects were ≥ 5 years older than all diagnostic groups reported by (Villemagne et al., 2011)), or possibly by small sample size effects when one considers that positive SUVR changes are driven by the fraction of subjects who are PiB positive. Nevertheless, these data suggest that, once initiated, the process of Aβ accumulation proceeds at a fixed rate independent of APOE genotype, i.e., APOE genotype appears to affect the seeding of Aβ deposition more than the propagation. Furthermore, the data suggest that the increased risk of AD conferred by APOE-ε4 is likely attributable to an earlier age-of-onset of amyloid pathology.
Lastly, consistent with previous observations (Jansen et al., 2015; Morris et al., 2010), our data shows APOE-ε2+ subjects to have a lower rate of PIB-positivity and lower baseline [11C]PiB retention compared to APOE-ε3ε3 or APOE-ε4+ subjects (Table 1), and a lower rate of incident Aβ positivity (12.5%) compared to other APOE classes (33.3%). The Aβ trajectory models in APOE-ε2+ subjects show a modestly delayed age of onset of Aβ positivity compared to APOE-ε3ε3 subjects (82.0 y vs 77.9 y). However, longitudinal [11C]PiB image data in APOE-ε2+ subjects were relatively sparse compared to other genotype classes (Figure 2), particularly so above the [11C]PiB positivity threshold. As such, our trajectory model in APOE-ε2+ subjects should be interpreted cautiously. However, our findings are consistent with earlier reports that describe uniformly low [11C]PiB retention in APOE-ε2+ subjects (Figure 2) and a delayed age of onset of Aβ positivity (Jansen et al., 2015; Morris et al., 2010).
In conclusion, the present work provides further support that APOE genotype is a key modulator of amyloidogenic processes. In our study, APOE-ε4 positivity was associated with increased Aβ load and an earlier age of onset of Aβ positivity compared to other APOE genotype classes, but a similar rate of amyloid accumulation. Our data also suggest a potential protective effect of APOE-ε2 positivity, although the effect is likely more modest than the deleterious effects associated with APOE-ε4 positivity. It is important to note that, while we and others have gathered evidence to show that APOE is an important modulator of amyloidogenesis, our data also suggests that there are likely other important factors that influence Aβ deposition. The variability in the age of onset of Aβ positivity within APOE-ε4 subjects and also the lack of detectible Aβ deposits in some APOE-ε4 positive subjects of advanced age are examples that support a more complex relationship between APOE and Aβ.
Supplementary Material
HIGHLIGHTS.
APOE-ε4 is associated with increased Aβ burden compared to other APOE genotypes.
Age-of-onset of Aβ positivity in APOE-ε4+ antecedes other genotypes by > 15 years.
APOE-ε2+ is associated with reduced Aβ burden and a delayed age-of-onset.
Within-genotype variability suggests additional factors influence Aβ trajectories.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institute on Aging: AG025204, AG005133, and AG025516
Study funding:
National Institute of Aging grants AG025204, AG005133, and AG025516
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Data:
Supplemental figure e-1, Supplemental Tables S1, S2
DISCLOSURE STATEMENT
Chester A. Mathis and William E. Klunk: GE Healthcare holds a license agreement with the University of Pittsburgh based on the technology described in this manuscript. Drs. William E. Klunk and Chester A. Mathis are co-inventors of PiB and, as such, have a financial interest in this license agreement. GE Healthcare provided no financial support for this study and had no role in the design or interpretation of results or preparation of this manuscript. All other authors have no conflicts of interest with this work and had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE, 2008. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 65(11), 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH, 2011. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7(3), 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris LM, Galloway NM, Millard S, Lockhart D, Li G, Galasko DR, Farlow MR, Clark CM, Quinn JF, Kaye JA, Schellenberg GD, Leverenz JB, Seubert P, Tsuang DW, Peskind ER, Yu CE, 2011. Amyloid precursor protein (APP) processing genes and cerebrospinal fluid APP cleavage product levels in Alzheimer’s disease. Neurobiol Aging 32(3), 556 e513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE, 2007. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39(1), 17–23. [DOI] [PubMed] [Google Scholar]
- Bilgel M, An Y, Zhou Y, Wong DF, Prince JL, Ferrucci L, Resnick SM, 2016. Individual estimates of age at detectable amyloid onset for risk factor assessment. Alzheimers Dement 12(4), 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TD, 2008. Genetic aspects of Alzheimer disease. Genet Med 10(4), 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacker D, Haines JL, Rodes L, Terwedow H, Go RC, Harrell LE, Perry RT, Bassett SS, Chase G, Meyers D, Albert MS, Tanzi R, 1997. ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology 48(1), 139–147. [DOI] [PubMed] [Google Scholar]
- Bonham LW, Geier EG, Fan CC, Leong JK, Besser L, Kukull WA, Kornak J, Andreassen OA, Schellenberg GD, Rosen HJ, Dillon WP, Hess CP, Miller BL, Dale AM, Desikan RS, Yokoyama JS, 2016. Age-dependent effects of APOE epsilon4 in preclinical Alzheimer’s disease. Ann Clin Transl Neurol 3(9), 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenstein AR, Copenhaver CI, Mortimer JA, 2006. Early-life risk factors for Alzheimer disease. Alzheimer Dis Assoc Disord 20(1), 63–72. [DOI] [PubMed] [Google Scholar]
- Butters MA, Klunk WE, Mathis CA, Price JC, Ziolko SK, Hoge JA, Tsopelas ND, Lopresti BJ, Reynolds CF 3rd, DeKosky ST, Meltzer CC, 2008. Imaging Alzheimer pathology in late-life depression with PET and Pittsburgh Compound-B. Alzheimer Dis Assoc Disord 22(3), 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Mowrey W, Weissfeld LA, Aizenstein HJ, McDade E, Mountz JM, Nebes RD, Saxton JA, Snitz B, Dekosky S, Williamson J, Lopez OL, Price JC, Mathis CA, Klunk WE, 2013. Classification of amyloid-positivity in controls: comparison of visual read and quantitative approaches. Neuroimage 71, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, Nebes RD, Saxton JA, Snitz BE, Aizenstein HA, Wolk DA, Dekosky ST, Mathis CA, Klunk WE, 2009. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci 29(47), 14770–14778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA, 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123), 921–923. [DOI] [PubMed] [Google Scholar]
- Durmaz A, Kumral E, Durmaz B, Onay H, Aslan GI, Ozkinay F, Pehlivan S, Orman M, Cogulu O, 2019. Genetic factors associated with the predisposition to late onset Alzheimer’s disease. Gene 707, 212–215. [DOI] [PubMed] [Google Scholar]
- Eichner JE, Kuller LH, Ferrell RE, Meilahn EN, Kamboh MI, 1990. Phenotypic effects of apolipoprotein structural variation on lipid profiles. III. Contribution of apolipoprotein E phenotype to prediction of total cholesterol, apolipoprotein B, and low density lipoprotein cholesterol in the healthy women study. Arteriosclerosis 10(3), 379–385. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM, 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278(16), 1349–1356. [PubMed] [Google Scholar]
- Fleisher AS, Chen K, Liu X, Ayutyanont N, Roontiva A, Thiyyagura P, Protas H, Joshi AD, Sabbagh M, Sadowsky CH, Sperling RA, Clark CM, Mintun MA, Pontecorvo MJ, Coleman RE, Doraiswamy PM, Johnson KA, Carpenter AP, Skovronsky DM, Reiman EM, 2013. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging 34(1), 1–12. [DOI] [PubMed] [Google Scholar]
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL, 2006. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63(2), 168–174. [DOI] [PubMed] [Google Scholar]
- Grimmer T, Tholen S, Yousefi BH, Alexopoulos P, Forschler A, Forstl H, Henriksen G, Klunk WE, Mathis CA, Perneczky R, Sorg C, Kurz A, Drzezga A, 2010. Progression of cerebral amyloid load is associated with the apolipoprotein E epsilon4 genotype in Alzheimer’s disease. Biol Psychiatry 68(10), 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Holtzman DM, 2013. Biomarker modeling of Alzheimer’s disease. Neuron 80(6), 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Wiste HJ, Lesnick TG, Weigand SD, Knopman DS, Vemuri P, Pankratz VS, Senjem ML, Gunter JL, Mielke MM, Lowe VJ, Boeve BF, Petersen RC, 2013. Brain beta-amyloid load approaches a plateau. Neurology 80(10), 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Wiste HJ, Weigand SD, Knopman DS, Vemuri P, Mielke MM, Lowe V, Senjem ML, Gunter JL, Machulda MM, Gregg BE, Pankratz VS, Rocca WA, Petersen RC, 2015. Age, Sex, and APOE epsilon4 Effects on Memory, Brain Structure, and beta-Amyloid Across the Adult Life Span. JAMA Neurol 72(5), 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Amyloid Biomarker Study G, Aalten P, Aarsland D, Alcolea D, Alexander M, Almdahl IS, Arnold SE, Baldeiras I, Barthel H, van Berckel BN, Bibeau K, Blennow K, Brooks DJ, van Buchem MA, Camus V, Cavedo E, Chen K, Chetelat G, Cohen AD, Drzezga A, Engelborghs S, Fagan AM, Fladby T, Fleisher AS, van der Flier WM, Ford L, Forster S, Fortea J, Foskett N, Frederiksen KS, Freund-Levi Y, Frisoni GB, Froelich L, Gabryelewicz T, Gill KD, Gkatzima O, Gomez-Tortosa E, Gordon MF, Grimmer T, Hampel H, Hausner L, Hellwig S, Herukka SK, Hildebrandt H, Ishihara L, Ivanoiu A, Jagust WJ, Johannsen P, Kandimalla R, Kapaki E, Klimkowicz-Mrowiec A, Klunk WE, Kohler S, Koglin N, Kornhuber J, Kramberger MG, Van Laere K, Landau SM, Lee DY, de Leon M, Lisetti V, Lleo A, Madsen K, Maier W, Marcusson J, Mattsson N, de Mendonca A, Meulenbroek O, Meyer PT, Mintun MA, Mok V, Molinuevo JL, Mollergard HM, Morris JC, Mroczko B, Van der Mussele S, Na DL, Newberg A, Nordberg A, Nordlund A, Novak GP, Paraskevas GP, Parnetti L, Perera G, Peters O, Popp J, Prabhakar S, Rabinovici GD, Ramakers IH, Rami L, Resende de Oliveira C, Rinne JO, Rodrigue KM, Rodriguez-Rodriguez E, Roe CM, Rot U, Rowe CC, Ruther E, Sabri O, Sanchez-Juan P, Santana I, Sarazin M, Schroder J, Schutte C, Seo SW, Soetewey F, Soininen H, Spiru L, Struyfs H, Teunissen CE, Tsolaki M, Vandenberghe R, Verbeek MM, Villemagne VL, Vos SJ, van Waalwijk van Doorn LJ, Waldemar G, Wallin A, Wallin AK, Wiltfang J, Wolk DA, Zboch M, Zetterberg H, 2015. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313(19), 1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboh MI, Aston CE, Hamman RF, 1995. The relationship of APOE polymorphism and cholesterol levels in normoglycemic and diabetic subjects in a biethnic population from the San Luis Valley, Colorado. Atherosclerosis 112(2), 145–159. [DOI] [PubMed] [Google Scholar]
- Kenward MG, Roger JH, 1997. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 53(3), 983–997. [PubMed] [Google Scholar]
- Kim YJ, Seo SW, Park SB, Yang JJ, Lee JS, Lee J, Jang YK, Kim ST, Lee KH, Lee JM, Lee JH, Kim JS, Na DL, Kim HJ, 2017. Protective effects of APOE e2 against disease progression in subcortical vascular mild cognitive impairment patients: A three-year longitudinal study. Sci Rep 7(1), 1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YY, Mormino EC, Alzheimer’s Disease Neuroimaging I, 2017. APOE genotype and early beta-amyloid accumulation in older adults without dementia. Neurology 89(10), 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez OL, Becker JT, Chang Y, Klunk WE, Mathis C, Price J, Aizenstein HJ, Snitz B, Cohen AD, DeKosky ST, Ikonomovic M, Kamboh MI, Kuller LH, 2018. Amyloid deposition and brain structure as long-term predictors of MCI, dementia, and mortality. Neurology 90(21), e1920–e1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis CA, Kuller LH, Klunk WE, Snitz BE, Price JC, Weissfeld LA, Rosario BL, Lopresti BJ, Saxton JA, Aizenstein HJ, McDade EM, Kamboh MI, DeKosky ST, Lopez OL, 2013. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol 73(6), 751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ, 2010. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330(6012), 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH, 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7(3), 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee RL, Yee SH, Price JC, Klunk WE, Rosario B, Weissfeld L, Ziolko S, Berginc M, Lopresti B, Dekosky S, Mathis CA, 2009. Consideration of optimal time window for Pittsburgh compound B PET summed uptake measurements. J Nucl Med 50(3), 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca AP, Barcelos NM, Wang S, Bruck A, Nabulsi N, Planeta-Wilson B, Nadelmann J, Benincasa AL, Ropchan J, Huang Y, Gelernter J, Van Ness PH, Carson RE, van Dyck CH, 2018. Cortical beta-amyloid burden, gray matter, and memory in adults at varying APOE epsilon4 risk for Alzheimer’s disease. Neurobiol Aging 61, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer CC, Leal JP, Mayberg HS, Wagner HN Jr., Frost JJ, 1990. Correction of PET data for partial volume effects in human cerebral cortex by MR imaging. J Comput Assist Tomogr 14(4), 561–570. [DOI] [PubMed] [Google Scholar]
- Mishra S, Blazey TM, Holtzman DM, Cruchaga C, Su Y, Morris JC, Benzinger TLS, Gordon BA, 2018. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE epsilon4 genotype. Brain 141(6), 1828–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CM, Benjamin R, Leake A, McArthur FK, Candy JM, Ince PG, Torvik A, Bjertness E, Edwardson JA, 1995. Effect of apolipoprotein E genotype on Alzheimer’s disease neuropathology in a cohort of elderly Norwegians. Neurosci Lett 201(1), 45–47. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA, 2010. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67(1), 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni NK, Tudorascu D, Campbell E, Snitz BE, Cohen AD, Halligan E, Mathis CA, Aizenstein HJ, Klunk WE, 2019. Association Between Amyloid-beta, Small-vessel Disease, and Neurodegeneration Biomarker Positivity, and Progression to Mild Cognitive Impairment in Cognitively Normal Individuals. J Gerontol A Biol Sci Med Sci 74(11), 1753–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Jobst KA, Johnston C, Litchfield S, Sim E, Smith AD, 1995. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience 69(3), 757–761. [DOI] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, DeKosky ST, Mathis CA, 2005. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab 25(11), 1528–1547. [DOI] [PubMed] [Google Scholar]
- Qian J, Wolters FJ, Beiser A, Haan M, Ikram MA, Karlawish J, Langbaum JB, Neuhaus JM, Reiman EM, Roberts JS, Seshadri S, Tariot PN, Woods BM, Betensky RA, Blacker D, 2017. APOE-related risk of mild cognitive impairment and dementia for prevention trials: An analysis of four cohorts. PLoS Med 14(3), e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall SC Jr., Weisgraber KH, Mahley RW, 1982. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem 257(8), 4171–4178. [PubMed] [Google Scholar]
- Resnick SM, Bilgel M, Moghekar A, An Y, Cai Q, Wang MC, Thambisetty M, Prince JL, Zhou Y, Soldan A, Wong DF, O’Brien RJ, Ferrucci L, Albert MS, 2015. Changes in Abeta biomarkers and associations with APOE genotype in 2 longitudinal cohorts. Neurobiol Aging 36(8), 2333–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda AR, Montoliu-Gaya L, Villegas S, 2019. The Role of Apolipoprotein E Isoforms in Alzheimer’s Disease. J Alzheimers Dis 68(2), 459–471. [DOI] [PubMed] [Google Scholar]
- Rosario BL, Weissfeld LA, Laymon CM, Mathis CA, Klunk WE, Berginc MD, James JA, Hoge JA, Price JC, 2011. Inter-rater reliability of manual and automated region-of-interest delineation for PiB PET. Neuroimage 55(3), 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, Saltvedt I, White LR, Lydersen S, Aasly JO, 2008. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, et al. , 1993. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43(8), 1467–1472. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD, 1993. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90(20), 9649–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PP, Singh M, Mastana SS, 2006. APOE distribution in world populations with new data from India and the UK. Ann Hum Biol 33(3), 279–308. [DOI] [PubMed] [Google Scholar]
- Snitz BE, Lopez OL, McDade E, Becker JT, Cohen AD, Price JC, Mathis CA, Klunk WE, 2015. Amyloid-beta Imaging in Older Adults Presenting to a Memory Clinic with Subjective Cognitive Decline: A Pilot Study. J Alzheimers Dis 48 Suppl 1, S151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang MX, Maestre G, Tsai WY, Liu XH, Feng L, Chung WY, Chun M, Schofield P, Stern Y, Tycko B, Mayeux R, 1996. Relative risk of Alzheimer disease and age-at-onset distributions, based on APOE genotypes among elderly African Americans, Caucasians, and Hispanics in New York City. Am J Hum Genet 58(3), 574–584. [PMC free article] [PubMed] [Google Scholar]
- Tudorascu DL, Anderson SJ, Minhas DS, Yu Z, Comer D, Lao P, Hartley S, Laymon CM, Snitz BE, Lopresti BJ, Johnson S, Price JC, Mathis CA, Aizenstein HJ, Klunk WE, Handen BL, Christian BT, Cohen AD, 2019. Comparison of longitudinal Abeta in nondemented elderly and Down syndrome. Neurobiol Aging 73, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier WM, Pijnenburg YA, Schoonenboom SN, Dik MG, Blankenstein MA, Scheltens P, 2008. Distribution of APOE genotypes in a memory clinic cohort. Dement Geriatr Cogn Disord 25(5), 433–438. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, Ackermann U, Jones G, Szoeke C, Salvado O, Martins R, O’Keefe G, Mathis CA, Klunk WE, Ames D, Masters CL, Rowe CC, 2011. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol 69(1), 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, Madison C, Ayakta N, Ghosh PM, La Joie R, Arthur-Bentil SK, Vogel JW, Marks SM, Lehmann M, Rosen HJ, Reed B, Olichney J, Boxer AL, Miller BL, Borys E, Jin LW, Huang EJ, Grinberg LT, DeCarli C, Seeley WW, Jagust W, 2015. Existing Pittsburgh Compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 138(Pt 7), 2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Bienias JL, Berry-Kravis E, Evans DA, Bennett DA, 2002. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry 73(6), 672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Villeneuve S, La Joie R, Marks SM, Jagust WJ, 2014. Gene-environment interactions: lifetime cognitive activity, APOE genotype, and beta-amyloid burden. J Neurosci 34(25), 8612–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk DA, Price JC, Madeira C, Saxton JA, Snitz BE, Lopez OL, Mathis CA, Klunk WE, DeKosky ST, 2012. Amyloid imaging in dementias with atypical presentation. Alzheimers Dement 8(5), 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Nho K, Del-Aguila JL, Wang X, Risacher SL, Fan KH, Snitz BE, Aizenstein HJ, Mathis CA, Lopez OL, Demirci FY, Feingold E, Klunk WE, Saykin AJ, Alzheimer’s Disease Neuroimaging I, Cruchaga C, Kamboh MI, 2018. Genome-wide association study of brain amyloid deposition as measured by Pittsburgh Compound-B (PiB)-PET imaging. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwan MD, Villemagne VL, Dore V, Buckley R, Bourgeat P, Veljanoski R, Salvado O, Williams R, Margison L, Rembach A, Macaulay SL, Martins R, Ames D, van der Flier WM, Ellis KA, Scheltens P, Masters CL, Rowe CC, 2016. Subjective Memory Complaints in APOEvarepsilon4 Carriers are Associated with High Amyloid-beta Burden. J Alzheimers Dis 49(4), 1115–1122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.