

Abstract
The majority of prostate cancer (PCa) patients treated with docetaxel develop resistance to it. In order to better understand the mechanism behind the acquisition of resistance, we conducted single cell RNA sequencing (scRNA-seq) of docetaxel sensitive and resistant variants of DU145 and PC3 PCa cell lines. Overall, sensitive and resistant cells clustered separately. Differential gene expression analysis between resistant and sensitive cells revealed 182 differentially expressed genes common to both PCa cell lines. A subset of these genes gave a gene expression profile in the resistant-transcriptome-like sensitive cells similar to the resistant cells. Exploration for functional gene pathways identified 218 common pathways between the two cell lines. Protein ubiquitination was the most differentially regulated pathway and was enriched in the resistant cells. Transcriptional regulator analysis identified potential 321 regulators across both cell lines. One of the top regulators identified was nuclear protein 1 (NUPR1). In contrast to the single cell analysis, bulk analysis of the cells did not reveal NUPR1 as a promising candidate. Knockdown and overexpression of NUPR1 in the PCa cells demonstrated that NUPR1 confers docetaxel resistance in both cell lines. Collectively, these data demonstrate the utility of scRNA-seq to identify regulators of drug resistance. Furthermore, NUPR1 was identified as a mediator of PCa drug resistance, which provides the rationale to explore NUPR1 and its target genes to for reversal of docetaxel resistance.
Keywords: single cell sequencing, prostate cancer, NUPR1
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer and the second leading cause of cancer-related death in men (1). Advanced disease is typically treated with androgen deprivation therapy (ADT); however, patients develop resistance to ADT in what is termed castration-resistant PCa (CRPC) (2). In 2004, docetaxel was approved for the treatment of CRPC (3,4). However, 50% of patients do not respond to docetaxel therapy and the majority of those that do respond relapse within three years of initiating therapy (3). Thus, understanding mechanisms through which docetaxel resistance develops is critical to improving therapy of CRPC.
Docetaxel has been shown to have two main mechanisms of anti-tumor activity; namely, microtubule depolymerization and inhibition of Bcl-2 expression (5,6). However, multiple mechanisms of resistance have previously been identified. Structural changes to β-tubulin block docetaxel from affecting microtubules (7,8). PCa cells can also up-regulate Bcl-2 in order to overcome docetaxel-induced apoptosis (9). However, these previous studies of docetaxel resistance were performed on bulk populations of PCa cells. It is unknown if the resistant cell population develops from a subset of resistant cells already present in the parental population or develop de novo subsequent to therapy. In order to study the process of acquisition of resistant characteristics, different methods of investigation are necessary.
Advances in next generation sequencing have allowed for the investigation at the single cell level. Single cell RNA sequencing (scRNA-seq) allows researchers to investigate the variability and complex gene expression across all the individual cells instead of a more homogeneous expression profile from traditional bulk RNA sequencing of tissues. In PCa, CTCs have been shown to have heterogeneous androgen resistance gene profiles within individual patients (10). This single cell heterogeneity may play a role in the clinical and therapeutic heterogeneous response (11). Additionally, with scRNA-seq it is possible to trace the origin of cancer cells to the cell type of origin (12). Similarly, scRNA-seq of docetaxel sensitive versus resistant cells may uncover mechanisms leading toward development of docetaxel resistance in PCa.
In this study, we conducted scRNA-seq of docetaxel sensitive and resistant variants of the DU145 and PC3 PCa lines. We analyzed the heterogeneity within and between the cell lines. We identified similar gene expression changes and functional gene pathways across both cell lines that are important to the acquisition of resistance. We also identified potential regulators of the resistant gene expression profile shared between both cell lines and identified a specific mediator of chemoresistance. Our findings uncover the heterogeneity in PCa as well as identifying signaling pathways important for the acquisition of docetaxel resistance.
Materials and Methods
Cell lines and Reagents
Parental (sensitive) DU145 and PC3 PCa cells were obtained from American Type Culture Collection (Manassas, VA). Docetaxel resistant variants of the DU145 and PC3 PCa cells have been previously described (13). Cell identification is confirmed every 6 months using short tandem repeat analysis. Cells are tested every three months for Mycoplasma. Cells were used within 3 passages from the time of thawing. Cells were cultured in RPMI 1640 (Invitrogen Co., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Life Technologies, Inc.). Resistant DU145 and PC3 cells were maintained with the addition of docetaxel in DMSO to a final concentration of10 nM while sensitive DU145 and PC3 cells were maintained with the addition of DMSO to a final level of 0.1%
Gene Expression Analysis
For one week, cells were transferred to docetaxel free media. Cells were trypsinized in 0.05% Trypsin EDTA for 5–10 minutes at 37°C and washed with media. The cell suspension was loaded into in the Fluidigm C1™ machine and processed into single cell cDNA libraries according to manufacturer protocol (PN 101–4981). Briefly, full length mRNA-seq libraries were generated from single cells captured using the Fluidigm C1™ Single Cell mRNA Seq IFC, 10–17μm (PN 100–5760) and Fluidigm C1™ Single-Cell Reagent Kit for mRNA Seq (PN 100–6201). Each chip was visually inspected to identify which wells contained cells. Wells containing one cell were included in library preparation ad sequencing. The capture rate was between 78% - 96% across all chips used in this study. Full length cDNA was converted into sequence ready libraries using SMART-seq v4 Ultra Low Input RNA kit for Fluidigm C1™ System (Takara Bio, Cat 635025) and SeqAmp™ DNA Polymerase (Takara Bio, Cat 638504). Library preparation was completed using Nextera XT DNA library prep kit (Illumina, Cat. FC-131–1096) and Nextera XT DNA Library Prep Index Kit (Illumina, Cat FC-131–1002). Samples following PCR reactions as called for in each kit’s manufacturer’s protocol was purified using Agencourt AMPure XP (Beckman Coulter, Item No A63880). Samples were sequenced on Illumina HiSeq 2500 Rapid for DU145 cell line variants and Illumina HiSeq4000 with single end option for PC3 cell line variants. Reads that were below the minimum quality controls were discarded. Each sample was aligned to the Human Genome hg38 (14) using bowtie alignment tool (15). We captured a total of 324 cells across all cell lines. Poor quality cells were removed based on low number of reads as determined using the Fluidigm Singular package (https://www.fluidigm.com/software/) (Supplementary Figure 1). A total of 12 cells were removed. 64 DU145 sensitive cells, 71 DU145 resistant cells, 89 PC3 sensitive cells and 88 PC3 resistant cells were included in all downstream analysis. To identify genes for downstream analysis, we used the Fluidigm Singular package. In brief, genes that were expressed in at less than 10 cells in each cell line were excluded. For the remaining genes, the lowest 15% of expressed genes were excluded. Lastly, genes need to be identified in all four cell line samples to be included in the final gene list. This resulted in 12,862 genes being included for all the subsequence downstream analyses.
Clustering Analysis
Dimensional reduction of cells were conducted using the t-distributed stochastic neighbor embedding method (tSNE) (16). All high-quality genes were included for clustering unless noted. Individual clusters were identified using the method proposed in the Seurat pipeline (17). In brief, the distance metric for identifying clusters is based on principal components (PCs) from principal component analysis. We next embedded the cells in a K-nearest neighbor graph structure and used the Louvain algorithm to iteratively group cells together. For the DU145 only cells, the top 10 PCs were used with a resolution of 0.1. For the PC3 only cells, the top 5 PCs were used with a resolution of 0.3. For all cells, the top 10 PCs were used with a resolution of 0.1.
Differential Expression Analysis
Differential expression analysis was conducted on cells using the scDE R package (18). All genes were included in differential expression analysis. Genes with a FDR adjusted p-value < 0.05 and a log fold change > 1 or < −1 were considered differentially expressed. To test for significance in overlap in Venn Diagrams, we used a hypergeometric probability.
Gene Ontology Analysis
Functional enrichment analysis for cellular location and molecular function ontologies was conducted using PANTHER protein classifications (19,20). Gene pathway analysis and upstream regulator analysis was conducted using Ingenuity Pathway Analysis (IPA) package (Qiagen) (21). Potential regulators must have a p-value less than 0.05 and an activation score above 2 or below −2 to be included in both analyses of DU145 and PC3 cell lines.
Oncomine analysis
We used the web-based database called ONCOMINE (www.oncomine.org) to analyze public microarray cancer data provided from over 700 independent datasets (22). We analyzed a prostate cancer dataset containing 10 primary and 21 hormone-refractory metastatic samples (23). Normalized intensities for NUPR1 for each sample was analyzed. Student t-test was used to determine statistical significance.
Transfection of siRNAs and Plasmid DNA
Plate 2 × 105 cells with 1ml of 10% RPMI medium without antibiotics in 12-well plate. PC3-R and DU145-R cells were transfected with human NUPR1 siRNAs (Catalog #AM16708, ThermoFisher Scientific) or Negative Control #1 siRNA (Catalog # AM4635, ThermoFisher Scientific) by using Lipofectamine RNAiMAX (Cat# 13778030, Thermo Fisher Scientific, USA). PC3-S and DU145-S cells were transfected with p8 (NUPR1) (NM_001042483) Human Tagged ORF Clone (CAT#: RG222237, ORIGENE, Rockville, MD) or pCMV6-AC-GFP vector (CAT#: PS100010, ORIGENE, Rockville, MD) by using Lipofectamine 2000 Reagent (Cat# 11668030, ThermoFisher Scientific, USA) while the cells grow to 80% confluent. 24–72 hours later, the validation of expected change in NUPR1 protein expression was followed by western blot.
Western blot analyses
The cells were lysed with RIPA lysis buffer (Millipore, Billerica, MA) containing a complete protease inhibitor tablet (Sigma). The protein concentration of tumor extracts was determined using BCA Protein Assay Kit (Pierce). Lysates were cleared by centrifugation at 14,000g for 20 min. The supernatant fractions were used for western blot. Protein extracts were resolved by 15% SDS-PAGE and probed with NUPR1(p8) Polyclonal Antibody (Cat #PA1–4177, ThermoFisher Scientific). Antibody to GAPDH (Millipore) was used as loading controls. The Ab binding was revealed using an HRP-conjugated anti rabbit IgG (1:3000, Cell Signaling), or anti-mouse IgG (1:3000, Amersham Pharmacia Biotech, Piscataway, New Jersey, USA). Antibody complexes were detected by SuperSignal West Pico Chemiluminescent Substrate or SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and ChemiDoc Imaging System (Bio-Rad, Hercules, CA).
Cell viability assay
Seed 2 × 104 PC3-S, PC3-R, DU145-S and DU145-R cells in 96-well plate. PC3-R and DU145-R cells were transfected with human NUPR1 siRNAs or Negative Control #1 by using Lipofectamine RNAiMAX. PC3-S and DU145-S cells were transfected with p8 (NUPR1) (NM_001042483) Human Tagged ORF Clone or pCMV6-AC-GFP vector by using Lipofectamine 2000 Reagent. After 24 hours, the transfected cells were treated with 0, 10, 20, 40, 80 and 160 nM Docetaxel. The cells were cultured for 2 days, and then the number of viable cells was measured by proliferation reagent MTS (Promega, Cambridge, MA) as directed by the manufacturer. Docetaxol IC50 was determined by GraphPad Software (San Diego, CA)
Real time PCR
Total RNA form prostate cancer cells was extracted using RNeasy Mini Kit (Qiagen), 2μg of total RNA were reversed-transcribed in a final 20 μl volume, Quantitative real time PCR was performed in triplicate using SYBR Green qPCR MasterMix (Qiagen) in a 10 μl reaction volume on Applied Biosystems QuantStudio 5 Real-Time PCR Systems (ThermoFisher Scientific). Human NUPR1 PKM2 primers (Cat. no. PPH19840F) was purchased from Qiagen. All analyses were performed in triplicate in two independent experiments. Measurements from triplicate Ct values were normalized to GAPDH (Invitrogen).
Data Availability
The accession numbers for the single cell RNA-seq dataset reported in this paper are GSE140440.
Results
Characterization of the Cell Line Response to Drug Resistance
We have previously created and characterized docetaxel resistant DU145 and PC3 prostate cancer cells (13). However, the underlying cellular heterogeneity associated with the development of drug resistance response was not investigated. To investigate the cellular heterogeneity of docetaxel resistance in PCa, we conducted single cell RNA sequencing (scRNA-seq) of both the resistant variant (resistant) and parental (sensitive) PCa cell lines (Fig 1A). Using the Fluidigm C1 system, we isolated 312 total high-quality cells across all the cell lines. Across all four cell lines, we observed genes with higher expression also detected in a higher number of cells (Supplementary Figure 2). The DU145 and PC3 variants the sensitive and resistant variants formed isolated clusters (Fig 1B). In the DU145 variants, four sensitive cells clustered with the resistant population (Fig 1B, right). In the few sensitive cells that clustered with the resistant cells in DU145, we observed similar number of total and mapped reads (Supplementary Fig 3A–C). We do note the few resistant-transcriptome-like sensitive DU145 cells did have a lower number of unique genes and a higher number of unique transcripts compared to the remaining sensitive and resistant cells (Supplementary Figure 3D–E). However, in the PC3 lines, none of the sensitive cells clustered with the resistant cells (Fig 1B, left). We did observe gene expression differences between the sensitive and resistant cell lines in both DU145 and PC3 (Fig 1C and Supplementary Table 1). To focus in on specific candidates that could promote resistance, we examined the top 12 differentially expressed genes in either the DU145 or PC3 sensitive and resistant cells. In the DU145 cells, we observed up-regulation of APOBEC3C expression in the resistant cells compared to sensitive cells (Fig 1D, left). Additionally, in KRT8, KYNU, and ICAM1 had down regulation in the resistant cells compared with the sensitive cells (Fig 1D, left). For the four DU145 sensitive that clustered with the DU145 resistant cells (DU145 resistant-transcriptome-like sensitive cells), we investigated which genes drove the clustering results. We identified 1,788 genes expressed in all four DU145 resistant-transcriptome-like sensitive cells, of which 436 were also up-regulated in the DU145 resistant cells in comparison with the DU145 sensitive cells indicating ~25% of the up-regulated genes in the resistant-transcriptome-like sensitive cells were genes also up-regulated in the DU145 resistant cells. This large concordance may account, in part, for the reason these four sensitive DU145 cells clustered with the resistant cells. We additionally investigated whether the 436 overlapping genes were highly expressed in the DU145 resistant cells. We ranked all 12,862 genes in the DU145 resistant cells based on overall single cell gene expression across all the DU145 resistant cells (Supplementary Table 2). We then noted where each of the 436 overlapping genes were located on the ranked list (Supplementary Figure 3F and Supplementary Table 2). The majority of genes were found in the top 30% of the ranked genes, indicating they would play major role in the clustering algorithm. These results suggest the presence of resistant-transcriptome-like sensitive cells are present in the sensitive population but that they do not fully recapitulate the gene expression profile of the drug resistant cells.
Figure 1: Identification of differentially expressed genes in two different docetaxel-resistant cell lines.
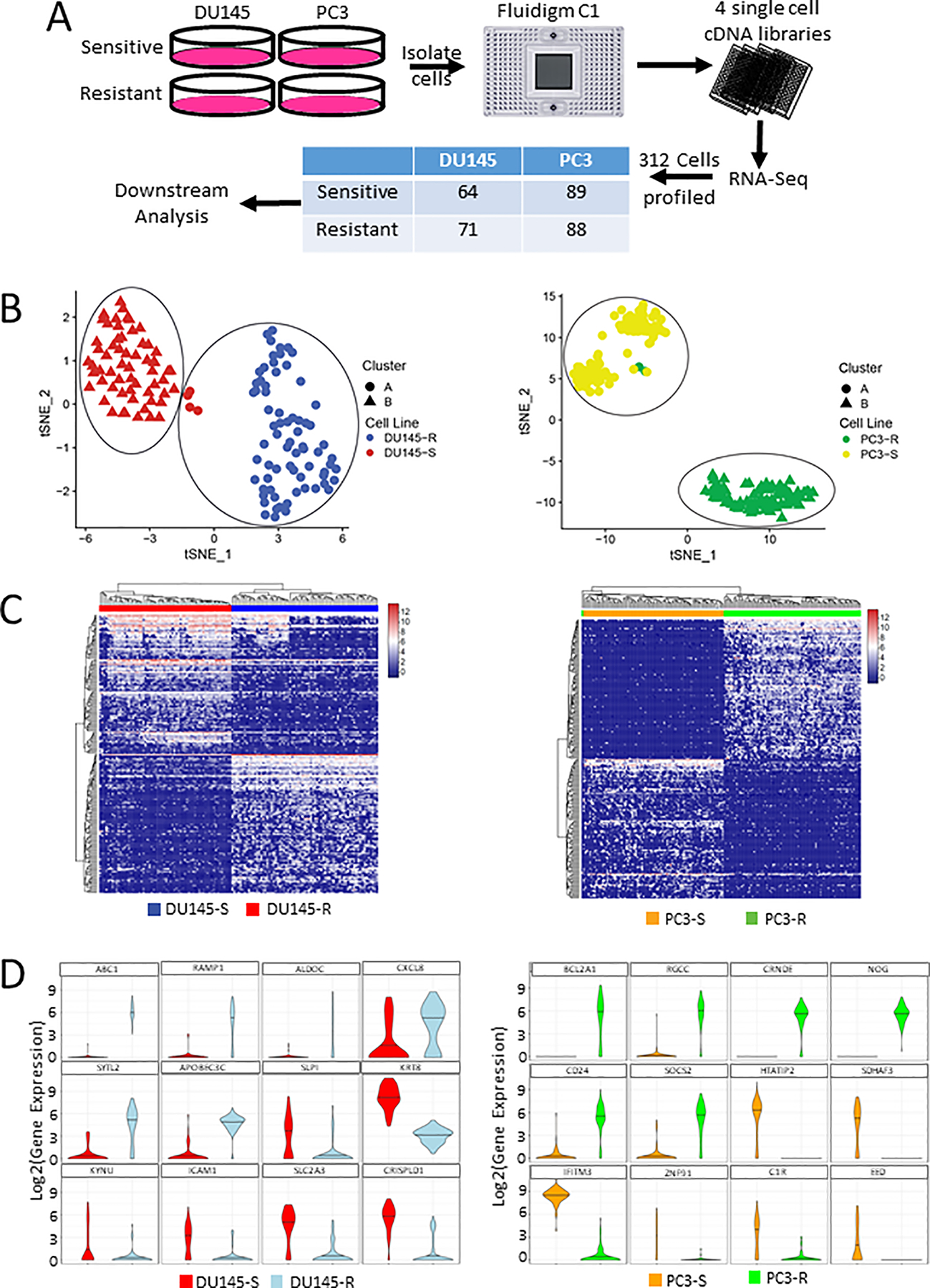
(A) Project workflow. (B) tSNE of all included single cells for (left) DU145 and (right) PC3. (C) Heatmap of top 50 upregulated genes in both sensitive and resistant cell lines. (left) DU145 and (right) PC3. (D) Violin plot of the top 12 differentially expressed genes in resistant vs sensitive cells for (left) DU145 and (right) PC3.
Gene Regulation Changes Across Both Cell Lines
The two cell lines, though both being PCa cell lines, expressed a different set of genes following development of drug resistance suggesting different genetic pathways of resistance (Fig 1). To determine if there were common changes in gene expression among the two different cell types at the single cell level we combined all 312 high quality cells and subjected them to clustering analysis. In the resulting tSNE plot, each the four cell lines clustered separately from each other (Fig 2A). As observed previously, several sensitive cells clustered with their respective resistant counterpart in the DU145 cell lines. We additionally visualized the gene expression of the top 25 expressed genes in each cluster (Fig 2B). To identify common changes in gene expression among all resistant cells compared to all the sensitive cells, we compared the differentially expressed genes in the DU145 and PC3 resistant cells versus their sensitive counter parts (Fig 2C). Among all the differentially expressed genes between the two cell types, 182 of the same genes were differentially expressed in both comparisons. Of these, 139 were changed in the same direction; namely, 4 were upregulated and 135 were downregulated in the resistant versus sensitive cells; whereas, 43 were altered in different directions between the Du145 and PC-3 resistant cells and their sensitive counterparts (Fig 2C, p-value < 4.8e-9). Using only those 139 genes, we clustered all the cells again to see if the cells would cluster based on their drug sensitivity as opposed to their cell line origin. The sensitive cells of both lines clustered together (Fig 2D). However, the resistant cells of DU145 and PC3 still clustered separately except for three PC3 resistant cells that clustered with the sensitive cells (Fig 2D). Since the changes in differential expression were in the same direction for both resistant cells lines, this observation suggests that there are still underlying differences in the gene expression other than regulation changes that result in the two resistant cell lines clustering separate from each other while the two sensitive cell lines cluster together. To investigate this further, we investigated the gene expression levels of the top 12 expressed genes. In the first six genes in the violin plot, both resistant variants had higher expression compared to their sensitive counterparts (Fig 2E). However, in the last six genes in the violin plots, the resistant variants had lower expression compared with their sensitive counterparts (Fig 2E). Together, this suggests that while similar gene directional changes are induced by docetaxel resistance in both cell lines, there are still variation in the cells lines due to the differences in gene expression magnitude.
Figure 2: Identification of differentially expressed genes common to two different docetaxel-resistant cell lines.

(A) tSNE of all four cell lines combined. (B) Heatmap of top 25 expressed genes for each cluster. (C) Venn diagram of differentially expressed genes from comparison of sensitive vs resistant DU145 and PC3 cell lines. (D) tSNE of four cell lines based on only differentially genes. (E) Violin plot of the 6 most upregulated and downregulated genes common between DU145 and PC3.
Gene Pathways Altered in Each Cell Line Following Induction of Drug Resistance
Having identified differentially expressed genes following docetaxel resistance, we investigated the functional implications of the differentially expression. To do this, we analyzed the cellular location and molecular functions of the differentially expressed genes in either DU145 or PC3 using the PANTHER database (19,20). The PANTHER database combines gene function classifications, pathways and statistical analysis tools to enable researchers to explore large-scale sequencing experimental data. There were little differences in the cellular location of the proteins encoded for by the differentially expressed genes across the DU145 and PC3 cell lines (Fig 3A, Supplementary Table 3). This suggests cellular location is similarly altered during drug resistance. In context of the molecular functions of the differentially expressed genes, we observed a higher percentage of genes related to catalytic activity in DU145 variant comparison than in the PC3 variant comparison (38% vs 32%, Fig 3B, Supplementary Table 4). However, a higher percentage of genes related to binding function were identified in the PC3 variant comparison than in the DU145 variant comparison (37% vs 41%, Fig 3B, Supplementary Table 4). Additionally, when we examine the sub-classes of the two largest molecular functions, we observe similar percentages (Fig 3B and Supplementary Table 4). However, there are large differences in the percentage of genes up-regulated or down-regulated in the sub-classes between the two cell lines. 1.3% of DU145 genes and 1.4% of PC3 genes are identified as having lyase activity. However, 2 of the 10 genes in DU145 were up-regulated in the resistant cells; whereas, 10 of the 12 PC3 genes are also up-regulated in the resistant cells (Fig 3B and Supplementary Table 4). This suggests there are some underlying gene function differences and similarities important to the acquisition of drug in resistance in either cell lines.
Figure 3: Functional analysis of genes involved in the acquisition of resistance.
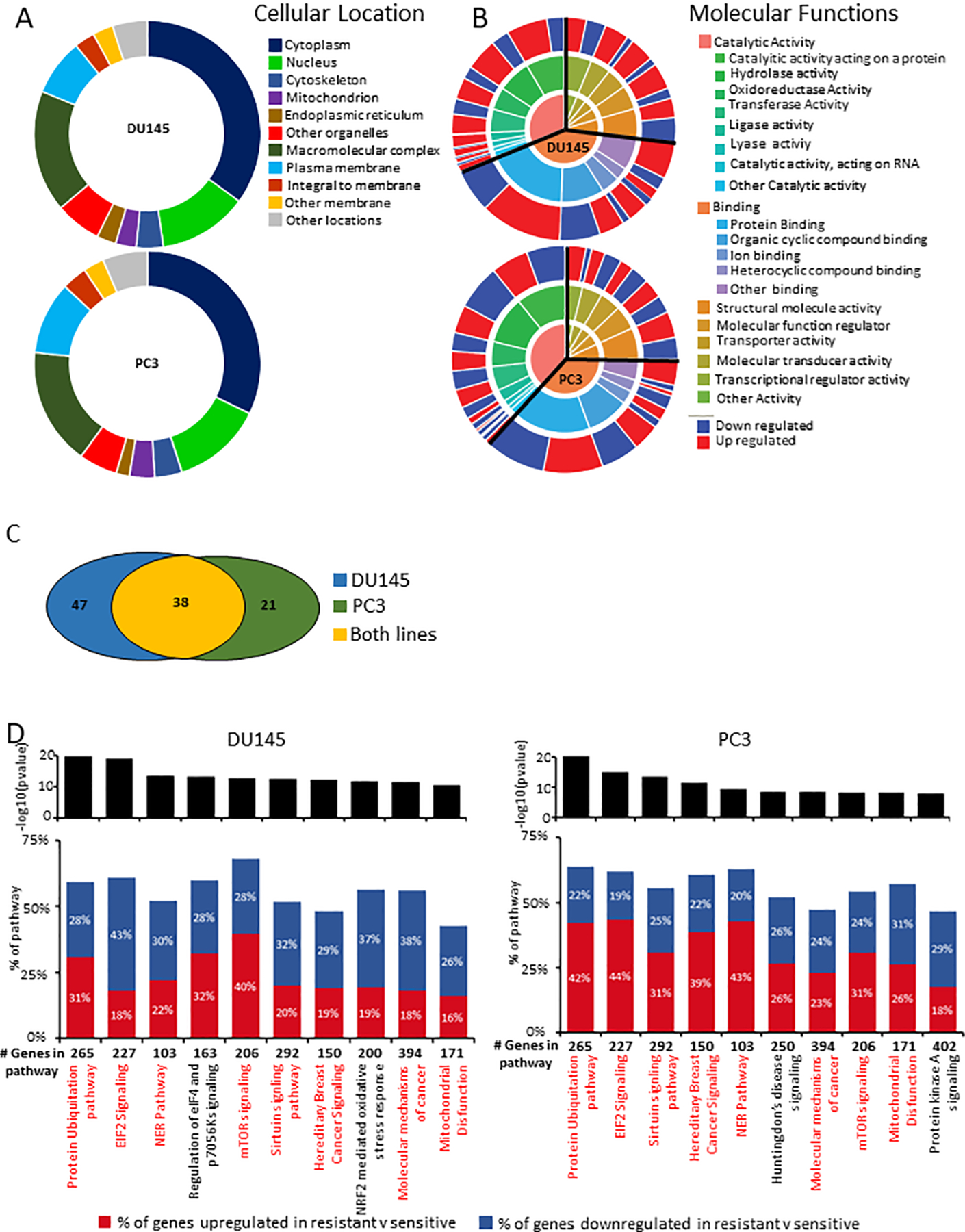
(A) Circle diagram of distribution of the cellular compartment for differentially expressed genes in (top) DU145 and (bottom) PC3. (B) Circle diagram of distribution of molecular function for differentially expressed genes in (top) DU145 and (bottom) PC3. (C) Venn diagram of differentially regulated pathways from comparison of sensitive and resistant cell lines. (D) Top 10 pathways identified through IPA pathway analysis of (left) DU145 and (right) PC3.
To investigate this further, we conducted causal network analysis using the Ingenuity Pathway Analysis (IPA) advanced analytics package (21) on the differentially expressed genes. One hundred and six pathways were identified across both cell lines (p-value < 0.05 in at least one comparison) as being differentially regulated in resistant versus sensitive cells lines of which 38 were common to both cell lines (Fig 3C). Of the top 10 pathways enriched in either cell line (based on p-value), 8 were common to both cell lines (Fig 3D, shared pathways marked in red). The top differentially regulated pathway in both cell lines was protein ubiquitination (Fig 3D). A total of 265 genes are associated with this pathway in which 157 were identified in the DU145 comparison and 169 were identified in the PC3 comparison (Supplementary Table 5). Ubiquitination is a post translational modification, that can mark a protein as target for degradation. Additionally, it is also associated with other functions such as cell signaling, cell cycle or DNA repair (24). Thus our finding provides strong evidence that our analytic method is capable of detecting pathways of resistance. However, this analysis is limited in determining the enrichment direction of each pathway. This suggests the poorly studied pathways we’ve identified are good candidates for further study in relation to docetaxel resistance in prostate cancer.
NUPR1 is a Driver of Docetaxel Resistance
With common gene pathways being dysregulated during drug resistance, we hypothesized these alterations were being driving by similar regulators. To test this, we conducted upstream regulator analysis using IPA to identify potential regulators (21). We identified 560 potential regulators with p-value < 0.05 in either both cell lines of which 471 were common to both cell lines (Fig 4A and Supplementary Table 6). A total of 121 were activated and 200 were inactivated in both comparisons (Fig 4A). To further investigate these putative regulators of docetaxel resistance, we focused on those with an absolute z-score greater than 2. A total of 42 regulators met this criterion (Supplementary Table 6). Of the top ten regulators (based on p-value), 5 are common to both cell lines (Fig 4B). Of these, 4 had activation in the same direction across both cell lines and one had different activation patterns in each cell line (Fig 4B). TP53 was the top regulator with the most significant p-value in both cell lines. This provides validation of our analysis method since TP53 has been shown to drive docetaxel resistance in PCa (25). The second most significant upregulated regulator (based on p-value) was nuclear protein 1 (NUPR1). NUPR1 has not previously been shown to play a role in docetaxel resistance in PCa; whereas, it has been shown to promote resistance in breast cancer (26). To determine if single cell analysis offered an advantage to nominating NUPR1 as a putative candidate over RNA-seq of bulk cell, we analyzed bulk samples of each cell line variant. While we found NUPR1to be upregulated in resistant cells, it was ranked as the 90th and 418th candidate gene in DU145 and PC3 cells, respectively, as opposed to 3rd and 2nd as found in single cell analysis (Supplementary Fig 4). This suggests even through NUPR1 was identified in both bulk and single cell samples, the single cell samples provided stronger evidence for nominating NUPR1 as a candidate in the development of docetaxel resistance. We investigated further why NUPR1 ranked significantly higher as a mediator of resistance using the single cell RNA-seq compared with the bulk sample RNA-seq. This analysis was based on IPA regulator analysis which does not look at the expression of the transcription regulators but rather the expression of the regulator’s targets to provide an approximation of the regulator’s functional state. In many instances, transcriptional regulation occurs independently of the regulator’s expression but rather regulator activity is based on the protein conformation (e.g., heat shock protein) or its localization (e.g. androgen receptor), which provides a rationale for IPA regulator analysis methodology. Thus, we looked at the average expression of NUPR1 pathway mediators in addition to the number of dropout events in both the single cell and bulk sequenced samples (Supplementary Figure 5A–B). In the single cell data, while not all gene targets were expressed in all the of the cells, we observed higher average expression in many of the resistant cells compared with the sensitive cells (Supplementary Figure 5A). However, in the bulk samples, although there was expression in all the samples there was minimal differential expression between the resistant and sensitive variants (Supplementary Figure 5B). This finding of higher overall differential expression of the NUPR1 pathway mediators in resistant versus sensitive cells in the single cell analysis compared to bulk analysis accounts, in part, for the higher ranking of NUPR1 in the single cell versus bulk analysis. The observations of inconsistency between pseudobulk constructed from the average of all of the cells from a single cell RNA-seq analysis versus true bulk is a common finding. In many cases, there is approximately an 80% coefficient of determination (R2) between pseudobulk and true bulk in static cell samples (27,28); however, in instances where a functional study is performed, such as a drug treatment, correlation may be as low as 2% (29). In our study, the R2 between single cell and bulk fold change is 44% for DU145 and 52% for PC3 (Supplementary Figure 5C–D). This level of fit may be due to the relatively small sample size of single cells used. Thus, the observation that the fit between bulk and single cell is only between 44 to 55% could contribute to differences in clustering or ranking of target genes. Finally, to provide one more strategy to account for differences between single cell and bulk; we evaluated all of the genes that were statistically significantly differentially expressed in the single cells and determined if they were also significantly differentially expressed in the bulk cells (Supplementary Figure 5E). We found that of 12 genes significantly differentially expressed in both cell lines based on the single cell data, 3 were not significant in both cell lines based on bulk; and that 5 were only significant in 5 of the cell lines; thus, overall there only 3 (or 25%) of the genes significantly expressed in both bulk cell lines compared to that observed in 100% of the genes when identified in the cell lines in which both genes were differentially expressed based on single cell analysis. These data provide another rationale as to why NUPR1 was shown to rank higher in the single cell versus bulk analysis.
Figure 4: Analysis of upstream regulators.
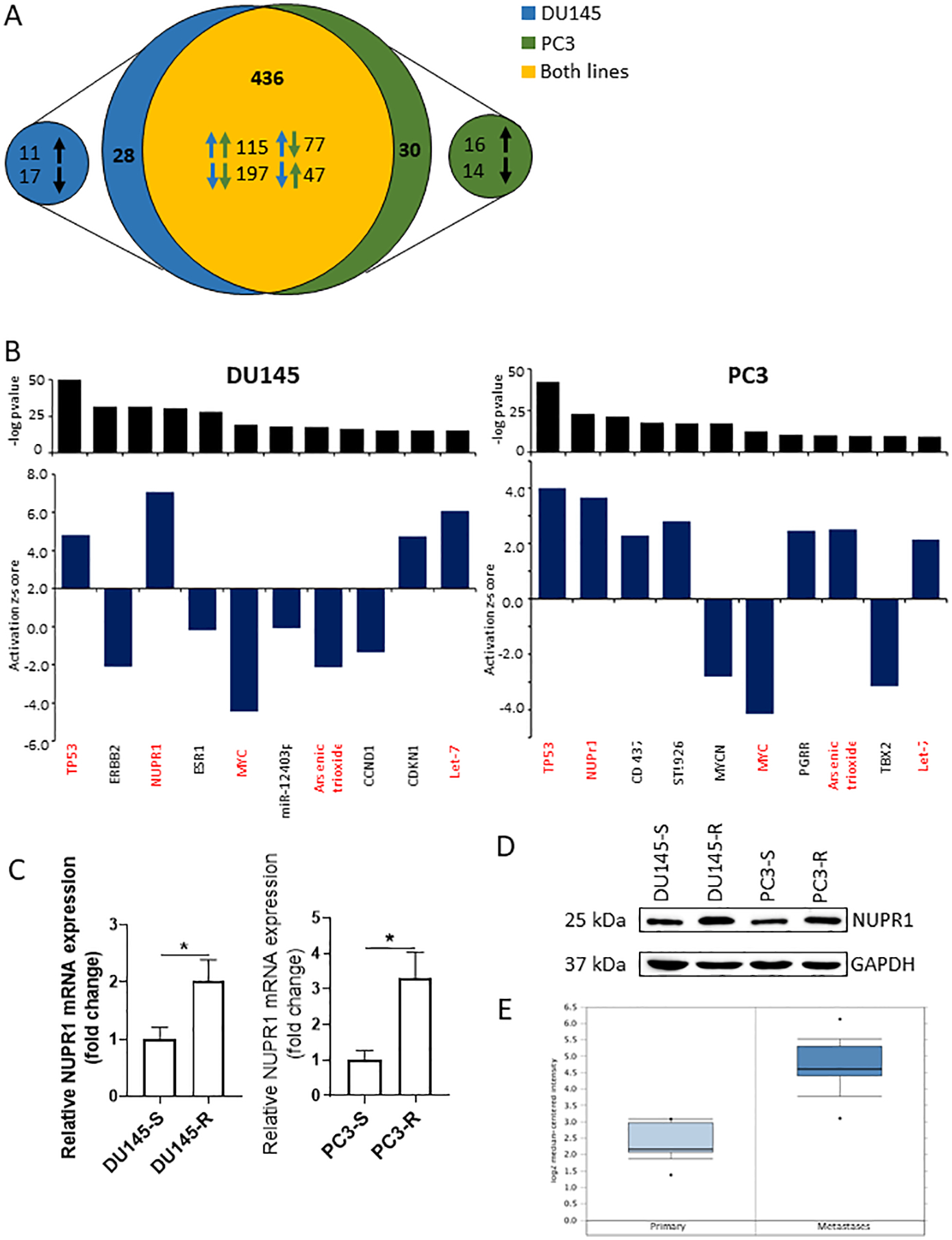
(A) Venn diagram of putative regulators in both cell lines. (B) Top 10 significant regulators in (left) DU145 and (right) PC3 cell line comparisons. (C) mRNA expression level of NUPR1 in DU145 and PC3 variants. (D) Western Blot of NUPR1 expression in DU145 and PC3 variants. (E) NUPR1 expression levels in primary or metastatic PCa patient samples.
To validate the prediction of the upstream regulator analysis, we analyzed mRNA and protein expression of NUPR1 in all cell lines. NUPR1 mRNA and protein expression were upregulated in both resistant cell lines compared to their sensitive counterparts (Fig 4C–D). These findings were consistent with the increased NUPR1 activity predicted by the upstream regulator analysis. To provide some clinical correlation to these findings, we examined for NUPR1 expression in prostate cancers in the Oncomine database. We found that NUPR1 was upregulated in prostate cancer metastases compared to primary tumors (Fig. 4E).
To determine the potential functional role for NUPR1 in docetaxel resistance, we initially determined the impact of knocking down NUPR1 in the DU145 resistant cells. We observed a significant decrease in NUPR1 protein and mRNA expression following knockdown compared with control cells (Fig 5A–B). We then evaluated the impact of NUPR1 knockdown on docetaxel resistance in the DU145 resistant cells. Knockdown of NUPR1 reduced the resistance to docetaxel of the DU145 resistant cells; albeit not to the level of DU145 sensitive cells (Fig. 5C). This was reflected as a decreased IC50 in the NUPR1 knockdown cell DU145 resistant cells compared to the control DU145 resistant cells, but not an IC50 as low as that of the DU145 sensitive cells (Fig 5D). We also evaluated the impact of knockdown of NUPR1 on docetaxel resistance in the PC-3 resistant cells and observed that similar to DU145 cells, knockdown of NUPR1 expression decreased the resistance and associated IC50 to docetaxel in the PC3 resistant cells (Supplementary Fig 6A–D). To provide further evidence for the ability of NUPR1 to promote docetaxel resistance, we overexpressed NUPR1 in the DU145 sensitive cells. Both NUPR1 protein and mRNA expression were increased in the DU145-S cells transfected with NUPR1 compared to the cells transfected with empty vector (Fig 5E–F). Overexpression of NUPR1 increased resistance to docetaxel in the DU145 cells; albeit, not to the level of DU145 resistant cells (Fig 5G). This was reflected as a higher IC50 for the DU145 sensitive cells transfected with the NUPR1 expression vector versus the empty vector, but not as high as the IC50 of the DU145 resistant cells (Fig 5H). We also evaluated the impact of overexpressing NUPR1 on docetaxel resistance in the PC-3 sensitive cells and observed that similar to DU145 cells, overexpression of NUPR1 expression increased the resistance and associated IC50 to docetaxel in the PC3 sensitive cells (Supplementary Fig 6E–H). Together, these data demonstrate that NUPR1 promotes docetaxel resistance in PCa.
Figure 5: NUPR1 necessary for docetaxel resistance.
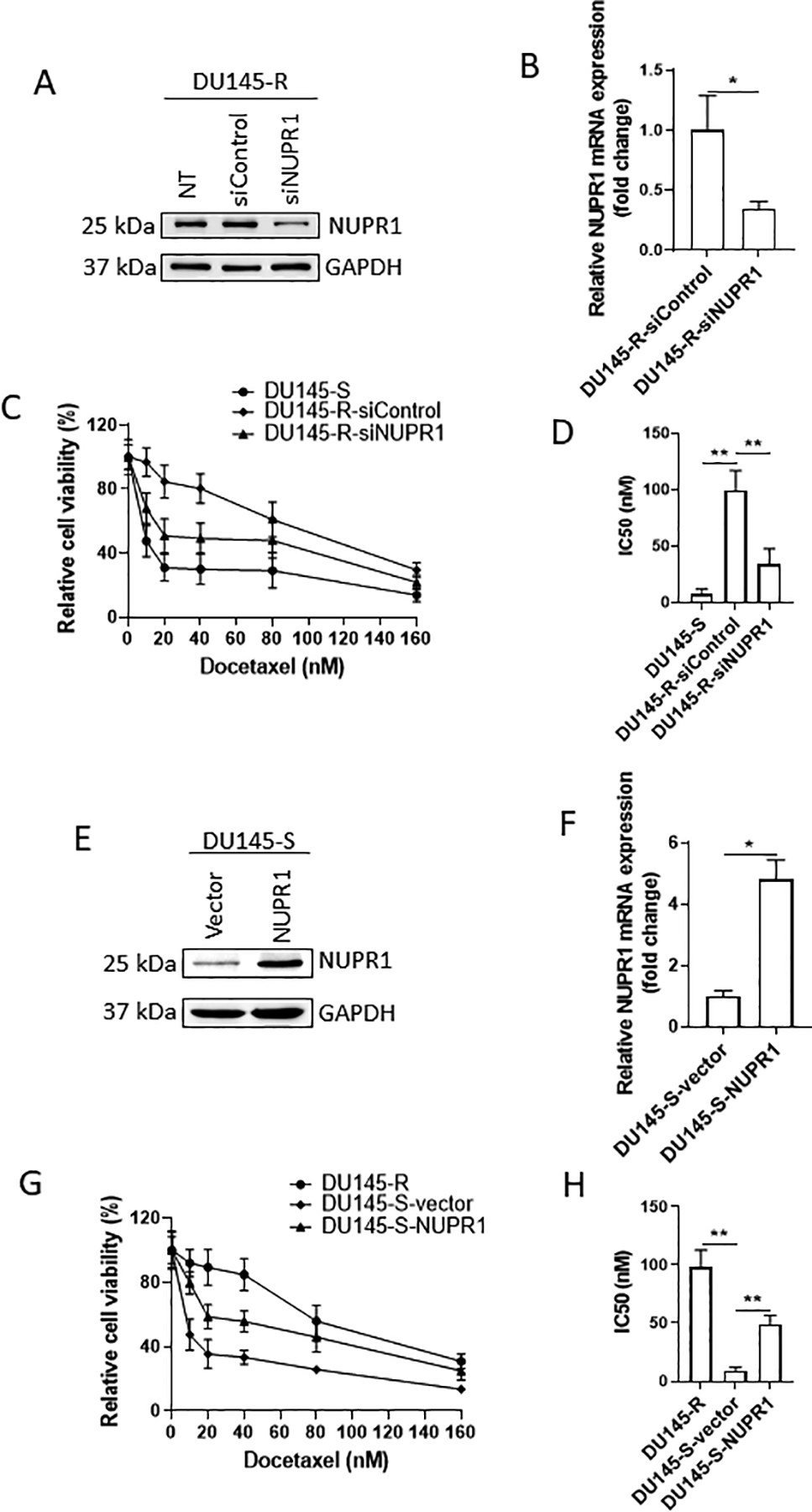
(A) Western Blot of NUPR1 knockdown in DU145 resistant cells. (B) mRNA expression level of NUPR1 in DU145-R cells after NUPR1 or control knockdown. (C) Cell viability in DU145 cells after docetaxel treatment after control or NUPR1 knockdown. (D) IC50 of DU145 cells following either control or NUPR1 knockdown. (E) Western Blot of NUPR1 overexpression in DU145 sensitive cells. (F) mRNA expression of NUPR1 in DU145-S cells following NUPR1 or vector overexpression. (G) Cell viability of DU145 cells after docetaxel treatment and NUPR1 or control overexpression. (H) IC50 of DU145 cells following either control of NUPR1 overexpression.
Discussion
Defining mechanisms of chemotherapeutic resistance provides novel opportunities to overcome this resistance and provide a significant therapeutic impact. In this study, we investigated docetaxel resistance in PCa at the single cell level. We conducted single cell RNA sequencing on two PCa cell lines, DU145 and PC3, and their docetaxel resistant variants. Our analysis revealed both independent and common gene changes in the two cell lines upon development of resistance. Furthermore, our strategy of using upstream regulator analysis using scRNA-seq data identified both a previously recognized mediator of PCa chemoresistance (i.e., TP53) and a novel mediator of PCa chemoresistance (NUPR1). We also identified that single cell analysis outperformed bulk analysis for nominating NUPR1 as a putative mediator of chemoresistance.
Clinically observable drug resistance may occur through selection of pre-existing resistant cells in the chemo-naïve tumor (30) or through development of de novo resistance through chemotherapy-induced genetic or epigenetic mechanisms (31,32). Evidence exists for both theories across multiple cancer types (33,34) and the mechanism may be due to the type of cancer and the type of chemotherapy. Our results suggest that a sensitive PCa cell populations contain a docetaxel resistant sub-population. However, we only observed in this one of the two cell lines we investigated. Additionally, we didn’t see a continuum of cell transitions between the sensitive and resistant cells. Instead, the two subpopulations were discretely separated. This may be due to the fact that the resistant cells were selected in vitro over time and only represent the final resistant cells. Furthermore, the number of cells analyzed were relatively small compared to tumors in vivo, thus limiting our ability to evaluate the full spectrum of cells that could be present in a tumor including limiting the ability to further determine these few cells were functionally resistant. To better understand how similar the resistant-transcriptome-like sensitive cells are to either treatment-naïve or drug resistant cells, more single cells would need to be sequenced (35)
By subjecting the gene expression alterations common to both cell lines to upstream regulator analysis, we identified potential global regulators driving docetaxel resistance in PCa. Some of the top regulators based on p-value in our analysis has previously been shown to play a role in PCa docetaxel resistance. For example, TP53 has been shown to drive docetaxel resistant in PCa (25). Additionally, MYC, the only regulator identified in both cell lines with negative activation (Fig 4B), has been shown to play a role in docetaxel-resistant PCa (36). The identification of a previously known regulator involved in docetaxel resistance suggests that the analytic method we applied to the scRNA-seq data is effective and may be useful to nominate additional putative regulators for further analysis. Furthermore, that NUPR1 was higher candidate on the list based on the scRNA-seq compared to the bulk analysis indicates that there may be complementary value to both analytic methods.
Based on the upstream regulatory analysis, we investigated a potential role for NUPR1 in docetaxel PCa resistance. Previously, NUPR1 was shown to overcome docetaxel treatment in breast cancer cells (37). However, while NUPR1 has been identified as a tumor suppressor in PCa (26) its role in development of docetaxel resistance in PCa was previously described. Our observation from the Oncomine database that NUPR1 is upregulated in advanced PCa provides support for the potential importance of NUPR1 in development of docetaxel resistance in clinical PCa. However, a limitation to these data is the treatment status of these patients is unknown. We hypothesized the primary cancer is not treated with docetaxel whereas, most hormone refractory metastatic cancers are treated with docetaxel. Thus, our finding that NUPR1 is higher in the metastatic vs primary is supportive but not conclusive. Regardless, the increase in advanced PCa patients indicates a potential role for NUPR1 in PCa. This observation combined with our functional studies demonstrating that NUPR1 promotes docetaxel resistance provides a degree of confidence that NUPR1 could contribute to docetaxel resistance in PCa patients. Further research work with clinical samples of pre- and post-docetaxel treatment could provide additional evidence of the role of NUPR1 in PCa.
Our in vitro studies demonstrated that NUPR1 contributed to docetaxel chemoresistance in two different cell lines. However, upon knockdown of NUPR1 activity, the resistant cells, which had diminished resistance, still had an IC50 greater than sensitive cells. Conversely, upon overexpression of NUPR1 activity, the sensitive cells, which gained resistance, still had an IC50 lower than the resistant cells. Taken together, these results demonstrate that NUPR1 contributes to docetaxel resistance, but indicates other factors also contribute to docetaxel resistance. NUPR1 has been previously implicated in multiple pathways that could aid in drug resistance. In breast cells, NUPR1 regulates p21 through formation of a complex of p53 to impart a cell growth and survival advantage (37). NUPR1 also plays a role in TFGβ signaling which can affect drug resistance (26). However, further research is necessary to determine the signaling pathways through which NUPR1 contributes to the development of resistance.
In conclusion, our study provides a view of the development of docetaxel resistance in PCa at the single cell level. Furthermore, it provides a strategy to exploit scRNA-seq data to nominate candidates for drug resistance. While each individual cancer may acquire resistance through independent means, it is likely there is a common subset of alterations that occurs across tumors from different patients that are important to the acquisition of resistance. Through focused analysis of these common genetic changes and the factors their regulatory factors, we identified a novel mediator of docetaxel resistance in PCa which offers a new therapeutic candidate to extend the effectiveness of docetaxel therapy for PCa patients.
Supplementary Material
Implications.
Using single cell sequencing of PCa, we show that NUPR1 plays a role in docetaxel resistance.
Acknowledgment
This work was supported by National Institutes of Health Grants P01093900 (E. Keller); P30CA046592 (E. Keller); UL1TR002240 (P. Schnepp); T32HG00040 (N. Wakim); and P30CA046592 (J. Hui). We would like to thank the University of Michigan’s Advanced Research Computing Technology Services for providing the computing resources. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA046592 by the use of the following Cancer Center Shared Resource that supported this study: Rogel Cancer Center Single Cell Analysis Shared Resource (P30 CA046592).
Footnotes
Conflict of interest: The authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: A Cancer Journal for Clinicians. 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Puente J, Grande E, Medina A, Maroto P, Lainez N, Arranz JA. Docetaxel in prostate cancer: a familiar face as the new standard in a hormone-sensitive setting. Ther Adv Med Oncol. 2017;9:307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrylak DP, Tangen CM, Hussain MHA, Lara PN, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. [DOI] [PubMed] [Google Scholar]
- 4.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. [DOI] [PubMed] [Google Scholar]
- 5.Haldar S, Basu A, Croce CM. Bcl2 is the guardian of microtubule integrity. Cancer Res. 1997;57:229–33. [PubMed] [Google Scholar]
- 6.Pienta KJ. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin Oncol. 2001;28:3–7. [DOI] [PubMed] [Google Scholar]
- 7.Berrieman HK, Lind MJ, Cawkwell L. Do beta-tubulin mutations have a role in resistance to chemotherapy? Lancet Oncol. 2004;5:158–64. [DOI] [PubMed] [Google Scholar]
- 8.Sève P, Dumontet C. Is class III beta-tubulin a predictive factor in patients receiving tubulin-binding agents? Lancet Oncol. 2008;9:168–75. [DOI] [PubMed] [Google Scholar]
- 9.Yamanaka K, Rocchi P, Miyake H, Fazli L, So A, Zangemeister-Wittke U, et al. Induction of apoptosis and enhancement of chemosensitivity in human prostate cancer LNCaP cells using bispecific antisense oligonucleotide targeting Bcl-2 and Bcl-xL genes. BJU Int. 2006;97:1300–8. [DOI] [PubMed] [Google Scholar]
- 10.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shoag J, Barbieri CE. Clinical variability and molecular heterogeneity in prostate cancer. Asian J Androl. 2016;18:543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barros-Silva JD, Linn DE, Steiner I, Guo G, Ali A, Pakula H, et al. Single-Cell Analysis Identifies LY6D as a Marker Linking Castration-Resistant Prostate Luminal Cells to Prostate Progenitors and Cancer. Cell Reports. 2018;25:3504–3518.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeda M, Mizokami A, Mamiya K, Li YQ, Zhang J, Keller ET, et al. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. The Prostate. 2007;67:955–67. [DOI] [PubMed] [Google Scholar]
- 14.Consortium IHGS. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 15.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Maaten L, Hinton G. Visualizing Data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–605. [Google Scholar]
- 17.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology. 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Meth. 2014;11:740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003;13:2129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia. 2004;6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi D, Grossman SR. Ubiquitin becomes ubiquitous in cancer. Cancer Biol Ther. 2010;10:737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C, Zhu Y, Lou W, Nadiminty N, Chen X, Zhou Q, et al. Functional p53 Determines Docetaxel Sensitivity in Prostate Cancer Cells. Prostate. 2013;73:418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chowdhury UR, Samant RS, Fodstad O, Shevde LA. Emerging role of nuclear protein 1 (NUPR1) in cancer biology. Cancer Metastasis Rev. 2009;28:225–32. [DOI] [PubMed] [Google Scholar]
- 27.Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol. Nature Publishing Group; 2019;20:915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collin J, Zerti D, Queen R, Santos-Ferreira T, Bauer R, Coxhead J, et al. CRX expression in pluripotent stem cell derived photoreceptors marks a transplantable subpopulation of early cones: CRX expression in PSC- derived photoreceptors. STEM CELLS. 2019;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Chen L, Chen Z, Zhang W. RNA-seq based transcriptomic analysis of single bacterial cells. Integr Biol (Camb). 2015;7:1466–76. [DOI] [PubMed] [Google Scholar]
- 30.Frank SA. Somatic mosaicism and cancer: inference based on a conditional Luria-Delbrück distribution. J Theor Biol. 2003;223:405–12. [DOI] [PubMed] [Google Scholar]
- 31.Coldman AJ, Goldie JH. A stochastic model for the origin and treatment of tumors containing drug-resistant cells. Bull Math Biol. 1986;48:279–92. [DOI] [PubMed] [Google Scholar]
- 32.Iwasa Y, Michor F, Nowak MA. Evolutionary dynamics of escape from biomedical intervention. Proc Biol Sci. 2003;270:2573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foo J, Michor F. Evolution of acquired resistance to anti-cancer therapy. Journal of Theoretical Biology. 2014;355:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature Reviews Cancer. 2013;13:714–26. [DOI] [PubMed] [Google Scholar]
- 35.Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med [Internet]. 2017. [cited 2019 May 15];9 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5561556/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatano K, Yamaguchi S, Nimura K, Murakami K, Nagahara A, Fujita K, et al. Residual Prostate Cancer Cells after Docetaxel Therapy Increase the Tumorigenic Potential via Constitutive Signaling of CXCR4, ERK1/2 and c-Myc. Mol Cancer Res. 2013;11:1088–100. [DOI] [PubMed] [Google Scholar]
- 37.Clark DW, Mitra A, Fillmore RA, Jiang WG, Samant RS, Fodstad O, et al. NUPR1 interacts with p53, transcriptionally regulates p21 and rescues breast epithelial cells from doxorubicin-induced genotoxic stress. Curr Cancer Drug Targets. 2008;8:421–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the single cell RNA-seq dataset reported in this paper are GSE140440.