

Abstract
Diamond-Blackfan anemia (DBA) was the first ribosomopathy described and is a constitutional inherited bone marrow failure syndrome. Erythroblastopenia is the major characteristic of the disease, which is a model for ribosomal diseases, related to a heterozygous allelic variation in 1 of the 20 ribosomal protein genes of either the small or large ribosomal subunit. The salient feature of classical DBA is a defect in ribosomal RNA maturation that generates nucleolar stress, leading to stabilization of p53 and activation of its targets, resulting in cell-cycle arrest and apoptosis. Although activation of p53 may not explain all aspects of DBA erythroid tropism, involvement of GATA1/HSP70 and globin/heme imbalance, with an excess of the toxic free heme leading to reactive oxygen species production, account for defective erythropoiesis in DBA. Despite significant progress in defining the molecular basis of DBA and increased understanding of the mechanistic basis for DBA pathophysiology, progress in developing new therapeutic options has been limited. However, recent advances in gene therapy, better outcomes with stem cell transplantation, and discoveries of putative new drugs through systematic drug screening using large chemical libraries provide hope for improvement.
Visual Abstract

Introduction
Diamond-Blackfan anemia (DBA)1-3 is a rare congenital intrinsic erythroid hypoplasia, identified in 20054 as the first human ribosomopathy.5,6 Mutations in 20 ribosomal protein (RP) genes associated with DBA have been identified to date.7 In all instances, the RP gene mutations lead to defective ribosomal RNA (rRNA) maturation, the signature feature of classical DBA.4,8 Indeed, the definition of DBA has become somewhat problematic. Rather than classical DBA with RP gene mutations, some patients exhibit erythroblastopenia resulting from non-RP mutated genes (GATA1, EPO, ADA2). Moreover, some patients without any history of anemia exhibit a pathogenous mutation in an RP gene associated with a malformative syndrome or preleukemic state.
Epidemiology
DBA is a rare congenital disease, with an incidence of 7 cases per million live births. Diagnosis is established at a median age of 2 to 3 months, with 95% of DBA cases diagnosed before 2 years of age and 99% before 5 years of age.9,10 DBA has also been diagnosed at birth in 13% to 16% of cases. However, progress in molecular diagnosis is enabling DBA diagnosis in more patients with unusual clinical presentation, such as unexplained anemia during fetal life or hydrops fetalis,11-13 and adult patients with mild anemia.
Clinical presentation
In infants, DBA is revealed by signs of anemia, including pallor, failure to thrive, and sucking difficulties during breast- or bottle feeding. In addition to anemia, various congenital malformations are reported in 50% of DBA-affected patients (Figure 1A-B).9,10 Premature birth occurs in 20% of cases, and hypotrophy at birth and growth retardation have been documented in 28% and 22%, respectively. Short stature noted during intrauterine life is part of the malformative syndrome and is reported in ∼22% of cases; this progressively worsens because of adverse effects of treatments such as steroid therapy and transfusion-induced iron overload.14 Malformations are mostly localized in the cephalic area (50% of patients) and extremities and include thumb anomalies (38%) with pathognomonic triphalangeal thumb. Moreover, the urogenital tract (39%) and heart (30%)15 are frequently affected (Figure 1A-B).9,10
Figure 1.

Congenital malformations in DBA. (A) Range of congenital anomalies observed in Diamond-Blackfan anemia. (B) Cephalic area malformations in detail.
Differential diagnosis
DBA should be differentiated from the acquired erythroblastopenia that follows parvovirus B19 infection with severe anemia in children with chronic hemolysis, during pregnancy or in neonates, or in immunosuppressed individuals and from transient erythroblastopenia of childhood, likely related to unidentified viruses, which has a favorable outcome.10 The other differential diagnoses with rarer occurrences include immune erythroblastopenia and other inherited bone marrow failure syndromes (IBMFSs), in which erythroid lineage is usually not the only lineage involved.
Management and treatment
DBA patients need a multifaceted approach to care that evolves with the age of the patient.16 For each age category, patient management must include hematological and extrahematological follow-up. Extrahematological care includes management of congenital anomalies (which will not be discussed in this review), growth retardation in children, treatment adverse effects (eg, iron overload in transfused patients, steroid toxicity), and long-term complications, mainly in adult patients, such as B-cell immunodeficiency and malignancies.
Treatment of anemia
At birth and during infancy, hemoglobin (Hb) level may be either normal or below normal, but the need for transfusion begins before the age of 1 year in 90% of patients.9 The international consensus is not to use glucocorticoids before the age of 1 year, and this may be further delayed in children with severe growth failure.10 The threshold for transfusion is 80 to 90 g/L.
In children >1 year of age, glucocorticoids must be tested at the standard dose of 2 mg/kg.10 In responders, the rise in reticulocytes is seen at day ∼10, and Hb value will reach normal values within 1 month. There is no clinical evidence to support prolonged treatment at this dose in responsive patients, and glucocorticoids must be tapered after reticulocyte increase to define the minimal active dose. Overall, 50% to 60% of patients are long-term responders to steroids.9 Nevertheless, the quality of response varies. Some patients achieve normal Hb level with a very low dose of steroid (<0.15 mg/kg per day), and some need a higher dose to maintain the Hb level just above the transfusion threshold. In countries with good transfusion support, the maximal tolerated dose of steroid is considered to be 0.3 mg/kg per day, and patients requiring a higher dose should enter an iterative transfusion program. This threshold may be increased to 0.5 mg/kg per day in countries in which safe red cell transfusions are not guaranteed. It should be noted that DBA is the only human disease in which steroids are administered for years, and both efficacy and adverse effects should be regularly evaluated. Discontinuation of steroids must be considered in instances such as the following: growth retardation in a preadolescent child, because stopping of steroids will allow pubertal growth and likely increase effectiveness of growth hormone (GH) therapy if indicated; during pregnancy, where the consensus is to revert to transfusions; and in patients, mainly adults, in whom long-term adverse effects of steroids are encountered. Steroid resistance occurs in 35% of DBA cases.9 These patients show no or limited reticulocyte response and nonclinically significant rises in reticulocyte counts with no significant changes in Hb levels. These individuals should receive red cell transfusion therapy and are candidates for hematopoietic stem cell transplantation (HSCT).
For patients receiving transfusion, the main challenge is the risk of iron overload,17 and a comparative study documented more severe iron overload in DBA patients compared with patients with thalassemia.18 Severe hemochromatosis has been documented in very young DBA patients, including severe cardiac iron overload.19 Therefore, effective and intense chelation therapy is warranted for DBA patients, even at a young age. In Europe, chelation is started early, usually before the age of 2 years, depending on the age at which the first transfusion was initiated and on the ferritin level (threshold to start, 500-1000 μg/L of ferritin). At this age, deferoxamine is the chelator of choice (maximal dose, 30 mg/kg) because of a lack of data on deferasirox use in infants and toddlers. Deferasirox may be used in children >2 years of age and if needed in association with deferoxamine. The goal is to achieve ferritin levels of <500 μg/L and normal liver iron status by magnetic resonance imaging, which may be performed as soon as possible (under general anesthesia if needed) in children with uncontrolled iron overload and regularly monitored thereafter.
Erythropoietin (EPO) treatment is not effective. Other drugs, such as ciclosporin, antithymocyte globulin, metoclopramide,20,21 leucine22,23 (registered at www.clinicaltrials.gov as #NCT01362595),24 and sotatercept (registered at www.clinicaltrials.gov as #NCT01464164) have either no or limited efficacy.
HSCT is currently the only option to cure the erythroid defect of DBA and must be considered early for children with transfusion dependency.25-28 A recent report from French and German groups included 68 children (median age, 5.2 years; range, 0.9-16.8 years); donors were matched sibling (n = 46), matched unrelated (n = 13), or mismatched unrelated donors (n = 7 [HLA 9/10, n = 6; HLA 8/10, n = 1]). In this cohort, overall survival was 91%, with no difference by donor type.29 In this study, as in a previous study from the Italian group,27 better outcomes were noted when HSCT was performed at a young age (<10 years). HSCT should be considered at the ages of 3 to 5 years, and current indications in children are mainly a lack of good response to steroids and more rarely steroid toxicity, failure of chelation, or severe associated neutropenia. HSCT should be considered with caution in adolescents and adults with no clonal evolution. Lastly, the oncogenic risk associated with transplantation remains to be established. HSCT is likely to reduce the risk of myelodysplastic syndrome and acute myelogenous leukemia; however, the possibility of increased incidence of solid tumors in engrafted patients, as reported for Fanconi anemia patients, cannot be excluded.
Gene therapy is a potential therapeutic option in DBA.30 As in other IBMFSs, somatic mosaicism associated with correction of hematological phenotype has been reported.31 Current gene therapy efforts are focused on the RPS19 gene, which has been successful in a mouse model,32 but currently, there are no active human trials. Under best circumstances, gene therapy will cure the erythropoietic defect at a lower cost compared with HSCT, if it can be performed without a myeloablative conditioning regimen, with no transplantation-related mortality, and no risk of graft-versus-host disease or oncogenic risk. However, this approach needs to be critically validated before implementation. Other approaches, such as use of induced pluripotent stem cells or genome editing, are also being considered.30
The availability of cell models, including fetal liver erythroid progenitors from mice models or reprogrammed hematopoietic progenitors, are making it possible to perform systematic drug screening using large-scale chemical libraries. Such approaches have identified different agents that are of potential clinical interest, such as SMER28 (a small-molecule inducer of autophagy),33 kinase inhibitors,34 and trifluoperazine, which is currently in a clinical trial (registered at www.clinicaltrials.gov as #NCT03966053).
Complication management
DBA patients need comprehensive multispecialty clinical care. In children, growth is the main concern. Therapeutic approaches include steroid use limitations; leucine, reported to be associated with growth acceleration; and GH. Treatment of 19 DBA children with GH was reported to be effective.35 GH administration should be considered for DBA children with major growth retardation. However, caution is warranted, because 3 cases of osteosarcoma were reported in DBA patients treated with GH.35
Adult DBA patients are at increased risk for severe complications. In the most recent analysis of data on 702 patients from the DBA Registry of North America, the cumulative incidence of death resulting from nonmalignant and malignant complications was noted to be 25% by the age of 50 years.36 DBA individuals are now formally recognized to have increased susceptibility for malignancies.36-38 The observed/expected (O/E) ratio for all cancers is 4.8 and the cumulative incidence of solid tumors and hematological malignancies is 13.7 by the age of 45 years.36 As expected for an IBMFS, the risk is very high for myelodysplastic syndromes (O/E ratio, 352.1) and myeloid leukemias (O/E ratio, 28.8). Among a spectrum of reported solid tumors, the most significant risks are for colon carcinoma (O/E ratio, 44.7) and osteogenic sarcoma (O/E ratio, 42.6).36,37 Solid tumors and myeloid malignancies rates increase at ages ∼30 and ∼40 years, respectively. These results support targeted cancer surveillance, such as colonoscopy for colon cancer.36
As in the other IBMFSs, immune deficiency is reported in DBA patients, mainly in adult patients. Immunoglobulin levels must be checked annually. In a cohort of 107 DBA patients, intrinsic quantitative defects were demonstrated in the lymphocyte count (mostly B and natural killer) and serum immunoglobulins.39
Finally, other concerns for adult patient include genetic counseling and reproductive choice. No hypofertility has been reported in male DBA patients. A study from French and German registries reported 42 (66%) complicated pregnancies among 64 total pregnancies in 26 women age >18 years.40 This and other reports show the importance of careful monitoring of pregnancies.40,41 The current consensus is that women undergo transfusion to maintain an Hb level >100 g/L.10
Biology and genetics
Complete blood count, reticulocytes, bone marrow, and fetal Hb
DBA is diagnosed by documenting specific and characteristic erythroblastopenia on bone marrow aspirates or bone marrow biopsies. Absence or <5% of erythroid precursors on the bone marrow smear (erythroblastopenia) or decreased erythroid cellularity on the bone marrow biopsy in otherwise normal bone marrow (no sign of dysplasia, no defect in other cell lineage) is diagnostic. Complete blood count and reticulocyte evaluation reveal moderate to severe anemia that is normochromic and usually macrocytic (mean corpuscular volume, >100 fL). Reticulocytopenia is a major feature of DBA, with absence or <20.109/L reticulocytes in patients with anemia. Neutrophil and platelet counts are usually normal, but transient thrombocytosis (young children) and mild thrombocytopenia and leukoneutropenia (25% of DBA cases) have been noted at diagnosis or during DBA evolution. However, severity of anemia is the most important feature, compared with other cytopenias.
Fetal Hb levels are elevated in DBA, as in other IBMFSs, and may be helpful for DBA diagnosis. To be relevant, it should be measured after the age of 6 months. Elevated EPO concentration is nonspecific, and it has been suggested that the lack of sufficient numbers of erythroid cells expressing EPO receptors in hypoplastic erythroid bone marrow may contribute to higher EPO levels.
eADA
Measurement of erythroid adenosine deaminase (eADA) activity is helpful in the diagnosis of DBA, but it is mandatory for genetic counseling and choice of a familial donor for HSCT to identify potential silent DBA phenotypes.42-45 eADA activity is elevated in 75%44 to 90%45 of nontransfused DBA patients, with a positive or negative predictive value of 91%,44 and elevation of eADA in 1 parent is supportive of a familial form, with implications for DBA genetic counseling.45 However, a link between eADA and DBA has not yet been defined. eADA activity should be measured before transfusion, and in the case of transfusion, it should be measured at least 3 months posttransfusion. Limited access is a challenge, with only a few laboratories performing eADA activity measurement. However, if feasible, eADA measurement should be accessible in at least 1 laboratory in every country.
Molecular diagnosis
Inheritance
DBA genotype, like phenotype, is very heterogeneous. Inheritance is autosomal dominant (45% of cases) and sporadic or familial, with seemingly different patterns of inheritance in the remaining DBA cases. Some recessive (EPO genes)46 or X-linked inheritances (GATA1,47 TSR248 genes) with DBA-like clinical phenotypes have been identified (Table 149; Figure 2).
Table 1.
Genes involved in DBA and DBA-like syndrome from the most to least frequently mutated: reported cases, incidence, and references
| Mutated gene | RP | Incidence in DBA population | Reference |
|---|---|---|---|
| Genes involved in DBA* | |||
| RPS19 | eS19 | 25%-30% | 7,51,53-55,102 |
| Large deletions | 10%-20% | 60,66-69 | |
| RPL5 | uL18 | 7%-12% | 7,57-59 |
| RPS26 | eS26 | 6.6%-9% | 7,64 |
| RPL11 | uL5 | 5%-7% | 7,57-59 |
| RPL35a | eL33 | 2%-3% | 7,60 |
| RPS10 | eS10 | 1%-3% | 7,64 |
| RPS24 | eS24 | 2.4%-3% | 7,61 |
| RPS17 | eS17 | 1%-3% | 7,62,63 |
| RPL15 | eL15 | 1 case | 12,73 |
| 6 cases | |||
| RPS28 | eS28 | 2 families | 48 |
| RPS29 | uS14 | 2 families | 71 |
| RPS7 | eS7 | 1 case | 58 |
| RPS15 | uS19 | 1 case | 58 |
| RPS27a | eS31 | 1 case | 58 |
| RPS27 | eS27 | 1 case | 70 |
| RPL9 | uL6 | 1 case | 58 |
| RPL18 | eL18 | 1 family | 71 |
| RPL26 | uL24 | 1 case | 74 |
| RPL27 | eL27 | 1 case | 70 |
| RPL31 | eL31 | 1 case | 75 |
| TSR2 (X linked)† | 1 family | 48 | |
| Genes involved in DBA-like diseases | |||
| GATA1 (X linked)‡ | 5 families | 47,77-80 | |
| EPO | 1 case | 46 | |
| ADA2§ | 9 individuals | 7 |
Data adapted.49
Defect in rRNA maturation as the signature of DBA.
Not an RP gene, but its protein product is involved in pre-rRNA maturation processing and binds to RPS26.
Whether GATA1 gene mutations are causative of DBA phenotypes is still being debated.
The ADA2 gene is not a classical DBA gene because of its involvement in DADA2 syndrome. Because erythroblastopenia is often a feature of patients with a mutated ADA2 gene, various groups in the DBA field choose to include it.
Figure 2.

Summary of DBA genotypes. Summary of the frequency of various mutant RP genes in DBA-affected patients compiled from published data from national registries. In ∼20% of the DBA cases, no genotype was identified after extensive sequencing (targeted next-generation sequencing) and screening for large deletions (quantitative polymerase chain reaction, multiplex ligation-dependent probe amplification, comparative genomic hybridization). It is anticipated that whole-exome sequencing or whole-genome sequencing (WGS) may fill this gap.
Penetrance of DBA is incomplete. Indeed, DBA exhibits varying clinical severity, ranging from silent phenotypes with no anemia to mild and severe DBA phenotypes with anemia. Silent phenotypes carry a mutation in an RP gene or exhibit limited biological features of the disease, such as isolated macrocytosis, increased fetal Hb, or elevated eADA activity but no anemia. Silent phenotype carriers have the same risk of transmission as DBA parental carriers and thus should be identified for genetic counseling or before selection as a stem cell donor.50 These patients are also at risk for hematological events (late onset of anemia or hematological malignancies) and solid tumors.
20 RP genes involved in DBA
The RPS19 gene was the first DBA gene identified,51 and subsequent global studies in very large cohorts of patients found its frequency to be ∼25% in DBA-affected patients.52-55 This gene was discovered in the late 1990s in the study of a typical case of DBA in a 7-year-old Swedish girl who carried a balanced chromosomal translocation [46, XX, (X;19)(p21;q13)] disrupting the RPS19 gene.51,56 Subsequent comprehensive work in the early 2000s led to the identification of mutations in a large set of ribosomal genes encoding protein constituents of both small and large ribosomal subunits. Indeed, the RPL5 (7% of DBA cases),57-59 RPL11 (5%),57-59 RPL35a (3.5%),60 RPS24 (2.4%),61 RPS17 (1%),62,63 RPS10 (3%), and RPS26 (7%)64 genes have been identified (Figure 2; Table 1). The RPS19 gene remains the most frequently mutated gene. All identified mutations are heterozygous, and homozygosity is considered to be lethal. Various kinds of mutations have been identified, with some particularities depending on the RP gene. Most mutations identified in RPS19 gene are missense, whereas nonsense mutations, small deletions or insertions, and splice site mutations are found in RPL5 and RPL11. Large deletions have been found in ∼20% of DBA cases, mostly in the RPS17, RPL35a, and RPS19 genes, using different techniques (quantitative polymerase chain reaction, multiplex ligation-dependent probe amplification, comparative genomic hybridization).65-69 Thus, at least 70% of DBA patients carry an allelic variation, including large deletions in only 6 RP genes: RPS19, RPL5, RPS26, RPL11, RPL35a, and RPS24. Multiple pathogenic RP mutations have not been reported in any DBA patients to date. Hotspot regions of mutations in DBA genes have been reported only for the RPS19 gene from codons 52 to 62.55 Mutations in other RP genes have been found in very small subsets of DBA-affected patients: RPS7, RPS15, RPS27,70 RPS27a,58 RPS28,48,71 RPS29,71 RPL9,72 RPL15,12,73 RPL18,71 RPL26,74 RPL27,70 and RPL3175 genes, each affecting <1% of DBA patients. In total, 20 RP genes have been identified in association with DBA, reinforcing the concept that DBA is a bona fide human ribosomal disease.49,76 Because defective maturation of rRNA is the signature feature of DBA, definition of DBA should be based on identification of an allelic variation that results in defective rRNA maturation and impairment of ribosomal biogenesis (Figure 3).
Figure 3.
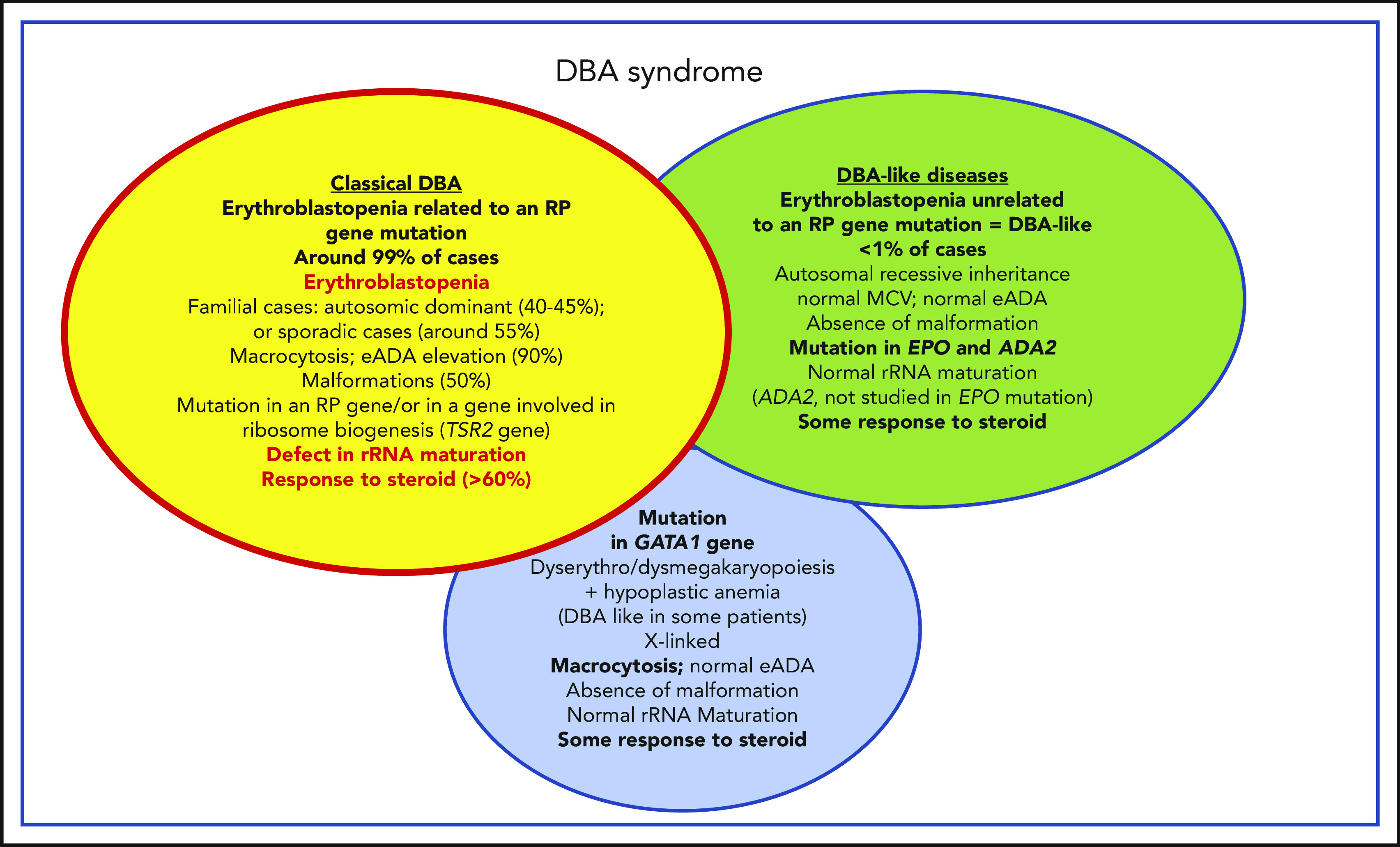
Phenotypic characteristics of patients affected by classical DBA, DBA-like conditions (erythroblastopenia unrelated to an RP gene mutation), and GATA1 DBA-related conditions. Summary of the phenotypic characteristics of DBA-affected patients who fit the DBA definition of congenital erythroblastopenia, with an allelic variation in a ribosomal gene leading to a defect in ribosomal biogenesis and in particular to a defect in rRNA maturation; DBA-like patients who exhibit a congenital erythroblastopenia with some response to steroid but exhibit no defects in ribosomal biogenesis or rRNA maturation (genes ascribed to this phenotype to date are EPO and ADA2); and DBA-like patients who carry a mutation in the GATA1 gene and exhibit some dyserythropoiesis/dysmegakaryopoiesis features in their hypocellular bone marrow. MCV, mean corpuscular volume.
Although some phenotype/genotype correlations have been described, our current insights are far from comprehensive, and the noted associations need to be critically validated. It has been noted that DBA-affected patients carrying a mutation in the RPS19 gene exhibit fewer malformations compared with other patients. In contrast, patients carrying a mutation in the RPL5 gene exhibit an important malformative syndrome that includes cleft palate and cardiac defects with multiple complications.58 A triphalangeal thumb is a common feature of DBA patients with a mutation in the RPL11 gene.58 Recently, congenital heart anomalies have been related to a mutation in the RPS24 gene.15
Non-RP genes
Non-RP genes identified in association with a DBA-like phenotype include GATA1,47,77-80 TSR2,48 EPO,46 and ADA2 (also known as CECR1).7 Although the TSR2 gene does not code for an RP, it plays a role in ribosomal biogenesis through its involvement in pre-rRNA processing and binds RPS26. The GATA1 gene is the major erythroid transcription factor, and <10 patients to date have been described with a mutation in the GATA1 gene and a DBA-like phenotype.47,77-80 The GATA1 mutation related to a DBA-like phenotype is located either in the translational initiation codon, ATG, or close vicinity or in the exon 2 splicing site, leading to exon 2 skipping and loss of the transactivating domain. This allelic variation has been previously described in association with congenital dyserythropoiesis/dysmegakaryopoiesis and not a classical DBA phenotype in the hypocellular bone marrow of a Brazilian proband.81 There is, however, strong evidence that GATA1 plays a major role in DBA pathophysiology, because it has been found that DBA patients carrying a mutation in an RP gene exhibit defects in GATA1 expression as a result of a specific defect in translation with a two- to threefold reduction in GATA1 messenger RNA (mRNA) abundance in polysomes.78
An EPO gene mutation in association with a DBA-like phenotype has been identified in a 1-year-old male infant from a consanguineous Turkish family by exome sequencing (homozygous missense allelic variation in exon 5, g.100 320,704G>A; p.Arg150Gln).46 The eADA activity was found to be normal, and the patient exhibited some response to steroid therapy (Figure 3).
Erythroblastopenia is a feature of patients with a mutant ADA2 (CECR1) gene, responsible for deficiency of adenosine deaminase 2 (DADA2).82 These individuals have normal stature, normal eADA, and no defect in rRNA maturation and exhibit some response to steroid therapy. The features of DADA2 also include hypogammaglobulinemia and risk for vascularitis and autoinflammatory disease. The prognosis is poor, and such patients require immediate HSCT.7,82 Immunoglobulin levels must be checked in DBA-like patients to establish diagnosis of DADA2 syndrome.82
There is continued debate regarding the issue of whether patients with erythroblastopenia phenotype resulting from genetic mutations who show no evidence of an rRNA maturation defect should be considered DBA patients. At a recent EURODBA consensus meeting (Freiburg, Germany, June 2017, EURODBA coordinator A. MacInnes), it was agreed that all erythroblastopenia phenotypes related to any gene mutation would be designated as DBA syndrome, and DBA designation would only be used for patients with a gene mutation leading to an rRNA maturation defect (Figure 3). Mutations identified in the ADA2 (P. E. Gleizes, M. F. O’Donohue, EURODBA Consortium, manuscript in preparation) and GATA1 genes (P. E. Gleizes, M. F. O’Donohue, H. Gazda, Université de Toulouse, personal communication, 15 December 2019) are not associated with defective rRNA maturation (Figure 3).
To date, no mutation could be identified in 20% of DBA patients by current screening strategies.76 In such cases, whole-exome sequencing or WGS should be performed to determine if promoter region or intronic mutations of known DBA genes (WGS) that are not presently screened for account for DBA and to identify new candidate genes.
Pathophysiology and research perspectives
Erythroid tropism in DBA
The pathophysiology of DBA has not yet been fully defined, and the major unresolved question is how a mutation in an RP gene, leading to a defect in rRNA maturation and ribosomal biogenesis, leads to a specific erythroid defect. Two hundred billion red blood cells are produced each day, and because RP genes are primarily involved in mRNA translation, it can be hypothesized that translation of some specific genes could be affected by a defect in ribosomal biogenesis. Indeed, ribosome amounts have been shown to be critical for cell lineage commitment. A global reduction in ribosome levels in DBA was documented, whereas ribosome composition was normal, leading to the alteration of the translation of specific RNA transcripts.83
Sankaran et al47 have shown that haploinsufficiency in some RP genes (RPL11, RPL5, RPS24) and a splice site mutation in the GATA1 gene are responsible for the disappearance of the short form of GATA1, either because of a specific translational defect in the GATA1 transcript78 or because of the direct effect of the GATA1 mutation on its mRNA translation and exon 2 skipping,47 respectively. In addition, in a nonexclusive and nonrestrictive manner, HSP70, the chaperon of GATA1, which protects GATA1 from caspase-3 cleavage during terminal erythroid differentiation upon EPO stimulation, has been shown to be involved as well (Figure 4).84 Using an in vitro erythroid culture system, depending on the mutated RP gene, 2 different erythroid DBA phenotypes could be characterized based on differences in extent of erythroid proliferation, erythroid differentiation, and degree of apoptosis.85 HSP70, which plays a role in cell survival and apoptosis, was shown to account for these dual phenotypes (Figure 4).84
Figure 4.
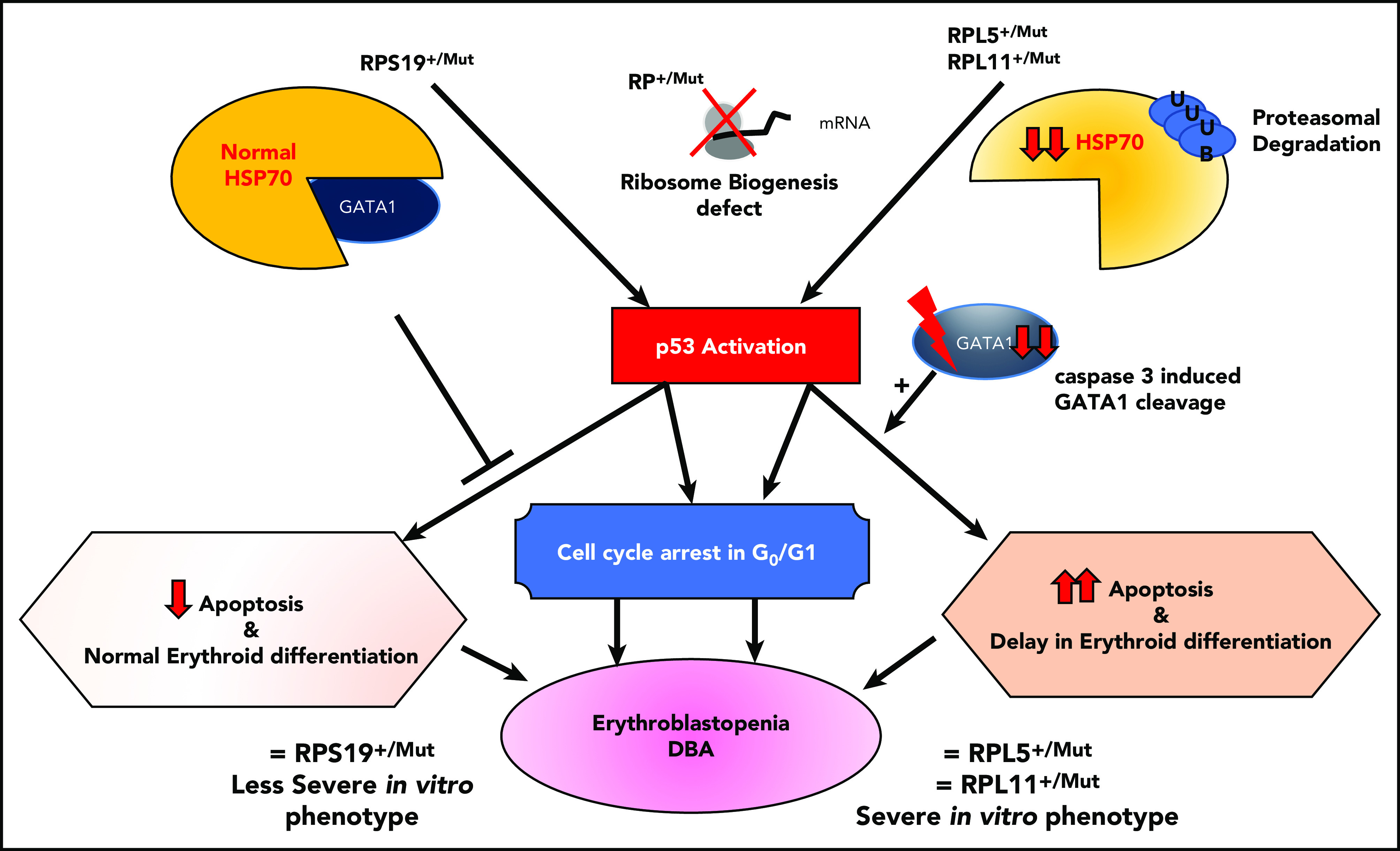
Summary of links between HSP70/GATA1 and p53 stabilization in DBA erythroid phenotype, depending on the RP gene mutation.84 Two DBA phenotypes have been identified in vitro from primary erythroid cells derived from DBA patients, depending on the mutated RP gene. Patients carrying a mutation in the RPS19 gene exhibit decreased erythroid proliferation but normal erythroid differentiation and no apoptosis. In contrast, DBA patients who carry a mutation in an RP gene other than RPS19 exhibit a large decrease in erythroid proliferation, delayed erythroid differentiation, and a large increase in apoptosis.85 In the case of haploinsufficiency of the RPS19, RPL5, or RPL11 gene, erythroblastopenia in DBA results at least in part from stabilization of p53 and activation of p53 targets (p21, Bax), which are responsible for activation of apoptosis and cell-cycle arrest in G0/G1 phases.85,92 HSP70 has been identified as the key protein responsible for the observed dual DBA phenotypes.84 Normal levels of HSP70 expression are noted in patients carrying a mutation in the RPS19 gene, whereas a large decrease in HSP70 expression is seen in patients who carry a mutation in the RPL5, RPL11, or RPS24 gene and even in patients with no identified genotype. HSP70 has been shown to be proteasomally degraded after ubiquitinylation. Decreased expression of HSP70 leads to the cleavage of GATA1 by caspase-3, which also leads to p53 stabilization, induction of apoptosis, and delayed erythroid differentiation. Apoptosis, delayed erythroid differentiation, and cell-cycle arrest lead to the decreased erythroid proliferation and characteristic erythroblastopenia seen in DBA.
Any factor that affects GATA1 or HSP70 expression would be deleterious in DBA. Indeed, it has been shown that as a result of RP haploinsufficiency, some genes are translationally downregulated. One of them is ribonuclease inhibitor 1 (RNH1),86 which binds to the 40S ribosome small subunit and plays a role in ribosomal biogenesis. It also plays a role in translational control of GATA1 mRNA.83 The HSP70/GATA1 complex and translational regulation lie at the center of DBA pathophysiology.
p53 stabilization
p53 also plays an important role in DBA pathophysiology. Findings from various model systems, including zebrafish,87-89 mice,90,91 and primary human cells,85,92 have shown that haploinsufficiency in the RPS19, RPL5, or RPL11 gene leads to stabilization of p53, with increased phosphorylation of p53 and activation of its transcriptional targets, including p21, bax, and noxa. It has been shown that activation of p53 in DBA is related to nucleolar stress and overexpression of some RPs (RPS3, RPS7, RPS27, RPS27a, RPL5, RPL11, and RPL23), which are able to directly bind MDM2 and release p53.93 However, some p53-independent effects have also been reported in mouse embryonic stem cells.94 The relationship between p53 and GATA1 is well established, with GATA1 inhibiting p53.95 Overexpression of wild-type HSP70, by restoring GATA1, decreased p53 activation in RPL5- and RPL11-depleted erythroid cells.84 Decreased expression of GATA1 could thus also account for p53 activation in DBA (Figure 4). In addition, 2 different gain-of-function mutations in exon 10 of TP53 gene (NM_001126112; p.Ser362Alafs*8) were recently reported in 2 DBA-like individuals with a borderline phenotype between DBA and dyskeratosis congenita.96 These findings reinforce the role of p53 in DBA pathophysiology.
Unbalanced globin/heme synthesis and cell metabolism
Cell metabolism and cell toxicity may also play important roles, as exemplified by the identification of unbalanced globin/heme synthesis in DBA97; this imbalance results in excess free heme, leading to production of reactive oxygen species, which is toxic to cells and leads to cell death and apoptosis.98 The identification of the deleterious effect of excess free heme in DBA was further strengthened by the following findings: the increased production of reactive oxygen species in primary cells from DBA patients, the extent of excess being dependent on the RP mutation; the importance of the GATA1/HSP70 complex in primary DBA erythroid cells, controlling globin synthesis and limiting heme synthesis by decreasing ALAS2 and ferrochelatase to adjust the level of heme synthesis in the context of decreased globin synthesis; and the attempt by erythroid DBA cells to increase globin synthesis by repressing EIF2α phosphorylation and thereby limiting free heme toxicity through increased expression of cell membrane heme exporter FLVCR1 (Figures 5 and 6).97,98 In addition, a recent study demonstrated autoregulated crosstalk between GATA1 and heme that induced normal erythroid differentiation; GATA1 initiated heme (and globin) synthesis during normal erythroid maturation, but heme suppressed GATA1 expression. Indeed, in Flvcr1 knockout mice, heme increased RP transcripts in early erythroid cells to generate effective ribosomal machinery and restore globin/heme balance. In more mature erythroid cells, however, excess heme decreased expression of GATA1 and its targets by decreasing RP transcripts.99 Free heme excess and globin/heme disequilibrium resulting from a GATA1 defect, generated either by its specific decreased translation or after caspase-3 cleavage caused by the degradation of its chaperone, HSP70, may indeed be key factors in DBA pathophysiology and modulators of DBA phenotypes. Autophagy33,100 and cell metabolism101 may also play important roles in modulating DBA phenotypes.
Figure 5.

Regulation of heme synthesis and heme scavengers in erythroid precursors from DBA-affected patients to restore globin/heme imbalance.98 GATA1 expression has been found to be decreased in DBA-affected patients because of an impairment of its translation47,78,99 or as a consequence of caspase-3 cleavage during terminal erythroid differentiation resulting from decreased levels of its chaperon, HSP70.84 As a consequence, there is decreased expression of GATA1 transcriptional targets, including globin chains. There is also decreased expression of ALAS2, a GATA1 target, and ferrochelatase, the first and last heme biosynthesis enzymes, respectively, in the mitochondria to limit excess heme and thereby restore globin/heme balance. At the same time, there is decreased expression of TfR1 to limit iron uptake. In DBA, particularly DBA resulting from non-RPS19 mutations, there is also increased expression of heme exporter FLVCR1 (feline leukemia virus C receptor type 1), inactivation of HRI, and proteasomal degradation of BACH1 after fixation of free heme to limit excess heme in the erythroid cells. Along with limiting excess free heme, DBA erythroid cells also attempt to increase the level of globin chain translation by NRF2/Maf transcriptional activation of globin mRNA after BACH1 degradation and decreased EIF2α phosphorylation following inactivation and hypophosphorylation of HRI. The failure of these regulatory mechanisms leads to unbalanced globin/heme equilibrium, with the resultant excess of toxic heme with increased reactive oxygen species production leading to apoptosis and worsening the intrinsic defect of erythropoiesis in DBA.
Figure 6.
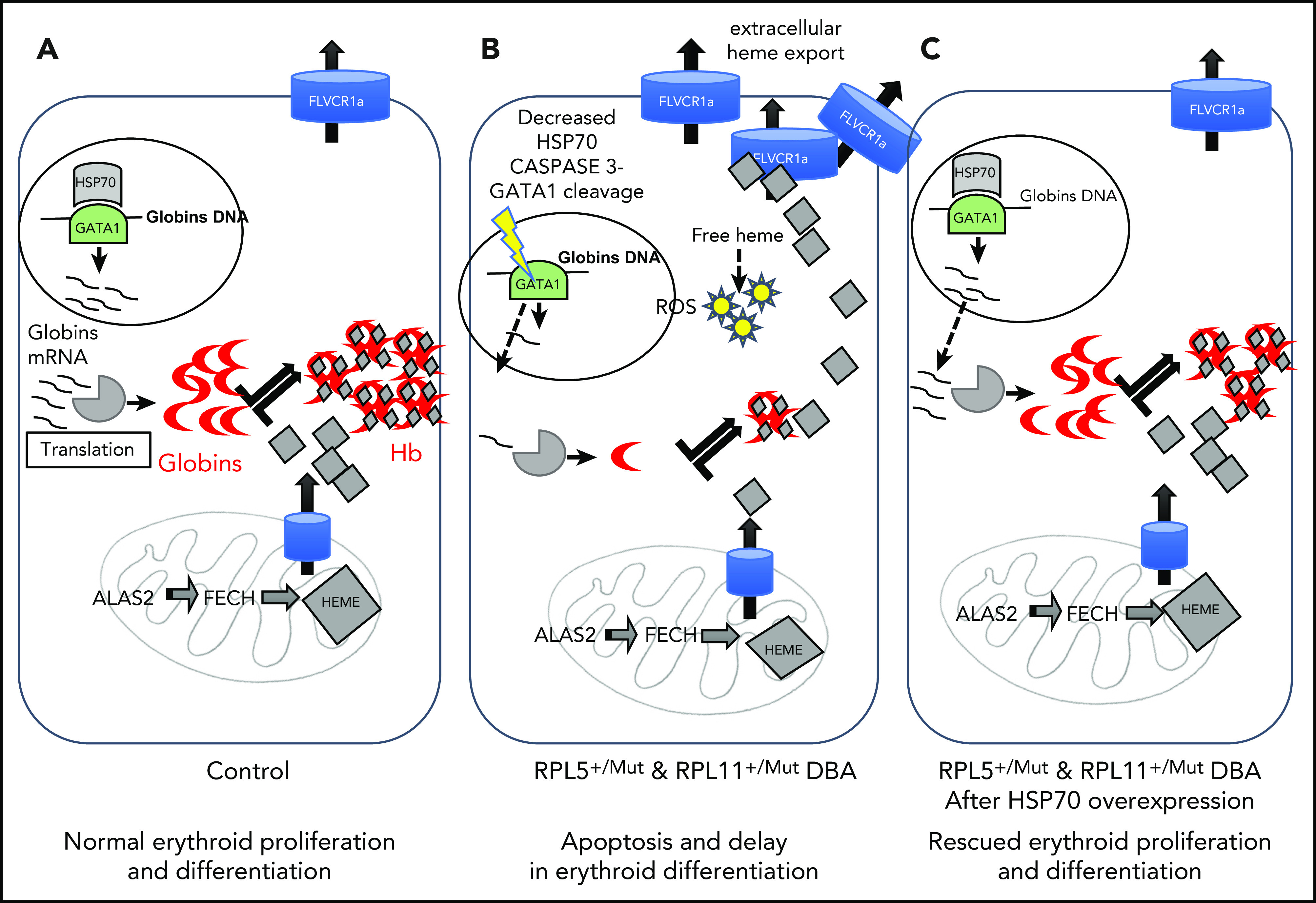
Summary of the unbalanced globin/heme equilibrium in DBA and the rescue of erythroid proliferation and differentiation by overexpression of HSP70.98 (A) During normal erythropoiesis, globin chains and heme are stoichiometrically expressed to produce Hb with no excess heme. (B) In DBA-affected patients who carry a mutation in the RPL5 or RPL11 gene, there is disequilibrium between globin and heme synthesis, resulting in excess free heme in erythroid cells. The excess free heme results in increased reactive oxygen species (ROS) production, contributing to cell death in DBA. (C) Overexpression of HSP70 rescues the DBA phenotype by protecting GATA1 and restoring globin/heme balance and thereby rescuing the erythroid proliferation and differentiation by decreasing apoptosis and erythroid cell death.
Discussion
DBA is an IBMFS that is characterized by erythroid tropism, with macrocytic aregenerative anemia and high eADA levels. Mutations in a large number of RP genes, leading to defective rRNA maturation, have been shown to account for erythroblastopenia. However, recent descriptions of patients with mutations in non-RP genes exhibiting DBA-like erythroblastopenia and normocytic hyporegenerative anemia with normal eADA levels and no defects in rRNA maturation, a signature feature of DBA with RP gene mutations, implies that DBA phenotypes are more complicated than previously understood. Significant progress has been made in DBA diagnosis using new molecular diagnostic tools (next-generation sequencing, whole-exome sequencing, WGS), which in turn has led to awareness of the higher prevalence of DBA than previously estimated. New genes are still being discovered. The GATA1/HSP70 complex, at least in part influenced by p53 stabilization and excess free heme, is a major factor involved in the DBA pathophysiology and erythroid tropism of the disease. In terms of treatment, there are continuous improvements in the appropriate use of well-established steroid therapy, transfusion support, and HSCT. Importantly, new therapeutic approaches, including targeted drugs and gene therapy, are being explored.
Acknowledgments
The authors thank the patients affected by Diamond-Blackfan anemia (DBA) and their families, the French DBA patients’ association AFMBD, and the Maria Daniella Arturi and DBA foundations; Gil Tchernia for his inspiration, scientific discussions, and perpetual interest in DBA; our colleagues and collaborators from EURODBA, particularly P.E. Gleizes and M.F. O’Donohue, for long-term collaboration and the opportunity to use their unpublished data; our colleagues and collaborators from US DBA groups and the DBA Registry; Isabelle Marie for her work with the French DBA registry; and Julie Galimand, Hélène Bourdeau, and Anaële Jaouen for their work in mutation screening analysis for DBA patients (Hematology Laboratory, Robert-Debré Hospital); and Régis Peffault De La Tour, Isabelle Brindel, and Amélie Marouane, who are in charge of the national reference center for aplastic anemia in which DBA is included and the National Network of Rare Diseases in Hematology and Immunology.
This work was supported by the French National Research Agency (ANR) through grants ANR-15-RAR3-0007-04 and ANR-12-RARE-0007-02 from EuroDBA, ANR-HSPathies-ANR-2012-BLAN-SVSE1, ANR-2015-AAP générique-CE12, and ANR Avenir-11-LABX-0005-02 from the Laboratory of Excellence for Red Cells; the French National PHRC OFABD (DBA registry and molecular biology) 2008-2012-2016; the Fondation pour la Recherche médicale; the Fondation ARC pour la recherche contre le cancer; and grant DK32094 from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
Authorship
Contribution: L.D.C., T.L., and N.M. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lydie Da Costa, Hôpital Robert Debré, Service d’Hématologie Biologique, 48 Boulevard Sérurier, 75019 Paris, France; e-mail: lydie.dacosta@aphp.fr.
REFERENCES
- 1.Diamond LK. Congenital hypoplastic anemia: Diamond-Blackfan syndrome. Historical and clinical aspects. Blood Cells. 1978;4(1-2):209-213. [PubMed] [Google Scholar]
- 2.Diamond LK, Blackfan KD. Hypoplastic anemia. Am J Dis Child. 1938;56:464-467. [Google Scholar]
- 3.Josephs HW. Anaemia of infancy and early childhood. Medicine (Baltimore). 1936;15(3):307-451. [Google Scholar]
- 4.Léger-Silvestre I, Caffrey JM, Dawaliby R, et al. . Specific role for yeast homologs of the Diamond Blackfan anemia-associated Rps19 protein in ribosome synthesis. J Biol Chem. 2005;280(46):38177-38185. [DOI] [PubMed] [Google Scholar]
- 5.Lipton JM, Ellis SR. Diamond Blackfan anemia 2008-2009: broadening the scope of ribosome biogenesis disorders. Curr Opin Pediatr. 2010;22(1):12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu JM, Ellis SR. Ribosomes and marrow failure: coincidental association or molecular paradigm? Blood. 2006;107(12):4583-4588. [DOI] [PubMed] [Google Scholar]
- 7.Ulirsch JC, Verboon JM, Kazerounian S, et al. . The genetic landscape of Diamond-Blackfan anemia [published correction appears in Am J Hum Genet. 2019;104(2):356]. Am J Hum Genet. 2018;103(6):930-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choesmel V, Bacqueville D, Rouquette J, et al. . Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood. 2007;109(3):1275-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willig TN, Niemeyer CM, Leblanc T, et al. . Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Société d’Hématologie et d’Immunologie Pédiatrique (SHIP), Gesellshaft für Pädiatrische Onkologie und Hämatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI). Pediatr Res. 1999;46(5):553-561. [DOI] [PubMed] [Google Scholar]
- 10.Vlachos A, Ball S, Dahl N, et al. ; Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference . Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Da Costa L, Chanoz-Poulard G, Simansour M, et al. . First de novo mutation in RPS19 gene as the cause of hydrops fetalis in Diamond-Blackfan anemia [published correction appears in Am J Hematol. 2013;88(2):160]. Am J Hematol. 2013;88(4):340-341. [DOI] [PubMed] [Google Scholar]
- 12.Wlodarski MW, Da Costa L, O’Donohue MF, et al. . Recurring mutations in RPL15 are linked to hydrops fetalis and treatment independence in Diamond-Blackfan anemia. Haematologica. 2018;103(6):949-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maisonneuve E, Ben M’Barek I, Leblanc T, et al. . Managing the unusual causes of fetal anemia. Fetal Diagn Ther. 2020;47(2):156-164. [DOI] [PubMed] [Google Scholar]
- 14.Chen S, Warszawski J, Bader-Meunier B, et al. ; Société Française d’Hématologie et d’Immunologie Pédiatrique . Diamond-Blackfan anemia and growth status: the French registry. J Pediatr. 2005;147(5):669-673. [DOI] [PubMed] [Google Scholar]
- 15.Vlachos A, Osorio DS, Atsidaftos E, et al. . Increased prevalence of congenital heart disease in children with Diamond Blackfan anemia suggests unrecognized Diamond Blackfan anemia as a cause of congenital heart disease in the general population: a report of the Diamond Blackfan Anemia Registry. Circ Genom Precis Med. 2018;11(5):e002044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010;116(19):3715-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porter JB, Walter PB, Neumayr LD, et al. . Mechanisms of plasma non-transferrin bound iron generation: insights from comparing transfused Diamond Blackfan anaemia with sickle cell and thalassaemia patients. Br J Haematol. 2014;167(5):692-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roggero S, Quarello P, Vinciguerra T, Longo F, Piga A, Ramenghi U. Severe iron overload in Blackfan-Diamond anemia: a case-control study. Am J Hematol. 2009;84(11):729-732. [DOI] [PubMed] [Google Scholar]
- 19.Berdoukas V, Nord A, Carson S, et al. . Tissue iron evaluation in chronically transfused children shows significant levels of iron loading at a very young age. Am J Hematol. 2013;88(11):E283-E285. [DOI] [PubMed] [Google Scholar]
- 20.Abkowitz JL, Schaison G, Boulad F, et al. . Response of Diamond-Blackfan anemia to metoclopramide: evidence for a role for prolactin in erythropoiesis. Blood. 2002;100(8):2687-2691. [DOI] [PubMed] [Google Scholar]
- 21.Leblanc TM, Da Costa L, Marie I, Demolis P, Tchernia G. Metoclopramide treatment in DBA patients: no complete response in a French prospective study. Blood. 2007;109(5):2266-2267. [DOI] [PubMed] [Google Scholar]
- 22.Pospisilova D, Cmejlova J, Hak J, Adam T, Cmejla R. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica. 2007;92(5):e66-e67. [DOI] [PubMed] [Google Scholar]
- 23.Narla A, Payne EM, Abayasekara N, et al. . L-Leucine improves the anaemia in models of Diamond Blackfan anaemia and the 5q- syndrome in a TP53-independent way. Br J Haematol. 2014;167(4):524-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vlachos A, Atsidaftos E, Muir E, et al. . Leucine for the treatment of transfusion dependence in patients with Diamond Blackfan anemia [abstract]. Blood. 2018;132(suppl 1). Abstract 755.
- 25.Alter BP. Inherited bone marrow failure syndromes: considerations pre- and posttransplant. Hematology Am Soc Hematol Educ Program. 2017;2017:88-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dietz AC, Duncan CN, Alter BP, et al. . The Second Pediatric Blood and Marrow Transplant Consortium International Consensus Conference on Late Effects after Pediatric Hematopoietic Cell Transplantation: defining the unique late effects of children undergoing hematopoietic cell transplantation for immune deficiencies, inherited marrow failure disorders, and hemoglobinopathies. Biol Blood Marrow Transplant. 2017;23(1):24-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fagioli F, Quarello P, Zecca M, et al. . Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol. 2014;165(5):673-681. [DOI] [PubMed] [Google Scholar]
- 28.Peffault de Latour R, Peters C, Gibson B, et al. ; Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation . Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50(9):1168-1172. [DOI] [PubMed] [Google Scholar]
- 29.Strahm B, Niemeyer C, Wlodarski M, et al. : Allogeneic hematopoietic stem cell transplantation in Diamond-Blackfan anemia: report from the German and French DBA registry [abstract]. Bone Marrow Transplant. 2018;53:19-144. Abstract O150. [Google Scholar]
- 30.Aspesi A, Borsotti C, Follenzi A. Emerging therapeutic approaches for Diamond Blackfan anemia. Curr Gene Ther. 2018;18(6):327-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garelli E, Quarello P, Giorgio E, et al. . Spontaneous remission in a Diamond-Blackfan anaemia patient due to a revertant uniparental disomy ablating a de novo RPS19 mutation. Br J Haematol. 2019;185(5):994-998. [DOI] [PubMed] [Google Scholar]
- 32.Debnath S, Jaako P, Siva K, et al. . Lentiviral vectors with cellular promoters correct anemia and lethal bone marrow failure in a mouse model for Diamond-Blackfan anemia. Mol Ther. 2017;25(8):1805-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doulatov S, Vo LT, Macari ER, et al. . Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med. 2017;9(376):eaah5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siva K, Ek F, Chen J, et al. . A phenotypic screening assay identifies modulators of Diamond Blackfan anemia. SLAS Discov. 2019;24(3):304-313. [DOI] [PubMed] [Google Scholar]
- 35.Howell JC, Joshi SA, Hornung L, Khoury J, Harris RE, Rose SR. Growth hormone improves short stature in children with Diamond-Blackfan anemia. Pediatr Blood Cancer. 2015;62(3):402-408. [DOI] [PubMed] [Google Scholar]
- 36.Vlachos A, Rosenberg PS, Atsidaftos E, et al. . Increased risk of colon cancer and osteogenic sarcoma in Diamond-Blackfan anemia. Blood. 2018;132(20):2205-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipton JM, Federman N, Khabbaze Y, et al. ; Diamond-Black Anemia Registry . Osteogenic sarcoma associated with Diamond-Blackfan anemia: a report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol. 2001;23(1):39-44. [DOI] [PubMed] [Google Scholar]
- 38.Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012;119(16):3815-3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iskander D, Roberts I, Rees C, et al. . Impaired cellular and humoral immunity is a feature of Diamond-Blackfan anaemia; experience of 107 unselected cases in the United Kingdom. Br J Haematol. 2019;186(2):321-326. [DOI] [PubMed] [Google Scholar]
- 40.Faivre L, Meerpohl J, Da Costa L, et al. . High-risk pregnancies in Diamond-Blackfan anemia: a survey of 64 pregnancies from the French and German registries. Haematologica. 2006;91(4):530-533. [PubMed] [Google Scholar]
- 41.Giri N, Stratton P, Savage SA, Alter BP. Pregnancies in patients with inherited bone marrow failure syndromes in the NCI cohort. Blood. 2017;130(14):1674-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glader BE, Backer K. Elevated red cell adenosine deaminase activity: a marker of disordered erythropoiesis in Diamond-Blackfan anaemia and other haematologic diseases. Br J Haematol. 1988;68(2):165-168. [DOI] [PubMed] [Google Scholar]
- 43.Glader BE, Backer K, Diamond LK. Elevated erythrocyte adenosine deaminase activity in congenital hypoplastic anemia. N Engl J Med. 1983;309(24):1486-1490. [DOI] [PubMed] [Google Scholar]
- 44.Narla A, Davis NL, Lavasseur C, Wong C, Glader B. Erythrocyte adenosine deaminase levels are elevated in Diamond Blackfan anemia but not in the 5q- syndrome. Am J Hematol. 2016;91(12):E501-E502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willig TN, Pérignon JL, Gustavsson P, et al. . High adenosine deaminase level among healthy probands of Diamond Blackfan anemia (DBA) cosegregates with the DBA gene region on chromosome 19q13. The DBA Working Group of Société d’Immunologie Pédiatrique (SHIP). Blood. 1998;92(11):4422-4427. [PubMed] [Google Scholar]
- 46.Kim AR, Ulirsch JC, Wilmes S, et al. . Functional selectivity in cytokine signaling revealed through a pathogenic EPO mutation. Cell. 2017;168(6):1053-1064.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sankaran VG, Ghazvinian R, Do R, et al. . Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122(7):2439-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gripp KW, Curry C, Olney AH, et al. ; UW Center for Mendelian Genomics . Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am J Med Genet A. 2014;164A(9):2240-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Da Costa L, Narla A, Mohandas N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000 Res. 2018;7:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orfali KA, Wynn RF, Stevens RF, Chopra R, Ball SE. Failure of red cell production following allogenic BMT for Diamond Blackfan anaemia (DBA) illustrates functional significance of high erythrocyte adenosine deaminase (eADA) activity in the donor [abstract]. Blood. 1994. Abstract 414a. [Google Scholar]
- 51.Draptchinskaia N, Gustavsson P, Andersson B, et al. . The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21(2):169-175. [DOI] [PubMed] [Google Scholar]
- 52.Ball S, Orfali K. Molecular diagnosis of Diamond-Blackfan anemia. Methods Mol Med. 2004;91:19-30. [DOI] [PubMed] [Google Scholar]
- 53.Campagnoli MF, Ramenghi U, Armiraglio M, et al. . RPS19 mutations in patients with Diamond-Blackfan anemia. Hum Mutat. 2008;29(7):911-920. [DOI] [PubMed] [Google Scholar]
- 54.Cmejla R, Blafkova J, Stopka T, et al. . Ribosomal protein S19 gene mutations in patients with diamond-blackfan anemia and identification of ribosomal protein S19 pseudogenes. Blood Cells Mol Dis. 2000;26(2):124-132. [DOI] [PubMed] [Google Scholar]
- 55.Willig TN, Draptchinskaia N, Dianzani I, et al. . Mutations in ribosomal protein S19 gene and diamond blackfan anemia: wide variations in phenotypic expression. Blood. 1999;94(12):4294-4306. [PubMed] [Google Scholar]
- 56.Gustavsson P, Skeppner G, Johansson B, et al. . Diamond-Blackfan anaemia in a girl with a de novo balanced reciprocal X;19 translocation. J Med Genet. 1997;34(9):779-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cmejla R, Cmejlova J, Handrkova H, et al. . Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemia. Hum Mutat. 2009;30(3):321-327. [DOI] [PubMed] [Google Scholar]
- 58.Gazda HT, Sheen MR, Vlachos A, et al. . Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83(6):769-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quarello P, Garelli E, Carando A, et al. . Diamond-Blackfan anemia: genotype-phenotype correlations in Italian patients with RPL5 and RPL11 mutations. Haematologica. 2010;95(2):206-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farrar JE, Nater M, Caywood E, et al. . Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112(5):1582-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gazda HT, Grabowska A, Merida-Long LB, et al. . Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79(6):1110-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28(12):1178-1182. [DOI] [PubMed] [Google Scholar]
- 63.Song MJ, Yoo EH, Lee KO, et al. . A novel initiation codon mutation in the ribosomal protein S17 gene (RPS17) in a patient with Diamond-Blackfan anemia. Pediatr Blood Cancer. 2010;54(4):629-631. [DOI] [PubMed] [Google Scholar]
- 64.Doherty L, Sheen MR, Vlachos A, et al. . Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2010;86(2):222-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farrar JE, Vlachos A, Atsidaftos E, et al. . Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood. 2011;118(26):6943-6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gustavsson P, Garelli E, Draptchinskaia N, et al. . Identification of microdeletions spanning the Diamond-Blackfan anemia locus on 19q13 and evidence for genetic heterogeneity. Am J Hum Genet. 1998;63(5):1388-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuramitsu M, Sato-Otsubo A, Morio T, et al. . Extensive gene deletions in Japanese patients with Diamond-Blackfan anemia. Blood. 2012;119(10):2376-2384. [DOI] [PubMed] [Google Scholar]
- 68.Quarello P, Garelli E, Brusco A, et al. . High frequency of ribosomal protein gene deletions in Italian Diamond-Blackfan anemia patients detected by multiplex ligation-dependent probe amplification assay. Haematologica. 2012;97(12):1813-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quarello P, Garelli E, Brusco A, et al. . Multiplex ligation-dependent probe amplification enhances molecular diagnosis of Diamond-Blackfan anemia due to RPS19 deficiency. Haematologica. 2008;93(11):1748-1750. [DOI] [PubMed] [Google Scholar]
- 70.Wang R, Yoshida K, Toki T, et al. . Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond-Blackfan anaemia. Br J Haematol. 2015;168(6):854-864. [DOI] [PubMed] [Google Scholar]
- 71.Mirabello L, Macari ER, Jessop L, et al. . Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood. 2014;124(1):24-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lezzerini M, Penzo M, O’Donohue MF, et al. . Ribosomal protein gene RPL9 variants can differentially impair ribosome function and cellular metabolism. Nucleic Acids Res. 2020;48(2):770-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Landowski M, O’Donohue MF, Buros C, et al. . Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond-Blackfan anemia. Hum Genet. 2013;132(11):1265-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gazda HT, Preti M, Sheen MR, et al. . Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in Diamond-Blackfan anemia. Hum Mutat. 2012;33(7):1037-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Farrar JE, Quarello P, Fisher R, et al. . Exploiting pre-rRNA processing in Diamond Blackfan anemia gene discovery and diagnosis. Am J Hematol. 2014;89(10):985-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Da Costa L, O’Donohue MF, van Dooijeweert B, et al. . Molecular approaches to diagnose Diamond-Blackfan anemia: The EuroDBA experience. Eur J Med Genet. 2018;61(11):664-673. [DOI] [PubMed] [Google Scholar]
- 77.Klar J, Khalfallah A, Arzoo PS, Gazda HT, Dahl N. Recurrent GATA1 mutations in Diamond-Blackfan anaemia. Br J Haematol. 2014;166(6):949-951. [DOI] [PubMed] [Google Scholar]
- 78.Ludwig LS, Gazda HT, Eng JC, et al. . Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med. 2014;20(7):748-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parrella S, Aspesi A, Quarello P, et al. . Loss of GATA-1 full length as a cause of Diamond-Blackfan anemia phenotype. Pediatr Blood Cancer. 2014;61(7):1319-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abdulhay NJ, Fiorini C, Verboon JM, et al. . Impaired human hematopoiesis due to a cryptic intronic GATA1 splicing mutation. J Exp Med. 2019;216(5):1050-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hollanda LM, Lima CS, Cunha AF, et al. . An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006;38(7):807-812. [DOI] [PubMed] [Google Scholar]
- 82.Lee PY. Vasculopathy, immunodeficiency, and bone marrow failure: the intriguing syndrome caused by deficiency of adenosine deaminase 2. Front Pediatr. 2018;6:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khajuria RK, Munschauer M, Ulirsch JC, et al. . Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell. 2018;173(1):90-103.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gastou M, Rio S, Dussiot M, et al. ; French Society of Immunology and Hematology (SHIP) . The severe phenotype of Diamond-Blackfan anemia is modulated by heat shock protein 70. Blood Adv. 2017;1(22):1959-1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moniz H, Gastou M, Leblanc T, et al. ; DBA Group of Société d’Hématologie et d’Immunologie Pédiatrique-SHIP . Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012;3(7):e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chennupati V, Veiga DF, Maslowski KM, et al. . Ribonuclease inhibitor 1 regulates erythropoiesis by controlling GATA1 translation. J Clin Invest. 2018;128(4):1597-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chakraborty A, Uechi T, Higa S, Torihara H, Kenmochi N. Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS One. 2009;4(1):e4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008;112(13):5228-5237. [DOI] [PubMed] [Google Scholar]
- 89.Torihara H, Uechi T, Chakraborty A, Shinya M, Sakai N, Kenmochi N. Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond-Blackfan anaemia. Br J Haematol. 2011;152(5):648-654. [DOI] [PubMed] [Google Scholar]
- 90.Devlin EE, Dacosta L, Mohandas N, Elliott G, Bodine DM. A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood. 2010;116(15):2826-2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McGowan KA, Li JZ, Park CY, et al. . Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40(8):963-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dutt S, Narla A, Lin K, et al. . Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117(9):2567-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fumagalli S, Di Cara A, Neb-Gulati A, et al. . Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11(4):501-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singh SA, Goldberg TA, Henson AL, et al. . p53-Independent cell cycle and erythroid differentiation defects in murine embryonic stem cells haploinsufficient for Diamond Blackfan anemia-proteins: RPS19 versus RPL5. PLoS One. 2014;9(2):e89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trainor CD, Mas C, Archambault P, Di Lello P, Omichinski JG. GATA-1 associates with and inhibits p53. Blood. 2009;114(1):165-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Toki T, Yoshida K, Wang R, et al. . De novo mutations activating germline TP53 in an inherited bone-marrow-failure syndrome. Am J Hum Genet. 2018;103(3):440-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang Z, Keel SB, Shimamura A, et al. . Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci Transl Med. 2016;8(338):338ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rio S, Gastou M, Karboul N, et al. . Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood. 2019;133(12):1358-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Doty RT, Yan X, Lausted C, et al. . Single-cell analyses demonstrate that a heme-GATA1 feedback loop regulates red cell differentiation. Blood. 2019;133(5):457-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heijnen HF, van Wijk R, Pereboom TC, et al. . Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway. PLoS Genet. 2014;10(5):e1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Antunes AT, Goos YJ, Pereboom TC, et al. . Ribosomal protein mutations result in constitutive p53 protein degradation through impairment of the AKT pathway. PLoS Genet. 2015;11(7):e1005326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ramenghi U, Garelli E, Valtolina S, et al. . Diamond-Blackfan anaemia in the Italian population. Br J Haematol. 1999;104(4):841-848. [DOI] [PubMed] [Google Scholar]