

Abstract
Background.
The key characteristics (KCs) of human carcinogens provide a uniform approach to evaluating mechanistic evidence in cancer hazard identification. Refinements to the approach were requested by organizations and individuals applying the KCs.
Methods.
We assembled an expert committee with knowledge of carcinogenesis and experience in applying the KCs in cancer hazard identification. We leveraged this expertise and an examination of the literature to more clearly describe each KC; identify current and emerging assays and in vivo biomarkers that can be used to measure them; and, make recommendations for future assay development.
Results.
We found that the KCs are clearly distinct from the Hallmarks of Cancer, that interrelationships among the KCs can be leveraged to strengthen the KC approach (and an understanding of environmental carcinogenesis), and that the KC approach is applicable to the systematic evaluation of a broad range of potential cancer hazards in vivo and in vitro. We identified gaps in coverage of the KCs by current assays.
Conclusion.
Future efforts should expand the breadth, specificity and sensitivity of validated assays and biomarkers that can measure the 10 KCs.
Impact.
Refinement of the KC approach will enhance and accelerate carcinogen identification, a first step in cancer prevention.
Keywords: environmental risk factors, biomarkers, risk assessment, key characteristics, hallmarks of cancer
Introduction
Carcinogenesis is a multi-step process in which normal cells are transformed into cancer cells by acquiring various properties that allow them to form tumors. These acquired properties of cancer cells that distinguish them from normal cells have been classified as a series of Hallmarks by Hanahan and Weinberg (1) (Figure 1A). Originally, six Hallmarks were described in 2000 (1,2) along with two enabling characteristics (genome instability and inflammation) and two emerging Hallmarks (deregulated metabolism and immune system evasion) were added in 2011 (2). By considering cancer as an accumulation of multiple Hallmarks, research has been guided to understand the origins of cancer, to identify targets for prevention and to design strategies to reverse various Hallmarks, individually as well as collectively, to treat cancer. Carcinogens (i.e., agents that induce cancer) are thought to act by inducing multiple Hallmarks in normal cells, thereby transforming them into cancer cells through a variety of mechanisms.
Figure 1. The Hallmarks of Cancer and the Key Characteristics of Carcinogens.
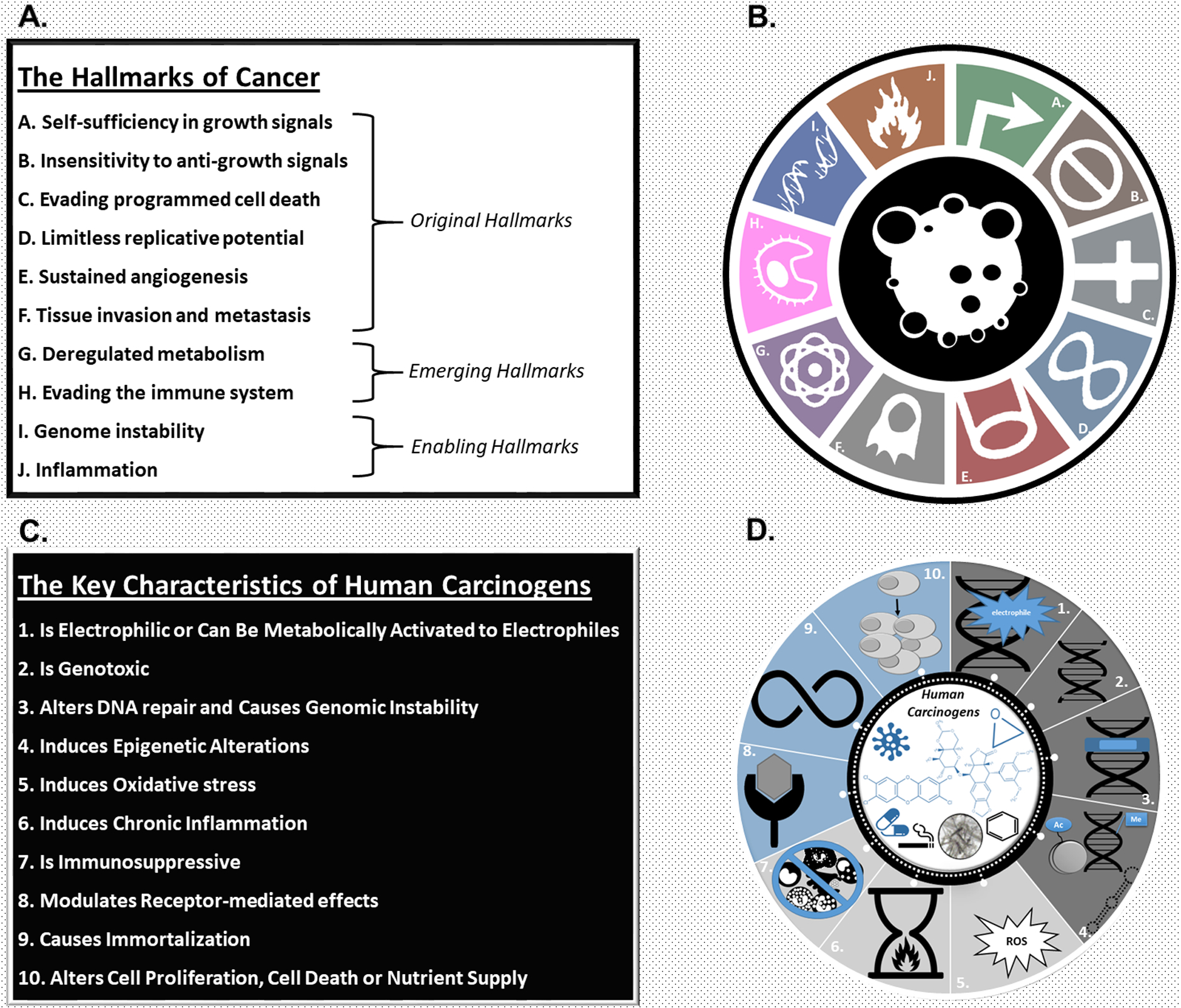
A. List of the Hallmarks of cancer. The Hallmarks of cancer, acquired properties of cancer cells that distinguish them from normal cells, have been described by Hanahan and Weinberg (1,2). Six Hallmarks were described in 2000 (1) and two emerging hallmarks (deregulated metabolism and immune system evasion) and two enabling characteristics (genome instability and inflammation) were added in 2011 (2).
B. Symbolic illustration of the Hallmarks of cancer.
C. List of the key characteristics of human carcinogens. The 10 key characteristics of human carcinogens describe the properties of human carcinogens that induce cancer (5). Expert participants at two IARC-led workshops initiated the development of the key characteristics based on empirical observations of the chemical and biological properties associated with the human carcinogens identified by the IARC Monographs program up to and including Volume 100.
D. Symbolic illustration of the key characteristics of human carcinogens. Reprinted with permission from (203) Guyton KZ, Rieswijk L, Wang A, Chiu W, Smith MT (2018). Key characteristics approach to carcinogenic hazard identification. Chemical Research in Toxicology. 31(12):1290–1292. Copyright (2018) American Chemical Society
In 2009, Guyton et al. (3) described how carcinogenic chemicals act through multiple pathways, mechanisms and/or modes-of-action to induce cancer. They identified 15 types of ‘key events’ associated with carcinogenesis and documented how known human carcinogens, such as benzene and arsenic, can cause many of these key events (e.g., genotoxicity, immunosuppression). Similarly, Kleinstreuer et al. (4) described how chemicals that target multiple Hallmark processes in vitro are more likely to be rodent carcinogens in vivo. In 2012, participants at two workshops organized by the International Agency for Research on Cancer (IARC) noted that human carcinogens, while operating individually through distinct mechanisms, often share one or more characteristics related to the multiple mechanisms by which agents cause cancer. This led to the identification of 10 key characteristics (KCs) of human carcinogens (Figure 1B) (5). These KCs, such as “is genotoxic”, “is immunosuppressive”, or “modulates receptor-mediated effects”, are based on empirical observations of the chemical and biological properties associated with the human carcinogens identified by the IARC Monographs program up to and including Volume 100. Hence, whereas the Hallmarks are the properties of cancer cells, the KCs are the properties of human carcinogens that induce cancer. Thus, while the Hallmarks describe what biology exists in a cancer, the KCs describe the actions of carcinogens that can cause those Hallmarks to become acquired.
Relationship of the Key Characteristics to the Hallmarks of Cancer
Some of the KCs produce effects analogous to the Hallmarks, e.g. carcinogens that induce genome instability can produce this Hallmark in both normal and cancer cells and those that induce immunosuppression produce an effect analogous to cancer cells evading immune destruction by suppressing immune surveillance mechanisms. However, as Stewart recently noted (6), there often is not a one-to-one relationship. Indeed, several of the KCs can produce genetic and epigenetic alterations that could cause almost all of the Hallmarks, whereas other Hallmarks, such as ‘reprogramming energy metabolism,’ which occur in many cancer cells, have no real equivalent in the KCs. It is therefore generally not possible to identify individual relationships between specific KCs of carcinogens and a single Hallmark of cancer.
Use of the Key Characteristics in Cancer Hazard Identification
Mechanistic evidence is an important contributor to hazard identification, the first step in human health risk assessment. For example, it can add biological plausibility to epidemiological findings, thereby strengthening causal inference, and contribute to understanding the human relevance of findings in experimental animal studies. Prior to the introduction of the key characteristics approach, however, there was no widely accepted method to systematically search for and organize relevant mechanistic evidence. The KCs now provide a common basis for assembling and evaluating mechanistic evidence to support cancer hazard identification and are increasingly being used by multiple authoritative bodies including the California EPA (CalEPA) OEHHA, who sponsored this project. One important advantage of using the KCs to assemble data relevant to carcinogenic mechanisms is that an a priori hypothesis about a specific mechanism of action is not required. Instead, as noted by a recent National Academies report, the KCs are based on the empirically observed, common properties of human carcinogens, thus avoiding “a narrow focus on specific pathways and hypotheses” and instead “providing for a broad, holistic consideration of the mechanistic evidence” (7). The key characteristics approach therefore presents a comprehensive and inclusive approach to using mechanistic data in cancer hazard identification, in contrast to more narrow, reductionist approaches such as adverse outcome pathway and mode of action frameworks that focus on singular events.
IARC has applied the KCs in mechanistic data evaluations for more than 50 diverse chemicals and complex exposures since 2015 (8), and have now formally incorporated them into the January 2019 Preamble of the IARC Monographs (9). Other authoritative bodies are increasingly using the KCs, such as the National Toxicology Program’s Report on Carcinogens (NTP, RoC), who used them in recent evaluations of antimony trioxide and haloacetic acids (10,11). As the KCs of carcinogens have been applied by IARC, CalEPA OEHHA, NTP and the U.S. EPA in their hazard identification and evidence integration efforts, several opportunities for refinement have been identified (8). For instance, some members of various IARC Monograph Working Groups and the Carcinogen Identification Committee of the State of California and several participants at the AACR conference, Environmental Carcinogenesis: Potential Pathway to Cancer Prevention, requested that they be more clearly defined and asked how they relate to the Hallmarks of Cancer. In addition, specific questions have arisen about which assays and biomarkers measure each of the KCs, e.g., whether certain measures of hematotoxicity belong under KC 7 (immunosuppression) or 10 (altered cell proliferation).
Materials and Methods
With funds provided by the CalEPA OEHHA, we assembled an expert working committee consisting of academics, regulators, scientists in the government and pharmaceutical industry, from the U.S., Canada, France, Netherlands and Japan. IARC, NTP, U.S. Environmental Protection Agency (U.S. EPA) and CalEPA were all represented. The committee was charged with: 1) more clearly describing each KC based on their knowledge and experience in applying them in cancer hazard identification; 2) describing the endpoints that best define each KC and the current and emerging assays and in vivo biomarkers that can be used to measure these endpoints; and, 3) making recommendations for future assay development to improve how agents (e.g., chemicals, therapeutics) can be systematically evaluated for cancer hazard in vitro and in vivo. A comprehensive list of all assays that measure each of the KCs and Hallmarks to differing degrees is being compiled in a separate project, and we note that Menyhart et al. (12) recently catalogued the functional assays which measure most of the Hallmarks of Cancer. Here, we describe each of the KCs and document representative assays that can be used to measure them in vitro, in vivo in experimental animals, and as biomarkers in humans. The committee selected assays and biomarkers that are well-validated or widely-used and identified emerging assays and biomarkers that could be used now or in the future (Tables 1 and 2).
Table 1.
Representative in silico and in vitro assays to measure the key characteristics of carcinogens
| Endpoint | In silico or non-human in vitro assay | Human in vitro assay |
|---|---|---|
| KC 1: Is Electrophilic | ||
| Electrophilic reactivity | In silico prediction (111) | |
| Protein adducts | Fluorescence-based (MSTI) assay (18) Glutathione depletion assay (112) *Chemoproteomics (113) |
|
| DNA adducts | DNA adduct measurement by HPLC (114) | |
| KC 2: Is Genotoxic | ||
| Mutation/single nucleotide variants | In silico prediction (115) Bacterial reverse mutation (Ames) (OECD 471) |
|
| Mammalian mutation assay (mouse lymphoma assay, human HPRT mutation assay) (OECD 476) | ||
| *Error-corrected Next-Generation Sequencing (116) | ||
| Structural chromosome alterations/DNA strand breaks (clastogenicity); aneugenicity | Chromosome aberration assay (OECD 473/475) Micronucleus assay (OECD 487) Comet assay (OECD 489) |
|
| *TGX-DDI biomarker in Human TK6 cells* (117) | ||
| KC 3: Alters DNA Repair or Causes Genomic Instability | ||
| Copy Number Variations (Duplications, Deletions, Amplifications, Insertions) | Comparative Genome Hybridization (CGH, Array based) (118) Next Generation High-throughput Sequencing for somatic mutation detection (43) |
|
| Inter-/intra-chromosomal translocations | Spectral Karyotyping (SKY), Karyotyping (50) Next Generation High-throughput Sequencing for somatic mutation detection (43) |
|
| Microsatellite instability | Fluorescent Multiplex PCR-based method using DNA (119) | |
| DNA repair capacity | Unscheduled DNA synthesis (120) Host cell reactivation for evaluation of nucleotide excision repair, mismatch repair, base excision repair, nonhomologous end joining, homologous recombination, and methylguanine methyltransferase (121,122) Topoisomerase I and II enzymatic activity analysis using gel electrophoresis (123) |
|
| KC 4: Induces Epigenetic Alterations | ||
| Global and locus-specific DNA methylation | High-performance liquid chromatography and ELISA-based methods (124) Enzyme activity assays for “writers, erasers, editors and readers” (125) Bisulfite sequencing (BS-seq) (126) |
|
| Histone modifications | ChIP-Seq (61) Stable isotope labeling by amino acid in culture (SILAC) (127) High throughput histone mapping (HiHiMap) (128) |
|
| Chromatin remodeling | Transposase-accessible chromatin using sequencing (ATAC-seq) (129) | |
| Changes in non-coding RNAs | RNA-Seq (130) in situ / FISH detection of small RNAs (131) and long non-coding RNAS (132,133) |
|
| KC 5: Induces Oxidative Stress | ||
| Oxidative DNA damage | 8-OHdG adducts via HPLC-electrochemical detection (134,135) Comet assay modified with lesion-specific repair endonucleases (e.g., 8-oxoguanine DNA glycosylase [OGG1], formamidopyrimidine (fapy)-DNA glycosylase [FPG], Endonuclease III [Nth]) (136) |
|
| Reactive oxygen species (ROS) formation | Electron paramagnetic resonance (137) | |
| Glutathione oxidation | Measurement of GSH/GSSG ratio (138) | |
| Nrf2-ARE-dependent gene expression response | Antioxidant enzyme activity (superoxide dismutase, catalase, glutathione reductase, glutathione peroxidase) (139)) Nrf2/ARE-dependent gene expression (140) |
|
| Lipid peroxidation | Thiobarbituric acid reactive substances assay for detection of malondialdehyde, 4-Hydroxynonenal hydroperoxides and isoprostanes (141) | |
| KC 6: Induces Chronic Inflammation | ||
| Inflammatory signaling | *Colonic organoid-based cell transformation assay (142) | |
| KC 7: Is Immunosuppressive | ||
| T cell activation and proliferation | Mitogen and/or antibody mediated proliferation (143) | |
| Cytotoxic T-lymphocyte (CTL) activity | *BiTE®-mediated CTL assay (144) | |
| Natural killer cell activity | Missing-self cytotoxicity assay (145) | |
| KC 8: Modulates Receptor-mediated Effects | ||
| Interacts with receptors | Androgen and estrogen receptor binding assay ((146), OECD TG 493) | |
| Receptor activation | Estrogen receptor Transactivation (147) Androgen receptor Transactivation (148) Aryl hydrocarbon receptor transactivation (149) |
|
| Alters ligand synthesis | Aromatase enzyme activity (US EPA 890.1200) H295R steroidogenesis assay (OECD TG 456) |
|
| KC 9: Causes Immortalization | ||
| Alters in vitro transformation activity | Cell transformation assays (150) | |
| Alters cellular senescence markers | Changes in B-galactosidase, p16, p21, and p53 protein levels (151) | |
| Telomere length and telomerase activity | Telomerase activity assay (152) | |
| Alterations in stem cell genes | Expression of MYC, Oct3/4, Klf4, Sox-2 (153,154) | |
| KC 10: Alters Cell Proliferation, Cell Death or Nutrient Supply | ||
| Cell proliferation | DNA labeling (e.g., EdU, 3H-thymidine) (155) Cell cycle markers (e.g., Ki-67, propidium iodide) (155) Metabolic activity (e.g., MTT) (155) Cell number/microscopy (e.g., Hemocytometer) (155) Colony formation (156) |
|
| Evasion or reduction of apoptosis | Evasion of apoptosis (157) by TUNEL, Annexin-V, PARP cleavage or others *Changes in expression of pro and anti-apoptotic factors (158) |
|
| Angiogenesis | Endothelial cell proliferation, migration and differentiation (159) Transwell cell invasion (Boyden) assay (160) Aortic ring assay (161) |
|
| Glycolytic (Warburg) shift | Cellular respiration and acidification (Seahorse) assay (162) | |
Emerging assay or biomarker. Abbreviations: 8-OHdG, 8-hydroxydeoxyguanosine; BITE, Bispecific T-cell Engager; ChIP-Seq, chromatin immunoprecipitation-sequencing; ELISA, enzyme-linked immunosorbent assay; GSH, glutathione; GSSG, glutathione-S-S-glutathione; HPLC, high-performance liquid-chromatography; Klf4, Krüppel-like factor 4; LC, liquid chromatography; MSTI, (E)-2-(4-mercaptostyryl)-1,3,3-trimethyl-3H-indol1-ium; nrf2/ARE, nuclear erythroid 2-related factor 2/antioxidant response element; Oct3/4, octamer-binding transcription factor 3 or 4; PARP, poly (ADP-ribose) polymerase; Sox-2, SRY-Box 2; TGx-DDI, toxicogenomics-DNA damage-inducing; TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Table 2.
Representative endpoints/biomarkers and corresponding examples of in vivo assays and biomarkers that can be used to measure the key characteristics of carcinogens
| Endpoint | In vivo assay in experimental animals | In vivo biomarker in humans |
|---|---|---|
| KC 1: Is Electrophilic | ||
| Protein adducts | Protein adduct measurement by LC/Mass spectrometry (163) *Chemoproteomics (113) Hemoglobin or albumin adducts in blood (164) *Protein adductomics (165) |
|
| DNA adducts | DNA adductomics (166,167) Nuclease P1-enhanced (32)P-postlabeling method (168) Mass spectrometry (21) |
|
| KC 2: Is Genotoxic | ||
| Mutation/single nucleotide variants | Transgenic rodent assay (e.g., Big Blue®) (OECD 488) Pig-a assay (169) |
Hypoxanthine-guanine phosphoribosyltransferase (HPRT) mutation assay (170) Glycophorin A (GPA) assay (171) |
| Next Generation High-throughput Sequencing for somatic mutation detection (43) | ||
| Structural chromosome alterations/DNA strand breaks (clastogenicity); aneugenicity |
Micronucleus assay (OECD 474) Chromosomal aberration test (OECD 475) |
Micronucleus assay (172,173) OctoChrome FISH (174) |
| Alkaline comet assay (OECD 489; (175)) Chromosomal aberration (44) Interphase and metaphase FISH (176) | ||
| KC 3: Alters DNA Repair or Causes Genomic Instability | ||
| Copy Number Variations (Duplications, Deletions, Amplifications, Insertions) | Comparative Genome Hybridization (CGH, Array based) (118) Next Generation High-throughput Sequencing for somatic mutation detection (43) |
|
| Inter-/intra-chromosomal translocations | Spectral Karyotyping (SKY), Karyotyping (50) Next Generation High-throughput Sequencing for somatic mutation detection (43) |
|
| Microsatellite instability | Fluorescent Multiplex PCR-based method using DNA (119) | |
| DNA repair capacity | Unscheduled DNA synthesis (120) Host cell reactivation for evaluation of nucleotide excision repair, mismatch repair, base excision repair, nonhomologous end joining, Homologous recombination and methylguanine methyltransferase (121,122) Topoisomerase I and II enzymatic activity analysis using gel electrophoresis (123) |
|
| Increased expression of activation-induced cytidine deaminase (AID) | Western blotting using antibodies (177) | |
| KC 4: Induces Epigenetic Alterations | ||
| Global and locus-specific DNA methylation | Illumina Methylation EPIC 850k Beadchip (178) High-performance liquid chromatography and ELISA-based methods (124) Enzyme activity assays for “writers, erasers, editors and readers” (125) Bisulfite sequencing (BS-seq) (126) |
|
| Histone modifications | ChIP-Seq (61) Stable isotope labeling by amino acid in culture (SILAC) (127) High throughput histone mapping (HiHiMap) (128) |
|
| Chromatin remodeling | Transposase-accessible chromatin using sequencing (ATAC-seq) (129) | |
| Changes in non-coding RNAs | RNA-Seq (130) In situ / FISH detection of small RNAs (131) and long non-coding RNAS (132,133) |
|
| KC 5: Induces Oxidative Stress | ||
| Oxidative DNA damage | 8-OHdG adducts via HPLC-electrochemical detection (PMID: 30992736) Comet assay modified with lesion-specific repair endonucleases (e.g., 8-oxoguanine DNA glycosylase [OGG1], formamidopyrimidine (fapy)-DNA glycosylase [FPG], Endonuclease III [Nth]) (136) |
|
| Reactive oxygen species (ROS) formation | Electron spin resonance imaging (179) | |
| Glutathione depletion | NMR-based non-targeted global metabolomics (180) | Magnetic resonance spectroscopy (181) |
| Nrf2-ARE-dependent gene expression response | Antioxidant enzyme activity (superoxide dismutase, catalase, glutathione reductase, glutathione peroxidase) (139) Nrf2/ARE-dependent gene expression (140) |
|
| Lipid peroxidation | Thiobarbituric acid reactive substances assay for detection of malondialdehyde, 4-Hydroxynonenal hydroperoxides and isoprostanes (141) | |
| KC 6: Induces Chronic Inflammation | ||
| Tissue inflammation | Histological examination (limited in human) | |
| KC 7: Is Immunosuppressive | ||
| Hematology | White blood cell counts and examination of lymphoid tissues (182) | |
| Immunophenotyping of T cells and NK cells | Enumeration of NK cell, CD4+ and CD8+ T-cells (183) | |
| T cell activation and proliferation | Mitogen and/or antibody mediated proliferation (143) | |
| Cytotoxic T-lymphocyte (CTL) activity | BiTE®-mediated CTL assay (144) | |
| Virus-specific CTL function in mouse (184) | ||
| Generation of antigen-specific CD8+ T cells | *Measurement of endogenous or vaccination-induced anti-viral immunity (185) | |
| Natural killer cell activity | Missing-self cytotoxicity assay (145) | |
| Systems immunology | Mass cytometry (186) | |
| KC 8: Modulates Receptor-mediated Effects | ||
| Activates or antagonizes receptors | Posttranslational and/or transcriptional changes associated with the estrogen, androgen and aryl hydrocarbon receptor activity (187) | |
| Alters receptor expression | Immunohistochemistry or western blotting in animal or human tissue (187) | |
| Alters ligand synthesis, clearance, distribution or levels | Circulating steroid hormone levels (188–190) Alteration in sex-hormone binding globulins (188,191,192) |
|
| KC 9: Causes Immortalization | ||
| Alters cellular senescence markers | Changes in B-galactosidase, p16, p21, and p53 levels (151) | |
| Telomere length and telomerase activity | Telomere length by real-time PCR (193,194) Telomerase activity assay (152) |
|
| Alterations in stem cell genes | Expression of MYC, Oct3/4, Klf4, Sox-2 (153,195) | |
| KC 10: Alters Cell Proliferation, Cell Death or Nutrient Supply | ||
| Proliferation/hyperplasia | Histology/microscopy DNA labeling (e.g., EdU, 3H-thymidine) (196) Cell cycle/cell number markers (e.g., Ki-67, propidium iodide) (197) Cell number/microscopy (155) |
|
| Evasion or reduction of apoptosis | Histology/microscopy *Changes in expression of pro and anti-apoptotic factors (198) |
|
| Angiogenesis | Factor VIII stains for capillary basement membrane (199) Tissue vascular permeability by magnetic resonance imaging (200) |
|
| Glycolytic (Warburg) shift | F-18-fluorodeoxyglucose (FDG) by computed tomography (CT) and positron emission tomography (PET) (201) Magnetic resonance imaging (MRI) and spectroscopy (MRS) (202) |
|
Emerging assay or biomarker. Abbreviations: 8-OHdG, 8-hydroxydeoxyguanosine; BITE, Bispecific T-cell Engager; ChIP-Seq, chromatin immunoprecipitation-sequencing; EdU, 5-ethynyl-2’-deoxyuridine; Klf4, Krüppel-like factor 4; LC, liquid chromatography; NMR, nuclear magnetic resonance; Nrf2/ARE, nuclear erythroid 2-related factor 2/antioxidant response element; Oct3/4, octamer-binding transcription factor 3 or 4; Sox-2, SRY-Box 2.. .
Results
Descriptions and Assays to Measure the Key Characteristics of Carcinogens
Key Characteristic 1: Is Electrophilic or Can Be Metabolically Activated to an Electrophile
Electrophiles are reactive, electron-seeking molecules capable of binding to electron-rich cellular macromolecules including DNA, RNA, lipids, and proteins, forming covalent adducts. The measurement of covalent adducts on DNA and proteins is the most common method of assessing electrophilic activity both in vitro (Table 1) and in vivo (Table 2). Electrophiles and their nucleophilic targets can be described by their strength, which can help predict their reactivity. “Hard” electrophiles, so called due to the relatively greater polarizability of their electrophilic center, are of high concern, having electron-withdrawing groups capable of binding nucleophilic N and O sites in DNA (e.g., epoxides) (13). “Soft” electrophiles primarily target nucleophilic sites in proteins, such as thiol groups, rather than DNA. This can cause glutathione depletion and functional inhibition of critical proteins, such as tubulin (14).
Computational chemistry tools can be used to calculate characteristics of chemical structures that can aid in identifying hard electrophiles (15). Structural alerts for reactive organic functional groups requiring metabolic activation have also been developed (16). While in silico calculations can identify potential DNA-reactive electrophiles, in vitro approaches can provide confirmation as well as information on chemical potency and/or reactivity. The direct measurement of electrophilicity involves approaches that determine a rate constant (k) as a measure of reactivity. This can be carried out through adduct measurement of deoxyribonucleosides, although this requires a high performance liquid chromatography method that limits throughput (17). High-throughput, in chemico assays are available, but these target primarily soft electrophiles (18) and there is a need for equivalent, high-throughput formats for hard electrophiles.
Biomarkers of reactive, electrophilic chemicals in humans have consisted primarily of measuring protein adducts reflecting the parent chemical structure through sampling of the readily accessible blood proteins hemoglobin and albumin (19). Various analytical techniques have been employed to measure adducts at known, susceptible amino acid residues in these proteins. Measurement of adducts on DNA is now less frequent but new approaches can determine DNA adducts in human biopsy samples by liquid chromatography-mass spectrometry, requiring only several milligrams of tissue (20,21). Methods for protein and DNA adductomics, which identify multiple adducts, are emerging (22,23), as is chemoproteomics, which assesses the sites on proteins adducted by electrophilic small molecules (24).
Key Characteristic 2: Is Genotoxic
Genotoxicity is generally defined as the capability of an external agent to cause DNA damage, alteration to the genome (mutation), or both. The link between genotoxicity and chemical carcinogenesis is well established and has shaped standardized testing batteries of carcinogens for decades (25). Genotoxicity can arise from, for example, DNA strand breaks, DNA adducts, DNA-DNA crosslinks, and DNA-protein crosslinks, as well as from oxidative damage to DNA (see also KC5, below); all of these types of damage may give rise to permanent changes in the nucleotide sequence (mutation) as the cell attempts to repair the damage. Genotoxicity can give rise to single nucleotide variants (point mutations), or it can manifest over a larger span of the genome at the chromosome level, e.g., structural (clastogenicity) or numerical chromosomal aberrations (aneuploidy). For practical purposes, we define KC2 as encompassing these forms of genotoxicity, which are commonly evaluated using classical in vitro assays referenced in some regulatory testing guidelines (Table 1) and can be measured preclinically using biomarkers in experimental animals and in humans (Table 2). Though measurements of endpoints such as DNA cross-links can provide valuable mechanistic information, more apical endpoints such as mutation or chromosome loss, which reflect the consequence of damage to daughter cells, are considered more relevant and useful for hazard identification. Indeed, chromosome aberrations and micronuclei have been shown to be associated with future cancer risk (26,27). A large proportion of established human Group 1 carcinogens are genotoxic (IARC Monographs Volume 100 A–F).
There is clear overlap between the genotoxic effects described here in KC2 and other KCs that can lead to genotoxicity: for example “KC1: is electrophilic,” for agents that can covalently bind DNA and form adducts; “KC3: alters DNA repair or causes genomic instability” for agents that inhibit DNA repair or induce error prone repair pathways; and “KC5: induces oxidative stress” which can cause oxidative DNA damage. The intent of considering multiple distinct KCs with seemingly similar effects on genome integrity is to functionally distinguish the mechanisms by which the agent can cause genotoxicity, which in turn may facilitate a more biologically accurate understanding of how the agent causes cancer. For example, benzene, an established human leukemogen, has all four of these characteristics (see also (28): its reactive metabolites are electrophilic (23); it is genotoxic, causing both structural and numerical chromosomal changes in humans (29); it inhibits topoisomerase II (30) causing genomic instability (31); and its metabolites induce oxidative DNA damage (8-hydroxydeoxyguanosine, 8-OHdG) and micronuclei from oxidative stress (32).
With the rapid development of next-generation sequencing (NGS) technologies, it is becoming possible to directly evaluate the result of genotoxicity at the nucleotide level with high resolution. However, further work is needed to validate these systems for detecting rare somatic mutations that approach the background level of mutations in humans (33). DNA sequencing-based measurements are broadly applicable to in vitro and in vivo model systems, and human-based investigations, providing an adequate amount of target tissue DNA, or a valid surrogate, can be obtained.
Key Characteristic 3: Alters DNA Repair or Causes Genomic Instability
Genomic instability is a Hallmark of Cancer and is an enabling characteristic of tumorigenesis (2). DNA damage response (DDR) pathways maintain genomic stability by ensuring the fidelity of DNA replication and by activating cell cycle checkpoints and/or DNA repair pathways in response to DNA damage caused by endogenous processes and exogenous agents (34). Disruptions in DDR pathways and DNA repair can lead to elevations in mutagenesis and genomic instability, defined by an increasing rate of the accumulation and clonal expansion of genomic alterations, and characterized by gene mutation, microsatellite instability (MSI), and genomic/chromosomal instability (CIN) (35).
Genomic instability has been hypothesized to result from the expression of a mutator phenotype driven by mutations in DNA repair genes (36). Altered DNA repair and/or induction of genomic instability can result from agents that are genotoxic (KC2), induce oxidative stress (KC5), or induce epigenetic alterations (KC4) and thereby disrupt the expression of genes involved in DNA repair. Several chemical and physical agents have been shown to impede or inhibit high-fidelity DNA repair and/or activate error-prone DNA repair pathways leading to genomic instability and cancer, including metals (37), aldehydes (38), and ionizing radiation (39).
DNA repair activity is a useful human biomarker for mutagenic agents. Repair activity can be detected by measuring the expression, location, or recruitment of DNA repair proteins, but these measurements may not reflect the end outcome or consequence of a change in mutagenic repair capacity. The comet assay (i.e., single-cell gel electrophoresis) can be used to measure the rate or capacity of DNA repair, including high-throughput applications (40). Other high-throughput approaches include molecular beacon assays that can detect multiple DNA repair enzyme activities (41), and fluorescence-based, multiplex flow-cytometric host cell reactivation assays that have been used to measure inter-individual differences in DNA repair capacity in humans (42). Newer higher-resolution NGS-based methodologies can measure the consequences of activated mutagenic DNA damage repair (e.g., large deletions/amplifications and inter-/intra-chromosomal translocations) which provides more direct evidence of the repair outcome itself (43,44).
Other potential contributors to genomic instability have been identified. Oncogene-induced DNA replication stress, generated by sustained cell proliferation, may confer a selective advantage that drives tumorigenesis (45). In addition, genomic instability can be induced by chronic inflammation (KC6), independently of DNA damage, following exposure to pathogens, chemicals, direct-/indirect DNA mutagens, radiation, hypoxia, or starvation (39,46–48). Several inflammatory mediators involved in pro-inflammatory signaling and activation of NF-κB have been implicated, including reactive oxygen species (ROS), cytokines, TNF-α, and aberrant expression of activation-induced cytidine deaminase (AID), a DNA mutator enzyme (39,49).
Spectral karyotyping (SKY) is a useful technique for identifying and characterizing genomic instability exhibited by CIN (50). Higher resolution NGS technologies have been established that can measure genomic instability at the DNA level (Tables 1 and 2), although they have been largely restricted to studying tumor heterogeneity (51). Further characterization with expanded numbers of drugs/xenobiotics is needed to validate these genomic endpoints and for the purposes of nonclinical and clinical safety studies.
Key Characteristic 4: Induces epigenetic alterations
Epigenetic modifications occur without changing DNA sequence and lead to stable and mitotically heritable changes in gene expression. Epigenetic modifications include alterations to DNA (e.g., DNA methylation), non-coding RNAs (e.g., altered expression of microRNA), chromatin (e.g., histone modifications), and 3D structures (e.g., nucleosome positioning) (52). These alterations are surprisingly common (53), and some cancer types show wide-spread losses in DNA methylation with small gains in specific genomic areas and genes (54). The downstream effects depend on the type of epigenetic modification and the location in the genome where they impact (e.g., promoter/exon/intron/inter-/intra-genic region of oncogene or tumor suppressor genes). Epigenetics marks are hypothesized to serve as mediators of cancer etiology and progression, in many cases preceding cancer (55).
A variety of methods exist to measure DNA methylation status at the global and locus-specific level. In human samples and cell cultures, the most commonly used method is to measure locus-specific methylation using Illumina 450K and 850K bead chip arrays. A more comprehensive but costly method is bisulfite sequencing, usually in the form of reduced representation bisulfite sequencing (RRBS) which can generate genome-wide methylation profiles at a single nucleotide level. Total genomic 5‐methylcytosine can be quantified by using a variety of antibody kits with ELISA, high‐performance liquid chromatography, or liquid chromatography/mass spectrometry. Since more than one‐third of DNA methylation occurs in repetitive elements, analyzing the methylation of repetitive elements can also serve as a surrogate marker for global genomic DNA methylation. Many carcinogens alter DNA methylation status. For example, arsenic exposure is associated with global DNA hypomethylation and increased DNA methylation of proto-oncogenes such as RAS (56), p53, and p16 (57).
Mass spectrometry is the gold-standard method for analyzing histone modifications, as it enables the quantification of specific modifications with high resolution (58). However, it is quite difficult to apply in practice and many researchers have used antibody-based methods. A variety of carcinogenic metals, including arsenic, chromium and nickel have been shown to cause significant modifications of histone proteins (59). The Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq), as the name suggests uses a transposase to insert sequencing adapters into accessible regions of chromatin, followed by sequencing, to assess genome-wide DNA accessibility (60). It is a rapid and sensitive alternative to DNase-Seq and has been used with multiple cell types and species. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) is extensively used to map epigenetic proteins such as histone proteins, chromatin regulators, and transcription factors in the genome (61).
Changes in non-coding RNA expression are now usually measured by small RNA sequencing, but a variety of arrays also exist. In situ hybridization/FISH are also used to detect both small RNAs and long-non-coding RNAs. Chemical carcinogens alter the expression of multiple microRNAs and this has been postulated to play a role in chemical carcinogenesis (62,63).
The difficulty with interpreting the KC “induces epigenetic modifications” is that the relevance of a particular epigenetic change for carcinogenesis by a specific agent may not be clear and causality is hard to establish. More validation is clearly needed to fully understand which epigenetic endpoints are most indicative of carcinogenic risk.
Key Characteristic 5: Induces Oxidative Stress
The induction of oxidative stress and subsequent injury is a characteristic of diverse carcinogens, including radiation, asbestos, some chemicals metabolized to quinones, and carcinogenic infectious agents (64). Specifically, these carcinogens are capable of influencing redox balance within target tissues, leading to an imbalance favoring formation of ROS at the expense of their detoxification, and may also be accompanied by the production of reactive nitrogen species. Oxidative stress can lead to oxidative damage to DNA, including base modification, DNA-protein crosslinks, and other lesions (65,66). For instance, 8-OHdG is a DNA adduct that is considered a critical biomarker for carcinogens that cause oxidative stress. Other biomolecules, including proteins and lipids, are also subject to oxidative damage. Thus, oxidative stress is directly related to many other KCs, notably KCs 2 and 3 via DNA damage leading to genotoxicity and alteration of DNA repair, as well as others including chronic inflammation (KC 6) and altered cell proliferation (KC 10). For example, oxidative stress affects cellular proliferation (e.g., EGFR regulation, ERK1/2 and MAPK activation), evasion of apoptosis (e.g., Src, NF-κB and PI3K/Akt activation), tissue invasion and metastasis (e.g., MMP secretion, Met overexpression, and Rho-Rac interaction), and angiogenesis (e.g., VEGF release) (67,68).
A wide variety of assay systems, exemplified in Tables 1 and 2, are available to measure an increase in ROS formation, changes in oxidative enzymatic activity, defects in the antioxidant defense system, lipid peroxidation, protein oxidation, and oxidative damage to DNA. These methods and associated biomarkers are widely used across in vivo studies in humans and experimental animals (69), small model organisms like C. elegans (70), and in vitro cell-based assays. This KC is often non-specific, as non-carcinogens can also induce oxidative stress. Therefore, oxidative stress is most informative in hazard identification when there is concordance among increases in oxidative stress markers and additional KCs. For instance, such concordance has been shown previously when oxidative stress was accompanied by genotoxicity in strains of yeast and bacteria known to be more sensitive to oxidative DNA damage (8). Furthermore, evidence is strengthened when these effects can be shown to be attenuated with co-exposure to antioxidants or in knockout animals (8). Overall, it appears that ROS production alone is not specific to carcinogens in the absence of other KCs. Thus, future efforts should be focused on further identifying ROS endpoints and markers that are specifically associated with induction of other KCs, such as DNA damage and inflammation.
Key Characteristic 6: Induces chronic inflammation
Chronic inflammation associated with the development of cancer is a prolonged response to persistent infections or irritants that inflict cell death and tissue injury followed by deregulated compensatory cell proliferation and aberrant repair (71). The type, intensity, and timing of the inflammatory response varies depending on the host immune status, the initiating stimulus, and the target organ. For example, infection of the gastric mucosa with the bacterium Helicobacter pylori induces epithelial cell death and acute and chronic inflammation leading to atrophic gastritis. Eventually, an adaptive response characterized by intestinal metaplasia of the gastric epithelium occurs and in ~3% of persistently-infected patients, intestinal-type gastric adenocarcinoma develops after a long latent period (72). In contrast to this complex inflammatory response, Schistosoma haematobium infection leads to chronic irritation and granulomatous inflammation in response to the helminth eggs in the lining of the urinary bladder. Over years or following repeated helminth infections, these eggs induce metaplasia and hyperplasia of the transitional epithelial lining of the bladder, potentially leading to squamous cell carcinoma (73). During these chronic inflammatory reactions, production of reactive oxygen and nitrogen species by infiltrating inflammatory cells contributes to oxidative stress and epigenetic alterations (KCs 4, 5). In addition, repeated episodes of cell necrosis or immune-mediated apoptosis (e.g., persistent hepatitis B infection in the liver) and altered parenchymal cell differentiation and proliferation links chronic inflammation with other KCs (e.g., KC 10). Sustained release of growth factors, cytokines, angiogenic factors, and matrix metalloproteinases perpetuate the chronic inflammatory response and facilitate tumor growth, progression, and invasion (74).
The best measure of chronic inflammation is by histopathology following repeated in vivo exposures (Table 2). Studies in rodents have provided strong evidence for the involvement of chronic inflammation in cancers induced by welding fumes, malathion, tetrachloroazobenzene, indium tin oxide and melamine in recent IARC evaluations (8). Due to the short-term nature of most in vitro assays, it is difficult to assess persistent, chronic inflammatory responses in these systems. Some ‘long-term’ in vitro co-culture models better mimic chronic inflammation, especially in regard to cytokine/chemokine profiles, but it is difficult to model inflammatory cell recruitment. It is also difficult to establish a causal relationship between inflammatory biomarkers and development of cancer, especially in epidemiological studies (75,76). Proinflammatory cytokines such as TNF-alpha or IL-6, C-reactive protein, fibrinogen, and high mobility group box 1 (HMGB1) have been investigated as potential circulating biomarkers for chronic inflammation in a variety of diseases, including asbestos-related diseases (77,78). Unfortunately, these biomarkers are not very specific for diagnosis of chronic inflammation due to poor sensitivity, confounding exposures, and variability in individual patients over time (76,79). Because of its stability IL-6 is perhaps the best current biomarker available (80).
Key Characteristic 7: Is Immunosuppressive
Immunosuppression is a reduction in the capacity of the immune system to respond effectively to foreign antigens, including antigens on tumor cells. Epidemiological data from patients with congenital immunodeficiencies, virally-induced immunodeficiencies (e.g., HIV-mediated), and from patients treated with immunosuppressive therapies (e.g., organ transplant rejection prevention therapies) indicate that profound immunosuppression is associated with an increased cancer risk. Immunosuppression-associated cancer types can include hematological malignancies and solid tumors and these cancers are often but not always associated with an oncogenic virus etiology. Examples include cyclosporine-induced non-Hodgkin lymphoma (81) and lung cancer from welding fumes (82), agents which are both immunosuppressive. The interplay between immunity and tumorigenesis is complex and different components of the immune system are either pro- or anti-tumorigenic. While profoundly altered immunity, such as that achieved during organ transplant can promote cancer risk, not all components of the immune system are equally important in defense against or promotion of cancer and a similar cancer hazard for all immunomodulatory molecules (e.g., for anti-inflammatory agents) should not be assumed (83).
Immunosuppressive agents are molecules that interfere with key steps of the “cancer-immunity cycle” which can be divided into several processes, starting with the release of antigens from the cancer cell, followed by the presentation of cancer antigens to immune effector cells, the priming / activation of these effector cells (e.g., T lymphocytes), infiltration / migration of immune effector cells into tumors, and ending with the killing of cancer cells (84). T cell function plays a particularly important role in anti-tumor immunity and some components of innate immunity. Natural Killer (NK) cells can also play an important role. Several immunosuppressive agents (such as cyclosporine A or dimethylbenzanthracene) directly inhibit T cell activation leading to decreased immune surveillance of pre-cancerous cells/lesions.
Rodent tumor models or rodent bioassays are generally poor models to assess the cancer hazard associated with immunosuppression (85). Endpoints or assays that can assess the degree of interference with anti-tumor immunity should ideally interrogate various key steps of the cancer-immunity cycle. Certain phenotyping and functional assays and endpoints listed in Tables 1 and 2 are representative of assays that can inform that hazard. These include standard hematology and anatomic pathology (e.g., evidence of marked lymphodepletion and hematotoxicity), lymphocyte population enumeration by flow cytometry, and NK cell activity. There are also some less routine but important endpoints such as T helper and cytotoxic T cell functions (including endpoints relating to factors that govern killing mechanisms such as cytokine production and evidence of degranulation). Other assays or models such as host resistance models may detect immunomodulation (e.g., decreased resistance to bacterial infection or even certain viruses) but these effects may not translate to decreased immune surveillance of tumors. Efforts are ongoing to identify and develop new approaches and methods to evaluate systemic immune status in vivo. Systems immunology approaches can leverage methods such as mass cytometry to interrogate multiple cell types and functions concomitantly (Table 2).
Key Characteristic 8: Modulates Receptor-Mediated Effects
Receptor-mediated effects can occur at the cell surface (through ligand-binding) or intracellularly (via the disruption of signaling cascades or actions on nuclear/cytosolic receptors), all of which can modulate transcriptional changes in the nucleus. Outcomes of transcriptional changes are varied and often regulate critical cellular pathways. Some receptors promote cell proliferation (e.g., hormone nuclear receptors such as estrogen (ER) (86), androgen (AR) (87), and progesterone (PR) (86) receptors and growth factor receptors such as EGFR/erbB (88) and HER2/neu (89)). Activation of the aryl hydrocarbon receptor (AhR) (KC8) can lead to immunosuppression (KC7), in addition to effects on cell proliferation and survival (KC10) (90–92). Finally, the activation of certain nuclear receptors, including peroxisome proliferator activated receptor alpha (93) and constitutive androstane receptor (94), is associated with rodent liver carcinogenesis (94,95).
Nuclear and cytosolic receptors are generally regulated by small molecules and are thus of higher priority for evaluating xenobiotics as carcinogens compared to peptide-regulated growth factor receptors. Binding of drugs/xenobiotics to receptors is readily measurable but does not completely inform how downstream signaling activity is modulated. Hence, the best approach is to evaluate both receptor binding and receptor functional activity. Radioligand binding and luciferase reporter gene assays in human cells are the current respective gold standard assays. For example, internationally harmonized guideline assays exist for binding and transcriptional activation of both ER and AR, although not all use human cells. Other binding and reporter assays are available for several aforementioned receptors although they generally lack extensive validation. As many of these receptors have been the target of pharmaceutical research, there are contract research organizations that provide testing services for many of them. Research efforts are still needed in some cases, such as with the AhR where distinguishing between effects that promote carcinogenesis from those that inhibit it cannot be readily determined through binding and transcriptional activation assays and is likely crucial for the identification of carcinogens (96).
Agents that modulate a ligand’s synthesis, transport, distribution, biotransformation, and clearance can indirectly modulate receptor mediated effects. We are only aware of one guideline assay investigating ligand synthesis (Table 1), and this is an area where many assays can be developed. The recent identification of the 10 KCs of endocrine disrupting chemicals also highlighted the need for additional assays to assess many aspects of endocrine disruption that may be useful in evaluating agents for this KC of carcinogens (i.e., KC8) (97).
Key Characteristic 9: Causes Immortalization
Cancer cells are immortal, and therefore have limitless replicative potential. Normal cells have a limited lifespan that has been described as the Hayflick limit, as measured in vitro by cell doublings of normal human fibroblasts. Several mechanisms can influence immortalization: cellular pathways that regulate stemness versus senescence, telomere length and telomerase activity, and Alternative Lengthening of Telomeres (ALT), break-induced telomere synthesis and mitotic DNA synthesis (MiDAS) at so-called common fragile sites and telomeres (98–100). Telomeres are the protective ends of chromosomes that are necessary to prevent chromosomal instability and are maintained through telomerase and other gene products. Cancers frequently have elevated telomerase activity and maintain or extend their telomeres through genetic and epigenetic mechanisms. Carcinogens have been shown to activate telomerase and/or extend telomeres (101–103).
Immortalization is associated with stemness, the ability of cells to self-replicate indefinitely. With the exception of normal stem cells, whose behavior relies on signals from stem cell niches (104), most normal cells differentiate and lose the capacity for self-replication (101–103). However, cancers generally appear to maintain a small subpopulation of their cells that are the cancer stem cells, which are required for unlimited replication. For example, the MYC oncogene, which is upregulated by some carcinogens, can promote the stemness of cancer stem cells (101–103). The opposite of immortalization and stemness is cellular senescence, a cellular program that results in terminal differentiation of cells that have undergone irreparable cellular stress, including DNA damage. Immortalization is regulated through many gene products including cell cycle checkpoint inhibitors such as p16 and DNA repair gene products such as p53 (101–103). Carcinogens including human DNA and RNA viruses, such as human papillomaviruses, Epstein-Barr virus, Kaposi sarcoma–associated herpes virus, hepatitis B virus, hepatitis C virus, HIV, Merkel cell polyomavirus (MCPyV), and human T-lymphotropic virus type 1 (HTLV-1), are carcinogenic through effects on cellular immortalization and senescence (105). Similarly, chemical carcinogens including tobacco, PCBs and asbestos have been shown to impede cellular responses to DNA damage that promote immortalization and inhibit senescence (106).
A carcinogen’s influence on immortalization and senescence can be measured through a number of biochemical endpoints in cultured cells, including: transformation assays, as well as more specific assays such as telomerase activity, telomere length, and regulation of certain genes in stem cells and cancer stem cells (e.g., MYC, p16 and p53) (101–103,105,106). Some of these endpoints can also be evaluated in vivo and in human biomarker studies to facilitate translation, however more validation is needed in this area. In the future, the development of imaging-based reporters for telomerase, cell cycle checkpoints, and senescence markers such as beta-galactosidase activity may be useful for assessing carcinogenic activity.
Key Characteristic 10: Alters Cell Proliferation, Cell Death or Nutrient Supply
Tumor size is regulated by cell proliferation (growth), cell loss by apoptosis (programmed cell death) or necrosis, and the vascular supply that provides oxygen and other nutrients. Cancer cells also often have different cellular energetics than normal cells, e.g., using glycolysis for energy under aerobic conditions under the Warburg effect. Carcinogens may impact these processes by stimulating uncontrolled cell proliferation, angiogenesis to increase vascularity and the evasion of apoptosis, rather than apoptosis induction. The resultant altered cell proliferation and/or cell-cycle control can contribute to carcinogenesis in three ways: 1. predispose replicating cells to propagate unrepaired DNA damage and cancer-causing mutations; 2. sustained replication may be an independent mechanistic event; and 3. abnormal proliferation may allow transformed cells to evade usual checkpoints and to continue replication. Such scenarios foster evasion of apoptosis or other terminal programming, e.g., autophagy (107).
Measures of cell proliferation include microscopic identification of mitotic cells on histology, measurement of S-phase cells by incorporation of 3H-thymidine or bromodeoxyuridine, stains for proliferating cell nuclear antigen, and identification of the growth fraction with stains such as MIB1 for Ki-67. In vitro assays for cell proliferation can be challenging owing to the potential influence of paracrine actions and other homeostatic responses that may be lost in simple 2-dimensional cell cultures or by use of transformed cell lines commonly used in the laboratory. Static measures of apoptosis include identification of apoptotic cells on histology (sometimes with TUNEL assay or Annexin-V), and expression of pro- and anti-apoptotic genes. Evasion of apoptosis is determined as a change in apoptosis in response to a known therapeutic agent, e.g., tamoxifen, compared to controls after exposure to a chemical of interest (108,109). The vascularity of a tumor can be assessed by visual observation of blood vessel density, sometimes employing stains for capillary basement membrane.
Discussion
Our primary goal was to clearly describe each KC, based on the latest scientific developments and from experience in applying them in cancer hazard identification. In demonstrating the applicability of the KCs to identify measurable, proximal effects of carcinogens rather than endpoints that may be a consequence of tumorigenesis, this exercise showed that the KCs are clearly distinct from the Hallmarks. It also showed the applicability of KCs to a broad range of agents, not just environmental chemicals but also drugs/xenobiotics, defined mixtures, and complex exposures as well as viruses, fibers and engineered nanomaterials.
A second goal was to describe some of the endpoints that best define each key characteristic and the current and emerging assays and in vivo biomarkers that can be used to measure these endpoints. This exercise revealed that the KCs not only vary in complexity, but also in the number of endpoints and available assays and useful model systems. Representative assays and biomarkers listed in Tables 1 and 2 were compiled from literature sources to illustrate the types of endpoints that reflect each KC and current approaches for measuring them. The assays and biomarkers presented in the Tables are by no means an exhaustive list and were compiled from literature sources. They represent a range of states of validation and experience in applying them. It would be valuable to understand the sensitivities and specificities for each of these as well as domains of applicability in order to utilize them most effectively. This could perhaps be accomplished through development and testing of a panel of known carcinogens and non-carcinogens covering a wide range of mechanisms across these assay panels.
A third goal was to make recommendations for future assay development and validation to improve how chemical agents (e.g. xenobiotics, therapeutics) can be systematically evaluated for cancer hazard in vitro and in vivo. In this regard, the compilation of assays revealed specific challenges for certain KCs. For KC7 (is immunosuppressive) in vivo approaches remain paramount, but existing rodent and other experimental tumor models have limitations for detecting cancer hazards associated with immunomodulators. New methods are needed to evaluate systemic immune status based on systems immunology approaches, leveraging methods such as mass cytometry to interrogate multiple cell types and functions concomitantly (e.g., evidence of hematotoxicity affecting leukocytes, impact on cytotoxic T cells and/or NK cells). For some of these endpoints, assays still need to be optimized, and there remains a very limited understanding of how changes in such endpoints relate to cancer hazard. For KC6 (induces chronic inflammation), chronic in vivo exposures (in experimental animals or in humans) also remain more pertinent than in vitro co-cultures, followed by more simplistic in vitro studies. Development and validation of in vivo alternatives, e.g., the colonic organoid-based cell transformation assay (see Table 1), could be prioritized to fill these gaps.
Another important issue that emerged is the interrelationship of KCs and the impact on testing strategies and interpretation. For instance, multiple KCs consider seemingly similar effects on genome integrity. However, this complementarity makes it possible to functionally distinguish among genotoxic mechanisms, providing a more biologically accurate understanding of how the agent may cause cancer. Thus, each of the implicated KCs can be seen to have an important, but distinct role. At the other extreme, KC5 (induces oxidative stress) does not have a stand-alone role in causal identification of carcinogens, and is best seen in the context of its impact on other KCs (such as KC2, is genotoxic). Endpoints and markers such as oxidative damage to DNA and inflammation that specifically probe KC5 (induces oxidative stress) in concert with KC2 (is genotoxic) and KC6 (induces chronic inflammation), may be informative for interpretation. Similarly, for KC8 (modulates receptor-mediated effects), which broadly encompasses a range of different ligands and receptors with varying relation to cancer causation, related efforts on the KCs of endocrine disrupting chemicals (97) may aid interpretation. As multiple KCs may be additive or synergistic, further work is merited to understand how to best integrate endpoints and biomarkers impacted by multiple KCs, as well as to define minimal set(s) of KCs that could by themselves characterize certain carcinogens. Such efforts could, in concert with in silico predictions, inform the order of testing and attendant conclusions.
This exercise also raised questions about whether the KCs as currently defined capture all the pertinent mechanisms of carcinogens. For instance, carcinogens may impact the tumor microenvironment, enhance invasiveness and promote metastasis, but there are very few specific examples. These impacts may be investigated using organoids or models on chips and as such may currently be encompassed in KC10 (alters cell proliferation, cell death or nutrient supply), but further focus on these critical aspects of carcinogen action may be warranted.
Despite the identified challenges, this compilation of assays and biomarkers is useful in various ways. For one, it can inform priority-setting for funding and development of new assays, and to fill the identified gaps. The updated definitions and lists of associated assays will also advance application of the KCs in cancer hazard identification, as the basis for improving searches for existing mechanistic data relevant to the KCs, and in prioritizing the findings informative for evaluation. This compilation can also be used to design and conduct a battery of tests to probe carcinogenic activity. This could address all KCs, for a complete assessment, or target specific assays based on agent properties, predictive modeling (e.g., computational test for electrophilicity), or outstanding research gaps. Ultimately the choice of assay and extent of testing would be dependent on the application of the data, from screening and prioritization of agents for further testing, to exploring specific hypotheses, supporting product registration, or performing hazard identification and risk assessment. In some cases, standard assays are already available (e.g., with guidance from the Organization for Economic Co-operation and Development, OECD) and amenable to study in vitro. In others, there is a possibility for investigation in populations exposed to carcinogens, such as in occupational settings, to provide data especially relevant in hazard identification exercises (110). It would be especially useful if the mutational signatures of tumors from individuals exposed to specific carcinogens could be identified. These in vivo biomarkers and signatures would then be of great value in the translation of in vitro hazards and assess dose-response in experimental animals or humans.
To support these future applications, a number of short- and long-term steps can be envisioned. In addition to the outstanding assay development needs for various KCs, as noted above in the sections on KC5 (induces oxidative stress), KC6 (induces chronic inflammation), and KC7 (is immunosuppressive), assay validation work is relevant for several KCs. For instance, error-corrected NGS systems for detecting rare somatic mutations that approach the background level of mutations in humans are especially relevant for KC2 (is genotoxic) (33). It would be beneficial to develop standard lists of carcinogens and non-carcinogens to support assay qualification and validation, as well as future efforts toward evidence-based KC grouping and weighting. Publicly available resources such as open-source bioinformatic tools, detailed compilations of assays and the associated best practices for study design, conduct and reporting could also be useful. Further work could outline the priorities for such assay development, validation, and guidance for reporting and interpretation. In all, these future efforts will help to improve current methods for hazard screening and testing, and thereby advance carcinogen identification, a first step in cancer prevention.
Acknowledgements
This project was supported by contract #17-E0023 from the Office of Environmental Health Hazard Assessment of California EPA and the Research Translation Core of the NIEHS Superfund Research Program under NIH grant P42ES004705 (M.T. Smith).
Footnotes
Conflict of Interest Statement: MTS has received consulting fees from attorneys representing plaintiffs in cases involving exposure to benzene, glyphosate and other chemical agents. DWF has received consulting fees from attorneys in cases involving exposure to asbestos, PCBs, TCE and other chemical agents. SM and HL are employees of Amgen. MF was an employee of Amgen during the initial meetings and is currently an employee of Expansion Therapeutics. The other authors declare no competing interests. The views expressed in this publication are those of the authors and do not necessarily represent the decisions, policy or views of their respective institutions. Reference to commercial products or services does not constitute endorsement.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Guyton KZ, Kyle AD, Aubrecht J, Cogliano VJ, Eastmond DA, Jackson M, et al. Improving prediction of chemical carcinogenicity by considering multiple mechanisms and applying toxicogenomic approaches. Mutat Res 2009;681(2–3):230–40 doi 10.1016/j.mrrev.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Kleinstreuer NC, Dix DJ, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, et al. In vitro perturbations of targets in cancer hallmark processes predict rodent chemical carcinogenesis. Toxicol Sci 2013;131(1):40–55 doi 10.1093/toxsci/kfs285. [DOI] [PubMed] [Google Scholar]
- 5.Smith MT, Guyton KZ, Gibbons CF, Fritz JM, Portier CJ, Rusyn I, et al. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environ Health Perspect 2016;124(6):713–21 doi 10.1289/ehp.1509912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart BW. Mechanisms of carcinogenesis: from initiation and promotion to the hallmarks In: Baan RA, Stewart BW, Straif K, editors. Tumour Site Concordance and Mechanisms of Carcinogenesis. Volume No. 165 Lyon, France: IARC Scientific Publications; 2019. p 93–106. [Google Scholar]
- 7.National Academies of Sciences E, Medicine. Using 21st Century Science to Improve Risk-Related Evaluations. Washington, DC: The National Academies Press; 2017. 200 p. [PubMed] [Google Scholar]
- 8.Guyton KZ, Rusyn I, Chiu WA, Corpet DE, van den Berg M, Ross MK, et al. Application of the key characteristics of carcinogens in cancer hazard identification. Carcinogenesis 2018;39(4):614–22 doi 10.1093/carcin/bgy031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.IARC. Preamble, IARC Monographs on the evaluation of carcinogenic risks to humans. Lyon, France: IARC, Amended January; 2019. Lyons, France2019. [Google Scholar]
- 10.National Toxicology Program. Report on Carcinogens Monograph on Antimony Trioxide. Research Traingle Park, NC:: U.S. Department of Health and Human Services, Public Health Service; 2018. [Google Scholar]
- 11.National Toxicology Program. Report on Carcinogens Monograph on Haloacetic Acids Found as Water Disinfection By-Products. Research Traingle Park, NC:: U.S. Department of Health and Human Services, Public Health Service; 2018. [PubMed] [Google Scholar]
- 12.Menyhart O, Harami-Papp H, Sukumar S, Schafer R, Magnani L, de Barrios O, et al. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim Biophys Acta 2016;1866(2):300–19 doi 10.1016/j.bbcan.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Schwobel JA, Koleva YK, Enoch SJ, Bajot F, Hewitt M, Madden JC, et al. Measurement and estimation of electrophilic reactivity for predictive toxicology. Chem Rev 2011;111(4):2562–96 doi 10.1021/cr100098n. [DOI] [PubMed] [Google Scholar]
- 14.Kuriyama R, Sakai H. Role of tubulin-SH groups in polymerization to microtubules. Functional-SH groups in tubulin for polymerization. J Biochem 1974;76(3):651–4 doi 10.1093/oxfordjournals.jbchem.a130609. [DOI] [PubMed] [Google Scholar]
- 15.Schultz TW, Carlson RE, Cronin MT, Hermens JL, Johnson R, O’Brien PJ, et al. A conceptual framework for predicting the toxicity of reactive chemicals: modeling soft electrophilicity. SAR QSAR Environ Res 2006;17(4):413–28 doi 10.1080/10629360600884371. [DOI] [PubMed] [Google Scholar]
- 16.Mekenyan OG, Dimitrov SD, Pavlov TS, Veith GD. A systematic approach to simulating metabolism in computational toxicology. I. The TIMES heuristic modelling framework. Curr Pharm Des 2004;10(11):1273–93. [DOI] [PubMed] [Google Scholar]
- 17.McCarthy TJ, Hayes EP, Schwartz CS, Witz G. The reactivity of selected acrylate esters toward glutathione and deoxyribonucleosides in vitro: structure-activity relationships. Fundam Appl Toxicol 1994;22(4):543–8. [DOI] [PubMed] [Google Scholar]
- 18.McCallum MM, Nandhikonda P, Temmer JJ, Eyermann C, Simeonov A, Jadhav A, et al. High-throughput identification of promiscuous inhibitors from screening libraries with the use of a thiol-containing fluorescent probe. J Biomol Screen 2013;18(6):705–13 doi 10.1177/1087057113476090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrenberg L, Granath F, Tornqvist M. Macromolecule adducts as biomarkers of exposure to environmental mutagens in human populations. Environ Health Perspect 1996;104 Suppl 3:423–8 doi 10.1289/ehp.96104s3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J, Villalta PW, Weight CJ, Bonala R, Johnson F, Rosenquist TA, et al. Targeted and Untargeted Detection of DNA Adducts of Aromatic Amine Carcinogens in Human Bladder by Ultra-Performance Liquid Chromatography-High-Resolution Mass Spectrometry. Chem Res Toxicol 2018. doi 10.1021/acs.chemrestox.8b00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwa Yun B, Guo J, Bellamri M, Turesky RJ. DNA adducts: Formation, biological effects, and new biospecimens for mass spectrometric measurements in humans. Mass Spectrom Rev 2018. doi 10.1002/mas.21570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang YJ, Cooke MS, Hu CW, Chao MR. Novel approach to integrated DNA adductomics for the assessment of in vitro and in vivo environmental exposures. Arch Toxicol 2018;92(8):2665–80 doi 10.1007/s00204-018-2252-6. [DOI] [PubMed] [Google Scholar]
- 23.Grigoryan H, Edmands WMB, Lan Q, Carlsson H, Vermeulen R, Zhang L, et al. Adductomic signatures of benzene exposure provide insights into cancer induction. Carcinogenesis 2018;39(5):661–8 doi 10.1093/carcin/bgy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ford B, Bateman LA, Gutierrez-Palominos L, Park R, Nomura DK. Mapping Proteome-wide Targets of Glyphosate in Mice. Cell Chem Biol 2017;24(2):133–40 doi 10.1016/j.chembiol.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Cimino MC. Comparative overview of current international strategies and guidelines for genetic toxicology testing for regulatory purposes. Environ Mol Mutagen 2006;47(5):362–90 doi 10.1002/em.20216. [DOI] [PubMed] [Google Scholar]
- 26.Bonassi S, El-Zein R, Bolognesi C, Fenech M. Micronuclei frequency in peripheral blood lymphocytes and cancer risk: evidence from human studies. Mutagenesis 2011;26(1):93–100 doi 10.1093/mutage/geq075. [DOI] [PubMed] [Google Scholar]
- 27.Bonassi S, Norppa H, Ceppi M, Stromberg U, Vermeulen R, Znaor A, et al. Chromosomal aberration frequency in lymphocytes predicts the risk of cancer: results from a pooled cohort study of 22 358 subjects in 11 countries. Carcinogenesis 2008;29(6):1178–83 doi 10.1093/carcin/bgn075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Benzene; Lyons, France: 2018. [Google Scholar]
- 29.Zhang L, Eastmond DA, Smith MT. The nature of chromosomal aberrations detected in humans exposed to benzene. Crit Rev Toxicol 2002;32(1):1–42 doi 10.1080/20024091064165. [DOI] [PubMed] [Google Scholar]
- 30.Eastmond DA, Mondrala ST, Hasegawa L. Topoisomerase II inhibition by myeloperoxidase-activated hydroquinone: a potential mechanism underlying the genotoxic and carcinogenic effects of benzene. Chem Biol Interact 2005;153–154:207–16 doi 10.1016/j.cbi.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Gowans ID, Lorimore SA, McIlrath JM, Wright EG. Genotype-dependent induction of transmissible chromosomal instability by gamma-radiation and the benzene metabolite hydroquinone. Cancer Res 2005;65(9):3527–30 doi 10.1158/0008-5472.CAN-04-4242. [DOI] [PubMed] [Google Scholar]
- 32.Kolachana P, Subrahmanyam VV, Meyer KB, Zhang L, Smith MT. Benzene and its phenolic metabolites produce oxidative DNA damage in HL60 cells in vitro and in the bone marrow in vivo. Cancer Res 1993;53(5):1023–6. [PubMed] [Google Scholar]
- 33.Salk JJ, Loubet-Senear K, Maritschnegg E, Valentine CC, Williams LN, Higgins JE, et al. Ultra-Sensitive TP53 Sequencing for Cancer Detection Reveals Progressive Clonal Selection in Normal Tissue over a Century of Human Lifespan. Cell Rep 2019;28(1):132–44 e3 doi 10.1016/j.celrep.2019.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 2017;58(5):235–63 doi 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen Z Genomic instability and cancer: an introduction. J Mol Cell Biol 2011;3(1):1–3 doi 10.1093/jmcb/mjq057. [DOI] [PubMed] [Google Scholar]
- 36.Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nature reviews Cancer 2011;11(6):450–7 doi 10.1038/nrc3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langie SA, Koppen G, Desaulniers D, Al-Mulla F, Al-Temaimi R, Amedei A, et al. Causes of genome instability: the effect of low dose chemical exposures in modern society. Carcinogenesis 2015;36 Suppl 1:S61–88 doi 10.1093/carcin/bgv031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan SLW, Chadha S, Liu Y, Gabasova E, Perera D, Ahmed K, et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017;169(6):1105–18 e15 doi 10.1016/j.cell.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mukherjee D, Coates PJ, Lorimore SA, Wright EG. Responses to ionizing radiation mediated by inflammatory mechanisms. J Pathol 2014;232(3):289–99 doi 10.1002/path.4299. [DOI] [PubMed] [Google Scholar]
- 40.Sykora P, Witt KL, Revanna P, Smith-Roe SL, Dismukes J, Lloyd DG, et al. Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci Rep 2018;8(1):2771 doi 10.1038/s41598-018-20995-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Svilar D, McClellan S, Kim JH, Ahn EE, Vens C, et al. DNA Repair Molecular Beacon assay: a platform for real-time functional analysis of cellular DNA repair capacity. Oncotarget 2018;9(60):31719–43 doi 10.18632/oncotarget.25859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaim IA, Nagel ZD, Jordan JJ, Mazzucato P, Ngo LP, Samson LD. In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proc Natl Acad Sci U S A 2017;114(48):E10379–E88 doi 10.1073/pnas.1712032114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dou Y, Gold HD, Luquette LJ, Park PJ. Detecting Somatic Mutations in Normal Cells. Trends in genetics : TIG 2018;34(7):545–57 doi 10.1016/j.tig.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouraoui S, Brahem A, Tabka F, Mrizek N, Saad A, Elghezal H. Assessment of chromosomal aberrations, micronuclei and proliferation rate index in peripheral lymphocytes from Tunisian nurses handling cytotoxic drugs. Environ Toxicol Pharmacol 2011;31(1):250–7 doi 10.1016/j.etap.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol 2015;10:425–48 doi 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- 46.Fitzgerald DM, Hastings PJ, Rosenberg SM. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu Rev Cancer Biol 2017;1:119–40 doi 10.1146/annurev-cancerbio-050216-121919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Compagno M, Wang Q, Pighi C, Cheong TC, Meng FL, Poggio T, et al. Phosphatidylinositol 3-kinase delta blockade increases genomic instability in B cells. Nature 2017;542(7642):489–93 doi 10.1038/nature21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018;553(7687):171–7 doi 10.1038/nature25154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimizu T, Marusawa H, Endo Y, Chiba T. Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci 2012;103(7):1201–6 doi 10.1111/j.1349-7006.2012.02293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Imataka G, Arisaka O. Chromosome analysis using spectral karyotyping (SKY). Cell Biochem Biophys 2012;62(1):13–7 doi 10.1007/s12013-011-9285-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris KL, Myers MB, McKim KL, Elespuru RK, Parsons BL. Rationale and Roadmap for Developing Panels of Hotspot Cancer Driver Gene Mutations as Biomarkers of Cancer Risk. Environ Mol Mutagen 2019. doi 10.1002/em.22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods Mol Biol 2015;1238:3–25 doi 10.1007/978-1-4939-1804-1_1. [DOI] [PubMed] [Google Scholar]
- 53.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science (New York, NY) 2017;357(6348) doi 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feinberg AP. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N Engl J Med 2018;378(14):1323–34 doi 10.1056/NEJMra1402513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016;17(5):284–99 doi 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okoji RS, Yu RC, Maronpot RR, Froines JR. Sodium arsenite administration via drinking water increases genome-wide and Ha-ras DNA hypomethylation in methyl-deficient C57BL/6J mice. Carcinogenesis 2002;23(5):777–85 doi 10.1093/carcin/23.5.777. [DOI] [PubMed] [Google Scholar]
- 57.Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, Lahiri S, et al. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol Sci 2006;89(2):431–7 doi 10.1093/toxsci/kfj030. [DOI] [PubMed] [Google Scholar]
- 58.Volker-Albert MC, Schmidt A, Forne I, Imhof A. Analysis of Histone Modifications by Mass Spectrometry. Curr Protoc Protein Sci 2018;92(1):e54 doi 10.1002/cpps.54. [DOI] [PubMed] [Google Scholar]
- 59.Cheng TF, Choudhuri S, Muldoon-Jacobs K. Epigenetic targets of some toxicologically relevant metals: a review of the literature. J Appl Toxicol 2012;32(9):643–53 doi 10.1002/jat.2717. [DOI] [PubMed] [Google Scholar]
- 60.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 2015;109:21 9 1–9 doi 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan H, Tian S, Slager SL, Sun Z. ChIP-seq in studying epigenetic mechanisms of disease and promoting precision medicine: progresses and future directions. Epigenomics 2016;8(9):1239–58 doi 10.2217/epi-2016-0053. [DOI] [PubMed] [Google Scholar]
- 62.Li M, Huo X, Davuljigari CB, Dai Q, Xu X. MicroRNAs and their role in environmental chemical carcinogenesis. Environ Geochem Health 2019;41(1):225–47 doi 10.1007/s10653-018-0179-8. [DOI] [PubMed] [Google Scholar]
- 63.Pogribny IP, Beland FA, Rusyn I. The role of microRNAs in the development and progression of chemical-associated cancers. Toxicol Appl Pharmacol 2016;312:3–10 doi 10.1016/j.taap.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 64.IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans.. Lyons, France: 2012. [Google Scholar]
- 65.Berquist BR, Wilson DM 3rd Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett 2012;327(1–2):61–72 doi 10.1016/j.canlet.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klaunig JE, Wang Z, Pu X, Zhou S. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol Appl Pharmacol 2011;254(2):86–99 doi 10.1016/j.taap.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 67.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 2010;49(11):1603–16 doi 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, ME LL. Oxidative stress and cancer: an overview. Ageing Res Rev 2013;12(1):376–90 doi 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 69.Schneider CD, Bock PM, Becker GF, Moreira JCF, Bello-Klein A, Oliveira AR. Comparison of the effects of two antioxidant diets on oxidative stress markers in triathletes. Biol Sport 2018;35(2):181–9 doi 10.5114/biolsport.2018.74194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roh JY, Kim PG, Kwon JH. Comparative study of oxidative stress caused by anthracene and alkyl-anthracenes in Caenorhabditis elegans. Environ Health Toxicol 2018;33(1):e2018006 doi 10.5620/eht.e2018006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature 2016;529(7586):307–15 doi 10.1038/nature17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diaz-Beveridge R, Bruixola G, Lorente D, Caballero J, Rodrigo E, Segura A, et al. An internally validated new clinical and inflammation-based prognostic score for patients with advanced hepatocellular carcinoma treated with sorafenib. Clin Transl Oncol 2018;20(3):322–9 doi 10.1007/s12094-017-1720-4. [DOI] [PubMed] [Google Scholar]
- 73.Honeycutt J, Hammam O, Fu CL, Hsieh MH. Controversies and challenges in research on urogenital schistosomiasis-associated bladder cancer. Trends Parasitol 2014;30(7):324–32 doi 10.1016/j.pt.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 2014;15(12):786–801 doi 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allin KH, Bojesen SE, Nordestgaard BG. Inflammatory biomarkers and risk of cancer in 84,000 individuals from the general population. Int J Cancer 2016;139(7):1493–500 doi 10.1002/ijc.30194. [DOI] [PubMed] [Google Scholar]
- 76.Brenner DR, Scherer D, Muir K, Schildkraut J, Boffetta P, Spitz MR, et al. A review of the application of inflammatory biomarkers in epidemiologic cancer research. Cancer Epidemiol Biomarkers Prev 2014;23(9):1729–51 doi 10.1158/1055-9965.EPI-14-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meirow Y, Baniyash M. Immune biomarkers for chronic inflammation related complications in non-cancerous and cancerous diseases. Cancer Immunol Immunother 2017;66(8):1089–101 doi 10.1007/s00262-017-2035-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mesaros C, Worth AJ, Snyder NW, Christofidou-Solomidou M, Vachani A, Albelda SM, et al. Bioanalytical techniques for detecting biomarkers of response to human asbestos exposure. Bioanalysis 2015;7(9):1157–73 doi 10.4155/bio.15.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med 2019;25(12):1822–32 doi 10.1038/s41591-019-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gong Y, Liang S, Zeng L, Ni Y, Zhou S, Yuan X. Effects of blood sample handling procedures on measurable interleukin 6 in plasma and serum. J Clin Lab Anal 2019;33(7):e22924 doi 10.1002/jcla.22924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Opelz G, Henderson R. Incidence of non-Hodgkin lymphoma in kidney and heart transplant recipients. Lancet 1993;342(8886–8887):1514–6 doi 10.1016/s0140-6736(05)80084-4. [DOI] [PubMed] [Google Scholar]
- 82.Honaryar MK, Lunn RM, Luce D, Ahrens W, t Mannetje A, Hansen J, et al. Welding fumes and lung cancer: a meta-analysis of case-control and cohort studies. Occup Environ Med 2019;76(6):422–31 doi 10.1136/oemed-2018-105447. [DOI] [PubMed] [Google Scholar]
- 83.Lebrec H, Brennan FR, Haggerty H, Herzyk D, Kamperschroer C, Maier CC, et al. HESI/FDA workshop on immunomodulators and cancer risk assessment: Building blocks for a weight-of-evidence approach. Regul Toxicol Pharmacol 2016;75:72–80 doi 10.1016/j.yrtph.2015.12.018. [DOI] [PubMed] [Google Scholar]
- 84.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39(1):1–10 doi 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 85.Bugelski PJ, Volk A, Walker MR, Krayer JH, Martin P, Descotes J. Critical review of preclinical approaches to evaluate the potential of immunosuppressive drugs to influence human neoplasia. Int J Toxicol 2010;29(5):435–66 doi 10.1177/1091581810374654. [DOI] [PubMed] [Google Scholar]
- 86.Yip CH, Rhodes A. Estrogen and progesterone receptors in breast cancer. Future Oncol 2014;10(14):2293–301 doi 10.2217/fon.14.110. [DOI] [PubMed] [Google Scholar]
- 87.Hoang DT, Iczkowski KA, Kilari D, See W, Nevalainen MT. Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles. Oncotarget 2017;8(2):3724–45 doi 10.18632/oncotarget.12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat 2012;136(2):331–45 doi 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cole KD, He HJ, Wang L. Breast cancer biomarker measurements and standards. Proteomics Clin Appl 2013;7(1–2):17–29 doi 10.1002/prca.201200075. [DOI] [PubMed] [Google Scholar]
- 90.Allan LL, Sherr DH. Constitutive activation and environmental chemical induction of the aryl hydrocarbon receptor/transcription factor in activated human B lymphocytes. Mol Pharmacol 2005;67(5):1740–50 doi 10.1124/mol.104.009100. [DOI] [PubMed] [Google Scholar]
- 91.Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nature reviews Cancer 2014;14(12):801–14 doi 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vos JG, Moore JA, Zinkl JG. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the immune system of laboratory animals. Environ Health Perspect 1973;5:149–62 doi 10.1289/ehp.7305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holden PR, Tugwood JD. Peroxisome proliferator-activated receptor alpha: role in rodent liver cancer and species differences. J Mol Endocrinol 1999;22(1):1–8. [DOI] [PubMed] [Google Scholar]
- 94.Lake BG. Human relevance of rodent liver tumour formation by constitutive androstane receptor (CAR) activators. Toxicol Res (Camb) 2018;7(4):697–717 doi 10.1039/c8tx00008e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Felter SP, Foreman JE, Boobis A, Corton JC, Doi AM, Flowers L, et al. Human relevance of rodent liver tumors: Key insights from a Toxicology Forum workshop on nongenotoxic modes of action. Regul Toxicol Pharmacol 2018;92:1–7 doi 10.1016/j.yrtph.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Safe S, Lee SO, Jin UH. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol Sci 2013;135(1):1–16 doi 10.1093/toxsci/kft128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.La Merrill MA, Vandenberg LN, Smith MT, Goodson W, Browne P, Patisaul HB, et al. Key Characteristics of Endocrine-Disrupting Chemicals as a Basis for Hazard Identification. Nature Reviews Endocrinology In Press 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer discovery 2018;8(5):537–55 doi 10.1158/2159-8290.cd-17-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016;539:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, et al. Replication stress activates DNA repair synthesis in mitosis. Nature 2015;528(7581):286–90 doi 10.1038/nature16139. [DOI] [PubMed] [Google Scholar]
- 101.Carnero A, Blanco-Aparicio C, Kondoh H, Lleonart ME, Martinez-Leal JF, Mondello C, et al. Disruptive chemicals, senescence and immortality. Carcinogenesis 2015;36 Suppl 1:S19–37 doi 10.1093/carcin/bgv029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nahta R, Al-Mulla F, Al-Temaimi R, Amedei A, Andrade-Vieira R, Bay SN, et al. Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression. Carcinogenesis 2015;36 Suppl 1:S2–18 doi 10.1093/carcin/bgv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004;23(16):2919–33 doi 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 104.Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res 2006;66(9):4553–7 doi 10.1158/0008-5472.CAN-05-3986. [DOI] [PubMed] [Google Scholar]
- 105.Iacovides D, Michael S, Achilleos C, Strati K. Shared mechanisms in stemness and carcinogenesis: lessons from oncogenic viruses. Front Cell Infect Microbiol 2013;3:66 doi 10.3389/fcimb.2013.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002;21(48):7435–51 doi 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 107.Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol 2014;2014:502676 doi 10.1155/2014/502676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dairkee SH, Luciani-Torres G, Moore DH, Jaffee IM, Goodson WH 3rd. A Ternary Mixture of Common Chemicals Perturbs Benign Human Breast Epithelial Cells More Than the Same Chemicals Do Individually. Toxicol Sci 2018;165(1):131–44 doi 10.1093/toxsci/kfy126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dairkee SH, Luciani-Torres MG, Moore DH, Goodson WH 3rd. Bisphenol-A-induced inactivation of the p53 axis underlying deregulation of proliferation kinetics, and cell death in non-malignant human breast epithelial cells. Carcinogenesis 2013;34(3):703–12 doi 10.1093/carcin/bgs379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Samet JM, Chiu WA, Cogliano V, Jinot J, Kriebel D, Lunn RM, et al. The IARC Monographs: Updated procedures for modern and transparent evidence synthesis in cancer hazard identification. J Natl Cancer Inst 2019. doi 10.1093/jnci/djz169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ford KA. Role of electrostatic potential in the in silico prediction of molecular bioactivation and mutagenesis. Mol Pharm 2013;10(4):1171–82 doi 10.1021/mp3004385. [DOI] [PubMed] [Google Scholar]
- 112.Rebecca CEMJGJRF. Glutathione depletion in a liver microsomal assay as an in vitro biomarker for reactive metabolite formation. Biomarkers 2000;5(4):285–94 doi 10.1080/135475000413836. [DOI] [PubMed] [Google Scholar]
- 113.Counihan JL, Ford B, Nomura DK. Mapping proteome-wide interactions of reactive chemicals using chemoproteomic platforms. Curr Opin Chem Biol 2016;30:68–76 doi 10.1016/j.cbpa.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Klaene JJ, Sharma VK, Glick J, Vouros P. The analysis of DNA adducts: the transition from (32)P-postlabeling to mass spectrometry. Cancer Lett 2013;334(1):10–9 doi 10.1016/j.canlet.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wichard JD. In silico prediction of genotoxicity. Food Chem Toxicol 2017;106(Pt B):595–9 doi 10.1016/j.fct.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 116.Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet 2018;19(5):269–85 doi 10.1038/nrg.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li HH, Chen R, Hyduke DR, Williams A, Frotschl R, Ellinger-Ziegelbauer H, et al. Development and validation of a high-throughput transcriptomic biomarker to address 21st century genetic toxicology needs. Proc Natl Acad Sci U S A 2017;114(51):E10881–E9 doi 10.1073/pnas.1714109114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shinawi M, Cheung SW. The array CGH and its clinical applications. Drug Discov Today 2008;13(17–18):760–70 doi 10.1016/j.drudis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 119.Deschoolmeester V, Baay M, Wuyts W, Van Marck E, Van Damme N, Vermeulen P, et al. Detection of microsatellite instability in colorectal cancer using an alternative multiplex assay of quasi-monomorphic mononucleotide markers. J Mol Diagn 2008;10(2):154–9 doi 10.2353/jmoldx.2008.070087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Madle S, Dean SW, Andrae U, Brambilla G, Burlinson B, Doolittle DJ, et al. Recommendations for the performance of UDS tests in vitro and in vivo. Mutat Res 1994;312(3):263–85 doi 10.1016/0165-1161(94)00013-1. [DOI] [PubMed] [Google Scholar]
- 121.Mendez P, Taron M, Moran T, Fernandez MA, Requena G, Rosell R. A modified host-cell reactivation assay to quantify DNA repair capacity in cryopreserved peripheral lymphocytes. DNA Repair (Amst) 2011;10(6):603–10 doi 10.1016/j.dnarep.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 122.Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, et al. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A 2014;111(18):E1823–32 doi 10.1073/pnas.1401182111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nitiss JL, Soans E, Rogojina A, Seth A, Mishina M. Topoisomerase assays. Curr Protoc Pharmacol 2012;Chapter 3:Unit 3 doi 10.1002/0471141755.ph0303s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chowdhury B, Cho IH, Irudayaraj J. Technical advances in global DNA methylation analysis in human cancers. J Biol Eng 2017;11:10 doi 10.1186/s13036-017-0052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Parfett CL, Desaulniers D. A Tox21 Approach to Altered Epigenetic Landscapes: Assessing Epigenetic Toxicity Pathways Leading to Altered Gene Expression and Oncogenic Transformation In Vitro. Int J Mol Sci 2017;18(6) doi 10.3390/ijms18061179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen YR, Yu S, Zhong S. Profiling DNA Methylation Using Bisulfite Sequencing (BS-Seq). Methods Mol Biol 2018;1675:31–43 doi 10.1007/978-1-4939-7318-7_2. [DOI] [PubMed] [Google Scholar]
- 127.Cuomo A, Soldi M, Bonaldi T. SILAC-Based Quantitative Strategies for Accurate Histone Posttranslational Modification Profiling Across Multiple Biological Samples. Methods Mol Biol 2017;1528:97–119 doi 10.1007/978-1-4939-6630-1_7. [DOI] [PubMed] [Google Scholar]
- 128.Zane L, Chapus F, Pegoraro G, Misteli T. HiHiMap: single-cell quantitation of histones and histone posttranslational modifications across the cell cycle by high-throughput imaging. Mol Biol Cell 2017;28(17):2290–302 doi 10.1091/mbc.E16-12-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murano K, Iwasaki YW, Siomi H. Profiling Open Chromatin Structure in the Ovarian Somatic Cells Using ATAC-seq. Methods Mol Biol 2018;1680:165–77 doi 10.1007/978-1-4939-7339-2_11. [DOI] [PubMed] [Google Scholar]
- 130.Yamada A, Yu P, Lin W, Okugawa Y, Boland CR, Goel A. A RNA-Sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Sci Rep 2018;8(1):575 doi 10.1038/s41598-017-18407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Urbanek MO, Nawrocka AU, Krzyzosiak WJ. Small RNA Detection by in Situ Hybridization Methods. Int J Mol Sci 2015;16(6):13259–86 doi 10.3390/ijms160613259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Salehi S, Taheri MN, Azarpira N, Zare A, Behzad-Behbahani A. State of the art technologies to explore long non-coding RNAs in cancer. J Cell Mol Med 2017;21(12):3120–40 doi 10.1111/jcmm.13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Soares RJ, Maglieri G, Gutschner T, Diederichs S, Lund AH, Nielsen BS, et al. Evaluation of fluorescence in situ hybridization techniques to study long non-coding RNA expression in cultured cells. Nucleic Acids Res 2018;46(1):e4 doi 10.1093/nar/gkx946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Katerji M, Filippova M, Duerksen-Hughes P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxid Med Cell Longev 2019;2019:1279250 doi 10.1155/2019/1279250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Koivisto P, Peltonen K. Analytical methods in DNA and protein adduct analysis. Anal Bioanal Chem 2010;398(6):2563–72 doi 10.1007/s00216-010-4217-3. [DOI] [PubMed] [Google Scholar]
- 136.Collins AR. Measuring oxidative damage to DNA and its repair with the comet assay. Biochim Biophys Acta 2014;1840(2):794–800 doi 10.1016/j.bbagen.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 137.Suzen S, Gurer-Orhan H, Saso L. Detection of Reactive Oxygen and Nitrogen Species by Electron Paramagnetic Resonance (EPR) Technique. Molecules 2017;22(1) doi 10.3390/molecules22010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Giustarini D, Colombo G, Garavaglia ML, Astori E, Portinaro NM, Reggiani F, et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic Biol Med 2017;112:360–75 doi 10.1016/j.freeradbiomed.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 139.Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc 2010;5(1):51–66 doi 10.1038/nprot.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Osburn WO, Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res 2008;659(1–2):31–9 doi 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol 2004;286(3):R431–44 doi 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 142.Hibiya S, Tsuchiya K, Hayashi R, Fukushima K, Horita N, Watanabe S, et al. Long-term Inflammation Transforms Intestinal Epithelial Cells of Colonic Organoids. J Crohns Colitis 2017;11(5):621–30 doi 10.1093/ecco-jcc/jjw186. [DOI] [PubMed] [Google Scholar]
- 143.Lebrec H, Roger R, Blot C, Burleson GR, Bohuon C, Pallardy M. Immunotoxicological investigation using pharmaceutical drugs. In vitro evaluation of immune effects using rodent or human immune cells. Toxicology 1995;96(2):147–56 doi 10.1016/0300-483x(94)02956-u. [DOI] [PubMed] [Google Scholar]
- 144.Frank B, Wei YL, Kim KH, Guerrero A, Lebrec H, Balazs M, et al. Development of a BiTE((R))-mediated CD8(+) cytotoxic T-lymphocyte activity assay to assess immunomodulatory potential of drug candidates in Cynomolgus macaque. J Immunotoxicol 2018;15(1):119–25 doi 10.1080/1547691X.2018.1486342. [DOI] [PubMed] [Google Scholar]
- 145.Li Q Natural Killer (NK) Cell Assays in Immunotoxicity Testing. Methods Mol Biol 2018;1803:231–41 doi 10.1007/978-1-4939-8549-4_15. [DOI] [PubMed] [Google Scholar]
- 146.Feau C, Arnold LA, Kosinski A, Guy RK. Ligand competition binding assay for the androgen receptor. Methods Mol Biol 2011;776:59–68 doi 10.1007/978-1-61779-243-4_4. [DOI] [PubMed] [Google Scholar]
- 147.Judson RS, Magpantay FM, Chickarmane V, Haskell C, Tania N, Taylor J, et al. Integrated Model of Chemical Perturbations of a Biological Pathway Using 18 In Vitro High-Throughput Screening Assays for the Estrogen Receptor. Toxicol Sci 2015;148(1):137–54 doi 10.1093/toxsci/kfv168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kleinstreuer NC, Ceger P, Watt ED, Martin M, Houck K, Browne P, et al. Development and Validation of a Computational Model for Androgen Receptor Activity. Chem Res Toxicol 2017;30(4):946–64 doi 10.1021/acs.chemrestox.6b00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Otarola G, Castillo H, Marcellini S. Aryl hydrocarbon receptor-based bioassays for dioxin detection: Thinking outside the box. J Appl Toxicol 2018;38(4):437–49 doi 10.1002/jat.3575. [DOI] [PubMed] [Google Scholar]
- 150.Creton S, Aardema MJ, Carmichael PL, Harvey JS, Martin FL, Newbold RF, et al. Cell transformation assays for prediction of carcinogenic potential: state of the science and future research needs. Mutagenesis 2012;27(1):93–101 doi 10.1093/mutage/ger053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Noren Hooten N, Evans MK. Techniques to Induce and Quantify Cellular Senescence. J Vis Exp 2017(123) doi 10.3791/55533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Skvortsov DA, Zvereva ME, Shpanchenko OV, Dontsova OA. Assays for detection of telomerase activity. Acta Naturae 2011;3(1):48–68. [PMC free article] [PubMed] [Google Scholar]
- 153.Wakao S, Kitada M, Kuroda Y, Ogura F, Murakami T, Niwa A, et al. Morphologic and gene expression criteria for identifying human induced pluripotent stem cells. PLoS One 2012;7(12):e48677 doi 10.1371/journal.pone.0048677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhu LF, Xiao M, Chen YQ, Wang LY, Luo XF, Yuan XH, et al. In vitro effects of reprogramming factors on the expressions of pluripotent genes and CD34 gene in human acute promyelocytic leukemia HL-60 cells. Genomics 2017;109(5–6):331–5 doi 10.1016/j.ygeno.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 155.Wiepz GJ, Edwin F, Patel T, Bertics PJ. Methods for determining the proliferation of cells in response to EGFR ligands. Methods Mol Biol 2006;327:179–87 doi 10.1385/1-59745-012-X:179. [DOI] [PubMed] [Google Scholar]
- 156.Borowicz S, Van Scoyk M, Avasarala S, Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK, et al. The soft agar colony formation assay. J Vis Exp 2014(92):e51998 doi 10.3791/51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sayeed A, Luciani-Torres G, Meng Z, Bennington JL, Moore DH, Dairkee SH. Aberrant regulation of the BST2 (Tetherin) promoter enhances cell proliferation and apoptosis evasion in high grade breast cancer cells. PLoS One 2013;8(6):e67191 doi 10.1371/journal.pone.0067191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lipponen P Apoptosis in breast cancer: relationship with other pathological parameters. Endocr Relat Cancer 1999;6(1):13–6. [DOI] [PubMed] [Google Scholar]
- 159.Eccles SA, Court W, Patterson L. In Vitro Assays for Endothelial Cell Functions Required for Angiogenesis: Proliferation, Motility, Tubular Differentiation, and Matrix Proteolysis. Methods Mol Biol 2016;1430:121–47 doi 10.1007/978-1-4939-3628-1_8. [DOI] [PubMed] [Google Scholar]
- 160.Marshall J Transwell((R)) invasion assays. Methods Mol Biol 2011;769:97–110 doi 10.1007/978-1-61779-207-6_8. [DOI] [PubMed] [Google Scholar]
- 161.Zippel N, Ding Y, Fleming I. A Modified Aortic Ring Assay to Assess Angiogenic Potential In Vitro. Methods Mol Biol 2016;1430:205–19 doi 10.1007/978-1-4939-3628-1_14. [DOI] [PubMed] [Google Scholar]
- 162.Pike Winer LS, Wu M. Rapid analysis of glycolytic and oxidative substrate flux of cancer cells in a microplate. PLoS One 2014;9(10):e109916 doi 10.1371/journal.pone.0109916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Koen YM, Galeva NA, Metushi IG, Uetrecht J, Hanzlik RP. Protein Targets of Isoniazid-Reactive Metabolites in Mouse Liver in Vivo. Chem Res Toxicol 2016;29(6):1064–72 doi 10.1021/acs.chemrestox.6b00098. [DOI] [PubMed] [Google Scholar]
- 164.Tornqvist M, Fred C, Haglund J, Helleberg H, Paulsson B, Rydberg P. Protein adducts: quantitative and qualitative aspects of their formation, analysis and applications. J Chromatogr B Analyt Technol Biomed Life Sci 2002;778(1–2):279–308 doi 10.1016/s1570-0232(02)00172-1. [DOI] [PubMed] [Google Scholar]
- 165.Carlsson H, Rappaport SM, Tornqvist M. Protein Adductomics: Methodologies for Untargeted Screening of Adducts to Serum Albumin and Hemoglobin in Human Blood Samples. High Throughput 2019;8(1) doi 10.3390/ht8010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yu Y, Wang P, Cui Y, Wang Y. Chemical Analysis of DNA Damage. Anal Chem 2018;90(1):556–76 doi 10.1021/acs.analchem.7b04247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Balbo S, Turesky RJ, Villalta PW. DNA adductomics. Chem Res Toxicol 2014;27(3):356–66 doi 10.1021/tx4004352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tang D, Phillips DH, Stampfer M, Mooney LA, Hsu Y, Cho S, et al. Association between carcinogen-DNA adducts in white blood cells and lung cancer risk in the physicians health study. Cancer Res 2001;61(18):6708–12. [PubMed] [Google Scholar]
- 169.Dobrovolsky VN, Miura D, Heflich RH, Dertinger SD. The in vivo Pig-a gene mutation assay, a potential tool for regulatory safety assessment. Environ Mol Mutagen 2010;51(8–9):825–35 doi 10.1002/em.20627. [DOI] [PubMed] [Google Scholar]
- 170.Olsen LS, Nielsen LR, Nexo BA, Wassermann K. Somatic mutation detection in human biomonitoring. Pharmacol Toxicol 1996;78(6):364–73 doi 10.1111/j.1600-0773.1996.tb00220.x. [DOI] [PubMed] [Google Scholar]
- 171.Grant SG. The GPA in vivo somatic mutation assay. Methods Mol Biol 2005;291:179–95 doi 10.1385/1-59259-840-4:179. [DOI] [PubMed] [Google Scholar]
- 172.Thomas P, Holland N, Bolognesi C, Kirsch-Volders M, Bonassi S, Zeiger E, et al. Buccal micronucleus cytome assay. Nat Protoc 2009;4(6):825–37 doi 10.1038/nprot.2009.53. [DOI] [PubMed] [Google Scholar]
- 173.Fenech M Cytokinesis-block micronucleus cytome assay. Nat Protoc 2007;2(5):1084–104 doi 10.1038/nprot.2007.77. [DOI] [PubMed] [Google Scholar]
- 174.Ji Z, Zhang L. Chromosomics: detection of numerical and structural alterations in all 24 human chromosomes simultaneously using a novel OctoChrome FISH assay. J Vis Exp 2012(60) doi 10.3791/3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Moller P The alkaline comet assay: towards validation in biomonitoring of DNA damaging exposures. Basic Clin Pharmacol Toxicol 2006;98(4):336–45 doi 10.1111/j.1742-7843.2006.pto_167.x. [DOI] [PubMed] [Google Scholar]
- 176.Cannizzaro LA. Fluorescent in situ hybridization of DNA probes in the interphase and metaphase stages of the cell cycle. Methods Mol Biol 2013;946:61–83 doi 10.1007/978-1-62703-128-8_5. [DOI] [PubMed] [Google Scholar]
- 177.Wang L, Deng Q, Hu H, Liu M, Gong Z, Zhang S, et al. Glyphosate induces benign monoclonal gammopathy and promotes multiple myeloma progression in mice. J Hematol Oncol 2019;12(1):70 doi 10.1186/s13045-019-0767-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jiao X, Zhang S, Jiao J, Zhang T, Qu W, Muloye GM, et al. Promoter methylation of SEPT9 as a potential biomarker for early detection of cervical cancer and its overexpression predicts radioresistance. Clin Epigenetics 2019;11(1):120 doi 10.1186/s13148-019-0719-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Togashi H, Aoyama M, Oikawa K. Imaging of reactive oxygen species generated in vivo. Magn Reson Med 2016;75(3):1375–9 doi 10.1002/mrm.25582. [DOI] [PubMed] [Google Scholar]
- 180.Chatterjee N, Jeong J, Yoon D, Kim S, Choi J. Global metabolomics approach in in vitro and in vivo models reveals hepatic glutathione depletion induced by amorphous silica nanoparticles. Chem Biol Interact 2018;293:100–6 doi 10.1016/j.cbi.2018.07.013. [DOI] [PubMed] [Google Scholar]
- 181.Rai S, Chowdhury A, Reniers R, Wood SJ, Lucas SJE, Aldred S. A pilot study to assess the effect of acute exercise on brain glutathione. Free Radic Res 2018;52(1):57–69 doi 10.1080/10715762.2017.1411594. [DOI] [PubMed] [Google Scholar]
- 182.Haley P, Perry R, Ennulat D, Frame S, Johnson C, Lapointe JM, et al. STP position paper: best practice guideline for the routine pathology evaluation of the immune system. Toxicol Pathol 2005;33(3):404–7; discussion 8 doi 10.1080/01926230590934304. [DOI] [PubMed] [Google Scholar]
- 183.Wang X, Lebrec H. Immunophenotyping: Application to Safety Assessment. Toxicol Pathol 2017;45(7):1004–11 doi 10.1177/0192623317736742. [DOI] [PubMed] [Google Scholar]
- 184.Burleson GR, Burleson FG, Dietert RR. The cytotoxic T lymphocyte assay for evaluating cell-mediated immune function. Methods Mol Biol 2010;598:195–205 doi 10.1007/978-1-60761-401-2_14. [DOI] [PubMed] [Google Scholar]
- 185.Kamperschroer C, O’Donnell LM, Schneider PA, Li D, Roy M, Coskran TM, et al. Measuring T-cell responses against LCV and CMV in cynomolgus macaques using ELISPOT: potential application to non-clinical testing of immunomodulatory therapeutics. J Immunotoxicol 2014;11(1):35–43 doi 10.3109/1547691X.2013.766287. [DOI] [PubMed] [Google Scholar]
- 186.Davis MM, Tato CM, Furman D. Systems immunology: just getting started. Nat Immunol 2017;18(7):725–32 doi 10.1038/ni.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Diel P, Schulz T, Smolnikar K, Strunck E, Vollmer G, Michna H. Ability of xeno- and phytoestrogens to modulate expression of estrogen-sensitive genes in rat uterus: estrogenicity profiles and uterotropic activity. J Steroid Biochem Mol Biol 2000;73(1–2):1–10 doi 10.1016/s0960-0760(00)00051-0. [DOI] [PubMed] [Google Scholar]
- 188.Dalvie MA, Myers JE, Lou Thompson M, Dyer S, Robins TG, Omar S, et al. The hormonal effects of long-term DDT exposure on malaria vector-control workers in Limpopo Province, South Africa. Environ Res 2004;96(1):9–19 doi 10.1016/j.envres.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 189.National Toxicology P Toxicology and carcinogenesis studies of 3,3’,4,4’-tetrachloroazobenzene (TCAB) (CAS No. 14047-09-7) in Harlan Sprague-Dawley rats and B6C3F1 mice (gavage studies). Natl Toxicol Program Tech Rep Ser 2010(558):1–206. [PubMed] [Google Scholar]
- 190.Keys B, Hlavinka M, Mason G, Safe S. Modulation of rat hepatic microsomal testosterone hydroxylases by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related toxic isostereomers. Can J Physiol Pharmacol 1985;63(12):1537–42 doi 10.1139/y85-253. [DOI] [PubMed] [Google Scholar]
- 191.Endogenous H, Breast Cancer Collaborative G, Key TJ, Appleby PN, Reeves GK, Roddam AW, et al. Circulating sex hormones and breast cancer risk factors in postmenopausal women: reanalysis of 13 studies. Br J Cancer 2011;105(5):709–22 doi 10.1038/bjc.2011.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hogervorst JG, Fortner RT, Mucci LA, Tworoger SS, Eliassen AH, Hankinson SE, et al. Associations between dietary acrylamide intake and plasma sex hormone levels. Cancer Epidemiol Biomarkers Prev 2013;22(11):2024–36 doi 10.1158/1055-9965.EPI-13-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liang G, Schernhammer E, Qi L, Gao X, De Vivo I, Han J. Associations between rotating night shifts, sleep duration, and telomere length in women. PLoS One 2011;6(8):e23462 doi 10.1371/journal.pone.0023462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Parks CG, DeRoo LA, Miller DB, McCanlies EC, Cawthon RM, Sandler DP. Employment and work schedule are related to telomere length in women. Occup Environ Med 2011;68(8):582–9 doi 10.1136/oem.2010.063214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Noguchi K, Eguchi H, Konno M, Kawamoto K, Nishida N, Koseki J, et al. Susceptibility of pancreatic cancer stem cells to reprogramming. Cancer Sci 2015;106(9):1182–7 doi 10.1111/cas.12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Riccardi A, Danova M, Wilson G, Ucci G, Dormer P, Mazzini G, et al. Cell kinetics in human malignancies studied with in vivo administration of bromodeoxyuridine and flow cytometry. Cancer Res 1988;48(21):6238–45. [PubMed] [Google Scholar]
- 197.Beresford MJ, Wilson GD, Makris A. Measuring proliferation in breast cancer: practicalities and applications. Breast Cancer Res 2006;8(6):216 doi 10.1186/bcr1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Elmore S Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35(4):495–516 doi 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nowak-Sliwinska P, Alitalo K, Allen E, Anisimov A, Aplin AC, Auerbach R, et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018;21(3):425–532 doi 10.1007/s10456-018-9613-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Li SP, Padhani AR, Taylor NJ, Beresford MJ, Ah-See ML, Stirling JJ, et al. Vascular characterisation of triple negative breast carcinomas using dynamic MRI. Eur Radiol 2011;21(7):1364–73 doi 10.1007/s00330-011-2061-2. [DOI] [PubMed] [Google Scholar]
- 201.Apostolova I, Wedel F, Brenner W. Imaging of Tumor Metabolism Using Positron Emission Tomography (PET). Recent Results Cancer Res 2016;207:177–205 doi 10.1007/978-3-319-42118-6_8. [DOI] [PubMed] [Google Scholar]
- 202.Lin G, Keshari KR, Park JM. Cancer Metabolism and Tumor Heterogeneity: Imaging Perspectives Using MR Imaging and Spectroscopy. Contrast Media Mol Imaging 2017;2017:6053879 doi 10.1155/2017/6053879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Guyton KZ, Rieswijk L, Wang A, Chiu WA, Smith MT. Key Characteristics Approach to Carcinogenic Hazard Identification. Chem Res Toxicol 2018;31(12):1290–2 doi 10.1021/acs.chemrestox.8b00321. [DOI] [PMC free article] [PubMed] [Google Scholar]