

Abstract
Background:
Microdeletions of the MEF2C gene are linked to a syndromic form of autism termed MEF2C Haploinsufficiency Syndrome (MCHS). MEF2C hypofunction in neurons is presumed to underlie most of the MCHS symptoms. However, it is unclear in which cell populations MEF2C functions to regulate neurotypical development.
Methods:
Multiple biochemical, molecular, electrophysiological, behavioral and transgenic mouse approaches were used to characterize MCHS-relevant synaptic, behavioral and gene expression changes in mouse models of MCHS.
Results:
We show here that MCHS-associated missense mutations cluster in the conserved DNA binding domain and disrupt MEF2C DNA binding. DNA binding-deficient global Mef2c heterozygous mice (Mef2c-Het) display numerous MCHS-related behaviors, including autism-related behaviors, changes in cortical gene expression, and deficits in cortical excitatory synaptic transmission. We detected hundreds of dysregulated genes in Mef2c-Het cortex, including significant enrichments of autism risk and excitatory neuron genes. In addition, we observe an enrichment of upregulated microglial genes, but not due to neuroinflammation in the Mef2c-Het cortex. Importantly, conditional Mef2c heterozygosity in forebrain excitatory neurons reproduces a subset of the Mef2c-Het phenotypes, while conditional Mef2c heterozygosity in microglia reproduces social deficits and repetitive behavior.
Conclusion:
Together, we show that mutations found in individuals with MCHS disrupt the DNA-binding function of MEF2C, and DNA binding-deficient Mef2c global heterozygous mice display numerous MCHS-related phenotypes, including excitatory neuron and microglia gene expression changes. Our findings suggest that MEF2C regulates typical brain development and function through multiple cell types, including excitatory neuronal and neuroimmune populations.
Keywords: Mef2c, mouse, autism, neuron, microglia, neurodevelopmental disorder
Introduction:
Myocyte Enhancer Factor 2 (MEF2) proteins are members of the MADS family of transcription factors that regulate gene expression during development and adulthood. In the brain, MEF2C is important for neuronal differentiation and synapse development (1). MEF2 proteins regulate numerous genes associated with synapse formation and function as well as multiple genes linked to neurodevelopmental disorders, including autism spectrum disorder (ASD) (2–4). Constitutively-active MEF2C can promote glutamatergic synapse elimination, a process requiring the RNA-binding function of the Fragile × Mental Retardation protein (FMRP) (5–8). Conditional knockout of Mef2c in neuronal populations within the mouse brain produces a myriad of severe behavioral and synaptic phenotypes, which emphasizes the importance of this gene in healthy brain development (2, 9–12).
MEF2C in the developing and mature brain is also expressed in microglia (13–15) – a population of macrophage-like cells throughout the brain that regulates synapse formation and pruning during early brain development (16–18). Microglia influence a number of brain functions, including synapse elimination, synapse formation, fasciculation of the corpus callosum, survival of oligodendrocyte precursor cells, and phagocytosis of other brain cells (16, 19–24). Microglia are recognized as not just responding to infection or injury, but as important regulators of brain development and function (25). In addition, microglial dysfunction might play an important role in disease pathology for other neurodevelopmental disorders, including Rett Syndrome (26–29).
Microdeletions on chromosome 5q14.3, that include the MEF2C gene, or point mutations within the protein-coding region of MEF2C are linked to a recently described neurodevelopmental disorder, termed MEF2C Haploinsufficiency Syndrome (MCHS) (30–40). Common symptoms of MCHS include autism spectrum disorder (ASD), absence of speech, stereotypical behaviors, hyperactivity, intellectual disability, hypotonia and motor abnormalities, high pain tolerance, sleep disturbances, and epilepsy. Individuals with MEF2C point mutations typically present with fewer and/or milder symptoms (30–40). Due to the abundance of neurological symptoms and neuronal-enriched expression of MEF2C, MEF2C haploinsufficiency within neurons is presumed to underlie most, if not all, of the MCHS symptoms. Interestingly, single-cell genomic profiling from cortical tissue of patients with idiopathic autism revealed that upper-layer excitatory neurons and microglia are preferentially affected in autism (41), and since both neurons and microglia express MEF2C, we sought to explore the possible cell type-specific effects of MEF2C hypofunction in MCHS-related behaviors in a construct-valid mouse model of human MCHS.
Methods and Materials:
Patients
Patients with developmental delay and a significant variant in the MEF2C gene were selected for this study. These patients were seen for clinical genetics evaluations at the Greenwood Genetic Center (Greenwood, SC) and data from these visits were gathered from records review. Internal informed consent to review and publish the data was obtained for each subject.
Animals
Mef2c+/− (Mef2c-Het) mice were generated by crossing Mef2c-flox mice (RRID:MGI:3719006) to Prm-Cre mice (Jackson Laboratory #003328). The Prm-Cre allele was subsequently removed during repeated backcrossing to C57BL/6J wild-type mice. Mef2c conditional heterozygous mice were generated by crossing Mef2c-flox mice with cell type-selective Cre-expressing transgenic mice (Emx1-Cre (Jackson Laboratory #005628 (42)), PV-Cre (Jackson Laboratory #017320), Pcp2-Cre (Jackson Laboratory #004146), or Cx3Cr1creER/creER (Jackson Laboratory #021160 (20)) to generate Mef2cfl/+; Cre+ conditional heterozygous (Mef2c cHet) mice that were compared to their Cre-negative or flox-negative littermates (Control). Experimenters were blinded to the mouse genotype during data acquisition and analysis. All procedures were conducted in accordance with the Medical University of South Carolina Institutional Animal Care and Use Committee (IACUC) and NIH guidelines.
Detailed Materials and Methods can be found within the supplemental information.
Results:
Patient MEF2C missense mutations cluster in DNA binding and dimerization domains and disrupt DNA binding
Deletions or mutations in MEF2C are assumed to create loss-of-function alleles that cause the symptoms of MCHS (30–40). Given that microdeletions of 5q14.3 often include additional genes beyond MEF2C, we identified individuals with mutations within the MEF2C protein-coding region, including an intragenic duplication (i.e. p.D40_C41dup) and two missense variants (i.e. p.K30N and p.I46T) (Table S1). We compared their clinical histories to those associated with two previously reported missense variants in the MEF2C gene (31). All five patients presented with global developmental delay and seizures. Common features of these individuals included absence of speech, repetitive movements, hypotonia, varied, but inconsistent, abnormalities on brain MRI, and breathing disturbances. High pain tolerance was noted in two of the patients. There were some minor facial dysmorphisms noted, though there did not seem to be a consistently recognizable gestalt. When a list of additional MCHS mutations was assembled (personal communications), several frameshift and premature stop codon mutations were identified – all of which, if stable, are predicted to produce a truncated MEF2C protein lacking its C-terminal nuclear localization sequence. We noted that all of the MEF2C missense (or small duplication) mutations were clustered within the highly conserved MADS (DNA binding) or MEF2 (dimerization) domains (Fig. 1A). In a MEF2 response element DNA binding assay, all five of the MADS domain patient mutations caused a loss of MEF2C DNA binding (Figs. 1B–C, S1A), and they did not appear to interfere with wild-type MEF2C DNA binding (Fig. S1B), suggesting a loss-of-function phenotype.
Figure 1.
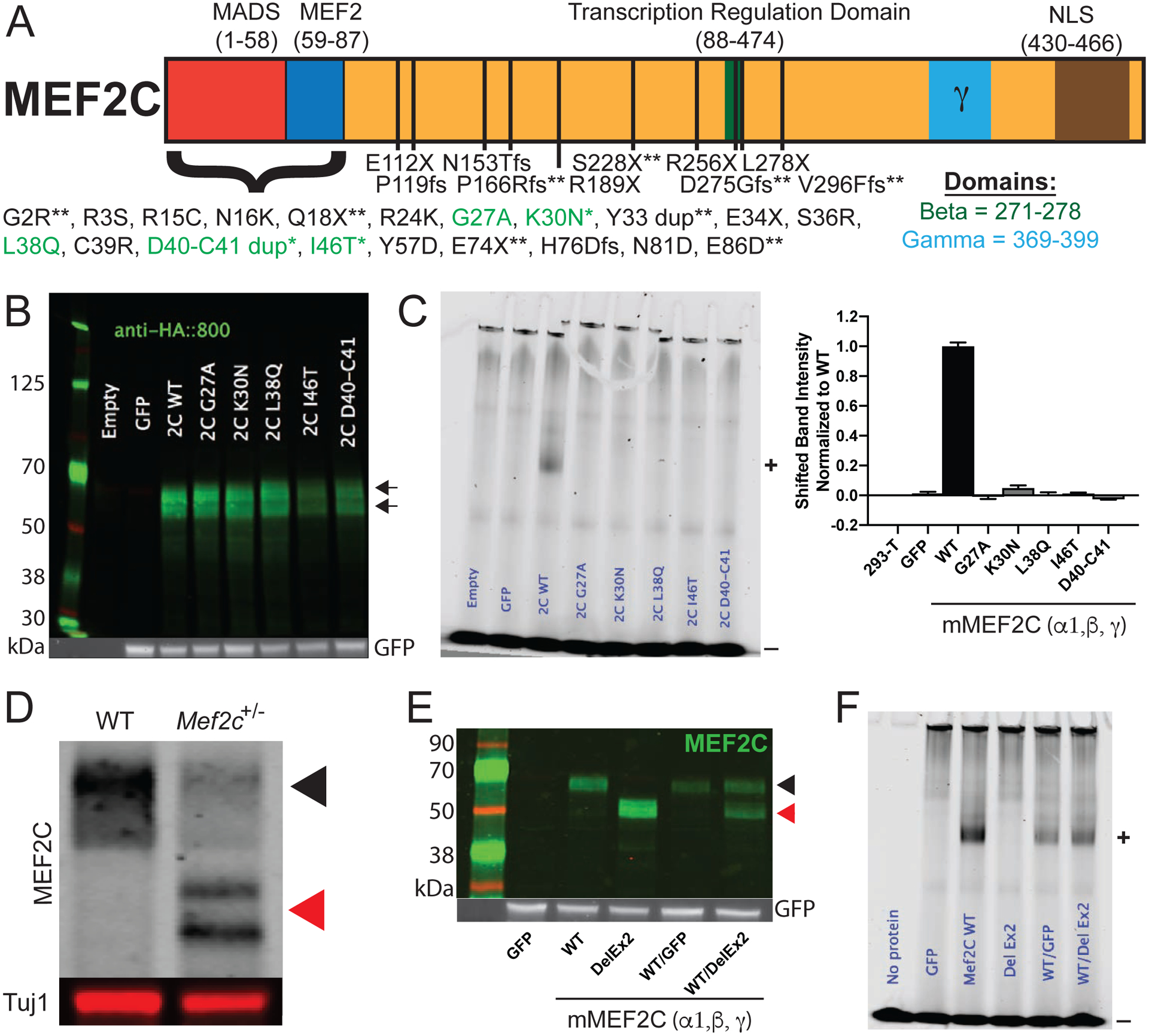
MCHS associated mutations in MEF2C disrupt DNA binding. (A) Schematic of the MEF2C protein with locations of MCHS mutations. MCHS mutations in green are further characterized (B-C). MCHS mutations that are newly described in this manuscript are denoted with “ * ”. MCHS mutations not previously reported (personal communications) are denoted by “ ** “. The alternatively spliced beta (green) and gamma (blue) domains are shown. All MEF2C transcripts contain a C-terminal Nuclear Localization Sequence (NLS) that is disrupted by the frame-shift (fs) mutations. (B) Western blot of MEF2C wild-type (WT) and MCHS mutations in 293-T cells show that all MCHS mutations lead to protein expression. Arrows denote WT and mutant protein MEF2C bands. (C) Electrophoretic mobility shift assay (EMSA) using fluorescently labeled MEF2 response element (MRE) probe and MEF2C protein lysates from 293-T cells containing MEF2C mutations. MEF2C bound probe is shifted in the gel (denoted by “+”). Unbound fluorescent probe is denoted with a “−“. Only MEF2C WT binds to the fluorescently labeled MRE, while MCHS mutant proteins fail to bind the MRE probe (C). Quantification of bound probe is included (C). (D) Western blot of MEF2C from cortical lysates of control and Mef2c-Het mice. The black arrow denotes MEF2C WT and red arrows denote MEF2C DelEx2 (D,E). (E) Western blot of MEF2C WT and MEF2C DelEx2 from 293-T cells. (F) MEF2C DelEx2 fails to bind the MRE probe and does not interfere with MEF2C WT binding to MRE probes. “+” is bound probe. “−“ is unbound probe. Data are reported as mean ± SEM. Also see Figure S1.
Mef2c heterozygous mouse model
To model the genetics of MCHS in mice, we generated a global heterozygous Mef2c mutant mouse lacking exon 2 (Mef2c+/ΔEx2 or Mef2c-Het) (Fig. 1D), which encodes a large portion of the MADS/MEF2 domains. The near full-length MEF2CΔEx2 protein had no detectable DNA binding affinity and did not reduce DNA binding affinity of wild-type MEF2C (Figs. 1E–F, S1C). We observed a non-Mendelian frequency of Mef2c-Hets, suggesting a partial embryonic lethality (Fig. S1D), similar to a previous report (43). To assess for gross morphological changes in Mef2c-Het brains, we measured the cortical thickness of the barrel cortex. We did not observe differences in cortical thickness of the barrel cortex between Mef2c-Hets and controls (Fig. S1E).
We examined whether male and female Mef2c-Het mice showed behavior phenotypes reminiscent of MCHS symptoms. Using a three-chamber social interaction (SI) test, we observed that Mef2c-Het mice have a lack of social preference with a novel same-sex mouse (Fig. 2A). We also found that Mef2c-Het male and female pups (P7-P10) produced significantly fewer ultrasonic vocalization (USV) calls during maternal separation (Fig. 2B), and young adult Mef2c-Het males produced significantly fewer USV calls (Fig. 2C) in the presence of an estrous female, suggesting that Mef2c-Hets have deficits in a putative species-appropriate form of social communication. Male Mef2c-Hets were hyperactive in a novel environment (Fig. 2E) and displayed an increase in jumping (Fig. 2F), a repetitive-type motor behavior; however, young adult Mef2c-Hets displayed normal performance on the accelerating rotarod test of motor coordination (Fig. 2D). In addition, Mef2c-Hets showed increased exploration of the open, unprotected arm of the elevated plus maze (Fig. 2G). Interestingly, Mef2c-Het mice showed a reduction in startle response to electrical foot-shocks (Fig. 2H). This phenotype might reflect reduced pain sensitivity, similar to MCHS subjects (31, 33), since startle responses to multiple white-noise intensities were indistinguishable from controls (Fig. S2A).
Figure 2.
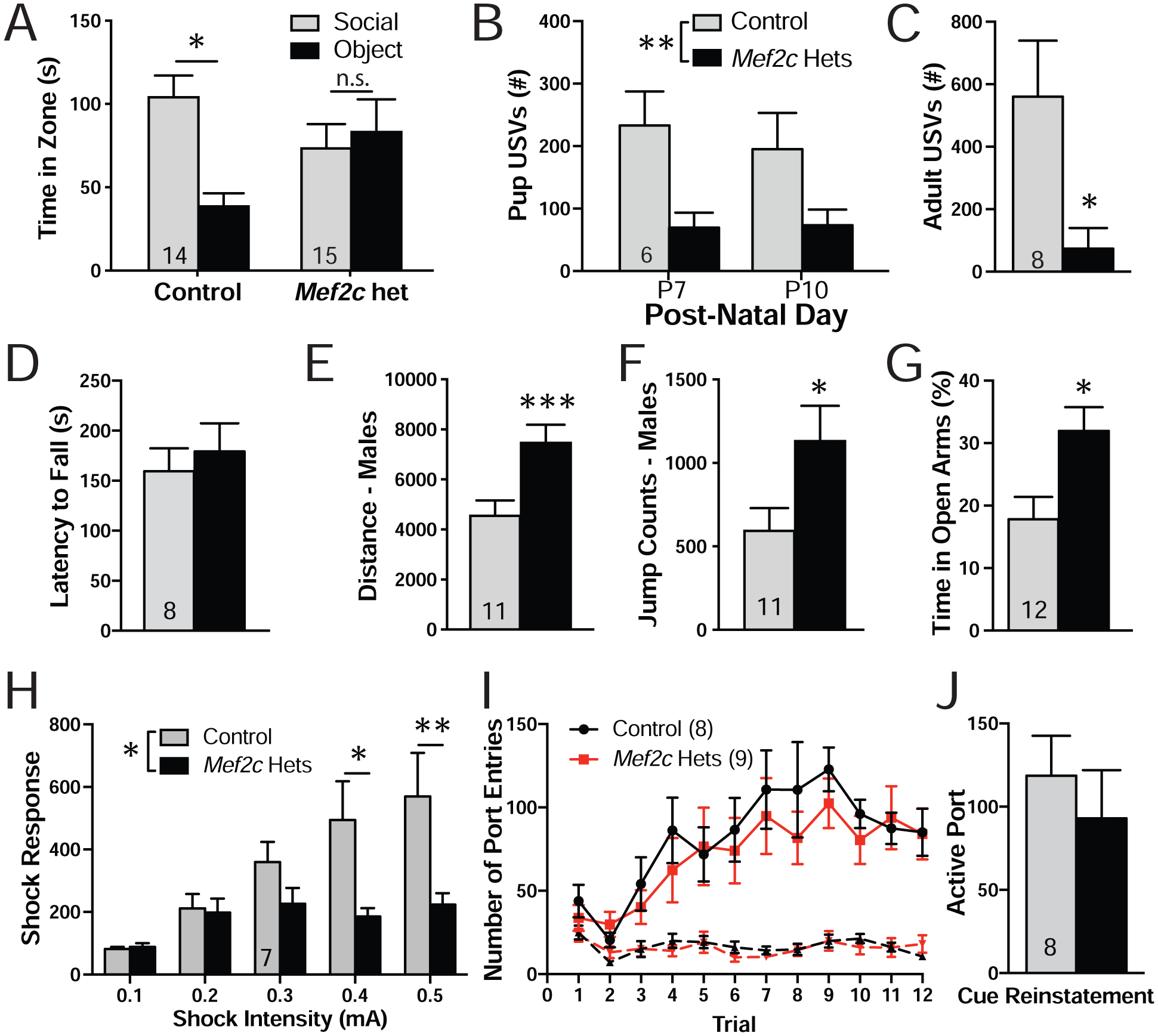
Mef2c-Het mice display multiple MCHS-relevant behaviors. (A) Three chamber social interaction test. Control mice spent significantly more time interacting with a novel animal over a novel object while the Mef2c-Het mice showed no preference for the novel object or the novel animal. (B) Mef2c-Het pups emitted fewer ultrasonic vocalizations (USVs) during maternal separation in early post-natal development. (C) Adult male Mef2c-Het mice produced fewer USVs that control mice in the presence of a female mouse in estrous. (D) Both control and Mef2c-Het mice have similar latencies to fall on an accelerating rotarod. (E,F) Male Mef2c-Het mice are hyperactive (E) and show increased jump counts (F). (G) Mef2c-Het mice spend significantly more time on the open arms of the elevated-plus maze. (H) Mef2c-Het mice have reduced response to shock. (I) Both control and Mef2c-Het mice increase the number of active port entries (solid line) during sucrose self-administration. Dashed line represents inactive port entries. (J) Both control and Mef2c-Hets show similar active port entries during cue-induced reinstatement of sucrose seeking. Data are reported as mean ± SEM. Statistical significance was determined by 2-way ANOVA (A,B,H,I) or unpaired t-test (C-G,J). *p<0.05, **p<0.01, ***p<0.005, n.s. = not significant. Number of animals (n) are reported in each graph for respective experiment. Also see Figure S2.
Despite a common occurrence of intellectual disability in MCHS, we failed to detect any clear learning and memory-related deficits in Pavlovian fear conditioning tests (Figs. S2B–D), the Barnes maze test for spatial learning and memory (Fig. S2E), and the Y-maze test for spatial working memory (Fig. S2F) in the Mef2c-Hets. These mice also showed a strong preference for the novel object in the novel object recognition test (Fig. S2G), and normal sucrose preference in a two-bottle choice test (Fig. S2H). In the cognitively-demanding operant sucrose self-administration (SA) assay, the Mef2c-Hets displayed wild-type levels of operant learning, operant discrimination (active vs. inactive port), context-related sucrose seeking after abstinence, extinction learning and cue-induced reinstatement (Figs. 2I–J, S2I–L). Taken together, our findings suggest that, unlike the conditional knockout of Mef2c in Emx1-lineage cells (2) or a related study (43), the global loss of one functional copy of Mef2c in mice is not sufficient to produce detectable deficits in learning and memory in the C57BL6/J genetic background.
Mef2c-Het mice display input-selective reductions in cortical excitatory synaptic transmission
In young Mef2c-Hets (p35-p40), gross structural organization of barrel fields within cortical layer 4 of the SSCtx appeared normal (Fig. 3A), and in SSCtx layer 2/3 pyramidal neurons, we detected no significant differences by genotype for intrinsic excitability (Fig. S3A), dendritic spine density or spine head diameter of apical or basal dendrites (Fig. S3B), or GABA-mediated inhibitory synaptic transmission (mIPSCs) (Fig. 3B). However, patch-clamp recordings of layer 2/3 neurons revealed an input-selective deficit in glutamatergic synaptic transmission. Electrical stimulation of horizontal fibers in layer 2/3 of a neighboring cortical column produced a significant reduction in the amplitude of evoked excitatory postsynaptic currents (eEPSCs) (Fig. 3C), suggesting a reduction in pre- and/or postsynaptic transmission. Paired-pulse facilitation (PPF) analysis (50 ms interstimulus interval) of local horizontal inputs revealed a significant increase in PPF ratio (Fig. 3C), indicating a decrease in presynaptic release probability (44). These effects were input-selective given that electrical stimulation of layer 4 (within the same cortical column) produced eEPSC and PPF responses in layer 2/3 neurons that were indistinguishable from controls (Fig. 3D). To examine if reductions in AMPA-mediated postsynaptic strength might also contribute to the reduced horizontal eEPSCs (Fig. 3C), we measured miniature EPSCs (mEPSCs) under conditions where action potentials are blocked pharmacologically. In layer 2/3 cells from Mef2c-Hets, we observed a significant reduction in mEPSCs amplitude (Fig. 3E), suggesting an overall reduction in AMPA-mediated postsynaptic strength. Similar to layer 2/3, we also observed a significant reduction of mEPSC amplitude in SSCtx layer 5 pyramidal neurons of Mef2c-Hets (Fig. 3F), suggesting that the reduction in glutamatergic postsynaptic strength is not limited to a specific cortical layer. Consistent with layer 2/3 pyramidal neurons, we did not observe any differences in dendritic spine density or dendritic spine head diameter in basal dendrites from layer 5 pyramidal neurons (Fig. S3C). There was no effect of genotype on layer 5 mEPSC frequency (Fig. 3F), but we observed a significant increase in the layer 2/3 mEPSCs frequency (Fig. 3E) that was not explained by an increase in dendritic spine density (Fig. S3B) or effects on presynaptic functions of local inputs (Figs. 3C,D), and might represent a compensatory effect of long-range connections (11).
Figure 3.
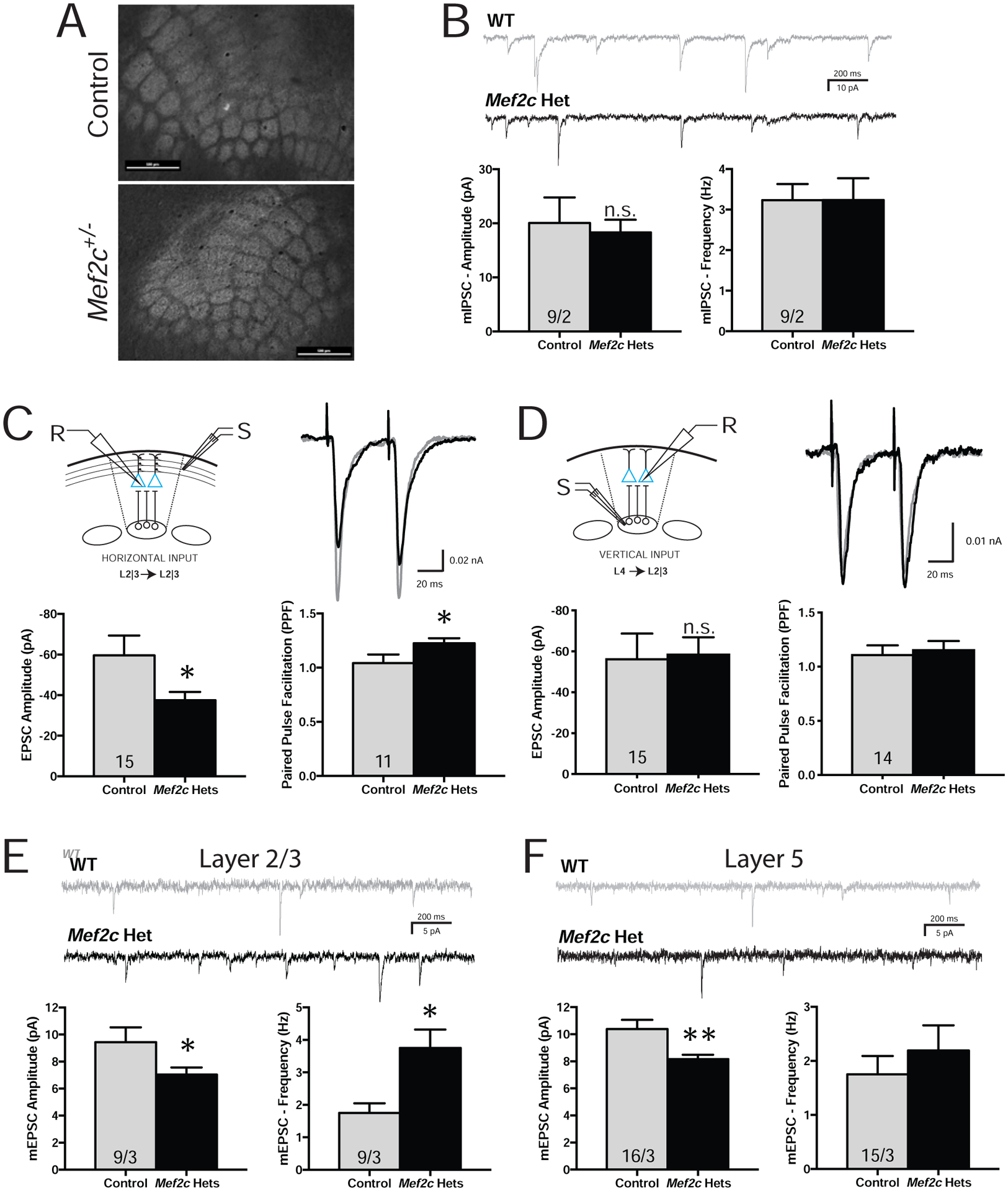
Mef2c-Het mice have alterations in cortical synaptic transmission. (A) Both control and Mef2c-Het mice have normal barrel fields in the somatosensory cortex, as reflected by VGlut2 staining. Scale bar=500 μm. (B-F) Ex vivo recordings from organotypic slices were collected from pyramidal neurons within the barrel cortex field. (B) No changes were observed in mIPSC amplitude or frequency in the Mef2c-Het layer 2/3 pyramidal neurons. (C) Reduced EPSC amplitude and increased paired pulse facilitation (PPF) were observed in layer 2/3 Mef2c-Het neurons after stimulating input neurons from neighboring layer 2/3 neurons in adjacent barrel fields (horizontal inputs). (D) No changes in evoked EPSC amplitude or PPF were observed in layer 2/3 pyramidal neurons after stimulating input neurons from layer 4 (vertical inputs). “R” is recording electrode. “S” is stimulating electrode. (E,F) Mef2c-Het cortical pyramidal neurons have reduced mEPSC amplitude in layer 2/3 (E) and layer 5 (F), and increased mEPSC frequency in layer 2/3 (E). Data are reported as mean ± SEM. Statistical significance was determined by unpaired t-test. *p<0.05. Number of cells and animals, respectively, are reported in each graph. Also see Figure S3.
Mef2c-Het mice display dysregulation of cortical genes associated with ASD risk, excitatory neurons and microglia
Using an unbiased RNA-sequencing (RNA-Seq) approach, we examined gene expression from whole cortex in control and Mef2c-Hets (p35-p40), and we identified 490 genes that were significantly dysregulated (FDR<0.05; Figs. 4A and S4A; Tables S2–S3). We confirmed the differential expression of select Mef2c-Het differentially expressed genes (DEGs) that are associated with ASD risk, microglia, and others by qRT-PCR (Fig. 4D). We also investigated the association of Mef2c-Het DEGs with sequencing data from various brain disorders. We found that the Mef2c-Het DEGs, particularly the downregulated genes, were overrepresented in genes associated with ASD risk and FMRP binding (Figs. 4B,D; Table S2). We also assessed enrichment for Mef2c-Het DEGs in genes that are dysregulated in a meta-analysis of transcriptomic data across neuropsychiatric disorders (45). Interestingly, Mef2c-Het DEGs, particularly the downregulated genes, were significantly enriched for a PsychENCODE excitatory neuron module of genes that are downregulated in ASD (versus other neuropsychiatric disorders) brains (geneM1; Fig. 4C; Table S2). Mef2c-Het DEGs, particularly the upregulated genes, were enriched in PsychENCODE module 6, which is a microglia module of genes upregulated in ASD, but downregulated in SCZ and BPD (geneM6; Fig. 4C; Table S2). Using single-cell RNA-seq data from mouse cortex (46), we observed that Mef2c-Het DEGs were strongly enriched for cortical excitatory neuron genes and microglia genes (Fig. S4B; Table S3), further supporting the importance of MEF2C in regulating gene expression in the two key brain populations with high MEF2C expression. Interestingly, enrichment for microglia genes was not detected on DEGs from Emx1-Cre Mef2c conditional knock-out mice (Mef2c cKOEmx1-cre) (2) (Fig. S4C), underscoring the specific association between microglia and Mef2c-Hets. To further support the role of Mef2c in regulation of the DEGs, we analyzed MEF2C ChIP-Seq data from an independent study (47). Notably, we found enrichment of Mef2c-Hets DEGs in genes bound by MEF2C in multiple genomic regions (Table S2; Fig. S4D). This result further validates the key role of MEF2C in regulating genes associated with microglia and synaptic etiologies.
Figure 4.
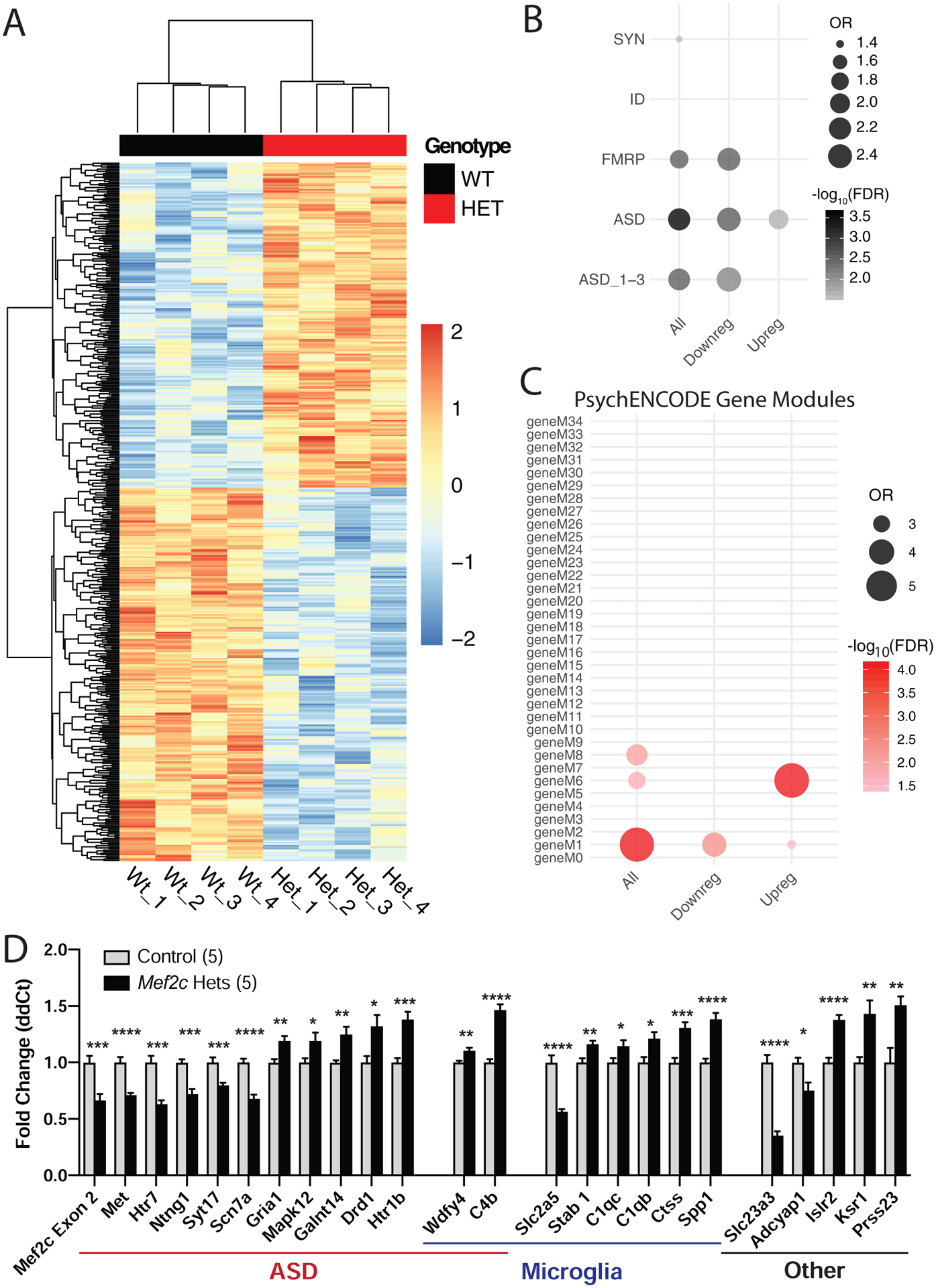
Differentially expressed genes in Mef2c-Het cortex. (A) Heatmap showing differentially expressed genes (DEGs) in Mef2c-Het cortex (p35-p40) compared with controls. In red, are genes with higher expression; in blue, are genes with lower expression. (B) Mef2c-Het DEGs are significantly enriched in genes associated with FMRP, ASD, or scored ASD (ASD_1–3; high-confidence ASD genes) (see Methods). (C) Mef2c-DEGs are enriched in gene modules dysregulated in neuropsychiatric disorders, specifically the M1 and M6 modules. (D) qPCR validation of select Mef2c-Het DEGs associated with autism, microglia, or other cellular functions. Data are reported as mean ± SEM (D). Statistical significance was determined by unpaired t-test (D). *p<0.05, **p<0.01, ***p<0.005, ****p<0.0005. See Methods for statistical analysis of A-C. Number of animals (n) is 4/genotype for RNA-Seq and 5/genotype for qPCR validation. Also see Figure S4.
Gene ontology analysis of the Mef2c-Het DEGs revealed significant enrichment of microglia proliferation genes, cell metabolism genes, and genes in a microglia subpopulation in the developing brain that is restricted to unmyelinated axon tracts (Fig. S5D). Since Mef2c-Hets showed significant dysregulation of microglial genes (Figs. 4C,D), and MEF2C is expressed in microglia in the developing and mature brain (Figs. S5A,B) (13–15), we analyzed the Mef2c-Het brain for possible upregulation of the microglia cell-type and neuroimmune activation marker, ionized calcium-binding adapter molecule 1 (Iba1) (48, 49). In both the cortex and hippocampus, we observed a significant increase in Iba1 expression (Figs. 5A–C,E) without a change in the density of microglia (Fig. S5C), suggesting possible microglial activation in the Mef2c-Het brain. This increase in Iba1 was present without an obvious change in microglial cell morphology or microglial cell soma volume (Figs. 5A,B,D,F). In addition, in the Mef2c-Het cortex, we observed no changes in classic- and alternative-pathway pro-inflammatory genes, including Cd68, Il6, Tnf, Il10, and several others (Fig. 5G). In addition, many cytokines in control and Mef2c-Het mice were undetectable by cytokine antibody array, and there was no difference in the level of IFN-g between genotypes (Fig. S5E). However, we did note a significant increase in the expression of several complement-related genes linked previously to synaptic pruning and/or ASD risk, including C1qb, C1qc and C4b (Fig. 4D) (16, 19, 50–52). Moreover, we observed significant enrichments of upregulated Mef2c-Het DEGs in scRNA-seq gene clusters associated with embryonic-like microglia, postnatal immature microglia, and homeostatic microglia (Fig. 5H). Taken together, these results reveal that the reduction of MEF2C levels has significant impacts on microglia gene expression programs.
Figure 5.
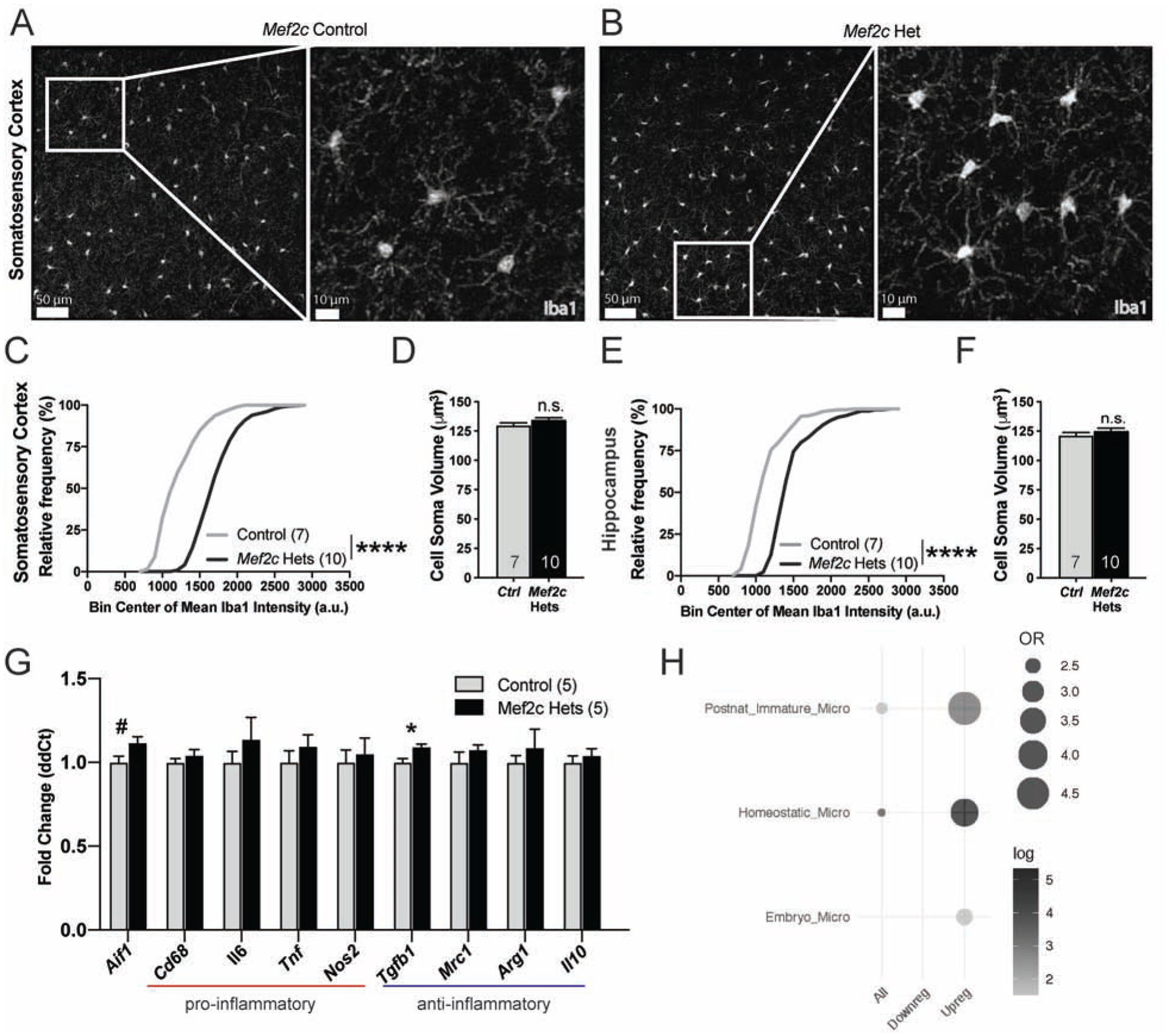
Mef2c-Het mice exhibit increased Iba1 expression levels. (A,B) Representative images of Iba1-positive microglia in the SSCtx in control (A) and Mef2c-Het mice (B). (C,E) Mef2c-Het mice have a right-shifted cumulative frequency distribution of mean Iba1 intensities in Iba1-positive cells (microglia) in the SSCtx (C) and hippocampus (E) compared to controls. Gray line represents distribution of control cells and black line represents distribution of Mef2c-Het cells. (D,F) There is no difference in the cell soma volume of Iba1 positive cells (microglia) in the SSCtx (D) or hippocampus (F) between controls and Mef2c-Het mice. (G) Fold changes of genes associated with microglial activation in controls and Mef2c-Hets. (H) Mef2c-Hets have an upregulation of genes expressed in postnatal immature, homeostatic, and embryonic microglia. Unless specified, data are reported as mean ± SEM. Statistical significance determined by Kolmogorov–Smirnov test (C,E) or unpaired two-tailed nested t-test (D,F), unpaired two-tailed t-test (G). ****<0.0001. Sample sizes for each genotype are denoted on bars of or above each graph unless otherwise specified. Images (A,B) have contrast and brightness enhanced for ease of viewing. Images are modified equally for both genotypes. Also see Figure S5.
MEF2C contributes to neurotypical behaviors through key roles in forebrain excitatory neurons and microglia
In the mouse brain, MEF2C is expressed in several neuronal cell types, including cortical excitatory pyramidal cells, parvalbumin-positive GABAergic inhibitory neurons, and cerebellar Purkinje cells, and in microglia (2, 9, 14, 15, 53–57). Since the Mef2c-Het mouse cortex showed robust changes in both excitatory neurons and microglia gene expression (Figs. 4,S4), we generated cell type-specific conditional Mef2c heterozygous mice to explore the contribution of neurons versus microglia for the development of MCHS-like phenotypes. We first generated mice heterozygous for Mef2c in Emx1-lineage cells (Mef2c-cHetEmx1-cre) (42), which represents ~85% of forebrain excitatory neurons throughout the cortex and hippocampus. Similar to the global Mef2c-Hets, the Mef2c-cHetEmx1-cre mice displayed altered anxiety-like behavior and male-selective increases in locomotion and repetitive jumping (Figs. 6A–C), but they showed no changes in social behavior or shock sensitivity (Figs. 6D, S6A). Interestingly, similar to global Mef2c-Hets (Fig. 3E) and Mef2c cKOEmx1-cre mice (2), we observed a reduction of mEPSC amplitude in layer 2/3 pyramidal neurons from Mef2c-cHetEmx1-cre mice (Fig. S6N). These findings suggest that Emx1-lineage excitatory forebrain neurons contribute to the development of some, but not all, of the behavior phenotypes observed in the global Mef2c-Hets. Interestingly, not all MEF2C-expressing populations are critical for MCHS-related behaviors since MEF2C hypofunction in PV-positive GABAergic interneurons (Mef2c-cHetPV-cre) or in cerebellar Purkinje cells (Mef2c-cHetPcp2-cre) showed behaviors indistinguishable from controls (Figs. S6B–K).
Figure 6.
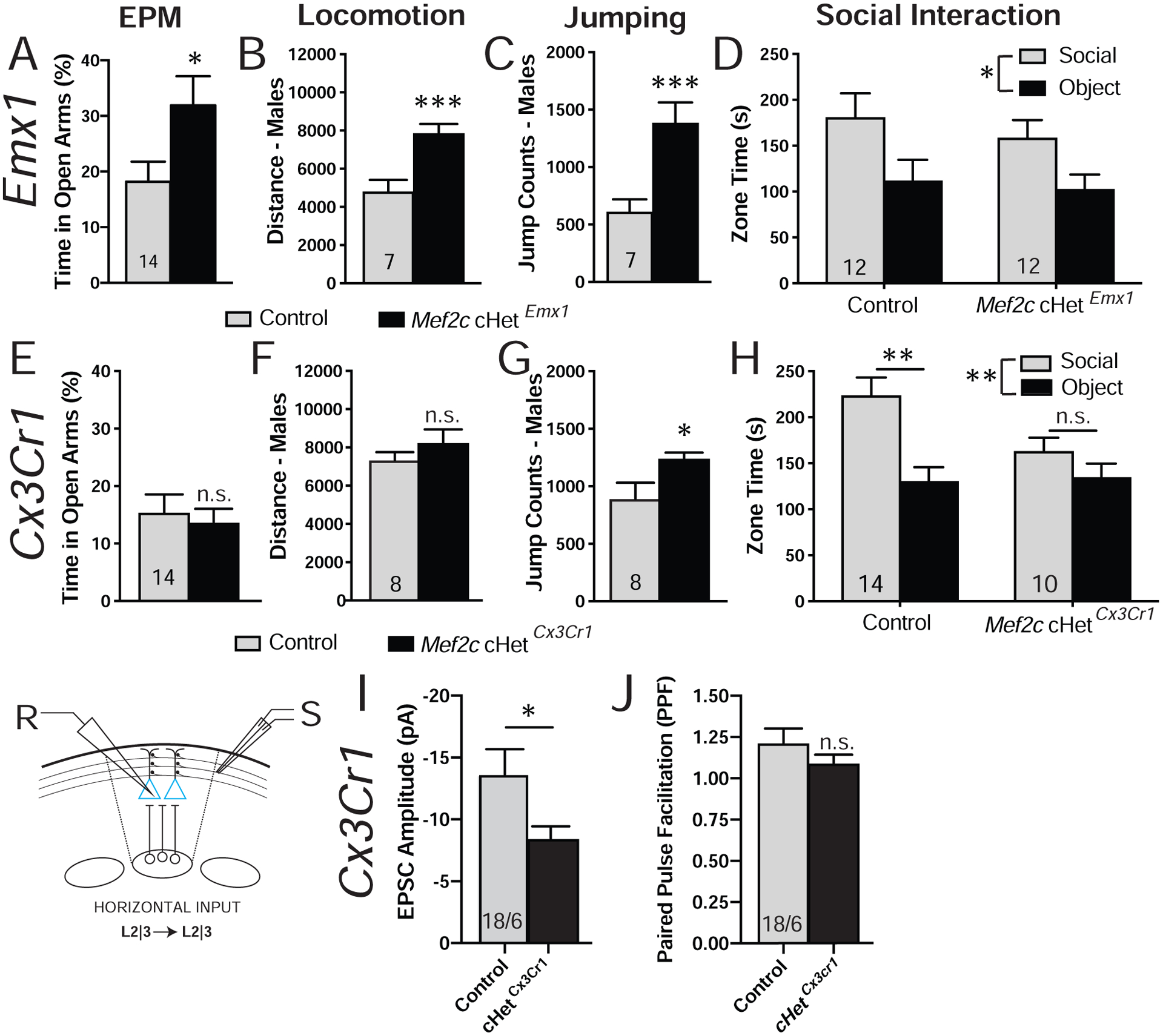
Cell type-selective phenotypes in Mef2c conditional heterozygous (Mef2c-cHet) mice. (A-D) Behaviors in Mef2c cHetEmx1 mice. (A) Mef2c cHetEmx1 mice spend more time on the open arms of the elevated plus maze. (B,C) Male Mef2c-cHet Emx1 mice are hyperactive (B) and show increased jump counts (C). (D) Mef2c cHetEmx1 mice have normal social interaction. (E-H) Behaviors in Mef2c cHetCx3cr1 mice. (E) Mef2c cHetCx3cr1 mice are similar to controls in elevated plus maze. (F,G) Male Mef2c cHetCx3cr1 mice have normal activity (F) but show increased jump counts (G) compared to control mice. (H) Mef2c cHetCx3cr1 mice have a lack of preference for interacting with a novel mouse (social) over the novel object. (I,J) Mef2c cHetCx3cr1 mouse layer 2/3 pyramidal neurons have decreased evoked EPSC amplitude (I) without a change in paired pulse facilitation (J). Data are reported as mean ± SEM. Statistical significance was determined by unpaired t-test (A-C, E-G, I-J) or 2-way ANOVA (D,H). n.s. = not significant, *p<0.05, **p<0.01, ***p<0.005, n.s. = not significant. Number of animals (A-H) or cells/animals (I-J), respectively, are reported in each graph for respective experiment. Also see Figure S6.
We next generated microglia-selective Mef2c heterozygous mice (Mef2c-cHetCx3cr1-cre) (Fig. S6L). The conditional mutant mice displayed social impairments in the 3-chamber social interaction test (Fig. 6H), similar to global Mef2c-Het mice. In addition, Mef2c-cHetCx3cr1-cre mice showed a significant increase in male-specific repetitive jumping (Fig. 6G), but with no discernable effects on exploratory activity (Fig. 6F), anxiety-like behavior or shock sensitivity (Figs. 6E,S6M). To investigate the possible influence of microglial MEF2C hypofunction on neuronal function, we recorded horizontally-evoked EPSCs in layer 2/3 of the somatosensory cortex of Mef2c-cHetCx3cr1-cre and controls. Similar to global Mef2c-Het mice, we observed a decrease in eEPSC amplitude in the Mef2c-cHetCx3cr1-cre mice (Fig. 6I). Interestingly, if we analyze by sex, the decrease in eEPSC amplitude is driven by males (Figs. S6P,Q). However, unlike the global Mef2c-Het mice, no deficits in presynaptic function were detected by paired-pulse ratio analysis in the Mef2c-cHetCx3cr1-cre mice (Fig. 6J). Taken together, our results suggest that: (1) Mef2c haploinsufficiency in early postnatal microglia is sufficient to produce autism-related behaviors, (2) MEF2C-deficient microglia can produce a non-cell autonomous influence on excitatory synaptic transmission of layer 2/3 pyramidal neurons, and (3) the majority of MCHS-like phenotypes in the global Mef2c-Hets can be recapitulated by MEF2C hypofunction in both forebrain excitatory neurons and microglia.
Discussion
We report here three new MEF2C mutations in individuals with MCHS-related symptoms, and all of these mutations disrupted MEF2C DNA binding. Interestingly, all of the known MCHS missense or duplication mutations cluster within the highly-conserved DNA binding and dimerization domains (Fig. 1A) (58). DNA binding-deficient Mef2c heterozygous mice displayed numerous behavioral phenotypes reminiscent of MCHS, including deficits in social interaction and communication (USVs), motor hyperactivity, repetitive behavior, anxiety-related behavior and reduced sensitivity to a painful stimulus (footshock). Unlike a previous study (43), the Mef2c-Hets did not show any clear learning and memory deficits, which might support a unique, primate-specific role of MEF2C (59) or reflect a background strain interaction. The Mef2c-Hets also possessed input-selective, pre- and postsynaptic deficits in glutamatergic excitatory synaptic transmission in the somatosensory cortex. Gene expression analysis of cortical tissue from Mef2c-Hets revealed significant enrichment of differentially-expressed genes linked to ASD risk, excitatory neurons and microglia, which is notable considering the enrichment of dysregulated genes linked to cortical excitatory neurons and microglia in brains of individuals with idiopathic ASD (41). Conditional Mef2c heterozygous mice in Emx1-lineage cells, which represent predominantly forebrain excitatory neurons, reproduced several of the global Mef2c-Het behaviors and cortical synaptic phenotypes. Consistent with the dysregulation of microglial genes in Mef2c-Het mice, early postnatal conditional Mef2c heterozygosity in Cx3cr1-lineage cells, which are almost exclusively microglia in the brain (48, 49, 60), produced offspring with social deficits, increased repetitive behavior, and reduced cortical glutamatergic synaptic transmission, suggesting a critical role for MEF2C in microglia during neurotypical development and behavior and supporting the growing view that microglial dysfunction in the developing brain can underlie ASD symptoms.
It’s interesting that we observed male-selective effects of Mef2c heterozygosity on hyperactivity and/or jumping behavior in Mef2c-Het and Mef2c-cHet mice (Figs. 2E,F; 6B,C,G), suggesting an interaction between sex-based mechanisms and MEF2C functions. Indeed, numerous studies show that both neuron and microglia functions can be differentially regulated in males and females (61–64). It is also interesting to note that Mef2c-Het DEGs linked to excitatory neurons show a preferential downregulation, whereas Mef2c-Het DEGs linked to microglia display a preferential upregulation. MEF2C is reported to function as both a transcriptional activator and a repressor, and there are cell type-specific signaling mechanisms that regulate MEF2C activity (2, 65, 66).
MEF2 proteins can regulate activity-dependent glutamatergic synapse elimination (5–7, 67), and MEF2C can function in cortical pyramidal neurons as a cell-autonomous transcriptional repressor to regulate dendritic spine density, synapse number and AMPA-mediated postsynaptic strength (2, 11). Conditional knockout of both Mef2c alleles in forebrain excitatory neurons produces mice with dramatic changes in cortical synapse functions, including decreased glutamatergic synaptic transmission, numerous alterations in typical mouse behaviors, and differential gene expression (2, 9, 10, 12). In the present study, we detected an input-selective reduction in glutamatergic synaptic strength in layer 2/3 pyramidal neurons from Mef2c-Hets, as well as a reduction in presynaptic release from local layer 2/3 inputs (Fig. 3C). Interestingly, we also observed an increase in mEPSC frequency in these neurons (Fig. 3E) that is possibly due to an increase in synaptic inputs from long-range cortico-cortical inputs that was observed in sparse cell-autonomous Mef2c cKO in layer 2/3 cortical neurons (11). Interestingly, Mef2c-Hets showed similar changes in basal glutamatergic synaptic transmission (i.e. reduced mEPSC amplitude and increased mEPSC frequency) in dentate gyrus granule neurons, and pharmacological manipulation of NMDA receptors rescued numerous phenotypes in the mutant mice (43). We also found that disruption of one copy of Mef2c in microglia (Mef2c cHetCx3Cr1-cre) (Fig. 6I) or in excitatory pyramidal neurons (Mef2c cHetEmx1-cre) (Fig. S6N,O) is sufficient to reduce glutamatergic strength in layer 2/3 pyramidal neurons, suggesting that MEF2C functions in both neuronal and nonneuronal populations to regulate glutamatergic synaptic development and transmission.
Since MCHS symptoms are reported predominantly from macro- and microdeletions that disrupt MEF2C and multiple neighboring genes, we sought to identify possible loss-of-function MEF2C mutations within its protein coding region to better understand the relationship between symptoms and MEF2C. By comparing multiple new MEF2C-related mutations from individuals with developmental delay and other MCHS-associated symptoms, we observed that all of the missense mutations concentrated within the MEF2C DNA binding and dimerization domains (MADS/MEF2). All tested mutations dramatically reduced MEF2C DNA binding (Fig. 1), suggesting loss-of-function mutant alleles; however, it remains possible that the mutated MEF2C proteins could influence cell function by titrating required MEF2C co-factors. In addition, there were multiple mutations that produced a premature stop codon or a frameshift predicted to produce a truncated MEF2C lacking the C-terminal nuclear localization sequence, again presumably causing a nuclear loss-of-function or a dominant-interfering form of the protein.
Most MCHS individuals (30–40) have robust physical and behavioral abnormalities and some show abnormal brain MRI scans, but no consistent effects are observed between subjects. Reported abnormalities are broad, including periventricular heterotopia, changes in corpus callosal thickness (thinned or thickened), ventricular changes (asymmetrical or enlarged), and changes in the gyral pattern of the cerebrum (30, 33, 34). Mef2c-Het mice did not have obvious changes in gross brain structures including cortical thickness (Fig. S1E), highlighting potential differences in brain development between humans and mice. Detailed analysis of Mef2c-Het brain structures will be important to determine if subtle morphological differences exist.
Developing and mature microglia play important roles in brain development, including synaptic phagocytosis (16, 17). Microglia also mediate synapse patterning, neurogenesis, myelinogenesis and cellular phagocytosis (18, 20, 24). MEF2C is expressed in both human and mouse microglia, and MEF2 proteins regulate microglia development (15). Microglia-enriched RNAs are dysregulated in human cortex from idiopathic ASD brains (41) and in the mouse Mef2c-Het cortex (Figs. 4, S4), and we find that Mef2c hypofunction in microglia is sufficient to produce autism-like behaviors in mice (Figs. 6G,H) and alter cortical glutamatergic signaling (Figs. 6I,J). Interestingly, despite a strong increase in the Mef2c-Het brain of the microglia cell type and activation marker, Iba1, (Fig. 5) as well as other microglia genes including several complement genes (e.g. C1qb, C1qc and C4b), osteopontin (Spp1), and Cathepsin S (Ctss) (Figs. 4C,D), we failed to detect a clear signature of basal neuroinflammation in the Mef2c-Het brains (Fig. 5G). Our findings suggest that loss of one Mef2c allele does not produce classic microglial “activation”, but rather that microglial development, function or maturation might be perturbed. Of note, Mef2c-Het DEGs showed enrichment for a scRNA-seq cluster of genes associated with embryonic and immature postnatal microglia, suggesting a possible delay in microglia maturation in the Mef2c-Het mice. Future studies will be important to determine the precise roles of MEF2C in microglial development and function, and whether Mef2c heterozygosity alters one or more of the numerous reported roles for microglia in brain development.
Taken together our findings reveal that MEF2C hypofunction throughout development produces numerous complex changes in cortical synaptic transmission, gene expression and behaviors reminiscent of MCHS and ASD. Specifically, the Mef2c-Het behaviors are associated with robust, input-selective deficits in cortical excitatory synaptic transmission, and disruption of excitatory neuronal and microglial gene expression. Importantly, our cell type-selective manipulations strongly suggest that MEF2C contributes to neurotypical development through critical roles in both neuron and neuroimmune subpopulations, including forebrain excitatory neurons (Emx1-lineage) and microglia (CX3CR1-lineage). Understanding the role of MEF2C in these cell populations in the body are likely to provide important new insights into effective treatment strategies for symptoms of MCHS.
Supplementary Material
Acknowledgments:
This work was supported in part by NIH grant R01 MH111464 (to C.W.C.), TL1 TR001451, UL1 TR001450 and F30 HD098893 (to C.M.B.), the Brain and Behavior Research Foundation NARSAD Young Investigator Award (to A.J.H.) and Simons Foundation SFARI Pilot grant #649452 (to C.W.C.). This work was conducted in a facility constructed with support from the NIH grant C06 RR015455. The authors would like to acknowledge Dr. Patrick J. Mulholland and the Shared Confocal Core (NIH grant S10 OD021532), Dr. Jeremy L. Barth and the MUSC Proteogenomics Facility for qPCR instrument (supported by NIGMS GM103499 and MUSC’s Office of the Vice President for Research), and Duncan Nowling for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
All authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Assali A, Harrington AJ, Cowan CW (2019): Emerging roles for MEF2 in brain development and mental disorders. Current opinion in neurobiology. 59:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrington AJ, Raissi A, Rajkovich K, Berto S, Kumar J, Molinaro G, et al. (2016): MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, et al. (2008): Identifying autism loci and genes by tracing recent shared ancestry. Science. 321:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, et al. (2008): Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 60:1022–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, et al. (2006): Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 311:1008–1012. [DOI] [PubMed] [Google Scholar]
- 6.Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, et al. (2010): Fragile × mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 66:191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, et al. (2012): Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 151:1581–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zang T, Maksimova MA, Cowan CW, Bassel-Duby R, Olson EN, Huber KM (2013): Postsynaptic FMRP bidirectionally regulates excitatory synapses as a function of developmental age and MEF2 activity. Molecular and cellular neurosciences. 56:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, et al. (2008): MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proceedings of the National Academy of Sciences of the United States of America. 105:9391–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Radford JC, Ragusa MJ, Shea KL, McKercher SR, Zaremba JD, et al. (2008): Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proceedings of the National Academy of Sciences of the United States of America. 105:9397–9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajkovich KE, Loerwald KW, Hale CF, Hess CT, Gibson JR, Huber KM (2017): Experience-Dependent and Differential Regulation of Local and Long-Range Excitatory Neocortical Circuits by Postsynaptic Mef2c. Neuron. 93:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adachi M, Lin PY, Pranav H, Monteggia LM (2015): Postnatal Loss of Mef2c Results in Dissociation of Effects on Synapse Number and Learning and Memory. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, Keeffe S, et al. (2014): An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. The Journal of Neuroscience. 34:11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, et al. (2017): Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 8:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, et al. (2017): An environment-dependent transcriptional network specifies human microglia identity. Science. 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. (2012): Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 74:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. (2011): Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science. 333:1456. [DOI] [PubMed] [Google Scholar]
- 18.Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. (2014): Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 17:400–406. [DOI] [PubMed] [Google Scholar]
- 19.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. (2007): The classical complement cascade mediates CNS synapse elimination. Cell. 131:1164–1178. [DOI] [PubMed] [Google Scholar]
- 20.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. (2013): Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pont-Lezica L, Beumer W, Colasse S, Drexhage H, Versnel M, Bessis A (2014): Microglia shape corpus callosum axon tract fasciculation: functional impact of prenatal inflammation. Eur J Neurosci. 39:1551–1557. [DOI] [PubMed] [Google Scholar]
- 22.Hagemeyer N, Hanft K-M, Akriditou M-A, Unger N, Park ES, Stanley ER, et al. (2017): Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 134:441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shigemoto-Mogami Y, Hoshikawa K, Goldman JE, Sekino Y, Sato K (2014): Microglia Enhance Neurogenesis and Oligodendrogenesis in the Early Postnatal Subventricular Zone. The Journal of Neuroscience. 34:2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sierra A, Encinas JM, Deudero JJP, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. (2010): Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell. 7:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q, Barres BA (2018): Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 18:225–242. [DOI] [PubMed] [Google Scholar]
- 26.Schafer DP, Heller CT, Gunner G, Heller M, Gordon C, Hammond T, et al. (2016): Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. Elife. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, et al. (2012): Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 484:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horiuchi M, Smith L, Maezawa I, Jin LW (2017): CX3CR1 ablation ameliorates motor and respiratory dysfunctions and improves survival of a Rett syndrome mouse model. Brain Behav Immun. 60:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Wegener JE, Huang T-W, Sripathy S, De Jesus-Cortes H, Xu P, et al. (2015): Wild-type microglia do not reverse pathology in mouse models of Rett syndrome. Nature. 521:E1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Meur N, Holder-Espinasse M, Jaillard S, Goldenberg A, Joriot S, Amati-Bonneau P, et al. (2010): MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. Journal of medical genetics. 47:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zweier M, Gregor A, Zweier C, Engels H, Sticht H, Wohlleber E, et al. (2010): Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Human mutation. 31:722–733. [DOI] [PubMed] [Google Scholar]
- 32.Vrecar I, Innes J, Jones EA, Kingston H, Reardon W, Kerr B, et al. (2017): Further Clinical Delineation of the MEF2C Haploinsufficiency Syndrome: Report on New Cases and Literature Review of Severe Neurodevelopmental Disorders Presenting with Seizures, Absent Speech, and Involuntary Movements. J Pediatr Genet. 6:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paciorkowski AR, Traylor RN, Rosenfeld JA, Hoover JM, Harris CJ, Winter S, et al. (2013): MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics. 14:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zweier M, Rauch A (2012): The MEF2C-Related and 5q14.3q15 Microdeletion Syndrome. Molecular syndromology. 2:164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, et al. (2011): Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. American journal of medical genetics Part A. 155A:2386–2396. [DOI] [PubMed] [Google Scholar]
- 36.Novara F, Beri S, Giorda R, Ortibus E, Nageshappa S, Darra F, et al. (2010): Refining the phenotype associated with MEF2C haploinsufficiency. Clinical genetics. 78:471–477. [DOI] [PubMed] [Google Scholar]
- 37.Engels H, Wohlleber E, Zink A, Hoyer J, Ludwig KU, Brockschmidt FF, et al. (2009): A novel microdeletion syndrome involving 5q14.3-q15: clinical and molecular cytogenetic characterization of three patients. Eur J Hum Genet. 17:1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berland S, Houge G (2010): Late-onset gain of skills and peculiar jugular pit in an 11-year-old girl with 5q14.3 microdeletion including MEF2C. Clin Dysmorphol. 19:222–224. [DOI] [PubMed] [Google Scholar]
- 39.Bienvenu T, Diebold B, Chelly J, Isidor B (2013): Refining the phenotype associated with MEF2C point mutations. Neurogenetics. 14:71–75. [DOI] [PubMed] [Google Scholar]
- 40.Tonk V, Kyhm JH, Gibson CE, Wilson GN (2011): Interstitial deletion 5q14.3q21.3 with MEF2C haploinsufficiency and mild phenotype: when more is less. American journal of medical genetics Part A. 155A:1437–1441. [DOI] [PubMed] [Google Scholar]
- 41.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, et al. (2019): Single-cell genomics identifies cell type-specific molecular changes in autism. Science. 364:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, Jones KR (2002): Cortical Excitatory Neurons and Glia, But Not GABAergic Neurons, Are Produced in the Emx1-Expressing Lineage. The Journal of Neuroscience. 22:6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tu S, Akhtar MW, Escorihuela RM, Amador-Arjona A, Swarup V, Parker J, et al. (2017): NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism. Nature communications. 8:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fioravante D, Regehr WG (2011): Short-term forms of presynaptic plasticity. Current opinion in neurobiology. 21:269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. (2018): Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, et al. (2018): Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell. 174:1015–1030 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Telese F, Ma Q, Perez PM, Notani D, Oh S, Li W, et al. (2015): LRP8-Reelin-Regulated Neuronal Enhancer Signature Underlying Learning and Memory Formation. Neuron. 86:696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998): Microglia-specific localisation of a novel calcium binding protein, Iba1. Molecular Brain Research. 57:1–9. [DOI] [PubMed] [Google Scholar]
- 49.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y (2001): Enhanced Expression of Iba1, Ionized Calcium-Binding Adapter Molecule 1, After Transient Focal Cerebral Ischemia In Rat Brain. Stroke. 32:1208–1215. [DOI] [PubMed] [Google Scholar]
- 50.Bialas AR, Stevens B (2013): TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nature Neuroscience. 16:1773. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Odell D, Maciulis A, Cutler A, Warren L, McMahon WM, Coon H, et al. (2005): Confirmation of the association of the C4B null allelle in autism. Human Immunology. 66:140–145. [DOI] [PubMed] [Google Scholar]
- 52.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. (2016): Schizophrenia risk from complex variation of complement component 4. Nature. 530:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kamath SP, Chen AI (2018): Myocyte Enhancer Factor 2c Regulates Dendritic Complexity and Connectivity of Cerebellar Purkinje Cells. Mol Neurobiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. (2014): An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, et al. (2018): Developmental diversification of cortical inhibitory interneurons. Nature. 555:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. (2019): Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron. 101:207–223 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. (2019): Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 50:253–271 e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKinsey TA, Zhang CL, Olson EN (2002): MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends in biochemical sciences. 27:40–47. [DOI] [PubMed] [Google Scholar]
- 59.Ataman B, Boulting GL, Harmin DA, Yang MG, Baker-Salisbury M, Yap EL, et al. (2016): Evolution of Osteocrin as an activity-regulated factor in the primate brain. Nature. 539:242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D (2015): Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation. 12:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wright-Jin EC, Gutmann DH (2019): Microglia as Dynamic Cellular Mediators of Brain Function. Trends Mol Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lenz KM, Nugent BM, Haliyur R, McCarthy MM (2013): Microglia Are Essential to Masculinization of Brain and Behavior. The Journal of Neuroscience. 33:2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith CJ, Bilbo SD (2019): Microglia Sculpt Sex Differences in Social Behavior. Neuron. 102:275–277. [DOI] [PubMed] [Google Scholar]
- 64.Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, et al. (2018): Sex-Specific Features of Microglia from Adult Mice. Cell Reports. 23:3501–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lyons MR, Schwarz CM, West AE (2012): Members of the myocyte enhancer factor 2 transcription factor family differentially regulate Bdnf transcription in response to neuronal depolarization. The Journal of neuroscience : the official journal of the Society for Neuroscience. 32:12780–12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang J, Gocke CB, Yu H (2006): Phosphorylation-facilitated sumoylation of MEF2C negatively regulates its transcriptional activity. BMC Biochem. 7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, et al. (2008): Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron. 59:621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.