

Abstract
While prevailing evidence supports that the amyloid cascade hypothesis is a key component of Alzheimer’s disease (AD) pathology, many recent studies indicate that the vascular system is also a major contributor to disease progression. Vascular dysfunction and reduced cerebral blood flow (CBF) occur prior to the accumulation and aggregation of amyloid-β (Aβ) plaques and hyperphosphorylated tau tangles. Although research has predominantly focused on the cellular processes involved with Aβ-mediated neurodegeneration, effects of Aβ on CBF and neurovascular coupling are becoming more evident. This review will describe AD vascular disturbances as they relate to Aβ, including chronic cerebral hypoperfusion, hypertension, altered neurovascular coupling, and deterioration of the blood-brain barrier. In addition, we will describe recent findings about the relationship between these vascular defects and Aβ accumulation with emphasis on in vivo studies utilizing rodent AD models.
Keywords: Amyloid-β peptide, amyloid cascade hypothesis, blood-brain barrier, cerebral amyloid angiopathy, chronic cerebral hypoperfusion, functional hyperemia, in vivo mouse model, in vivo rat model, neurovascular coupling, vascular hypothesis
INTRODUCTION: ALZHEIMER’S DISEASE AND THE AMYLOID CASCADE HYPOTHESIS
Alzheimer’s disease (AD) was first described by Alois Alzheimer in 1906 after examining the brain of Auguste Deter, a 51-year-old woman with aggressive and progressive memory loss. Alzheimer’s findings eventually became known as the pathological hallmarks of AD [1]. AD is a devastating illness characterized by a progressive decline in cognition, which is currently the 6th leading cause of death in the United States, and the primary cause of dementia [2]. Initially, AD was considered a middle-age disease whereas senile dementia was separately defined as progressive dementia in the elderly. However, since middle-age AD and senile dementia patients share indistinguishable postmortem neuropathology, including the abnormal accumulation of amyloid plaques, both conditions are now identified as AD [3, 4]. The amyloid cascade hypothesis describes the events leading to the development of plaques in AD brains by preferential overproduction of the amyloid-β (Aβ)42 isoform that is prone to aggregation compared to the Aβ40 isoform [5]. This accumulation of toxic Aβ42 isoforms begins when amyloid-β protein precursor (AβPP) is cleaved sequentially by β-secretase BACE1 (β-site APP-cleaving enzyme 1) and γ-secretase [6]. Additional processes that increase the amount of Aβ42 include impaired degradation by neprilysin or insulin degrading enzymes [7, 8], or reduced clearance across the blood-brain barrier (BBB) [9] through low density lipoprotein receptor related protein 1 (LRP1) and receptor for advanced glycation end products (RAGE) found on epithelial cells [10, 11]. Eventually accumulation reaches a threshold where Aβ42 begins to aggregate into low molecular weight oligomers followed by formation of insoluble plaques that are deposited throughout the brain [12–14]. While femtomolar to picomolar concentrations of Aβ42 play a role in the healthy brain [15, 16], higher nanomolar concentrations of soluble Aβ42 is known to increase neuronal activity that may create an excitotoxic environment [17–20]. Excitotoxicity includes an inflammatory response consisting of microglial and astrocytic activation [21, 22], progressive neuronal/synaptic injury, altered neuronal ionic homeostasis and oxidative injury, and aberrant tau protein hyperphosphorylation and aggregation [23]. The eventual neuronal dysfunction and cell death associated with this cascade of events leads to widespread cerebral atrophy [24] that is prominent throughout the neocortex including the medial temporal lobe, cingulate gyrus, parietal lobe, and frontal lobe [25–27]. Altered neurotransmission and the subsequent neurodegeneration are hypothesized to contribute to the cognitive and behavioral decline observed in AD [28, 29].
The identification of early-onset familial AD caused by genetic mutations of APP, presenilin-1, and presenilin-2 (the catalytic subunit of γ-secretase), strengthened the case for the amyloid cascade hypothesis of AD pathogenesis [30]. These mutations result in a higher ratio of Aβ42/Aβ40 production, increased rates of Aβ oligomerization, and senile plaque formation [28, 31]. Consequently, disease-modifying therapies targeting Aβ production were developed [32], but all clinical trials, to date, targeting abnormal Aβ accumulation have been unsuccessful at slowing or halting AD progression [33, 34]. However, these studies primarily focused on mild-to-moderate AD, presumably when pathological hallmarks and neurodegeneration were already present. It is currently unclear if clinical trials designed to treat AD prior to the development of severe pathological changes might improve patient outcome. However, obstacles exist to implementing this strategy in the human population, such as the lack of a reliable early biomarker for AD.
A surprising discovery came from ‘the Nun Study’ in which cognition in elderly nuns (aged 77–103 years) was assessed with tests including the Mini-Mental State Examination and postmortem neuropathological changes were quantified [35]. A subset of individuals with normal cognition had an abundance of diffuse plaques in the neocortex and hippocampus, indicating the involvement of additional unknown factors in the cognitive symptoms of AD. Findings such as these suggest that the amyloid cascade hypothesis is not the sole contributor to the etiology of AD and overlapping neuropathologies may contribute to disease progression.
CEREBRAL VASCULATURE AND THE TWO-HIT VASCULAR HYPOTHESIS OF ALZHEIMER’S DISEASE
Blood is delivered to the brain by cardiovascular circulation that is regulated by heart rate. The internal carotid artery supplies the frontal, parietal, and lateral temporal lobes, while the vertebral arteries supply the occipital lobe, brainstem, and cerebellum. Under physiological conditions, systemic blood flow delivery into the brain remains relatively constant and entry is regulated by the precise architecture of blood vessels branching into a vascular tree [36]. The large penetrating arteries break off and diverge into arterioles, precapillary arterioles, and finally into capillaries. The regulation of local cerebral blood flow (CBF) requires different cell types distributed along the vascular tree (Fig. 1). While all blood vessels are coated by endothelial cells, the presence of other cell types depends on the region of vascular branching. For example, penetrating arteries have 2–3 layers of vascular smooth muscle cells (VSMCs) whereas arterioles only have a single VSMC layer. Capillaries share a common basement membrane with cells called pericytes, and are covered by astrocytic endfeet that are innervated by local neurons [36]. Neurons, astrocytes, VSMCs, pericytes, and endothelial cells work in concert to form the neurovascular unit (NVU). The NVU regulates local CBF in response to brain activity, a process called neurovascular coupling (NVC) or functional hyperemia [37, 38].
Fig. 1.
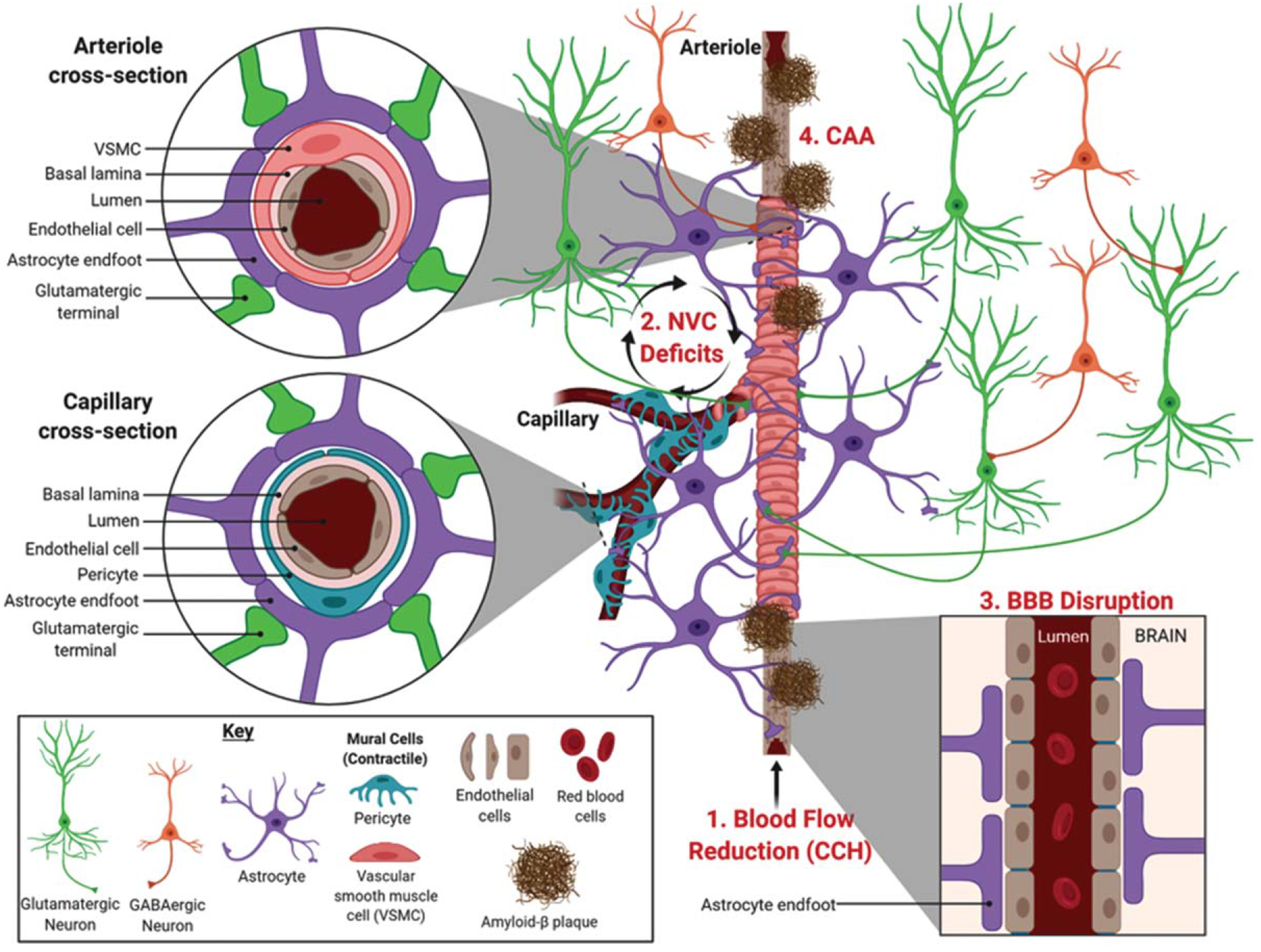
Neurovascular deficits linked to Alzheimer’s disease. The illustration shows the various cell types involved in regulating localized cerebral blood flow along an arteriole bifurcating into capillaries. Neurovascular deficits related to AD include (1) chronic cerebral hypoperfusion (CCH), a reduction in blood flow into the brain that occurs prior to symptomology, (2) neurovascular coupling (NVC) deficits, and (3) blood-brain barrier (BBB) disruption that can be related to (4) cerebral amyloid angiopathy (CAA), characterized by the deposition of Aβ in blood vessels. Image created with BioRender.com.
Historically, it was postulated that the increase in CBF in response to increased neuronal activity was associated with energetic costs and driven by metabolic byproducts, such as carbon dioxide [39], which was termed the ‘negative feedback’ hypothesis. This has been superseded by the ‘feedforward’ CBF hypothesis that involves vasoactive messengers including nitric oxide, which plays a role in neuronal signaling and regulating CBF responses [37, 40–43]. It is now well established that astrocytes also regulate CBF through release of vasoactive substances, such as arachidonic acid and its metabolites, prostaglandins and epoxyeicosatrienoic acids [44, 45]. Thus, the NVU is sometimes more aptly referred to as either the neurogliovascular or gliovascular unit. Astrocytes have perivascular endfeet that wrap around endothelial cells (Fig. 1, cross section) to regulate CBF entry, as well as establish and maintain the BBB integrity [44, 46, 47]. VSMCs and pericytes both regulate CBF by physically controlling vascu lar reactivity of arterioles and capillaries on which they respectively ensheathe [48]. In particular, pericytes possess the largest vascular resistance in the brain and are estimated to increase CBF by 84% in response to neuronal activity [49]. This makes pericytes promising targets to treat medical conditions involving neurovascular dysfunction [36, 50, 51].
Recent evidence indicates a causal relationship between cerebrovascular dysfunction and the development of AD [52]. Vascular abnormalities are present in at least 50% of AD cases and becomes more prevalent with increasing age of diagnosis [53]. Furthermore, vascular risk factors associated with AD, such as hypertension and atherosclerosis [54, 55], cause additional damage leading to progressive cerebral hypoperfusion [56]. Over time, homeostatic and hemodynamic disruption damages the cerebral vasculature that perturbs delivery of macromolecules necessary to maintain neuronal activity. The subsequent oxidative stress damages cellular membranes, leads to neuronal and astroglial cell death, and culminates in the cognitive decline observed in AD [57]. Other factors are known to correlate with cognitive deficits including stroke severity, hypertension, atherosclerosis, and arteriosclerosis [58, 59], while macroscopic cerebral infarcts increases the likelihood of AD diagnosis [60]. As such, evidence support that neurovascular dysregulation plays a role in determining the severity of the clinical symptoms of AD.
A neurovascular hypothesis that incorporates a vascular pathogenic component and excessive Aβ accumulation was initially proposed and developed into the two-hit hypothesis of AD [61]. This model hypothesizes that vascular damage, such as BBB disruption that concurrently impairs the machinery for Aβ clearance (hit 1) is followed by the accumulation of Aβ in the brain to vasculotoxic and neurotoxic levels (hit 2) [62, 63]. Supporting this hypothesis are early neuropathology studies that found AD patients had Aβ peptides in [64, 65] and along the walls of cerebral blood vessels [66, 67].
While it is difficult to address the most important question “What causes Alzheimer’s disease?”, and which hypothesis more precisely reflects its pathogenesis, traditional models have focused on the classic AD hallmarks of Aβ accumulation, tau hyperphosphorylation, and neuronal loss [68]. However, retrospective neuroimaging analysis of healthy controls, mild cognitive impairment (MCI), and AD patients indicated that vascular abnormalities occur first, followed by changes in Aβ deposition, metabolic dysregulation, functional impairment, and cerebral atrophy [69]. In addition, studies using arterial spin labeling magnetic resonance imaging (MRI) have detected changes in CBF years before AD symptoms appear [70]. Hence, a growing body of literature support neurovascular dysregulation is a critical factor underlying AD pathogenesis.
Although numerous vascular risk factors and conditions are associated with AD [71], in this review we focus on neurovascular processes observed in AD patients, including (1) chronic CBF reduction, (2) NVC dysfunction, and (3) BBB disruption related to cerebral amyloid angiopathy (CAA). We also discuss findings from recent studies that examine these neurovascular irregularities in AD amyloidogenic animal models. With the advent of transgenic mouse models that develop classical AD pathology, we are able to study how the progression of AD relates to vascular aberrations. While no model adequately recapitulates either familial or sporadic AD, many transgenic amyloidogenic AD mouse models have been developed by incorporating mutations from familial AD. All of the transgenic AD mouse models described in this review display parenchymal Aβ accumulation and some also exhibit Aβ accumulation in blood vessels. These models also vary according to the onset of Aβ accumulation, the rate and spatial distribution of Aβ accumulation, and the extent of cognitive decline. A descriptive table of the models mentioned in this review is presented in Table 1.
Table 1.
Transgenic amyloidogenic AD mouse models and an AD rat model have been developed incorporating mutations from familial AD that display progressive accumulation of Aβ in the brain. All of the transgenic AD mouse models described in this review display parenchymal Aβ accumulation and some exhibit Aβ accumulation in blood vessels
| Model | Neuropathological and behavioral characterization | References |
|---|---|---|
| Tg2576 | elevated Aβ plaques in cortical and limbic structures, learning and memory | [219] |
| impairments at 9–10 months of age | [220] | |
| PS1V97L | prominent Aβ accumulation in the brain at 6 months, concurrent memory deficits | [221] |
| APP23 | develops extensive parenchymal and cerebrovascular Ap deposits after 6 months of age, memory impairments begin at 3 months of age |
[222] [223] |
| TgSwDI | Aβ deposition in cerebral microvasculature and throughout the brain parenchyma starting at 6 months of age | [224] |
| APPSwInd or hAPPJ20 APP/PS1 |
high levels of Aβ accumulation throughout the brain, particularly in neocortex and hippocampus, by 2–4 months of age, memory deficits observed at 6 months of age develops Aβ plaques starting at 4 and 6 months in the cortex and hippocampus, respectively, progressive spatial memory impairment between 6–15 months of age |
[225] [226] [227] [228] |
| 3xTg-AD | neocortical and hippocampal Aβ accumulation by 4 and 6 months of age, respectively, memory deficits detected at 2 months of age |
[229] [230] |
| TgCRND8 | early-onset Aβ accumulation (starting at 2 months) and significant cognitive deficits observed by 11 weeks of age | [231] |
| 5xFAD | early-onset Aβ accumulation (starting at 1.5 months) and memory deficits are seen At 3 months of age |
[232] [233] |
| TgF344-AD* | displays broad pathological changes of AD, including progressive Aβ accumulation, neurodegeneration, and cognitive deficits at 6 months of age | [234] |
Transgenic rat model.
CHRONIC CEREBRAL BLOOD FLOW REDUCTION IN ALZHEIMER’S DISEASE
Early ultrastructural studies showed that AD brains with amyloid deposits had abnormally shaped blood vessels [72, 73]. These finding led to the hypothesis that amyloid fibrils caused blood vessel wall deformations and lumen stenosis resulting in reduced blood flow and restricted nutrient entry into the brain [74]. Subsequent studies supported this hypothesis by showing that people with MCI and AD exhibit hypoperfusion in many brain areas, especially those involved in learning and memory [75–79]. The gradual reduction in CBF observed during AD progression is referred to as chronic cerebral hypoperfusion (CCH). It is unclear whether hypoperfusion causes AD or if CBF deficits develop in response to AD neuropathological changes. However, evidence support that hypoperfusion is present at preclinical AD stages [70, 80]. In agreement, patients with chronic vertebra-basilar stenosis, resulting in CCH to the posterior brain regions, display severe cognitive impairment attributed to perfusion deficits [81]. While this study suggests that CCH in itself is detrimental to cognition, the relationship between CBF and cerebrovascular Aβ has been examined using florbetapir positron emission tomography (PET) imaging. The presence of Aβ in cognitively normal, older adults predicted an association between hippocampal hyperperfusion and diminished memory performance, whereas no association was observed in older adults negative for Aβ [82]. However, others have reported that increased Aβ was associated with hypoperfusion that diminished in respect to the stage of cognitive impairment [83]. Although these studies show discordant results regarding the relationship between Aβ and CBF, they suggest that Aβ presence does affect CBF in a spatially dependent manner.
Apolipoprotein E (ApoE) is a glycoprotein responsible for triglyceride and cholesterol transport, but the ε4 allele is less efficient at this transport and is the second biggest genetic risk factor for AD. Hyper-perfusion has been observed in several brain areas, including the medial temporal lobe of cognitively normal or MCI patients that are APOE ε4 carriers [84, 85]. This phenomenon has been attributed to a compensatory response in which increased neuronal activation required by APOE ε4 carriers during memory formation requires hyperperfusion [86]. However, as the disease progresses in these APOE ε4 carriers, reduced CBF is observed [85, 87], which could be the result of vascular damage related to neuropathological effects. Therefore, studies that reported hyperperfusion in the medial temporal lobe in early MCI or in non-amnestic MCI, might have seen skewed results by not controlling for APOE ε4 genotype status [88, 89]. The overall trend for the majority of sporadic AD patients is reduced CBF prior to MCI diagnosis that continues to decline with disease progression and is affected by Aβ accumulation.
ANIMAL STUDIES OF CHRONIC CEREBRAL BLOOD FLOW REDUCTION (OR CCH) IN ALZHEIMER’S DISEASE
Although some consider CCH to be ischemia, it is important to note that CCH implies a moderate reduction in CBF over a prolonged period of time. In contrast, ischemic insults completely eliminate CBF and delivery of metabolic substances, such as glucose and oxygen, to specific brain regions resulting in rapid damage to those areas lacking blood supply. Since some animal models that induce CCH result in different levels of brain hypoxia, they are considered to be viable models of ischemia.
The established animal CCH model was first developed in rats by performing bilateral common carotid artery occlusion (BCCAO), also called 2-vessel occlusion (2VO). This was performed by ligating the common carotid arteries (CCAs) while the vertebral arteries remained open resulting in a partial CBF reduction [90, 91]. The BCCAO CCH model reduced cortical and hippocampal CBF leading to impaired spatial learning, deficient memory performance, and damage to neurons in the hippocampal CA1 [92–95]. In addition, the CBF reduction induced by BCCAO is thought to disrupt the NVU leading to increased hippocampal astrocyte reactivity [96, 97]. Since inducing CCH in rats with the BCCAO technique can elicit cognitive deficiencies that recapitulate cognitive symptoms and pathology seen in AD [98], additional studies have examined the relationship between CCH and Aβ in the brain. CCH induced by BCCAO in rats resulted in increased Aβ accumulation in the hippocampus [99], which has been attributed to increased AβPP expression and enhanced β- and γ-secretase activity [100–102]. A recent imaging study in rats confirmed that BCCAO induced higher frontal cortex and hippocampal Aβ levels and a concurrent reduction in glucose metabolism in the hippocampus, entorhinal cortex, and amygdala [103]. Injecting Aβ into the brain of young rats that underwent CCH conferred synergistic spatial memory impairments and increased cortical and striatal AβPP levels compared to CCH or Aβ alone [104, 105], indicating that Aβ can influence CCH-induced changes.
While BCCAO has been employed in numerous studies in rats, ligating arteries is thought to produce a more profound and rapid reduction in CBF than the gradual, CCH observed in AD. Moreover, BCCAO is not amenable to study CCH in C57Bl/6 mice because chronically occluding both CCAs results in a high mortality rate [106]. Chronic unilateral right CCA occlusion (rCCAO) was developed to study CCH in mice. This model decreases CBF to ipsilateral brain regions, induces cognitive deficits, and results in ipsilateral white matter lesion (WML) development [107]. In two AD mouse models, rCCAO exacerbates learning and memory deficits compared to genetically- and age-matched controls without rCCAO. In the PS1V97 L mouse model, rCCAO exacerbated Aβ expression that was attributed to changes in expression of Aβ degrading enzymes and clearance proteins [108]. In the Tg2576 AD mouse model, learning and memory deficits caused by rCCAO-induced CCH were attributed to hypometabolism [109]. Although these studies suggest that CCH induced by BCCAO in rats or by rCCAO in mice can induce or exacerbate AD-like pathology, the CBF pattern does not fully represent the progressive CCH seen in AD.
Bilateral carotid artery stenosis (BCAS) is an alternative CCH model for mice in which microcoils of different sizes narrow bilateral CCAs, thereby decreasing CBF based on CCA constriction [110]. Effects of BCAS-induced CCH indicate global CBF reduction lasts for several weeks to months [111, 112]. Mice that underwent BCAS have memory impairment, WML formation, and astroglia and microglia activation [110, 113]. CCH induced in mice have decreased hippocampal metabolism and atrophy 6 to 8 months post BCAS, respectively [114], and increased endogenous Aβ accumulation throughout the hippocampus and cortex [115]. Studies using BCAS-induced CCH in amyloidogenic AD mouse models showed a synergistic effect of learning and memory impairment and Aβ accumulation in APPSwInd mice [116, 117], increased parenchymal and cerebrovascular Aβ accumulation in Tg-SwDI mice [118], and increased Aβ aggregation in APP/PS1 mice without increasing total Aβ levels [119]. Like the BCCAO CCH model in rats, the BCAS CCH model in mice is thought to model vascular cognitive impairment because it induces formation of WMLs leading to behavioral/cognitive deficits [120]. Although the BCAS model is popular to study vascular dementia, it provides a useful model to further study CCH in mouse models of AD.
Another approach to recapitulate CCH seen in AD is the 2-vessel/bilateral gradual CCA occlusion/stenosis (2VGO or BCCS or GCAS) model. Instead of ligating CCAs, ameroid constrictors are used to partially constrict the CCAs bilaterally that narrow overtime leading to progressive cerebral hypoperfusion. In rats, compared to the BCCAO model, using 2VGO improved survival while eliciting an attenuated CBF decrease and neuroinflammatory response. However, both CCH models showed comparable impaired working memory [121]. In mice, 2VGO elicits a distinct progressive CBF reduction compared to the rapid and profound decrease observed after BCAS [122, 123]. But, 2VGO does not consistently induce hippocampal neuronal loss. Although 2VGO in these mice impaired working memory, they were comparable to sham controls in regard to hippocampal-dependent reference learning [122]. The 2VGO model has been used extensively in the Koji Abe laboratory to examine the effect of CCH in APP23 mice. Overall, compared to ageand genotype-matched controls, CCH in APP23 mice enhances motor and cognitive deficits while increasing hippocampal cell death. [124–127]. CCH in APP23 mice also decreases LRP1 and increases RAGE expression in vascular endothelial cells, which would perturb Aβ clearance resulting in parenchymal and cerebrovascular Aβ deposition [125]. CCH also induced NVU dissociation as measured by decreased astrocyte to blood vessel immunofluorescent staining overlap that could be a consequence of Aβ accumulation [124].
Since 2VGO results in increased mortality after 28 days when ameroid constrictors close, another CCH model developed in mice is the asymmetrical gradual carotid artery occlusion. This method consists of one CCA progressively occluded with an ameroid constrictor while a microcoil is placed on the other CCA resulting in decreased CBF and hippocampal neuronal loss [128, 129]. However, despite the promising translational implications of this model, it has not been tested in AD mouse models yet.
The majority of studies using AD mouse models are in agreement that CCH exacerbates Aβ accumulation and increases neuronal death. When interpreting results from these studies, it is important to discern the differences in CCH and AD models employed. As mentioned, BCAS creates a prolonged CCH followed by CBF recovery making this suitable to model vascular dementia [114]. Still, since BCAS is a long-lasting CCH model, it might be the best model currently available to study CCH in mice. More studies using the BCAS CCH in AD mouse models are warranted to further understand the impact of CCH on Aβ accumulation and to identify potential therapeutic interventions to counteract the negative CCH effects. As stated, a recurring theme is that mice tend to be more vulnerable to CCH than rats. It is plausible that 2VGO in TgF344-AD rats might be a viable model to study the relationship between CCH and Aβ accumulation.
BLOOD PRESSURE AND ALZHEIMER’S DISEASE
Cerebral autoregulation refers to myogenic, autonomic, and metabolic mechanisms that maintain adequate blood perfusion to the brain despite changes in blood pressure [130, 131]. Although a single study of sporadic AD patients showed impaired cerebrovascular autoregulation [132], most studies have shown that cerebrovascular autoregulation is unaffected in AD patients [133–135]. However, it is established that having either low blood pressure (hypotension) or high blood pressure (hypertension) increases the likelihood of developing dementia and AD [136–138]. In particular, elevated systolic blood pressure (BP) is a major vascular risk factor for developing AD and is also associated with cerebrovascular disease, including stroke and cerebral infarcts [54, 139, 140]. Accordingly, hypertension-induced vascular changes, such as small vascular lesions and BBB damage [141], possibly contribute to the development of CCH and cognitive deficits [142]. A neuroimaging study showed AD patients with hypertension had worse cognitive function and reduced hippocampal metabolism compared to normotensive AD patients. Interestingly, no differences in Aβ were observed between these AD patients [143]. Another study showed hypertension in AD patients accelerated the rate of cognitive decline only in AD patients under the age of 65 [144]. A study in older patients (aged 71–85) with moderate AD found decreases in BP as the disease and cognition worsen [145], whereas others showed a decrease in BP years before cognitive decline [146]. Although a comprehensive report concluded that reducing BP in hypertensive people does not help prevent cognitive decline or the development of dementia, methodological variation of these studies resulted in self-reported problems with the analyses performed [147].
The discordant results of these studies may be attributed to the differential effect that CCH or BBB deterioration would have on BP in subjects, thereby skewing the selection criteria. For example, someone who was hypertensive prior to AD diagnosis could have developed a cardiovascular abnormality, such as CCH, which would result in lower BP readings and a subsequent “normotensive” categorization during the study design. Therefore, prospective, longitudinal studies are warranted to examine the effect of lowering BP on the development of dementia or AD that carefully factor in differences in BP and presence/absence of vascular irregularities, such as CCH. One study following MCI patients for 6 years found that higher plasma levels of atrial natriuretic peptide, involved in diuresis and lowering BP, were associated with conversion from MCI to dementia or probable AD. However, MCI patients with increased atrial natriuretic peptide levels that received antihypertensive treatment had a lower likelihood of converting to probable AD [148]. It is thought that elevated atrial natriuretic peptide is indicative of disturbed vascular function, which could be used as an early biomarker to determine the optimal therapeutic treatment window in MCI patients at risk for converting to AD. Overall, we know that hypertension is implicated with the development of dementia and AD, but the effect of using antihypertensive medications to halt the progression from early cognitive symptoms into AD is still inconclusive. In addition to continuing studies to test the effectiveness of antihypertensive treatments, we must identify and study alternative targets related to hypertension-induced effects, such as damaged blood vessels from microinfarcts.
ANIMAL STUDIES OF HYPERTENSION AND ALZHEIMER’S DISEASE
While many animal studies described above pertain to CCH, very few animal studies have explored the effects on Aβ and hypertension. It has been shown that intra-arterial infusion of Aβ40 into the right common carotid artery increased mean arterial blood pressure (MAP) in hypotensive rats (MAP < 100 mm Hg), but had no effect in normotensive (MAP = 100–129 mm Hg) or hypertensive rats (MAP > 139 mm Hg) [149–151]. The Arendash et al. study also showed that the Aβ42 isoform did not increase BP in hypotensive rats. However, after decreasing BP with the vasodilator sodium nitroprusside, both Aβ40 and Aβ42 infusion induced a return to baseline BP, supporting that Aβ can contribute to hypertension by constricting blood vessels. These studies support that increased soluble Aβ can exacerbate cerebrovascular dysfunction by inducing cerebral and peripheral vasoconstriction contributing to hypertension during the early disease stages. However, as amyloid aggregates in the brain, cerebrospinal fluid levels drop potentially resulting in a shift from a hyper to hypotensive state that reduces amyloid clearance through the BBB. As such, a feed-forward mechanism results contributing to reduced CBF, further Aβ accumulation, and the eventual cognitive and functional decline contributing to MCI and AD progression.
Inducing hypertension in wildtype mice increased Aβ levels in the cortex and hippocampus as well as surrounding blood vessels that led to BBB deterioration [152, 153]. An increased RAGE expression in capillaries was also observed, indicating hyper-tension might result in upregulated RAGE-mediated Aβ influx into the brain [153]. Inducing hyper-tension in APP/PS1 and TgSwDI mice led to increased brain Aβ accumulation, damage to blood vessels, and accelerated cognitive deficits compared to genotype-matched normotensive controls [154, 155]. Compared to wildtype mice, APP/PS1 mice were more responsive to hypertensive treatment, but less responsive to antihypertensive treatment. Reduced hippocampal CBF was also observed in these mice, signifying that hypertension coupled with hippocampal Aβ accumulation combines to disrupt cerebral hemodynamics [156]. In addition, inducing hypertension in APP/PS1 mice exacerbates cognitive performance compared to normotensive APP/PS1 mice, which was attributed to decreased functional connectivity in the brain measured by resting state blood oxygen-level dependent (BOLD) fMRI [157]. This study employed useful methodology in rodents that yielded deficits that are comparable to those found in people with cognitive decline in resting state fMRI studies. When examining age on vascular parameters, 16–18-month-old APP/PS1 mice had higher systolic BP, decreased cortical and thalamic CBF, and altered hippocampal vasoreactivity that contributed to their behavioral and cognitive impairments compared to age-matched control mice [158]. These studies support that hypertension has deleterious effects on AD-like neuropathology, and the potential benefits of treating hypertension to quell AD progression should be explored further.
NEUROVASCULAR COUPLING DYSFUNCTION IN ALZHEIMER’S DISEASE
Changes in CBF in humans are typically measured with imaging techniques, including BOLD fMRI and FDG-PET imaging. BOLD fMRI relies on the observation that CBF increases to areas with neuronal activation. In FDG-PET imaging, radiolabeled fluorodeoxyglucose accumulation in tissue reflects metabolic changes in response to neuronal activity [159, 160]. Recently, a noninvasive MRI alternative using arterial spin labeling perfusion to monitor CBF has been developed that matches metabolic patterns observed with FDG-PET and reflects CBF more precisely than BOLD fMRI [161]. These neuroimaging techniques that measure CBF changes are considered to reflect NVC. By measuring brain activity with BOLD fMRI, patients with MCI, mild AD, or AD exhibit disruptions in functional connectivity in a resting-state default mode network (DMN). The DMN is comprised of widespread cortical regions, including the prefrontal, parietal, and temporal cortices and the hippocampal formation [162–164] that are commonly affected in AD [165]. A study combining arterial spin labeling perfusion and resting-state fMRI in AD patients confirmed functional connectivity abnormalities in DMN regions, and also showed regional CBF disruptions in brain areas affected in AD [166]. By measuring Aβ levels with PET using Pittsburgh Compound-B and resting-state DMN with BOLD fMRI it was shown that disrupted functional connectivity in DMN regions is associated with increased Aβ deposition in AD patients even though deposition was also evident in cognitively normal subjects [167]. To measure changes in neuronal activity, electroencephalogram recordings have shown weaker alpha and beta rhythms, but enhanced delta and theta rhythms in the DMN of AD patients [168, 169]. Reduced associations between BOLD signaling and alpha band power was also observed in the DMN of probable AD patients [170]. While deficits in DMN connectivity have been linked to AD, these studies assess brain activity in a specific network during rest, which reflect global NVC anomalies. Incorporation of memory tasks or other sensory stimuli during BOLD fMRI measurements have found region-specific NVC deficiencies in AD patients [171–174]. For example, memory encoding tasks show decreased activation of the medial temporal lobe [172], while exposure to visual stimuli indicates reduced CBF in numerous brain regions of AD patients [175] compared to healthy elderly controls. Recent studies have verified these results in mild AD patients by showing normal CBF in the visual cortex at rest, but upon presentation of visual stimuli, hypoperfusion becomes evident [176]. Retinal NVC measurements are a newer, less invasive method for determining pathophysiological brain processes while providing information regarding the NVU integrity [177]. Retinal vessel reaction to a noninvasive flicker stimulation found that AD patients displayed more pronounced and delayed reactive dilation compared to MCI and healthy controls, which was attributed to a compromised NVU [178].
Many of these imaging studies have examined MCI or early-stage AD to understand how NVU disruption changes during the course of AD progression. With this information, a predictive biomarker can be developed for earlier probable dementia diagnosis allowing for improved therapies and patient outcome. Overall, these studies support a reduction in NVC of AD patients, which may hinder amyloid clearance leading to its accumulation and deposition particularly within the DMN. Additional prospective, longitudinal studies are needed to gain a better understanding the role of NVC in AD progression.
ANIMAL STUDIES OF NEUROVASCULAR COUPLING DYSFUNCTION IN ALZHEIMER’S DISEASE
Measuring NVC functionally requires simultaneous measurements of neuronal/astrocytic activity and vascular changes. Since this is technically challenging, there is a scarcity of research examining NVC in rodent models of AD, but most studies indicate NVU dysfunction. The TgF344-AD rat model has diminished cerebrovascular reactivity that correlated with increasing Aβ load in vessel walls as measured by changes in dilatory capacity induced by hypercapnia [179]. At 9 months TgF344-AD rats also have neuronal network dysfunction between the hippocampus and medial prefrontal cortex that is observed after onset of Aβ plaque deposition but prior to cognitive deficits [180]. Two-photon imaging of 3xTg-AD, TgSwDI, and Tg2576 mice that overexpress Aβ in cerebral blood vessels showed abnormalities in both astrocytic and vascular activity in response to whisker stimulation [181]. Moreover, 3xTg-AD mice display impaired neurovascular coupling induced by neuronal-derived nitric oxide signaling [182]. In agreement, hAPPJ20 mice showed NVU disruption characterized by astrocytic endfoot separation from blood vessels as a result of Aβ deposition [183]. Others have shown neutrophils cause CBF deficits in APP/PS1 and 5xFAD mice [184].
A recent study in human tissue and in rat brain slices showed that Aβ oligomers act specifically on pericytes to constrict capillaries [185]. Thus, pericytes have become a potential target to alleviate conditions characterized by blood flow deficiencies [186]. Pericyte deficient mice are a useful model for studying their contribution to NVC. These mice develop neurovascular uncoupling, diminished cerebral oxygen supply, and metabolic stress [187, 188]. Crosses of pericyte-deficient mice with amyloid mouse models of AD may elucidate the role of specific BBB cell types in AD progression.
BLOOD-BRAIN BARRIER, CEREBRAL AMYLOID ANGIOPATHY, AND ALZHEIMER’S DISEASE
The brain microvasculature supports the exchange of critical metabolic substrates between the brain and circulating blood [189]. Importantly, the BBB protects the brain from entry of toxic compounds and eliminates waste products, including Aβ, via LRP1 and RAGE [10, 11, 190]. It is thought that impaired Aβ clearance across the BBB is involved in the pathogenesis of AD [191–193], and deficient Aβ clearance, rather than Aβ overproduction, results in aggregation and plaque pathology [9, 69]. AD brains consistently display CAA, characterized by Aβ deposition along the walls of cerebral blood vessels and leptomeningeal blood vessels that are located between the subarachnoid and pia mater [67]. Although CAA is considered a clinically distinct phenomenon than AD, it shares common cerebrovascular and neurodegenerative properties suggesting a mechanistic link [194]. Originally, sporadic CAA was characterized according to the absence or presence of Aβ deposition in capillaries, the latter referred to as capillary cerebral amyloid angiopathy [195]. More recently, this has been reclassified to detail the cortical penetrance of accumulation along capillaries [196], which has been reviewed elsewhere [197]. Examination of autopsy-confirmed AD cases reported an overlap with CAA in which 82.9% of AD patients exhibited at least mild CAA in parenchymal or leptomeningeal vessels and 25.6% with moderate to severe CAA throughout various brain regions [66]. CAA is prevalent in postmortem AD brains and correlates with both Aβ deposition and dementia severity as assessed by the clinical dementia rating scale [198]. In agreement, CCA severity is associated with lower cognition proximal to death of the patient [199].
A recent study showed that microvessels from the parietal cortex of AD patients or those with advanced CAA contained higher concentrations of vascular Aβ40 and Aβ42 [200]. Vascular levels of P-glycoprotein and neprilysin, which are involved in Aβ degradation, were lower in persons with AD, positively correlated with cognitive function, and inversely correlated with vascular Aβ40 levels. In contrast, BACE1, a protein required to produce Aβ, was increased in AD, negatively correlated with cognitive function, and positively correlated with Aβ40 in microvessel extracts. These studies support the idea that CAA in AD reflects failed cerebral Aβ clearance along perivascular lymphatic drainage pathways and could be a factor in the etiology of AD [201].
It is important to know that CAA is the most common form of cerebral small vessel disease, which encompasses diseases affecting small arteries, arterioles, venules, and capillaries. Initial studies indicated no differences in BBB permeability between early AD and non-demented individuals [202]. However, advances in neuroimaging have allowed the assessment of BBB deterioration in patients with cerebral small vessel disease [203], but only a few studies have examined BBB integrity in AD patients. These studies indicate increased BBB permeability in early AD patients and a correlation between reduced CBF and increased BBB leakage rate [204, 205].
ANIMAL STUDIES OF CAA AND BBB DYSFUNCTION IN ALZHEIMER’S DISEASE
Several different animal models exhibit vascular Aβ deposition similar to observations in human CAA. Compared to control mice, 6–12-month-old TgCRND8 have cortical arterioles with increased tortuosity as well as decreased size and CBF [206]. These structural and functional deficiencies observed in TgCRND8 mice were rescued by inhibiting Aβ oligomerization. Similar to TgCRND8 mice, Tg-SwDI mice develop cerebral microvascular amyloid pathology with reduced CBF, vasoreactivity, and cognitive deficits that can all be prevented by pharmacologically blocking Aβ oligomerization [207]. Transplanting epithelial progenitor cells into the hippocampus of APP/PS1 mice repaired BBB damage and led to improved cognitive performance [208]. Removing Aβ from leptomeningeal and cerebral vessels with immunotherapy in transgenic CAA mouse models improved responsivity to vasodilators [209]. These studies strongly suggest that Aβ accumulation in cerebral blood vessels plays a critical role in cognitive decline, which highlights the importance of additional research further investigating CAA models. For example, the stroke-prone spontaneously hypertensive rats were crossed with TgF344-AD rats to develop a novel mixed vascular dementia and AD model. These rats have robust Aβ accumulation, gliosis, and behavioral alterations [210], which would be useful to study the relationship between Aβ accumulation and damage to cerebral blood vessels.
REVISITING THE VASCULAR HYPOTHESIS FOR SPORADIC ALZHEIMER’S DISEASE
Vascular risk factors for AD are known to alter CBF although the mechanisms associated with these changes have not been fully elucidated. As the vascular and two-hit hypothesis for AD stipulate, aging, genetic, and environmental factors compromise the cerebral vasculature and BBB integrity [62, 71]. Microinfarcts or microhemorrhages may be early contributors to BBB deterioration leading to CBF deficits (Fig. 2). Subsequently, BBB deterioration, NVC dysfunction, and reduced CBF impede physiological clearance of Aβ causing its accumulation and aggregation in the CNS and cerebral vasculature. A second alternative is that pathological Aβ accumulation occurs independently of neurovascular dysfunction. Since Aβ constricts the cerebral vasculature, its accumulation results in further CBF reductions [149, 211]. The scarcity of metabolic substrates needed by astrocytes and neurons causes oxidative stress and cell death that starts in regions requiring higher energy demands, such as the cortex and hippocampus. Over time, this extends to other cortical areas causing widespread neurodegeneration. The alarming part about the neurovascular complications associated with AD development, is that they affect similar cell populations within the NVU creating a vicious cycle of BBB deterioration. This could explain the rapid progression from MCI or early AD to late stage AD. Perhaps, the association between small vascular lesions and neurodegenerative diseases has been lacking due to visualization methods. Initially, these vascular lesions were only observed during postmortem analysis; however, recent advances in human neuroimaging methods are now able to detect cerebral microinfarcts [212]. By employing advanced imaging methods, we can identify ischemic small vessel disease on a macro and microstructural scale [213], allowing us to design prospective, longitudinal studies to better understand the role of small vascular lesions in AD pathogenesis and other neurodegenerative disorders.
Fig. 2.
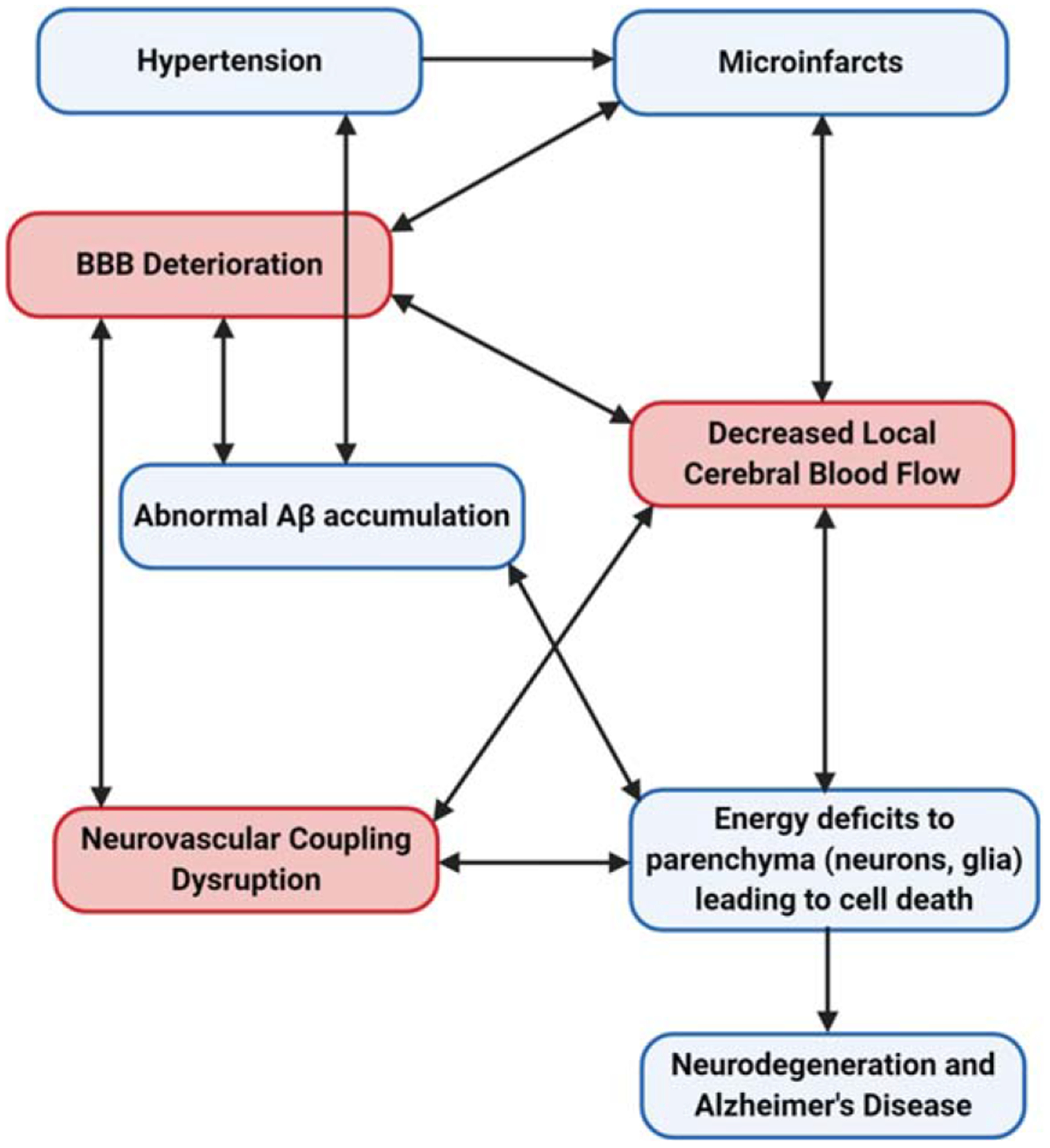
Vascular Theory of Alzheimer’s disease. Hypertension and other vascular risk factors contribute to the likelihood of cerebral blood vessels damage and microinfarcts. This causes blood-brain barrier (BBB) deterioration and diminishes blood supply to the affected cerebral regions. As the BBB breaks down, cells involved in neurovascular coupling are affected and consequently, are not able to properly regulate and supply local cerebral blood flow (CBF). Concurrently, the abnormal accumulation of amyloid-β (Aβ) in the brain parenchyma and in cerebral blood vessels contributes to the local CBF deficits. Whereas soluble Aβ can decrease CBF by directly constricting blood vessels, Aβ deposition is known to contribute to CBF reduction or chronic cerebral hypoperfusion. Over time, essential macromolecules to support neuronal network activity becomes limited leading to oxidative, neuronal and glial cell damage, and eventual neurodegeneration. Image created with BioRender.com.
SUMMARY: VASCULAR DYSREGULATION IN ALZHEIMER’S DISEASE
For nearly 30 years the majority of AD research has been conducted under the assumption that the cause of the disease is dependent upon accumulation of Aβ plaques and hyperphosphorylated tau tangles, which leads to a series of cellular events ultimately causing neurodegeneration and cognitive decline [214, 215]. Although the familial form of AD is caused by mutations in genes resulting in the abnormal accumulation and perturbed clearance of Aβ and hyperphosphorylated tau, the cause of sporadic AD that represents over 95% of all cases is still unclear [216–218]. While evidence supports that Aβ accumulation contributes to or exacerbates sporadic AD, many studies indicate neurovascular dysfunction occurs prior to Aβ accumulation and contributes to the development and/or progression of AD. Regardless of which hypothesis stands the test of time, a better understanding of the relationship between neurovascular dysfunction and AD is warranted. In addition to focusing on abnormal Aβ accumulation as the key event in the pathogenesis for AD, we need to identify alternative mechanisms involved in AD pathogenesis. However, since many rodent models of AD have been developed that over-express Aβ and tau pathology, much research has been devoted to understanding mechanisms related to these proteinopathies. Fortunately, over the past decade an increased interest in understanding how neurovascular dysfunction relates to AD and the relationship between Aβ accumulation and the NVU. The focus of this review was to describe neurovascular dysfunction linked to AD as a contributor to AD pathogenesis. The hope is that a deeper understanding of how the cellular components of the NVU are affected in early AD will lead to the identification of novel targets to prevent or repair neurovascular dysfunction thereby slowing or stopping AD progression. Understanding CBF dysregulation during the development and progression of AD could help identify novel targets for disease modifying therapies. The interrelatedness of CBF dysfunctions indicates the complexity of fully understanding individual contributions to dementia. But, it also suggests that multiple factors (including age, sex, environment, and genetic predilection) may initiate the neurodegeneration and cognitive decline observed in AD. As such, individualized treatment therapies meant to repair the NVU while clearing accumulated AD proteinopathies may prove useful in a subset of patients.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health [NIA R01AG057767 and NIA R01AG061937], from the SIU Foundation at the School of Medicine [Harriss and Fannie Belle Roe Malan Research Endowment and the Illinois Health Improvement Association Research Endowment], the Center for Alzheimer’s Disease and Related Disorders, and the Kenneth Stark Endowment.
We are grateful to the SIU SOM Medical Library staff, including Sheri Daniels, Ann Gonterman, Lydia Howes, Jaedyn Maltimore, and Mackenzie Sanner for their help with the literature search and locating articles.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0473r1).
REFERENCES
- [1].Strassnig M, Ganguli M (2005) About a peculiar disease of the cerebral cortex: Alzheimer’s original case revisited. Psychiatry (Edgmont) 2, 30–33. [PMC free article] [PubMed] [Google Scholar]
- [2].Alzheimer’s Association (2019) 2019 Alzheimer’s disease facts and figures. Alzheimers Dement 15, 321–387. [Google Scholar]
- [3].Swerdlow RH (2007) Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol Aging 28, 1465–1480. [DOI] [PubMed] [Google Scholar]
- [4].Katzman R (1976) Editorial: The prevalence and malignancy of Alzheimer disease. A major killer. Arch Neurol 33, 217–218. [DOI] [PubMed] [Google Scholar]
- [5].Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 13, 45–53. [DOI] [PubMed] [Google Scholar]
- [6].Cole SL, Vassar R (2008) The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J Biol Chem 283, 29621–29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kurochkin IV, Goto S (1994) Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett 345, 33–37. [DOI] [PubMed] [Google Scholar]
- [8].Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC (2000) Identification of the major Abeta1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat Med 6, 143–150. [DOI] [PubMed] [Google Scholar]
- [9].Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sagare AP, Bell RD, Zlokovic BV (2013) Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer’s disease. J Alzheimers Dis 33 Suppl 1, S87–S100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sita G, Hrelia P, Tarozzi A, Morroni F (2017) P-glycoprotein (ABCB1) and oxidative stress: Focus on Alzheimer’s disease. Oxid Med Cell Longev 2017, 7905486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lewis DA, Campbell MJ, Terry RD, Morrison JH (1987) Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer’s disease: A quantitative study of visual and auditory cortices. J Neurosci 7, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rogers J, Morrison JH (1985) Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer’s disease. J Neurosci 5, 2801–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex 1, 103–116. [DOI] [PubMed] [Google Scholar]
- [15].Palmeri A, Ricciarelli R, Gulisano W, Rivera D, Rebosio C, Calcagno E, Tropea MR, Conti S, Das U, Roy S, Pronzato MA, Arancio O, Fedele E, Puzzo D (2017) Amyloid-beta peptide is needed for cGMP-induced long-term potentiation and memory. J Neurosci 37, 6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O (2008) Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 28, 14537–14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Findley CA, Bartke A, Hascup KN, Hascup ER (2019) Amyloid beta-related alterations to glutamate signaling dynamics during Alzheimer’s disease progression. ASN Neuro 11, 1759091419855541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ferreira ST, Vieira MN, De Felice FG (2007) Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life 59, 332–345. [DOI] [PubMed] [Google Scholar]
- [19].Hascup KN, Hascup ER (2015) Altered neurotransmission prior to cognitive decline in AbetaPP/PS1 mice, a model of Alzheimer’s disease. J Alzheimers Dis 44, 771–776. [DOI] [PubMed] [Google Scholar]
- [20].Hascup KN, Hascup ER (2016) Soluble amyloid-beta42 stimulates glutamate release through activation of the alpha7 nicotinic acetylcholine receptor. J Alzheimers Dis 53, 337–347. [DOI] [PubMed] [Google Scholar]
- [21].Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, Shivers B, Walker D, McGeer P (1996) Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging 17, 681–686. [DOI] [PubMed] [Google Scholar]
- [22].Garwood CJ, Ratcliffe LE, Simpson JE, Heath PR, Ince PG, Wharton SB (2017) Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol Appl Neurobiol 43, 281–298. [DOI] [PubMed] [Google Scholar]
- [23].Naseri NN, Wang H, Guo J, Sharma M, Luo W (2019) The complexity of tau in Alzheimer’s disease. Neurosci Lett 705, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chan D, Fox NC, Jenkins R, Scahill RI, Crum WR, Rossor MN (2001) Rates of global and regional cerebral atrophy in AD and frontotemporal dementia. Neurology 57, 1756–1763. [DOI] [PubMed] [Google Scholar]
- [25].Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC (2002) Mapping the evolution of regional atrophy in Alzheimer’s disease: Unbiased analysis of fluid-registered serial MRI. Proc Natl Acad Sci U S A 99, 4703–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Baron JC, Chetelat G, Desgranges B, Perchey G, Landeau B, de la Sayette V, Eustache F (2001) In vivo mapping of gray matter loss with voxel-based morphometry in mild Alzheimer’s disease. Neuroimage 14, 298–309. [DOI] [PubMed] [Google Scholar]
- [27].Ballmaier M, O’Brien JT, Burton EJ, Thompson PM, Rex DE, Narr KL, McKeith IG, DeLuca H, Toga AW (2004) Comparing gray matter loss profiles between dementia with Lewy bodies and Alzheimer’s disease using cortical pattern matching: Diagnosis and gender effects. Neuroimage 23, 325–335. [DOI] [PubMed] [Google Scholar]
- [28].Barage SH, Sonawane KD (2015) Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 52, 1–18. [DOI] [PubMed] [Google Scholar]
- [29].Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8, 101–112. [DOI] [PubMed] [Google Scholar]
- [30].Lanoiselee HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, Quillard-Muraine M, de la Sayette V, Boutoleau-Bretonniere C, Etcharry-Bouyx F, Chauvire V, Sarazin M, le Ber I, Epelbaum S, Jonveaux T, Rouaud O, Ceccaldi M, Felician O, Godefroy O, Formaglio M, Croisile B, Auriacombe S, Chamard L, Vincent JL, Sauvee M, Marelli-Tosi C, Gabelle A, Ozsancak C, Pariente J, Paquet C, Hannequin D, Campion D, collaborators of the CNRMAJp (2017) APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med 14, e1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bertram L, Lill CM, Tanzi RE (2010) The genetics of Alzheimer disease: Back to the future. Neuron 68, 270–281. [DOI] [PubMed] [Google Scholar]
- [32].Cummings J, Lee G, Ritter A, Zhong K (2018) Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 4, 195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Panza F, Lozupone M, Dibello V, Greco A, Daniele A, Seripa D, Logroscino G, Imbimbo BP (2019) Are antibodies directed against amyloid-beta (Abeta) oligomers the last call for the Abeta hypothesis of Alzheimer’s disease? Immunotherapy 11, 3–6. [DOI] [PubMed] [Google Scholar]
- [34].Behl C (2017) Amyloid in Alzheimer’s disease: Guilty beyond reasonable doubt? Trends Pharmacol Sci 38, 849–851. [DOI] [PubMed] [Google Scholar]
- [35].Snowdon DA (1997) Aging and Alzheimer’s disease: Lessons from the Nun Study. Gerontologist 37, 150–156. [DOI] [PubMed] [Google Scholar]
- [36].Kisler K, Nelson AR, Montagne A, Zlokovic BV (2017) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lecrux C, Hamel E (2011) The neurovascular unit in brain function and disease. Acta Physiol (Oxf) 203, 47–59. [DOI] [PubMed] [Google Scholar]
- [38].Muoio V, Persson PB, Sendeski MM (2014) The neurovascular unit – concept review. Acta Physiol (Oxf) 210, 790–798. [DOI] [PubMed] [Google Scholar]
- [39].Roy CS, Sherrington CS (1890) On the regulation of the blood-supply of the brain. J Physiol 11, 85–158 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ma J, Ayata C, Huang PL, Fishman MC, Moskowitz MA (1996) Regional cerebral blood flow response to vibrissal stimulation in mice lacking type I NOS gene expression. Am J Physiol 270, H1085–1090. [DOI] [PubMed] [Google Scholar]
- [41].Yang G, Zhang Y, Ross ME, Iadecola C (2003) Attenuation of activity-induced increases in cerebellar blood flow in mice lacking neuronal nitric oxide synthase. Am J Physiol Heart Circ Physiol 285, H298–304. [DOI] [PubMed] [Google Scholar]
- [42].Lourenco CF, Santos RM, Barbosa RM, Cadenas E, Radi R, Laranjinha J (2014) Neurovascular coupling in hip pocampus is mediated via diffusion by neuronal-derived nitric oxide. Free Radic Biol Med 73, 421–429. [DOI] [PubMed] [Google Scholar]
- [43].Bartus K, Pigott B, Garthwaite J (2013) Cellular targets of nitric oxide in the hippocampus. PLoS One 8, e57292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Filosa JA, Iddings JA (2013) Astrocyte regulation of cerebral vascular tone. Am J Physiol Heart Circ Physiol 305, H609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Howarth C (2014) The contribution of astrocytes to the regulation of cerebral blood flow. Front Neurosci 8, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Koehler RC, Roman RJ, Harder DR (2009) Astrocytes and the regulation of cerebral blood flow. Trends Neurosci 32, 160–169. [DOI] [PubMed] [Google Scholar]
- [48].Grant RI, Hartmann DA, Underly RG, Berthiaume AA, Bhat NR, Shih AY (2019) Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. J Cereb Blood Flow Metab 39, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sweeney MD, Ayyadurai S, Zlokovic BV (2016) Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat Neurosci 19, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T (2016) What is a pericyte? J Cereb Blood Flow Metab 36, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Janota C, Lemere CA, Brito MA (2016) Dissecting the contribution of vascular alterations and aging to Alzheimer’s disease. Mol Neurobiol 53, 3793–3811. [DOI] [PubMed] [Google Scholar]
- [53].Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, Monsell SE, Kukull WA, Trojanowski JQ (2013) Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 136, 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Skoog I (1997) The relationship between blood pressure and dementia: A review. Biomed Pharmacother 51, 367–375. [DOI] [PubMed] [Google Scholar]
- [55].Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, van Duijn CN, Van Broeckhoven C, Grobbee DE (1997) Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 349, 151–154. [DOI] [PubMed] [Google Scholar]
- [56].Hays CC, Zlatar ZZ, Wierenga CE (2016) The utility of cerebral blood flow as a biomarker of preclinical Alzheimer’s disease. Cell Mol Neurobiol 36, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].de la Torre J (2018) The vascular hypothesis of Alzheimer’s disease: A key to preclinical prediction of dementia using neuroimaging. J Alzheimers Dis 63, 35–52. [DOI] [PubMed] [Google Scholar]
- [58].Kalaria RN (2000) The role of cerebral ischemia in Alzheimer’s disease. Neurobiol Aging 21, 321–330. [DOI] [PubMed] [Google Scholar]
- [59].Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277, 813–817. [PubMed] [Google Scholar]
- [60].Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA (2016) Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol 15, 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28, 202–208. [DOI] [PubMed] [Google Scholar]
- [62].Zlokovic BV (2011) Neurovascular pathways to neurode-generation in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12, 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA (2010) Low-density lipoprotein receptor-related protein-1: A serial clearance homeostatic mechanism controlling Alzheimer’s amyloid beta-peptide elimination from the brain. J Neurochem 115, 1077–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wong CW, Quaranta V, Glenner GG (1985) Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc Natl Acad Sci U S A 82, 8729–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ (1993) Beta-amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A 90, 10836–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, part XV. Neurology 46, 1592–1596. [DOI] [PubMed] [Google Scholar]
- [67].Soontornniyomkij V, Choi C, Pomakian J, Vinters HV (2010) High-definition characterization of cerebral beta-amyloid angiopathy in Alzheimer’s disease. Hum Pathol 41, 1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH (2011) Toward defining the pre-clinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC, Alzheimer’s Disease Neuroimaging I (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7, 11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wierenga CE, Hays CC, Zlatar ZZ (2014) Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer’s disease. J Alzheimers Dis 42 Suppl 4, S411–S419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].de la Torre JC (2010) Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res Rev 9, 218–225. [DOI] [PubMed] [Google Scholar]
- [72].Higuchi Y, Miyakawa T, Shimoji A, Katsuragi S (1987) Ultrastructural changes of blood vessels in the cerebral cortex in Alzheimer’s disease. Jpn J Psychiatry Neurol 41, 283–290. [DOI] [PubMed] [Google Scholar]
- [73].Kidd M (1964) Alzheimer’s Disease–an electron microscopical study. Brain 87, 307–320. [DOI] [PubMed] [Google Scholar]
- [74].de la Torre JC (1994) Impaired brain microcirculation may trigger Alzheimer’s disease. Neurosci Biobehav Rev 18, 397–401. [DOI] [PubMed] [Google Scholar]
- [75].Binnewijzend MA, Kuijer JP, Benedictus MR, van der Flier WM, Wink AM, Wattjes MP, van Berckel BN, Scheltens P, Barkhof F (2013) Cerebral blood flow measured with 3D pseudocontinuous arterial spin-labeling MR imaging in Alzheimer disease and mild cognitive impairment: A marker for disease severity. Radiology 267, 221–230. [DOI] [PubMed] [Google Scholar]
- [76].Miners JS, Palmer JC, Love S (2016) Pathophysiology of hypoperfusion of the precuneus in early Alzheimer’s disease. Brain Pathol 26, 533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nicolakakis N, Hamel E (2011) Neurovascular function in Alzheimer’s disease patients and experimental models. J Cereb Blood Flow Metab 31, 1354–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sabayan B, Jansen S, Oleksik AM, van Osch MJ, van Buchem MA, van Vliet P, de Craen AJ, Westendorp RG (2012) Cerebrovascular hemodynamics in Alzheimer’s disease and vascular dementia: A meta-analysis of transcranial Doppler studies. Ageing Res Rev 11, 271–277. [DOI] [PubMed] [Google Scholar]
- [79].Yan S, Qi Z, An Y, Zhang M, Qian T, Lu J (2019) Detecting perfusion deficit in Alzheimer’s disease and mild cognitive impairment patients by resting-state fMRI. J Magn Reson Imaging 49, 1099–1104. [DOI] [PubMed] [Google Scholar]
- [80].Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM (2005) Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam Study. Ann Neurol 57, 789–794. [DOI] [PubMed] [Google Scholar]
- [81].Deng Y, Wang L, Sun X, Liu L, Zhu M, Wang C, Sui B, Shen M, Gu W, Mo D, Ma N, Song L, Li X, Huo X, Miao Z, Chen D, Gao F (2018) Association between cerebral hypoperfusion and cognitive impairment in patients with chronic vertebra-basilar stenosis. Front Psychiatry 9, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bangen KJ, Clark AL, Edmonds EC, Evangelista ND, Werhane ML, Thomas KR, Locano LE, Tran M, Zlatar ZZ, Nation DA, Bondi MW, Delano-Wood L (2017) Cerebral blood flow and amyloid-beta interact to affect memory performance in cognitively normal older adults. Front Aging Neurosci 9, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mattsson N, Tosun D, Insel PS, Simonson A, Jack CR Jr., Beckett LA, Donohue M, Jagust W, Schuff N, Weiner MW, Alzheimer’s Disease Neuroimaging Initiative (2014) Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain 137, 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wierenga CE, Clark LR, Dev SI, Shin DD, Jurick SM, Rissman RA, Liu TT, Bondi MW (2013) Interaction of age and APOE genotype on cerebral blood flow at rest. J Alzheimers Dis 34, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kim SM, Kim MJ, Rhee HY, Ryu CW, Kim EJ, Petersen ET, Jahng GH (2013) Regional cerebral perfusion in patients with Alzheimer’s disease and mild cognitive impairment: Effect of APOE epsilon4 allele. Neuroradiology 55, 25–34. [DOI] [PubMed] [Google Scholar]
- [86].Bondi MW, Houston WS, Eyler LT, Brown GG (2005) fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology 64, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wierenga CE, Dev SI, Shin DD, Clark LR, Bangen KJ, Jak AJ, Rissman RA, Liu TT, Salmon DP, Bondi MW (2012) Effect of mild cognitive impairment and APOE genotype on resting cerebral blood flow and its association with cognition. J Cereb Blood Flow Metab 32, 1589–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Luckhaus C, Fluss MO, Wittsack HJ, Grass-Kapanke B, Janner M, Khalili-Amiri R, Friedrich W, Supprian T, Gaebel W, Modder U, Cohnen M (2008) Detection of changed regional cerebral blood flow in mild cognitive impairment and early Alzheimer’s dementia by perfusion-weighted magnetic resonance imaging. Neuroimage 40, 495–503. [DOI] [PubMed] [Google Scholar]
- [89].Dai W, Lopez OL, Carmichael OT, Becker JT, Kuller LH, Gach HM (2009) Mild cognitive impairment and Alzheimer disease: Patterns of altered cerebral blood flow at MR imaging. Radiology 250, 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].de la Torre JC, Fortin T, Park GA, Butler KS, Kozlowski P, Pappas BA, de Socarraz H, Saunders JK, Richard MT (1992) Chronic cerebrovascular insufficiency induces dementia-like deficits in aged rats. Brain Res 582, 186–195. [DOI] [PubMed] [Google Scholar]
- [91].Tsuchiya M, Sako K, Yura S, Yonemasu Y (1992) Cerebral blood flow and histopathological changes following permanent bilateral carotid artery ligation in Wistar rats. Exp Brain Res 89, 87–92. [DOI] [PubMed] [Google Scholar]
- [92].Pappas BA, de la Torre JC, Davidson CM, Keyes MT, Fortin T (1996) Chronic reduction of cerebral blood flow in the adult rat: Late-emerging CA1 cell loss and memory dysfunction. Brain Res 708, 50–58. [DOI] [PubMed] [Google Scholar]
- [93].Bennett SA, Tenniswood M, Chen JH, Davidson CM, Keyes MT, Fortin T, Pappas BA (1998) Chronic cerebral hypoperfusion elicits neuronal apoptosis and behavioral impairment. Neuroreport 9, 161–166. [DOI] [PubMed] [Google Scholar]
- [94].Wang Z, Fan J, Wang J, Li Y, Duan D, Du G, Wang Q (2016) Chronic cerebral hypoperfusion induces long-lasting cognitive deficits accompanied by long-term hippocampal silent synapses increase in rats. Behav Brain Res 301, 243–252. [DOI] [PubMed] [Google Scholar]
- [95].Cechetti F, Pagnussat AS, Worm PV, Elsner VR, Ben J, da Costa MS, Mestriner R, Weis SN, Netto CA (2012) Chronic brain hypoperfusion causes early glial activation and neuronal death, and subsequent long-term memory impairment. Brain Res Bull 87, 109–116. [DOI] [PubMed] [Google Scholar]
- [96].Kato H, Kanai Y, Watabe T, Ikeda H, Horitsugi G, Hatazawa J (2019) Quantitative measurement of regional cerebral blood flow and oxygen metabolism in a rat model of cerebral hypoperfusion. Brain Res 1719, 208–216. [DOI] [PubMed] [Google Scholar]
- [97].Vicente E, Degerone D, Bohn L, Scornavaca F, Pimentel A, Leite MC, Swarowsky A, Rodrigues L, Nardin P, de Almeida LM, Gottfried C, Souza DO, Netto CA, Goncalves CA (2009) Astroglial and cognitive effects of chronic cerebral hypoperfusion in the rat. Brain Res 1251, 204–212. [DOI] [PubMed] [Google Scholar]
- [98].Farkas E, Luiten PG, Bari F (2007) Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res Rev 54, 162–180. [DOI] [PubMed] [Google Scholar]
- [99].Wang X, Xing A, Xu C, Cai Q, Liu H, Li L (2010) Cerebrovascular hypoperfusion induces spatial memory impairment, synaptic changes, and amyloid-beta oligomerization in rats. J Alzheimers Dis 21, 813–822. [DOI] [PubMed] [Google Scholar]
- [100].Liu H, Xing A, Wang X, Liu G, Li L (2012) Regulation of beta-amyloid level in the brain of rats with cerebrovascular hypoperfusion. Neurobiol Aging 33, 826 e831–842. [DOI] [PubMed] [Google Scholar]
- [101].Cai Z, Liu Z, Xiao M, Wang C, Tian F (2017) Chronic cerebral hypoperfusion promotes amyloid-beta pathogenesis via activating beta/gamma-secretases. Neurochem Res 42, 3446–3455. [DOI] [PubMed] [Google Scholar]
- [102].Zhiyou C, Yong Y, Shanquan S, Jun Z, Liangguo H, Ling Y, Jieying L (2009) Upregulation of BACE1 and beta-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer’s disease. Neurochem Res 34, 1226–1235. [DOI] [PubMed] [Google Scholar]
- [103].Park JH, Hong JH, Lee SW, Ji HD, Jung JA, Yoon KW, Lee JI, Won KS, Song BI, Kim HW (2019) The effect of chronic cerebral hypoperfusion on the pathology of Alzheimer’s disease: A positron emission tomography study in rats. Sci Rep 9, 14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Choi BR, Lee SR, Han JS, Woo SK, Kim KM, Choi DH, Kwon KJ, Han SH, Shin CY, Lee J, Chung CS, Lee SR, Kim HY (2011) Synergistic memory impairment through the interaction of chronic cerebral hypoperfusion and amyloid toxicity in a rat model. Stroke 42, 2595–2604. [DOI] [PubMed] [Google Scholar]
- [105].Sun MK, Alkon DL (2004) Cerebral hypoperfusion and amyloid-induced synergistic impairment of hippocampal CA1 synaptic efficacy and spatial memory in young adult rats. J Alzheimers Dis 6, 355–366; discussion 443–449. [DOI] [PubMed] [Google Scholar]
- [106].Terashima T, Namura S, Hoshimaru M, Uemura Y, Kikuchi H, Hashimoto N (1998) Consistent injury in the striatum of C57BL/6 mice after transient bilateral common carotid artery occlusion. Neurosurgery 43, 900–907; discussion 907–908. [DOI] [PubMed] [Google Scholar]
- [107].Yoshizaki K, Adachi K, Kataoka S, Watanabe A, Tabira T, Takahashi K, Wakita H (2008) Chronic cerebral hypoper-fusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp Neurol 210, 585–591. [DOI] [PubMed] [Google Scholar]
- [108].Yang H, Hou T, Wang W, Luo Y, Yan F, Jia J (2018) The effect of chronic cerebral hypoperfusion on amyloid-beta metabolism in a transgenic mouse model of Alzheimer’s disease (PS1V97L). J Alzheimers Dis 62, 1609–1621. [DOI] [PubMed] [Google Scholar]
- [109].Lee JS, Im DS, An YS, Hong JM, Gwag BJ, Joo IS (2011) Chronic cerebral hypoperfusion in a mouse model of Alzheimer’s disease: An additional contributing factor of cognitive impairment. Neurosci Lett 489, 84–88. [DOI] [PubMed] [Google Scholar]
- [110].Shibata M, Ohtani R, Ihara M, Tomimoto H (2004) White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 35, 2598–2603. [DOI] [PubMed] [Google Scholar]
- [111].Temma T, Yamazaki M, Miyanohara J, Shirakawa H, Kondo N, Koshino K, Kaneko S, Iida H (2017) Sequential PET estimation of cerebral oxygen metabolism with spontaneous respiration of (15)O-gas in mice with bilateral common carotid artery stenosis. J Cereb Blood Flow Metab 37, 3334–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hattori Y, Enmi J, Iguchi S, Saito S, Yamamoto Y, Nagatsuka K, Iida H, Ihara M (2016) Substantial reduction of parenchymal cerebral blood flow in mice with bilateral common carotid artery stenosis. Sci Rep 6, 32179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R, Ihara M, Takahashi R, Tomimoto H (2007) Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 38, 2826–2832. [DOI] [PubMed] [Google Scholar]
- [114].Nishio K, Ihara M, Yamasaki N, Kalaria RN, Maki T, Fujita Y, Ito H, Oishi N, Fukuyama H, Miyakawa T, Takahashi R, Tomimoto H (2010) A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke 41, 1278–1284. [DOI] [PubMed] [Google Scholar]
- [115].Khan MB, Hoda MN, Vaibhav K, Giri S, Wang P, Waller JL, Ergul A, Dhandapani KM, Fagan SC, Hess DC (2015) Remote ischemic postconditioning: Harnessing endogenous protection in a murine model of vascular cognitive impairment. Transl Stroke Res 6, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Yamada M, Ihara M, Okamoto Y, Maki T, Washida K, Kitamura A, Hase Y, Ito H, Takao K, Miyakawa T, Kalaria RN, Tomimoto H, Takahashi R (2011) The influence of chronic cerebral hypoperfusion on cognitive function and amyloid beta metabolism in APP overexpressing mice. PLoS One 6, e16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kitaguchi H, Tomimoto H, Ihara M, Shibata M, Uemura K, Kalaria RN, Kihara T, Asada-Utsugi M, Kinoshita A, Takahashi R (2009) Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res 1294, 202–210. [DOI] [PubMed] [Google Scholar]
- [118].Salvadores N, Searcy JL, Holland PR, Horsburgh K (2017) Chronic cerebral hypoperfusion alters amyloid-beta peptide pools leading to cerebral amyloid angiopathy, microinfarcts and haemorrhages in Tg-SwDI mice. Clin Sci (Lond) 131, 2109–2123. [DOI] [PubMed] [Google Scholar]
- [119].Bannai T, Mano T, Chen X, Ohtomo G, Ohtomo R, Tsuchida T, Koshi-Mano K, Hashimoto T, Okazawa H, Iwatsubo T, Tsuji S, Toda T, Iwata A (2019) Chronic cerebral hypoperfusion shifts the equilibrium of amyloid beta oligomers to aggregation-prone species with higher molecular weight. Sci Rep 9, 2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Bink DI, Ritz K, Aronica E, van der Weerd L, Daemen MJ (2013) Mouse models to study the effect of cardiovascular risk factors on brain structure and cognition. J Cereb Blood Flow Metab 33, 1666–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Kitamura A, Fujita Y, Oishi N, Kalaria RN, Washida K, Maki T, Okamoto Y, Hase Y, Yamada M, Takahashi J, Ito H, Tomimoto H, Fukuyama H, Takahashi R, Ihara M (2012) Selective white matter abnormalities in a novel rat model of vascular dementia. Neurobiol Aging 33, 1012 e1025–e1035. [DOI] [PubMed] [Google Scholar]
- [122].Hattori Y, Enmi J, Iguchi S, Saito S, Yamamoto Y, Tsuji M, Nagatsuka K, Kalaria RN, Iida H, Ihara M (2016) Gradual carotid artery stenosis in mice closely replicates hypoperfusive vascular dementia in humans. J Am Heart Assoc 5, e002757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Hattori Y, Kitamura A, Nagatsuka K, Ihara M (2014) A novel mouse model of ischemic carotid artery disease. PLoS One 9, e100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Shang J, Yamashita T, Zhai Y, Nakano Y, Morihara R, Li X, Tian F, Liu X, Huang Y, Shi X, Sato K, Takemoto M, Hishikawa N, Ohta Y, Abe K (2019) Acceleration of NLRP3 inflammasome by chronic cerebral hypoperfusion in Alzheimer’s disease model mouse. Neurosci Res 143, 61–70. [DOI] [PubMed] [Google Scholar]
- [125].Shang J, Yamashita T, Tian F, Li X, Liu X, Shi X, Nakano Y, Tsunoda K, Nomura E, Sasaki R, Tadokoro K, Sato K, Takemoto M, Hishikawa N, Ohta Y, Abe K (2019) Chronic cerebral hypoperfusion alters amyloid-beta transport related proteins in the cortical blood vessels of Alzheimer’s disease model mouse. Brain Res 1723, 146379. [DOI] [PubMed] [Google Scholar]
- [126].Shang J, Yamashita T, Zhai Y, Nakano Y, Morihara R, Fukui Y, Hishikawa N, Ohta Y, Abe K (2016) Strong impact of chronic cerebral hypoperfusion on neurovascular unit, cerebrovascular remodeling, and neurovascular trophic coupling in Alzheimer’s disease model mouse. J Alzheimers Dis 52, 113–126. [DOI] [PubMed] [Google Scholar]
- [127].Zhai Y, Yamashita T, Nakano Y, Sun Z, Shang J, Feng T, Morihara R, Fukui Y, Ohta Y, Hishikawa N, Abe K (2016) Chronic cerebral hypoperfusion accelerates Alzheimer’s disease pathology with cerebrovascular remodeling in a novel mouse model. J Alzheimers Dis 53, 893–905. [DOI] [PubMed] [Google Scholar]
- [128].Quintana DD, Ren X, Hu H, Engler-Chiurazzi EB, Rellick SL, Lewis SE, Povroznik JM, Simpkins JW, Alvi M (2018) Gradual common carotid artery occlusion as a novel model for cerebrovascular Hypoperfusion. Metab Brain Dis 33, 2039–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hattori Y, Enmi J, Kitamura A, Yamamoto Y, Saito S, Takahashi Y, Iguchi S, Tsuji M, Yamahara K, Nagatsuka K, Iida H, Ihara M (2015) A novel mouse model of subcortical infarcts with dementia. J Neurosci 35, 3915–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Yang S, Liu R (2017) Cerebral autoregulation In Primer on Cerebrovascular Diseases: Second Edition, Caplan LR, Biller J, Leary MC, Lo EH, Thomas AJ, Yenari M, Zhang JH, eds. Elsevier Inc. [Google Scholar]
- [131].van Beek AH, Claassen JA, Rikkert MG, Jansen RW (2008) Cerebral autoregulation: An overview of current concepts and methodology with special focus on the elderly. J Cereb Blood Flow Metab 28, 1071–1085. [DOI] [PubMed] [Google Scholar]
- [132].den Abeelen AS, Lagro J, van Beek AH, Claassen JA (2014) Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr Alzheimer Res 11, 11–17. [DOI] [PubMed] [Google Scholar]
- [133].Zazulia AR, Videen TO, Morris JC, Powers WJ (2010) Autoregulation of cerebral blood flow to changes in arterial pressure in mild Alzheimer’s disease. J Cereb Blood Flow Metab 30, 1883–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Claassen JA, Diaz-Arrastia R, Martin-Cook K, Levine BD, Zhang R (2009) Altered cerebral hemodynamics in early Alzheimer disease: A pilot study using transcranial Doppler. J Alzheimers Dis 17, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Claassen JA, Zhang R (2011) Cerebral autoregulation in Alzheimer’s disease. J Cereb Blood Flow Metab 31, 1572–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Kennelly S, Collins O (2012) Walking the cognitive “minefield” between high and low blood pressure. J Alzheimers Dis 32, 609–621. [DOI] [PubMed] [Google Scholar]
- [137].Novak V, Hajjar I (2010) The relationship between blood pressure and cognitive function. Nat Rev Cardiol 7, 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Kennelly SP, Lawlor BA, Kenny RA (2009) Blood pressure and the risk for dementia: A double edged sword. Ageing Res Rev 8, 61–70. [DOI] [PubMed] [Google Scholar]
- [139].Skoog I, Gustafson D (2006) Update on hypertension and Alzheimer’s disease. Neurol Res 28, 605–611. [DOI] [PubMed] [Google Scholar]
- [140].Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A (2001) Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 322, 1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Liu Y, Dong YH, Lyu PY, Chen WH, Li R (2018) Hypertension-induced cerebral small vessel disease leading to cognitive impairment. Chin Med J (Engl) 131, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].de la Torre JC (2017) Are major dementias triggered by poor blood flow to the brain? Theoretical considerations. J Alzheimers Dis 57, 353–371. [DOI] [PubMed] [Google Scholar]
- [143].Moonga I, Niccolini F, Wilson H, Pagano G, Politis M, Alzheimer’s Disease Neuroimaging Initiative (2017) Hypertension is associated with worse cognitive function and hippocampal hypometabolism in Alzheimer’s disease. Eur J Neurol 24, 1173–1182. [DOI] [PubMed] [Google Scholar]
- [144].Bellew KM, Pigeon JG, Stang PE, Fleischman W, Gardner RM, Baker WW (2004) Hypertension and the rate of cognitive decline in patients with dementia of the Alzheimer type. Alzheimer Dis Assoc Disord 18, 208–213. [PubMed] [Google Scholar]
- [145].Hanon O, Latour F, Seux ML, Lenoir H, Forette F, Rigaud AS, Group RF (2005) Evolution of blood pressure in patients with Alzheimer’s disease: A one year survey of a French Cohort (REAL.FR). J Nutr Health Aging 9, 106–111. [PubMed] [Google Scholar]
- [146].Qiu C, von Strauss E, Winblad B, Fratiglioni L (2004) Decline in blood pressure over time and risk of dementia: A longitudinal study from the Kungsholmen project. Stroke 35, 1810–1815. [DOI] [PubMed] [Google Scholar]
- [147].McGuinness B, Todd S, Passmore P, Bullock R (2009) Blood pressure lowering in patients without prior cerebrovascular disease for prevention of cognitive impairment and dementia. Cochrane Database Syst Rev, CD004034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Schneider P, Buerger K, Teipel S, Uspenskaya O, Hart-mann O, Hansson O, Minthon L, Rujescu D, Moeller HJ, Zetterberg H, Blennow K, Ernst A, Bergmann A, Hampel H (2011) Antihypertensive therapy is associated with reduced rate of conversion to Alzheimer’s disease in midregional proatrial natriuretic peptide stratified subjects with mild cognitive impairment. Biol Psychiatry 70, 145–151. [DOI] [PubMed] [Google Scholar]
- [149].Suo Z, Su G, Placzek A, Kundtz A, Humphrey J, Crawford F, Mullan M (2000) A beta vasoactivity in vivo. Ann N Y Acad Sci 903, 156–163. [DOI] [PubMed] [Google Scholar]
- [150].Arendash GW, Su GC, Crawford FC, Bjugstad KB, Mullan M (1999) Intravascular beta-amyloid infusion increases blood pressure: Implications for a vasoactive role of beta-amyloid in the pathogenesis of Alzheimer’s disease. Neurosci Lett 268, 17–20. [DOI] [PubMed] [Google Scholar]
- [151].Suo Z, Humphrey J, Kundtz A, Sethi F, Placzek A, Crawford F, Mullan M (1998) Soluble Alzheimers beta-amyloid constricts the cerebral vasculature in vivo. Neurosci Lett 257, 77–80. [DOI] [PubMed] [Google Scholar]
- [152].Gentile MT, Poulet R, Di Pardo A, Cifelli G, Maffei A, Vecchione C, Passarelli F, Landolfi A, Carullo P, Lembo G (2009) Beta-amyloid deposition in brain is enhanced in mouse models of arterial hypertension. Neurobiol Aging 30, 222–228. [DOI] [PubMed] [Google Scholar]
- [153].Carnevale D, Mascio G, D’Andrea I, Fardella V, Bell RD, Branchi I, Pallante F, Zlokovic B, Yan SS, Lembo G (2012) Hypertension induces brain beta-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 60, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Cifuentes D, Poittevin M, Dere E, Broqueres-You D, Bonnin P, Benessiano J, Pocard M, Mariani J, Kubis N, Merkulova-Rainon T, Levy BI (2015) Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension 65, 218–224. [DOI] [PubMed] [Google Scholar]
- [155].Kruyer A, Soplop N, Strickland S, Norris EH (2015) Chronic hypertension leads to neurodegeneration in the TgSwDI mouse model of Alzheimer’s disease. Hypertension 66, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Wiesmann M, Capone C, Zerbi V, Mellendijk L, Heerschap A, Claassen JA, Kiliaan AJ (2015) Hypertension impairs cerebral blood flow in a mouse model for Alzheimer’s disease. Curr Alzheimer Res 12, 914–922. [DOI] [PubMed] [Google Scholar]
- [157].Wiesmann M, Roelofs M, van der Lugt R, Heerschap A, Kiliaan AJ, Claassen JA (2017) Angiotensin II, hyper-tension and angiotensin II receptor antagonism: Roles in the behavioural and brain pathology of a mouse model of Alzheimer’s disease. J Cereb Blood Flow Metab 37, 2396–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Wiesmann M, Zerbi V, Jansen D, Lutjohann D, Veltien A, Heerschap A, Kiliaan AJ (2017) Hypertension, cerebrovascular impairment, and cognitive decline in aged AbetaPP/PS1 mice. Theranostics 7, 1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Cabral C, Morgado PM, Campos Costa D, Silveira M, Alzheimers Disease Neuroimaging I (2015) Predicting conversion from MCI to AD with FDG-PET brain images at different prodromal stages. Comput Biol Med 58, 101–109. [DOI] [PubMed] [Google Scholar]
- [160].Reivich M, Kuhl D, Wolf A, Greenberg J, Phelps M, Ido T, Casella V, Fowler J, Hoffman E, Alavi A, Som P, Sokoloff L (1979) The [18F]fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ Res 44, 127–137. [DOI] [PubMed] [Google Scholar]
- [161].Haller S, Zaharchuk G, Thomas DL, Lovblad KO, Barkhof F, Golay X (2016) Arterial spin labeling perfusion of the brain: Emerging clinical applications. Radiology 281, 337–356. [DOI] [PubMed] [Google Scholar]
- [162].Sorg C, Riedl V, Muhlau M, Calhoun VD, Eichele T, Laer L, Drzezga A, Forstl H, Kurz A, Zimmer C, Wohlschlager AM (2007) Selective changes of resting-state networks in individuals at risk for Alzheimer’s disease. Proc Natl Acad Sci U S A 104, 18760–18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Greicius MD, Srivastava G, Reiss AL, Menon V (2004) Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: Evidence from functional MRI. Proc Natl Acad Sci U S A 101, 4637–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Zhong Y, Huang L, Cai S, Zhang Y, von Deneen KM, Ren A, Ren J, Alzheimer’s Disease Neuroimaging I (2014) Altered effective connectivity patterns of the default mode network in Alzheimer’s disease: An fMRI study. Neurosci Lett 578, 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Wu X, Li R, Fleisher AS, Reiman EM, Guan X, Zhang Y, Chen K, Yao L (2011) Altered default mode network connectivity in Alzheimer’s disease–a resting functional MRI and Bayesian network study. Hum Brain Mapp 32, 1868–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Zheng W, Cui B, Han Y, Song H, Li K, He Y, Wang Z (2019) Disrupted regional cerebral blood flow, functional activity and connectivity in Alzheimer’s disease: A combined ASL perfusion and resting state fMRI study. Front Neurosci 13, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, Mintun MA (2010) Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry 67, 584–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Babiloni C, Del Percio C, Caroli A, Salvatore E, Nicolai E, Marzano N, Lizio R, Cavedo E, Landau S, Chen K, Jagust W, Reiman E, Tedeschi G, Montella P, De Stefano M, Gesualdo L, Frisoni GB, Soricelli A (2016) Cortical sources of resting state EEG rhythms are related to brain hypometabolism in subjects with Alzheimer’s disease: An EEG-PET study. Neurobiol Aging 48, 122–134. [DOI] [PubMed] [Google Scholar]
- [169].Hsiao FJ, Wang YJ, Yan SH, Chen WT, Lin YY (2013) Altered oscillation and synchronization of default-mode network activity in mild Alzheimer’s disease compared to mild cognitive impairment: An electrophysiological study. PLoS One 8, e68792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Brueggen K, Fiala C, Berger C, Ochmann S, Babiloni C, Teipel SJ (2017) Early changes in alpha band power and DMN BOLD activity in Alzheimer’s disease: A simultaneous resting state EEG-fMRI study. Front Aging Neurosci 9, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Machulda MM, Ward HA, Borowski B, Gunter JL, Cha RH, O’Brien PC, Petersen RC, Boeve BF, Knopman D, Tang-Wai DF, Ivnik RJ, Smith GE, Tangalos EG, Jack CR Jr. (2003) Comparison of memory fMRI response among normal, MCI, and Alzheimer’s patients. Neurology 61, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Rombouts SA, Barkhof F, Veltman DJ, Machielsen WC, Witter MP, Bierlaagh MA, Lazeron RH, Valk J, Scheltens P (2000) Functional MR imaging in Alzheimer’s disease during memory encoding. AJNR Am J Neuroradiol 21, 1869–1875. [PMC free article] [PubMed] [Google Scholar]
- [173].Remy F, Mirrashed F, Campbell B, Richter W (2005) Verbal episodic memory impairment in Alzheimer’s disease: A combined structural and functional MRI study. Neuroimage 25, 253–266. [DOI] [PubMed] [Google Scholar]
- [174].Peters F, Collette F, Degueldre C, Sterpenich V, Majerus S, Salmon E (2009) The neural correlates of verbal short-term memory in Alzheimer’s disease: An fMRI study. Brain 132, 1833–1846. [DOI] [PubMed] [Google Scholar]
- [175].Mentis MJ, Alexander GE, Krasuski J, Pietrini P, Furey ML, Schapiro MB, Rapoport SI (1998) Increasing required neural response to expose abnormal brain function in mild versus moderate or severe Alzheimer’s disease: PET study using parametric visual stimulation. Am J Psychiatry 155, 785–794. [DOI] [PubMed] [Google Scholar]
- [176].Janik R, Thomason LA, Chaudhary S, Dorr A, Scouten A, Schwindt G, Masellis M, Stanisz GJ, Black SE, Stefanovic B (2016) Attenuation of functional hyperemia to visual stimulation in mild Alzheimer’s disease and its sensitivity to cholinesterase inhibition. Biochim Biophys Acta 1862, 957–965. [DOI] [PubMed] [Google Scholar]
- [177].Ravi Teja KV, Tos Berendschot T, Steinbusch H, Carroll Webers AB, Praveen Murthy R, Mathuranath PS (2017) Cerebral and retinal neurovascular changes: A biomarker for Alzheimer’s disease. J Gerontol Geriatr Res 6, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Kotliar K, Hauser C, Ortner M, Muggenthaler C, Diehl-Schmid J, Angermann S, Hapfelmeier A, Schmaderer C, Grimmer T (2017) Altered neurovascular coupling as measured by optical imaging: A biomarker for Alzheimer’s disease. Sci Rep 7, 12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Joo IL, Lai AY, Bazzigaluppi P, Koletar MM, Dorr A, Brown ME, Thomason LA, Sled JG, McLaurin J, Stefanovic B (2017) Early neurovascular dysfunction in a transgenic rat model of Alzheimer’s disease. Sci Rep 7, 46427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Bazzigaluppi P, Beckett TL, Koletar MM, Lai AY, Joo IL, Brown ME, Carlen PL, McLaurin J, Stefanovic B (2018) Early-stage attenuation of phase-amplitude coupling in the hippocampus and medial prefrontal cortex in a transgenic rat model of Alzheimer’s disease. J Neurochem 144, 669–679. [DOI] [PubMed] [Google Scholar]
- [181].Takano T, Han X, Deane R, Zlokovic B, Nedergaard M (2007) Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann N Y Acad Sci 1097, 40–50. [DOI] [PubMed] [Google Scholar]
- [182].Lourenco CF, Ledo A, Barbosa RM, Laranjinha J (2017) Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: Impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp Neurol 291, 36–43. [DOI] [PubMed] [Google Scholar]
- [183].Kimbrough IF, Robel S, Roberson ED, Sontheimer H (2015) Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 138, 3716–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Cruz Hernandez JC, Bracko O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, Park L, Vinarcsik LK, Ivasyk I, Rivera DA, Kang Y, Cortes-Canteli M, Peyrounette M, Doyeux V, Smith A, Zhou J, Otte G, Beverly JD, Davenport E, Davit Y, Lin CP, Strickland S, Iadecola C, Lorthois S, Nishimura N, Schaffer CB (2019) Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat Neurosci 22, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H, Attwell D (2019) Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365, eaav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Fernandez-Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U (2010) Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci U S A 107, 22290–22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, Lazic D, Tsai PS, Zhao Z, Zhou Y, Boas DA, Sakadzic S, Zlokovic BV (2017) Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci 20, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Gosselet F, Saint-Pol J, Candela P, Fenart L (2013) Amyloid-beta peptides, Alzheimer’s disease and the blood-brain barrier. Curr Alzheimer Res 10, 1015–1033. [DOI] [PubMed] [Google Scholar]
- [190].Ueno M, Chiba Y, Matsumoto K, Nakagawa T, Miyanaka H (2014) Clearance of beta-amyloid in the brain. Curr Med Chem 21, 4085–4090. [DOI] [PubMed] [Google Scholar]
- [191].Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Yamazaki Y, Kanekiyo T (2017) Blood-brain barrier dys-function and the pathogenesis of Alzheimer’s disease. Int J Mol Sci 18, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Deane R, Zlokovic BV (2007) Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 4, 191–197. [DOI] [PubMed] [Google Scholar]
- [194].Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM (2017) Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 140, 1829–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del Tredici K, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61, 282–293. [DOI] [PubMed] [Google Scholar]
- [196].Allen N, Robinson AC, Snowden J, Davidson YS, Mann DM (2014) Patterns of cerebral amyloid angiopathy define histopathological phenotypes in Alzheimer’s disease. Neuropathol Appl Neurobiol 40, 136–148. [DOI] [PubMed] [Google Scholar]
- [197].Attems J, Jellinger K, Thal DR, Van Nostrand W (2011) Review: Sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37, 75–93. [DOI] [PubMed] [Google Scholar]
- [198].Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer disease: Correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62, 1287–1301. [DOI] [PubMed] [Google Scholar]
- [199].Brenowitz WD, Nelson PT, Besser LM, Heller KB, Kukull WA (2015) Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol Aging 36, 2702–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Bourassa P, Tremblay C, Schneider JA, Bennett DA, Calon F (2019) Beta-amyloid pathology in human brain microvessel extracts from the parietal cortex: Relation with cerebral amyloid angiopathy and Alzheimer’s disease. Acta Neuropathol 137, 801–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Weller RO, Preston SD, Subash M, Carare RO (2009) Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res Ther 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Starr JM, Farrall AJ, Armitage P, McGurn B, Wardlaw J (2009) Blood-brain barrier permeability in Alzheimer’s disease: A case-control MRI study. Psychiatry Res 171, 232–241. [DOI] [PubMed] [Google Scholar]
- [203].Thrippleton MJ, Backes WH, Sourbron S, Ingrisch M, van Osch MJP, Dichgans M, Fazekas F, Ropele S, Frayne R, van Oostenbrugge RJ, Smith EE, Wardlaw JM (2019) Quantifying blood-brain barrier leakage in small vessel disease: Review and consensus recommendations. Alzheimers Dement 15, 840–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, Hofman PA, Verhey FR, Backes WH (2017) Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 282, 615. [DOI] [PubMed] [Google Scholar]
- [205].van de Haar HJ, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Wong SM, Hofman PAM, Burgmans S, Verhey FRJ, Backes WH (2016) Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging 45, 190–196. [DOI] [PubMed] [Google Scholar]
- [206].Dorr A, Sahota B, Chinta LV, Brown ME, Lai AY, Ma K, Hawkes CA, McLaurin J, Stefanovic B (2012) Amyloid-beta-dependent compromise of microvascular structure and function in a model of Alzheimer’s disease. Brain 135, 3039–3050. [DOI] [PubMed] [Google Scholar]
- [207].Saito S, Yamamoto Y, Maki T, Hattori Y, Ito H, Mizuno K, Harada-Shiba M, Kalaria RN, Fukushima M, Takahashi R, Ihara M (2017) Taxifolin inhibits amyloid-beta oligomer formation and fully restores vascular integrity and memory in cerebral amyloid angiopathy. Acta Neuropathol Commun 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Zhang S, Zhi Y, Li F, Huang S, Gao H, Han Z, Ge X, Li D, Chen F, Kong X, Lei P (2018) Transplantation of in vitro cultured endothelial progenitor cells repairs the blood-brain barrier and improves cognitive function of APP/PS1 transgenic AD mice. J Neurol Sci 387, 6–15. [DOI] [PubMed] [Google Scholar]
- [209].Bales KR, O’Neill SM, Pozdnyakov N, Pan F, Caouette D, Pi Y, Wood KM, Volfson D, Cirrito JR, Han BH, Johnson AW, Zipfel GJ, Samad TA (2016) Passive immunotherapy targeting amyloid-beta reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain 139, 563–577. [DOI] [PubMed] [Google Scholar]
- [210].Denver P, D’Adamo H, Hu S, Zuo X, Zhu C, Okuma C, Kim P, Castro D, Jones MR, Leal C, Mekkittikul M, Ghadishah E, Teter B, Vinters HV, Cole GM, Frautschy SA (2019) A novel model of mixed vascular dementia incorporating hypertension in a rat model of Alzheimer’s disease. Front Physiol 10, 1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Thomas T, Thomas G, McLendon C, Sutton T, Mullan M (1996) Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380, 168–171. [DOI] [PubMed] [Google Scholar]
- [212].Smith EE, Schneider JA, Wardlaw JM, Greenberg SM (2012) Cerebral microinfarcts: The invisible lesions. Lancet Neurol 11, 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Reijmer YD, van Veluw SJ, Greenberg SM (2016) Ischemic brain injury in cerebral amyloid angiopathy. J Cereb Blood Flow Metab 36, 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Hardy JA, Higgins GA (1992) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- [215].Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12, 383–388. [DOI] [PubMed] [Google Scholar]
- [216].Makin S (2018) The amyloid hypothesis on trial. Nature 559, S4–S7. [DOI] [PubMed] [Google Scholar]
- [217].Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Kozlov S, Afonin A, Evsyukov I, Bondarenko A (2017) Alzheimer’s disease: As it was in the beginning. Rev Neurosci 28, 825–843. [DOI] [PubMed] [Google Scholar]
- [219].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- [220].Shin J, Yu SB, Yu UY, Jo SA, Ahn JH (2010) Swedish mutation within amyloid precursor protein modulates global gene expression towards the pathogenesis of Alzheimer’s disease. BMB Rep 43, 704–709. [DOI] [PubMed] [Google Scholar]
- [221].Wang W, Lu L, Wu QQ, Jia JP (2016) Brain amyloid-beta plays an initiating role in the pathophysiological process of the PS1V97L-Tg mouse model of Alzheimer’s disease. J Alzheimers Dis 52, 1089–1099. [DOI] [PubMed] [Google Scholar]
- [222].Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A 94, 13287–13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Van Dam D, D’Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP (2003) Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci 17, 388–396. [DOI] [PubMed] [Google Scholar]
- [224].Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE (2004) Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem 279, 20296–20306. [DOI] [PubMed] [Google Scholar]
- [225].Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Espana J, Valero J, Minano-Molina AJ, Masgrau R, Martin E, Guardia-Laguarta C, Lleo A, Gimenez-Llort L, Rodriguez-Alvarez J, Saura CA (2010) Beta-amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J Neurosci 30, 9402–9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Holscher C, Mathews PM, Jucker M (2006) Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7, 940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Minkeviciene R, Ihalainen J, Malm T, Matilainen O, Keksa-Goldsteine V, Goldsteins G, Iivonen H, Leguit N, Glennon J, Koistinaho J, Banerjee P, Tanila H (2008) Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J Neurochem 105, 584–594. [DOI] [PubMed] [Google Scholar]
- [229].Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. [DOI] [PubMed] [Google Scholar]
- [230].Stevens LM, Brown RE (2015) Reference and working memory deficits in the 3xTg-AD mouse between 2 and 15-months of age: A cross-sectional study. Behav Brain Res 278, 496–505. [DOI] [PubMed] [Google Scholar]
- [231].Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276, 21562–21570. [DOI] [PubMed] [Google Scholar]
- [232].Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Wei Z, Chen XC, Song Y, Pan XD, Dai XM, Zhang J, Cui XL, Wu XL, Zhu YG (2016) Amyloid beta protein aggravates neuronal senescence and cognitive deficits in 5XFAD mouse model of Alzheimer’s disease. Chin Med J (Engl) 129, 1835–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, Bholat Y, Vasilevko V, Glabe CG, Breunig JJ, Rakic P, Davtyan H, Agadjanyan MG, Kepe V, Barrio JR, Bannykh S, Szekely CA, Pechnick RN, Town T (2013) A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J Neurosci 33, 6245–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]