

Abstract
Background:
Genetic variants in SCN5A can result in channelopathies such as the long QT syndrome type 3 (LQT3), but the therapeutic response to Na+ channel blockers can vary. We previously reported a case of an infant with malignant LQT3 and a missense Q1475P SCN5A variant, who was effectively treated with phenytoin, but only partially with mexiletine. Here, we functionally characterized this variant and investigated possible mechanisms for the differential drug actions.
Methods:
Wild-type or mutant Nav1.5 cDNAs were examined in transfected HEK293 cells with patch clamping and biochemical assays. We used computational modelling to provide insights into altered channel kinetics and to predict effects on the action potential.
Results:
The Q1475P variant in Nav1.5 reduced the current density and channel surface expression, characteristic of a trafficking defect. The variant also led to positive shifts in the voltage dependence of steady-state activation and inactivation, faster inactivation and recovery from inactivation, and increased the “late” Na+ current. Simulations of Nav1.5 gating with a 9-state Markov model suggested that transitions from inactivated to closed states were accelerated in Q1475P channels, leading to accumulation of channels in non-inactivated closed states. Simulations with a human ventricular myocyte model predicted action potential prolongation with Q1475P, compared with wild type, channels. Patch clamp data showed that mexiletine and phenytoin similarly rescued some of the gating defects. Chronic incubation with mexiletine, but not phenytoin, rescued the Nav1.5-Q1475P trafficking defect, thus increasing mutant channel expression.
Conclusions:
The gain-of-function effects of Nav1.5-Q1475P predominate to cause a malignant long QT phenotype. Phenytoin partially corrects the gating defect without restoring surface expression of the mutant channel, whereas mexiletine restores surface expression of the mutant channel, which may explain the lack of efficacy of mexiletine when compared to phenytoin. Our data makes a case for experimental studies before embarking on a one-for-all therapy of arrhythmias.
Keywords: Long QT syndrome, Na+ channel, Gating, Trafficking, Mexiletine, Phenytoin
Subject terms: Arrhythmias, Na+ channels
INTRODUCTION
Congenital long QT syndrome (LQTS) is characterized by prolongation of the QT interval and manifests as episodes of recurrent syncope, seizure, Torsades de Pointes (TdP) and sudden death [1]. In a reappraisal of the 17 genes reported to cause LQTS in humans, an International, Multicentered LQTS ClinGen Working Group recently concluded that only 3 genes, KCNQ1, KCNH2, and SCN5A, had definitive evidence as a genetic cause for typical LQTS [2]. LQT3 is the third most common of the different types of long QT syndrome [3, 4]. SCN5A variants were first associated with LQT3 more than 20 years ago [5] and there are now scores of cases linking SCN5A variants with LQT3. The cardiac Na+ channel (Nav1.5), responsible for the rapid upstroke of the action potential, is encoded by SCN5A [6]. Most LQT3 SCN5A variants cause missense amino acid substitutions or early truncations within Nav1.5. Many LQT3 variants produce a persistent (late) inward current during the action potential plateau, resulting in action potential prolongation and abnormal early afterdepolarizations that can trigger TdP and sudden death [7]. In rare cases, however, LQT3 variants may prolong Na+ channel opening (leading to slower inactivation) or cause a positive shift in the voltage-dependence of steady-state inactivation, leading to an increased “window current” and enhanced channel availability at a given voltage [8, 9]. Even more rarely, for example with the Nav1.5-I1768V LQT3 variant, channel reopening results in an accelerated recovery from inactivation [10], which increases channel availability during subsequent action potentials. Thus, a variety of gain-of-function mechanisms are possible, and it is therefore important to understand the nature of channel defects when considering patient-specific therapy.
We have previously described an infant with malignant LQT3 syndrome presenting with fetal bradycardia, 2:1 AV block, episodes of TdP and left ventricular dysfunction. Genetic testing revealed a novel c.A4424C SCN5A missense variant that leads to a p.Q1475P amino acid change in Nav1.5 [4]. Treating the patient with several drugs, including the Na+ channel blocker mexiletine, only led to partial control of arrhythmias. Complete cessation of TdP was only achieved by the addition of phenytoin, a Na+ channel blocker that is often used to treat epilepsy. The purpose of this study was to investigate the mechanism by which Q1475P affects Nav1.5 function and to determine possible mechanisms by which phenytoin may offer therapeutic benefits over a standard class 1B agent such as mexiletine.
METHODS
An expanded Materials and Methods description is available in the Supplemental Materials.
Cell culture, cDNAs, transfections, Western blotting and surface biotinylation assays
Human embryonic kidney 293 (HEK-293) cells, cultured in Dulbecco’s Modified Eagle Media (Thermo Fisher Scientific, Waltham, MA), were transiently transfected with wild-type (WT) or mutant Nav1.5 cDNAs (0.9 μg), together with EGFP (0.1 μg) (Lipofectamine 2000, Life Technologies, Carlsbad, CA). For some experiments, surface proteins were biotinylated (EZ Link Sulfo-NHS-SS-Biotin, Thermo Fisher Scientific) and enriched with NeutrAvidin beads (Pierce, Appleton, WI). Cells were lysed in RIPA buffer (Sigma Aldrich, St. Louis, MO, USA), cleared by centrifugation and protein concentration was determined (Bio-Rad Laboratories). Proteins were fractionated by SDS-PAGE, using 4–15% polyacrylamide gradient gels, transferred to polyvinylidene difluoride membranes (Bio-Rad) and immunoblotted with anti-Nav1.5 antibodies (Sigma-Aldrich). Bands were visualized by chemiluminescence (SuperSignal West Dura, Thermo Fisher Scientific), exposed to film and quantified using ImageJ.
Patch Clamping
Whole-cell Nav1.5 currents were recorded 48 h after transfection at 22–24°C (Axopatch 200B; Molecular Devices, Sunnyvale, CA). Patch electrodes were pulled using borosilicate glass (1.5 mm OD; World Precision Instruments, Sarasota, FL) to yield tip resistances of 2–3 MΩ when filled with (in mmol/L): 10 KCl, 105 CsF, 10 NaCl, 10 HEPES, 10 EGTA, and 10 TEA-Cl and pH adjusted to 7.2 with 2 N CsOH. The bath solution consisted of (in mmol/L): 30 CsCl, 107 NaCl, 5.4 KCl, 10 HEPES, 0.5 MgCl2, 1.8 CaCl2 with pH 7.4 adjusted with 2 N NaOH. Data were not corrected for the liquid junction potential, calculated to be 7.4 mV. The whole-cell capacitance and series resistance were compensated to levels greater than 80%. Currents were corrected for cell size by dividing membrane current by the cell capacitance. Further details, including the voltage-clamp protocols used and the methods of analysis, are provided in the Supplemental Material.
Mathematical modeling of Nav1.5
A Markov model of Nav1.5 [11, 12], with three closed states, one open state, and five inactivated states, was employed to deduce the likely effects of variants and drugs on channel gating. To address the fact that parameters of Markov models may not be uniquely identifiable, we employed a population-based computational approach, namely a genetic algorithm, to find multiple sets of model parameters that could recapitulate data. The genetic algorithm, inspired by the principles of natural selection, generates an initial population of individuals, computes the “fitness” of each individual based on how well each reproduces data, then favors individuals with higher fitness when proceeding to the next generation. For this optimization problem, rate constants in the Markov model varied between individuals, and fitness was quantified as based on the squared differences between simulated and experimental values of steady-state activation, steady-state inactivation, and recovery from inactivation, summed over all voltages or intervals. The genetic algorithm was run multiple times (n=5) with random initial conditions to generate sets of parameters that are compatible with the experimental results. Three distinct populations of channels were identified that were consistent with data obtained with WT Nav1.5 channels, Q1475P Nav1.5 channels, and Q1475P Nav1.5 channels acutely treated with mexiletine or phenytoin. Full details are provided in the Supplementary Methods.
Modeling the effects of mutated Nav1.5 on human ventricular action potential
We incorporated the Nav1.5 channel variants into a model of the human ventricular action potential [13]. The original formulations for fast and late Na+ current were replaced by the Markov model, and each member of the 3 populations calibrated to voltage clamp data was simulated in an isolated endocardial cell. Maximal conductance for Na+ current (GNa) was reduced by 50% for untreated Q1475P Na+ channels. In drug-treated Q1475P groups, GNa was 50% of WT for phenytoin treatment, and 100% of WT for mexiletine treatment, reflecting the current-voltage plots recorded after 48 hour drug treatment (Figure 7C). Simulation and analysis details are provided in the Supplementary Materials.
Figure 7: Mexiletine, but not phenytoin, rescues the defective surface trafficking of Nav1.5-Q1475P.
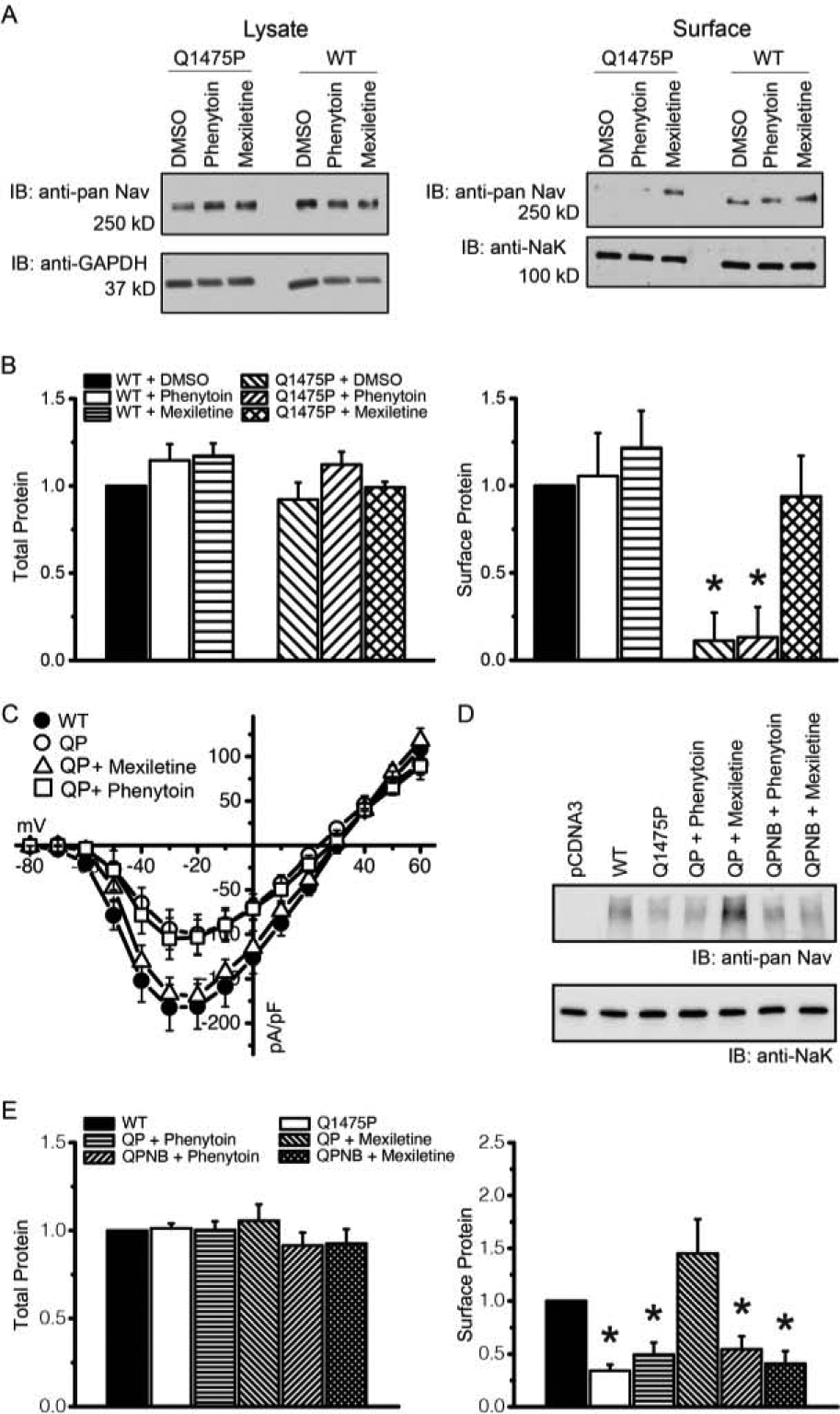
(A) HEK293 cells transfected with WT or Q1475P Nav1.5 were preincubated for 48h with 100 μM mexiletine, 100 μM phenytoin of with only solvent as a control (DMSO). Western blotting was performed with cell lysates (left) or with the surface biotinylated protein fractions (right). (B) Summary of the averaged band intensities of surface biotinylated Nav1.5, normalized to band intensities of surface biotinylated Na+/K+ ATPase. (C) Whole-cell current densities as a function of the test voltages for the various experimental groups (WT n=21; Q1475P n=18; Q1475P plus phenytoin n=15; Q1475P plus mexiletine n=5). (D) Mutations (F1760A and F7167A) were made in Q1475P to disrupt mexiletine binding (NB). HEK293 cells were transfected with Q1475P (QP) or Q1475P non-binding (QPNB) Nav1.5 constructs and were preincubated with 100 μM phenytoin or mexiletine for 48h. Shown is a Western blot of surface biotinylated and total Nav1.5 proteins. Vector pcDNA3 and WT Nav1.5 were used as antibody controls. Na+/K+ ATPase was used as a loading control. (E) Summary of the averaged band intensities of surface biotinylated Nav1.5, normalized to band intensities of surface biotinylated Na+/K+ ATPase. Representative blots are shown in panels A & D. Experiments were repeated three times and quantified, results are shown in panels B & E. Phenytoin and mexiletine were each applied at 100 μM. *p < 0.05 versus WT.
Statistics
Results are presented as mean ± standard error of the mean. Statistical comparisons were performed using one-way analysis of variance (ANOVA) followed by Dunnett’s t-test. Statistical significance was assumed when p < 0.05.
RESULTS
Treatment with phenytoin is sufficient for arrhythmia control
We have previously reported malignant ventricular arrhythmias in a female infant with a Q1475P SCN5A missense variant in in the DIII-DIV interdomain loop of Nav1.5 (Figure 1A) [4]. The arrhythmia burden was decreased with a combined regimen of mexiletine, ranolazine and propranolol, but control was only achieved with the addition of phenytoin [4]. The patient was subsequently weaned off ranolazine and mexiletine with no arrhythmia recurrence. Currently she is maintained on high dose phenytoin (8 mg/kg/day) and propranolol (2.5 mg/kg/day). Phenytoin dosing is adjusted to maintain a drug level of 20–25 μg/ml, or an estimated plasma drug concentration of 80 μM. Lower doses of phenytoin in this patient sometimes resulted in T wave alternans (calculated at 40–80 μM) or episodes of TdP (calculated at less than 40 μM).
Figure 1: Effects of Q1475P on current amplitudes.
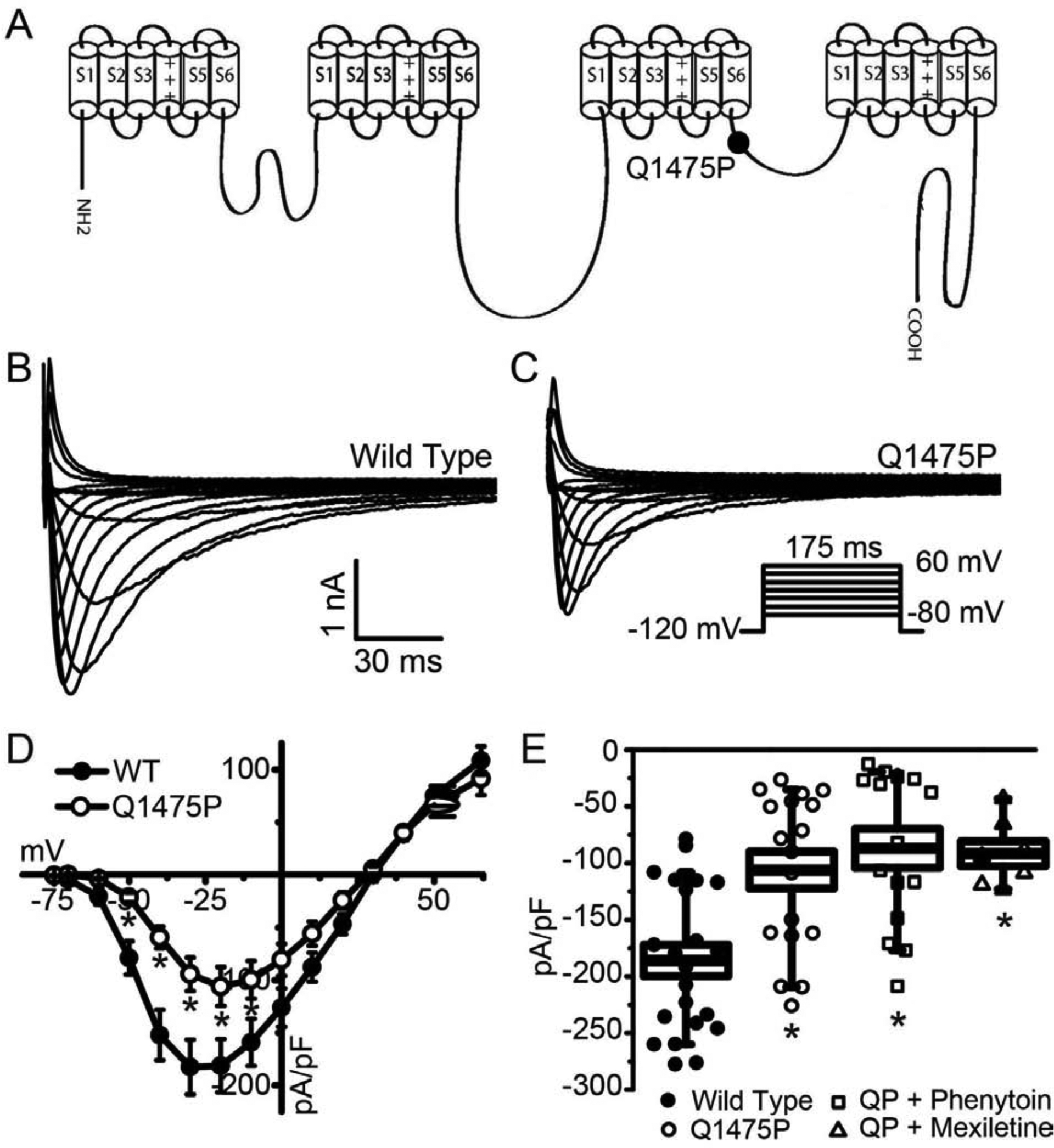
(A) Schematic representation showing the position of Q1475P in the Nav1.5 α-subunit. (B, C) Representative whole-cell current traces of WT and Q1475P Nav1.5 when expressed in HEK293 cells. The voltage protocol used is shown in the inset. (D) The peak current densities are averaged and plotted as a function of the test voltages. (E) Averaged values of the peak current density at −20 mV for the various experimental groups (WT n=21; Q1475P n=18; Q1475P plus phenytoin n=15; Q1475P plus mexiletine n=7). Phenytoin and mexiletine were each applied at 100 μM. *p < 0.05. versus WT.
Q1475P reduces Nav1.5 current density
We examined the functional effects of Q1475P in transiently transfected HEK-293 cells by whole-cell patch clamping. Representative recordings in Figure 1B & C show that Nav1.5-Q1475P current density was reduced by ~50% compared to WT, with the reduction approximately uniform across all voltages (Figure 1D). In order to investigate the effects of mexiletine and phenytoin on the mutant channels, we applied 100 μM of drug to the bath solution, which is close to the therapeutic dose used in this patient. Neither mexiletine nor phenytoin significantly affected the Nav1.5-Q1475P current density when applied acutely over a 3–10 min period (Figure 1E).
Q1475P accelerates Nav1.5 inactivation kinetics
The Q1475P variant is located in the DIII-DIV inter-domain loop, which participates in rapid Na+ channel inactivation [6]. We therefore investigated whether gating kinetics are altered by the variant. Representative current traces at −20 mV were superimposed and scaled to the maximum peak amplitude (Figure 2A). Q1475P mutant channels exhibit a trend towards faster time to peak current, although this did not reach statistical significance (data not shown). Inactivation, in contrast, was clearly accelerated. Curve fitting of data traces to a sum of two exponential functions show that both the fast and slow components of inactivation occurred more rapidly (Figure 2B). Neither mexiletine nor phenytoin rescued the defect in inactivation kinetics (Figure 2B).
Figure 2: Nav1.5-Q1475P inactivates more rapidly.
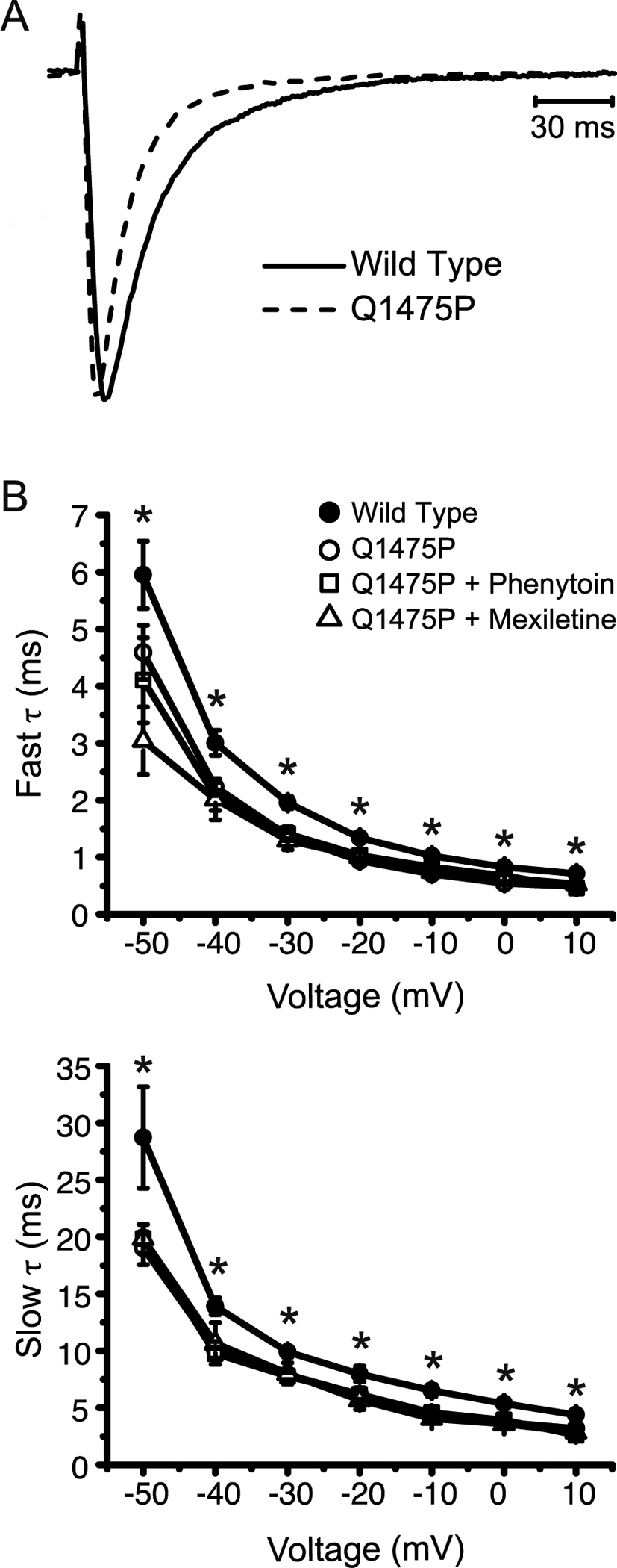
(A) Representative whole-cell recordings at −20 mV of WT and Q11475P Nav1.5 currents were normalized to their peak amplitudes and superimposed. (B) Individual current traces were subjected to curve fitting to a double exponential function. The slow and fast time constants are summarized and plotted against votlage (WT n=19; Q1475P n=16; Q1475P plus phenytoin n=15; Q1475P plus mexiletine n=5). Phenytoin and mexiletine were each applied at 100 μM. *p < 0.05 versus WT.
Q1475P causes a positive shift in the voltage dependence of the kinetic variables
The IV curve in Figure 1D suggests that Nav1.5-Q1475P currents may activate at more positive voltages. Steady-state activation curves were constructed from these data and fit to a Boltzmann equation (Figure 3A), which indeed showed that the voltage at which half-maximal activation occurs (V1/2) was positively shifted by Q1475P (−38.5 ± 1.5 mV, n=21 vs. −29.5 ± 1.70 mV, n=18 respectively for WT and Nav1.5-Q1475P; p<0.05). The slope factor (k) was not changed (respectively 9.8 ± 0.46 and 10.0 ± 0.39 for WT and Nav1.5-Q1475P; p=0.75). Neither mexiletine (−30.3 ± 1.4 mV, n=7) nor phenytoin (−32.1 ± 1.2 mV, n=15) restored the defect in the voltage dependence of activation (p<0.05 vs WT) (Figure 3C).
Figure 3: Voltage-dependence of activation and inactivation kinetic variables at steady-state.
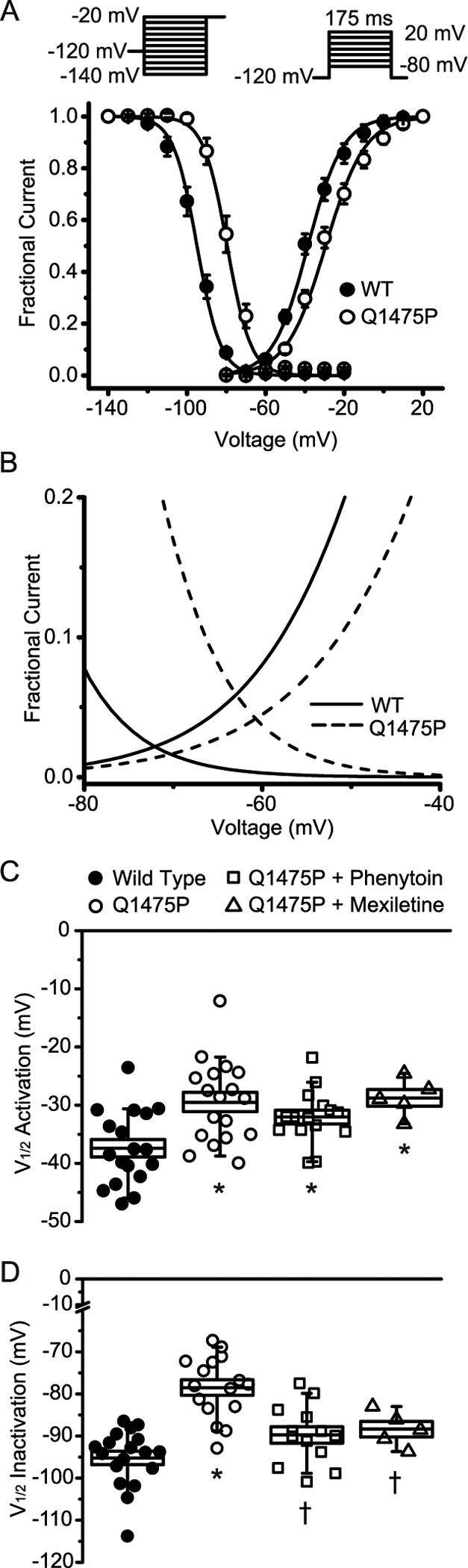
(A) Steady-state activation and inactivation curves for WT and Nav1.5-Q1475P obtained using the voltage protocols depicted as insets. Data points were subjected to curve fitting to a Boltzmann function and the solid lines were derived from the averaged fitted parameters. (B) The steady-state activation and inactivation curves are shown over a limited voltage range to depict the larger “window” current of Nav1.5-Q1475P. (C and D) The voltages of half-maximal (V½) activation and inactivation obtained from the curve fitting are summarized and depicted for the various experimental groups (WT n=18; Q1475P n=16; Q1475P plus phenytoin n=13; Q1475P plus mexiletine n=7). Phenytoin and mexiletine were each applied at 100 μM. *p < 0.05 versus WT; †p < 0.05 drug groups versus Q1475P.
We next investigated the voltage dependence of steady-state inactivation using a conditioning pulse between −140 mV and −20 mV, followed by a test pulse at −20 mV. Currents during the test pulse were normalized against those recorded during the pre-pulse potential and fit to a modified Boltzmann equation (Figure 3A). A depolarizing shift of ~17 mV was evident for Nav1.5-Q1475P, with a V1/2 of −78.5 ± 1.9 mV (n=16), compared to −95.2 ± 1.6 mV for WT (n=18; p<0.05), suggesting that the inactivated state of the channel may be destabilized by Q1475P. After acute application of mexiletine or phenytoin, the voltage dependence of the steady state inactivation of Nav1.5-Q1475P was not significantly different from WT (−88.2 ± 1.3 mV, n=7 and −89.8 ± 1.9 mV, n=13 for mexiletine and phenytoin respectively; p>0.05) (Figure 3D & S1).
Q1475P increases the persistent (late) Na+ current
Our data demonstrate that the Q1475P variant shifts the voltage dependence of both steady-state activation and inactivation, which results in a larger overlap of these curves (Figure 3B). This increased ‘window current’ may be sufficient to prolong the action potential. Certain LQT3 variants additionally increase the non-inactivating component of the cardiac Na+ channel (also referred to as the ‘late’ current), which contributes to action potential prolongation, the development of early after-depolarizations (EADs) and arrhythmias [14]. We investigated these sustained Na+ currents using a slow ramp voltage clamp protocol, which demonstrated a significantly larger sustained inward current in Q1475P channels (Figure 4). Note that currents were larger in the voltage range of the ‘window’ current (−80 mV to −50 mV) as well as at more positive potentials, indicating that ‘late’ Na+ current was increased by this mutation. Phenytoin effectively decreased the elevated ‘late’ current of the Q1475P channels.
Figure 4: Nav1.5-Q1475P persistent current is increased.
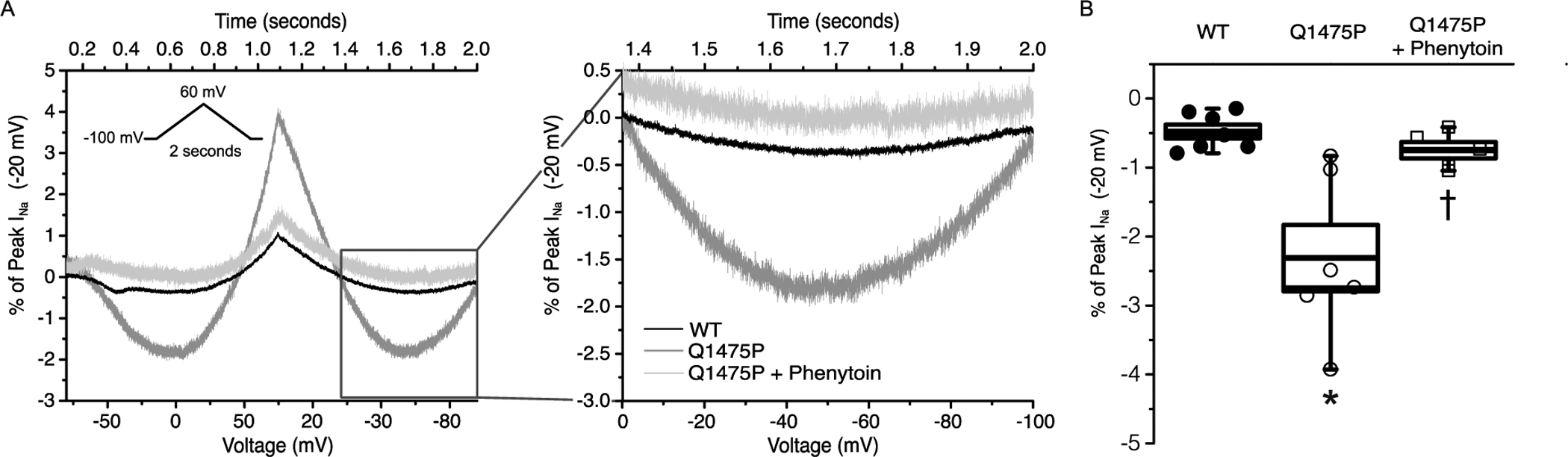
(A) Representative ramp current traces for WT, Q1475P and Q1374P + phenytoin in response to a depolarizing voltage ramp over 1 s from a holding potential of −100 mV to +60 mV, followed by a descending ramp back to −100 mV over 1 s. Ramp current amplitude is presented as a percentage of the peak transient current elicited with a standard I–V protocol, then yielding the percentage of peak current for each recording. (B) Bar graph interpretation of the percentage of peak ramp current to the transient peak current elicited with a standard I–V protocol (WT, n = 7; Q1475P, n = 7; Q475P + phenytoin, n = 5). Phenytoin and mexiletine were each applied at 100 μM. *p < 0.05 versus WT; †p < 0.05 drug group versus Q1475P.
Q1475P increases channel availability following inactivation
We next measured recovery from inactivation using a standard two-pulse protocol. WT Nav1.5 recovered to 50% of maximal availability in ~10–12 ms (Figure 5A). In contrast, there was a ~300% acceleration in inactivation recovery kinetics for Nav1.5-Q1475P, which needed only ~3 ms to recover to 50% of maximal availability. We quantified these data by fitting the data points to an exponential function, which showed that Nav1.5-Q1475P currents recovered significantly more rapidly from inactivation compared to WT (Figure 5B). This result suggests that the Q1475P mutation prevents the channel from entering deep inactivated states, thereby allowing the channel to more readily re-enter the open state. Although acute application of mexiletine or phenytoin had marginal effects on the recovery time constants of Nav1.5-Q1475P, it is worth noting that the recovery from activation still occurred almost twice faster than WT.
Figure 5: Recovery from inactivation of Nav1.5-Q1475P is accelerated.
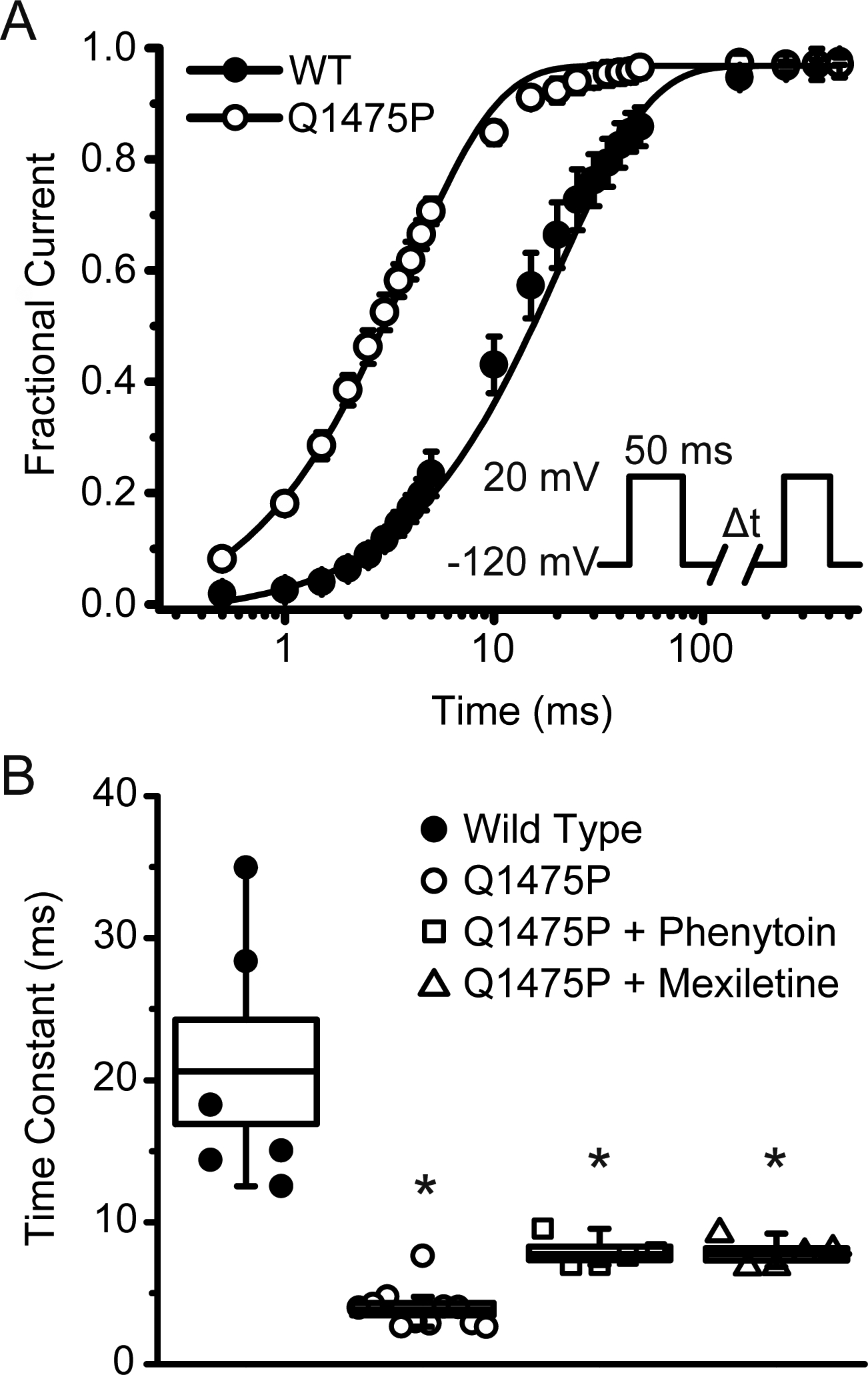
(A) Recovery from inactivation was measured using a double pulse protocol (see inset). The peak current amplitude during the second pulse was normalized relative to the first, averaged and plotted as a function of the inter-pulse duration. Individual data points were subjected to curve fitting to an exponential function and the solid lines were reconstructed from the averaged fitting parameters. (B) Shown are the averaged values of the exponential time constants for the different experimental groups (WT n=6; Q1475P n=11; Q1475P plus phenytoin n=5; Q1475P plus mexiletine n=7). Phenytoin and mexiletine were each applied at 100 μM. *p < 0.05 versus WT.
Modeling of Q1475P channel kinetics
To gain insight into the kinetic mechanisms by which Q1475P affects gating, we performed numerical analysis using a nine state Markov model [11, 12]. Using multiple runs with a genetic algorithm (see Methods), we determined parameter sets that were compatible with the experimentally obtained activation, inactivation, and recovery from inactivation curves for WT, Q1475P and drug-treated Q1475P channels (Figure 6A). Because mexiletine and phenytoin had similar effects on Nav1.5 gating (Figures 2, 3, and 5), the two drugs were considered equivalent in these simulations. To identify the gating transitions most likely to be modified, we compared rate constants in Q1475P channels with WT channels (Figure 6B), and rate constants in drug-treated Q1475P channels with untreated Q1475P channels (Figure 6C) This analysis suggests that, relative to WT channels, Q1475P channels exhibit an increase in α3, controlling the inactivated to closed transition, decreases in α11 and α12, regulating opening steps, and decreases in rate constants entering deep inactivated states (α4, α5 and β4). Interestingly, the analysis does not suggest that drug treatment induces a straightforward correction of these rates. Instead, treatment of Q1475P channels with phenytoin or mexiletine is predicted to cause an increase in α12 (an opening step) and an increase in β3 (a closed to inactivated transition).
Figure 6: Modeling to infer altered gating transitions in Q1475P and drug-treated Q1475P channels.
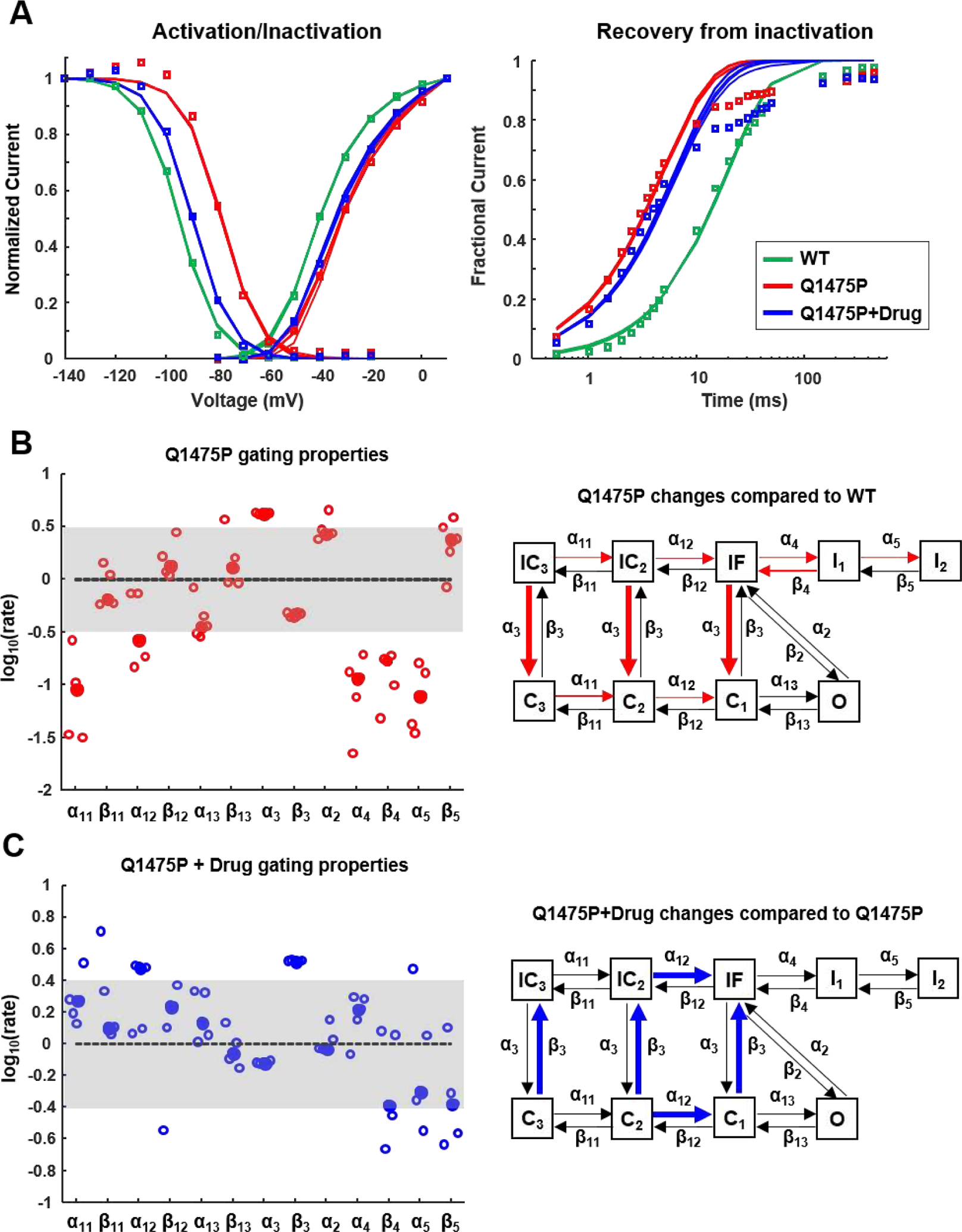
(A) Populations of steady-state activation, inactivation and recovery from inactivation curves produced by the different parameter sets obtained with the genetic algorithm (n=5) (see Methods). The symbols show the experimental data, the solid lines are the solutions from each population. (B) Predicted changes in rate constants in Q1475P channels, compared with median WT values, are shown at left. At right, the rate constants predicted to be increased (thick lines) or decreased (thin lines) are illustrated on the Nav1.5 gating scheme. (C) Rate constants in drug-treated Q1475P channels, compared with untreated channels, displayed as in (B).
Defective surface trafficking of Nav1.5-Q1475P: differential rescue by mexiletine and phenytoin
The results presented thus far demonstrate altered Nav1.5 gating in Q1475P channels, and partial correction of gating defects with drug treatment. These results cannot, however, explain the fact that phenytoin produced superior arrhythmia control compared to mexiletine in this patient. To explore potential mechanisms, we examined Nav1.5 trafficking with surface biotinylation assays. The total Nav1.5 protein in the cell lysates was somewhat reduced by the Q1475P mutation, suggesting a degree of protein instability, most likely due to misfolding and breakdown. The surface expression, in contrast, was markedly decreased (Figure 7A), suggesting a trafficking defect, which potentially explains the reduced Q1475P current density (Figure 1D). Since drugs can rescue trafficking defects in channelopathies [15], we tested the effects of chronic treatment with mexiletine or phenytoin. Surface biotinylation and patch clamp experiments demonstrated that the trafficking defects of the mutant channel was rescued by mexiletine, but not by phenytoin (Figure 7). The trafficking defect of an unrelated mutant channel (TRPM4-L519P) [16] was unaffected by mexiletine (Figure S3). When mutating residues involved in mexiletine binding (F1760A and F7167A) in the Q1475P background (Nav1.5-Q1475PNB), mexiletine neither affected steady-state inactivation nor restored the trafficking defect (Figures S4, 7D & 7E) demonstrating that mexiletine acted by binding to the Na+ channel itself. To investigate if this is a general mechanism (as opposed to an effect specific to this variant), we repeated the experiment using another trafficking-defective Nav1.5 (Q1832E), which we previously identified in a pediatric case of sudden death [17]. Mexiletine did not rescue Nav1.5-Q1832E trafficking, but surprisingly, phenytoin partially corrected the trafficking defect of this mutant channel (Figure S5). These data demonstrate that class 1B Na+ channel blockers can have differential effects on Na+ channels, and that the effect can be specific to the mutation that is present.
Action potential simulations and possible arrhythmia mechanisms
Finally, to predict the functional consequences of changes in Nav1.5 gating and/or trafficking caused by the Q1475P variant and drug treatment, we incorporated the populations of parameter sets from Figure 6 into a mathematical model of the human ventricular action potential [13]. In these simulations, we assumed that mexiletine and phenytoin caused identical alterations in Q1475P Na 1.5 gating, while cellular Nav+ conductance (GNa) reflected the rescue of the trafficking defect caused by mexiletine (GNa equal to WT) and the lack of changes to trafficking caused by phenytoin (GNa equal to untreated Q1475P, 50% of WT). These simulations showed that Q1475P parameter sets led to small reductions in action potential amplitude and upstroke velocity of the action potential (Figures S6 and S7), but mostly resulted in longer action potentials (Figures 8A & 8B), and substantially larger late INa (Figure 8C), compared with WT parameter sets. Thus, the gain-of-function phenotype (increased late Na+ current) predominated over loss-of-function defects (reduced conductance), and led to increased action potential duration in Q1475P cells. Importantly, these simulations predicted much larger therapeutic reductions in late INa and action potential duration in phenytoin-treated, compared with mexiletine-treated, Q1475P cells. To link these simulation results back to the changes in Nav1.5 gating identified in Figure 6, we performed action potential clamp simulations and quantified the state distribution of each population of Na+ channels, immediately before the last action potential in a sequence of 200. These results (Figure 8D) suggest that greater late INa is seen in Q1475P channels due to an accumulation of channels in the closed state compared with inactivated or deep inactivated states, thereby enabling more rapid reopening of channels during the action potential. These simulations further suggest that drug treatment with either phenytoin or mexiletine reduces the action potential duration by shifting channels into inactivated and deep inactivated states, leading to a reduction in late INa.
Figure 8: Simulation of human ventricular action potentials incorporating the Nav1.5 rate constants obtained through parameter optimization.
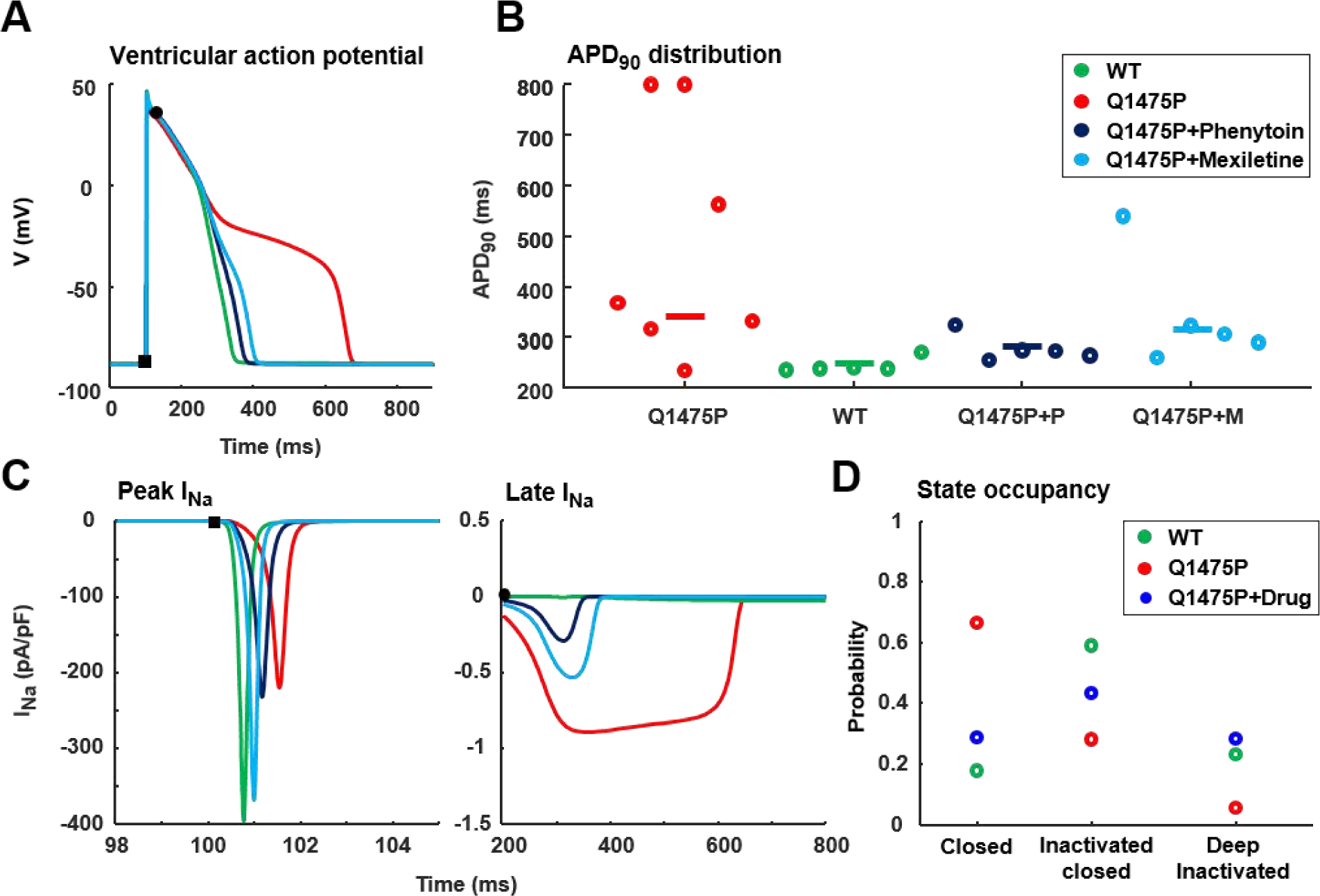
Simulations were performed with each of the genetic algorithm solutions, resulting in a population of outcomes for WT, Q1475P and Q1475P plus drug. Na+ current maximal conductance reflected the trafficking defect in the Q1475P channels, and the rescue of this trafficking defect by mexiletine, but not phenytoin. (A) Simulations of a ventricular myocyte model predict that Q1475P Na+ channels (red) produce longer action potentials than WT (green) or Q1475P channels treated with either phenytoin (dark blue) or mexiletine (light blue). For each type of channel, the action potential generated by the median genetic algorithm solution is shown. (B) Summary data quantifying action potential duration at 90% of repolarization (APD90). Two dots corresponding to short and long action potentials are shown for the two Q1475P solutions (out of 5) that resulted in action potential duration alternans. (C) Analysis of peak Na+ current and late Na+ current during action potential simulations. Median values in each group are plotted. (D) The occupancies of each state are calculated immediately before the last action potential in a train of 200 action potentials, obtained through action potential clamp simulations. States are grouped in closed, inactivated closed and deep inactivated states and the median occupancy of each type of state is shown for WT, Q1475P and drug treated Q1475P channels. WT is shown in green symbols, Q1475P is red and Q1475P plus drug is depicted in blue.
DISCUSSION
Population-based strategy to investigate ion channel gating defects
We have characterized the electrophysiological effects of a Nav1.5-Q1475P variant previously reported in an infant with LTQ3 [4]. Our data show that Q1475P reduces the current density, accelerates the rate of inactivation, shifts the steady-state activation and inactivation curves in a depolarizing direction, and accelerates recovery from inactivation. To gain a mechanistic understanding of these results, we performed simulations with a Markov model of Nav1.5 gating. Compared with simpler, Hodgkin-Huxley formulations, Markov schemes potentially offer greater insights into molecular-level changes caused by mutations or drugs [18–20], but with the disadvantage that standard experimental protocols may be insufficient to uniquely determine model parameters. To balance these tradeoffs, we used a genetic algorithm to constrain parameters in the model of Nav1.5 gating, and ran the algorithm repeatedly with different initial conditions to obtain multiple parameter sets for each condition (WT, Q1475P, drug-treated Q1475P). Such population-based modeling approaches [21–23] allow for general trends between groups to be observed while allowing for a lack of absolute confidence in the state-dependent effects of each drug and mutation. The parameter sets that we obtained (Figure 6) demonstrated that faster transitions from inactivated states to closed states (rate constant α3) in the Nav1.5 Q1475P channel led to greater availability of these channels (Figure 8D), which can account for channel reopenings during the action potential and increased late INa. Interestingly, the simulations predicted that drug treatment did not reverse the changes in the Q1475P channels, but instead accelerated transitions from closed to inactivated states, thereby decreasing the late INa (e.g. β3). While the results arising from our simulations represent valuable hypotheses on possible drug mechanisms, these would need to be further investigated in future studies for instance through single channels recordings.
Potential arrhythmia mechanisms in this patient
Although the LQT3 diagnosis in the patient strongly suggested that ventricular action potentials will be prolonged, our experimental data showed both gain-of-function and loss-of function changes in Q1475P compared with WT channels. It was unclear how the action potential would be affected when the Na+ current density is decreased by ~50% while there is positive shift of the voltage-dependence of steady state kinetic variables. To address this problem, we introduced the Nav1.5 Markov models into simulations of the human ventricular action potential. These simulations suggested that the altered kinetics in Q1475P predominate over the reduced channel density and are sufficient to prolong the action potential. Cells with simulated Q1475P channels generated substantially more ‘late’ INa than WT, due in part to faster recovery from inactivation in these channels, which was decreased with the addition of phenytoin or mexiletine. An additional contributing factor is that the voltage shifts in the steady-state activation and inactivation curves lead to an increased “window current” (defined as the overlap between these curves shown in Figure 3B), which can prolong action potentials by providing a persistent inward current within the plateau voltage range [24]. The combination of these kinetic effects promotes channel availability during phase 3 of an action potential, which may result in development of early afterdepolarizations and triggered arrhythmias.
Possible explanations for the superior efficacy of phenytoin to treat arrhythmias in this patient
The patient’s arrhythmias were controlled by phenytoin, but not by the more commonly used mexiletine. It should be noted that propranolol, which was reported to block Na+ channels at high micromolar concentrations [25], was co-administered before and after introduction of phenytoin and we cannot rule out the possibility that this β-receptor antagonist contributed via this mechanism. We initially investigated the possibility that mexiletine and phenytoin differentially rescued kinetic defects of the Nav1.5-Q1475P channel when applied acutely during patch clamp studies. We found that both drugs partially restored some of the gating defects, such as the positive shift in the voltage-dependence of steady state inactivation. Other defects were not improved, however, including the positive shift in the voltage-dependence of steady state activation and recovery from inactivation. These drugs can therefore only be partially anti-arrhythmic. Given the patient’s clinical profile, it was puzzling that there was no difference in the in vitro actions of mexiletine and phenytoin during the patch clamp experiments. Because one of the Nav1.5-Q1475P defects was a ~50% reduction in current density, we examined how the mutation and drugs influenced surface expression and trafficking. Interestingly, our surface biotinylation data confirmed a trafficking defect in Nav1.5-Q1475P channels, and demonstrated that preincubation with mexiletine, but not phenytoin, could rescue the defect. The effect of mexiletine was specific to the Na+ channel since mutagenesis of amino acid residues involved in the Na+ channel mexiletine binding eliminated rescue. Moreover, mexiletine did not rescue the trafficking defect of a TRPM4 channel. The most parsimonious conclusion from these data is that mexiletine treatment resulted in enhanced surface expression of the defective Nav1.5-Q1475P channel, which counteracted the partial correction of the gating defects and led to the only partial effectiveness of mexiletine in this patient. In contrast, phenytoin does not rescue the trafficking of the defective Na+ channels, yet is able to partially restore the kinetic defects, which is a possible reason for the beneficial therapeutic response of phenytoin in this patient.
General clinical implications
Based on studies demonstrating that mexiletine can effectively reduce life-threatening arrhythmic events in LQT3 patients [26–28], Na+ channel blockers are often used for treatment. In the present case, however, the patient was partially responsive to mexiletine, consistent with previous studies demonstrating that mexiletine is not equally effective in all LQT3 patients [29–31]. In a study aimed to predict the clinical response to mexiletine on the basis of the Na+ channel biophysical properties, it was suggested that the single most important factor that correlates with a positive response to therapy was a left-ward displacement of steady-state inactivation curve [29]. The concept that the response to mexiletine is mutation-specific was underscored by an infant with LQT3 with a F1473S mutation, whose symptoms improved immediately after the onset of mexiletine treatment, but then progressively deteriorated over the next 10 days, resulting in the death of this young patient [29]. Another LQT3 infant with a different mutation in the same position (F1473C) was only partially responsive to mexiletine and was discharged at 3 months of age with an implantable cardioverter defibrillator/pacemaker [30]. Interestingly, these LQT3 F1473 mutations occurred two amino acids from the Q1475P mutation described in this study and caused a very similar electrophysiological phenotype, namely positive voltage shifts in the steady-state activation and inactivation curves, accelerated recovery from inactivation and a reduced current density due to trafficking defects that could be rescued by mexiletine. Similar to our conclusion and the prediction made in an editorial by Bezzina et al [32], these authors concluded that rescuing certain mutant channels with abnormal gating behavior can actually pose a proarrhythmic threat. Unlike the F1473S LQT3 infant who died within two weeks after mexiletine treatment, we were able to control the arrhythmias of the Q1475P infant with phenytoin.
Limitations
An obvious limitation of the study is that experiments were performed in vitro using a mammalian heterologous expression system. Although drug concentrations used in the study exceed typical therapeutic concentrations, they are consistent with the high doses necessary to control arrhythmias in this patient. Another potential limitation is that we performed experiments in the absence of β subunits. Although β subunits can modify the kinetics and expression of Na+ channels, these proteins have diverse functions in addition to regulating the Na+ channel. Moreover, there are several different types of β subunits (products of different genes and as a result of alternative splicing), and it is not completely clear which combination of these beta subunits are relevant in the context of our studies. To avoid these complexities, we studied Nav1.5 α subunits in isolation, an approach that allows for straightforward comparisons with several other studies that characterized Nav1.5 variants in the absence of beta subunits; e.g. F1473S [31], F1486del [33] and Q1476R [34]. In addition to the SCN5A variant, this patient also has a common KCNH2 variant (ExAC allele frequency 0.1822) that causes a missense mutation (K897T) in hERG. Although this variant has been reported as a genetic modifier of latent congenital long QT syndrome [35], subsequent studies have shown that arrhythmias are not influenced by this KCNH2 polymorphism [36]. In future work, the generation of cardiomyocytes from patient-specific induced pluripotent stem cells can help to both validate the present findings and address the key issue of whether other genetic influences modulate the disease burden of this patient.
Conclusions
In conclusion, in this study we have shown how cellular electrophysiology can be combined with mathematical modeling to gain insight into why certain drugs may be more efficacious than others in a particular patient. There are prior reports demonstrating that phenytoin can effectively treat ventricular arrhythmias in young patients [37, 38] and its use needs to be considered in pediatric LQT3 cases when mexiletine is contraindicated or ineffective. Our data demonstrate that the effect of class 1B Na+ channel blockers can be dictated by the mutation that is present. Patch clamp studies such as this can be useful before embarking on a general “gene-specific” drug therapy of arrhythmias.
Supplementary Material
Highlights.
Arrhythmias in child with Nav1.5-Q1475P was better controlled by phenytoin but not mexilitine
Nav1.5-Q1475P had gain-of-function (e.g. increased late current) and trafficking defects
Numerical modeling shows the channel is trapped in a pre-open closed state with a longer APD
Mexiltine, but not phenytoin, rescued trafficking of the mutant channel
Phenytoin may be better since it partially rescues gating, but not trafficking defects
FUNDING SOURCES
These studies were supported in part by KiDS of NYU and partly by the Seventh Masonic District Association, Inc, and by U01 HL 136297 from the National Institutes of Health to EAS
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None of the authors have any significant affiliations or relationships with commercial enterprise or any other potential conflicts of interest.
DISCLOSURES
None.
REFERENCES
- [1].Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, Kass RS, Molecular Pathophysiology of Congenital Long QT Syndrome, Physiol. Rev 97(1) (2017) 89–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, Feilotter H, Amenta S, Mazza D, Bikker H, Sturm AC, Garcia J, Ackerman MJ, Hershberger RE, Perez MV, Zareba W, Ware JS, Wilde AAM, Gollob MH, An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome, Circulation 141(6) (2020) 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ruan Y, Liu N, Priori SG, Sodium channel mutations and arrhythmias, Nat. Rev. Cardiol 6(5) (2009) 337–48. [DOI] [PubMed] [Google Scholar]
- [4].Tan RB, Chakravarti S, Busovsky-McNeal M, Walsh A, Cecchin F, Complexity of ranolazine and phenytoin use in an infant with long QT syndrome type 3, HeartRhythm case reports 3(1) (2017) 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, Schwartz PJ, Keating MT, Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia, Hum. Mol. Genet 4(9) (1995) 1603–7. [DOI] [PubMed] [Google Scholar]
- [6].Veerman CC, Wilde AA, Lodder EM, The cardiac sodium channel gene SCN5A and its gene product NaV1.5: Role in physiology and pathophysiology, Gene 573(2) (2015) 177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bennett PB, Yazawa K, Makita N, George AL Jr., Molecular mechanism for an inherited cardiac arrhythmia, Nature 376(6542) (1995) 683–5. [DOI] [PubMed] [Google Scholar]
- [8].Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, Borggrefe M, Brinkmann B, Warnecke I, Funke H, Bhuiyan ZA, Wilde AA, Breithardt G, Haverkamp W, De novo mutation in the SCN5A gene associated with early onset of sudden infant death, Circulation 104(10) (2001) 1158–64. [DOI] [PubMed] [Google Scholar]
- [9].Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, Napolitano C, Priori SG, Kass RS, Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes, J. Biol. Chem 276(33) (2001) 30623–30. [DOI] [PubMed] [Google Scholar]
- [10].Clancy CE, Tateyama M, Liu H, Wehrens XH, Kass RS, Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia, Circulation 107(17) (2003) 2233–7. [DOI] [PubMed] [Google Scholar]
- [11].Clancy CE, Rudy Y, Na(+) channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism, Circulation 105(10) (2002) 1208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bondarenko VE, Szigeti GP, Bett GC, Kim SJ, Rasmusson RL, Computer model of action potential of mouse ventricular myocytes, Am. J. Physiol. Heart Circ. Physiol 287(3) (2004) H1378–403. [DOI] [PubMed] [Google Scholar]
- [13].O’Hara T, Virag L, Varro A, Rudy Y, Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation, PLoS Comput. Biol 7(5) (2011) e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, Kirsch GE, Multiple mechanisms of Na+ channel--linked long-QT syndrome, Circ Res 78(5) (1996) 916–24. [DOI] [PubMed] [Google Scholar]
- [15].Valdivia CR, Tester DJ, Rok BA, Porter CB, Munger TM, Jahangir A, Makielski JC, Ackerman MJ, A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs, Cardiovasc. Res 62(1) (2004) 53–62. [DOI] [PubMed] [Google Scholar]
- [16].Subbotina E, Williams N, Sampson BA, Tang Y, Coetzee WA, Functional characterization of TRPM4 variants identified in sudden unexpected natural death, Forensic Sci. Int 293 (2018) 37–46. [DOI] [PubMed] [Google Scholar]
- [17].Gando I, Morganstein J, Jana K, McDonald TV, Tang Y, Coetzee WA, Infant sudden death: Mutations responsible for impaired Nav1.5 channel trafficking and function, Pacing Clin. Electrophysiol 40(6) (2017) 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fink M, Noble D, Markov models for ion channels: versatility versus identifiability and speed, Philos Trans A Math Phys Eng Sci 367(1896) (2009) 2161–79. [DOI] [PubMed] [Google Scholar]
- [19].Moreno JD, Zhu ZI, Yang PC, Bankston JR, Jeng MT, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS, Clancy CE, A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms, Sci. Transl. Med 3(98) (2011) 98ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Clancy CE, Zhu ZI, Rudy Y, Pharmacogenetics and anti-arrhythmic drug therapy: a theoretical investigation, Am. J. Physiol. Heart Circ. Physiol 292(1) (2007) H66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sarkar AX, Christini DJ, Sobie EA, Exploiting mathematical models to illuminate electrophysiological variability between individuals, J. Physiol 590(11) (2012) 2555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Muszkiewicz A, Britton OJ, Gemmell P, Passini E, Sanchez C, Zhou X, Carusi A, Quinn TA, Burrage K, Bueno-Orovio A, Rodriguez B, Variability in cardiac electrophysiology: Using experimentally-calibrated populations of models to move beyond the single virtual physiological human paradigm, Prog. Biophys. Mol. Biol 120(1–3) (2016) 115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ni H, Morotti S, Grandi E, A Heart for Diversity: Simulating Variability in Cardiac Arrhythmia Research, Frontiers in physiology 9 (2018) 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Horvath B, Bers DM, The late sodium current in heart failure: pathophysiology and clinical relevance, ESC Heart Fail 1(1) (2014) 26–40. [DOI] [PubMed] [Google Scholar]
- [25].Wang DW, Mistry AM, Kahlig KM, Kearney JA, Xiang J, George AL Jr., Propranolol blocks cardiac and neuronal voltage-gated sodium channels, Frontiers in pharmacology 1 (2010) 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Funasako M, Aiba T, Ishibashi K, Nakajima I, Miyamoto K, Inoue Y, Okamura H, Noda T, Kamakura S, Anzai T, Noguchi T, Yasuda S, Miyamoto Y, Fukushima Kusano K, Ogawa H, Shimizu W, Pronounced Shortening of QT Interval With Mexiletine Infusion Test in Patients With Type 3 Congenital Long QT Syndrome, Circ. J 80(2) (2016) 340–5. [DOI] [PubMed] [Google Scholar]
- [27].Hu RM, Tester DJ, Li R, Sun T, Peterson BZ, Ackerman MJ, Makielski JC, Tan BH, Mexiletine rescues a mixed biophysical phenotype of the cardiac sodium channel arising from the SCN5A mutation, N406K, found in LQT3 patients, Channels (Austin) 12(1) (2018) 176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M, Novelli V, Baiardi P, Bagnardi V, Etheridge SP, Napolitano C, Priori SG, Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3, J. Am. Coll. Cardiol 67(9) (2016) 1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG, Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients, Circulation 116(10) (2007) 1137–44. [DOI] [PubMed] [Google Scholar]
- [30].Bankston JR, Yue M, Chung W, Spyres M, Pass RH, Silver E, Sampson KJ, Kass RS, A novel and lethal de novo LQT-3 mutation in a newborn with distinct molecular pharmacology and therapeutic response, PLoS ONE 2(12) (2007) e1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ruan Y, Denegri M, Liu N, Bachetti T, Seregni M, Morotti S, Severi S, Napolitano C, Priori SG, Trafficking defects and gating abnormalities of a novel SCN5A mutation question gene-specific therapy in long QT syndrome type 3, Circ. Res 106(8) (2010) 1374–83. [DOI] [PubMed] [Google Scholar]
- [32].Bezzina CR, Tan HL, Pharmacological rescue of mutant ion channels, Cardiovasc. Res 55(2) (2002) 229–32. [DOI] [PubMed] [Google Scholar]
- [33].Song W, Shou W, Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia, Pediatr. Cardiol 33(6) (2012) 943–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Moreau A, Krahn AD, Gosselin-Badaroudine P, Klein GJ, Christe G, Vincent Y, Boutjdir M, Chahine M, Sodium overload due to a persistent current that attenuates the arrhythmogenic potential of a novel LQT3 mutation, Front Pharmacol 4 (2013) 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL Jr., Schwartz PJ, KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome, Circulation 112(9) (2005) 1251–8. [DOI] [PubMed] [Google Scholar]
- [36].Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH, Lu JT, Jean JC, Omari A, Sampson KJ, Kotton DN, Keller G, Kass RS, Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics, J. Gen. Physiol 141(1) (2013) 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kavey RE, Blackman MS, Sondheimer HM, Phenytoin therapy for ventricular arrhythmias occurring late after surgery for congenital heart disease, Am. Heart J 104(4 Pt 1) (1982) 794–8. [DOI] [PubMed] [Google Scholar]
- [38].Garson A Jr., Gillette PC, Treatment of chronic ventricular dysrhythmias in the young, Pacing Clin. Electrophysiol 4(6) (1981) 658–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.