

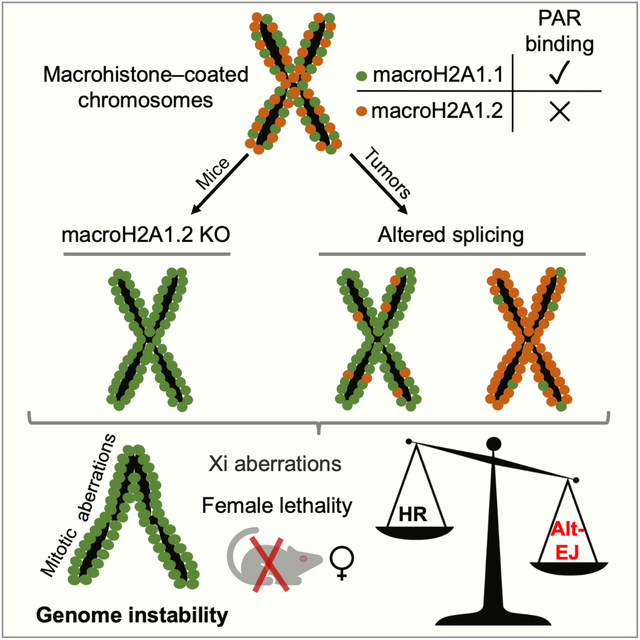
Summary
The inactive X chromosome (Xi) is inherently susceptible to genomic aberrations. Replication stress (RS) has been proposed as an underlying cause, but the mechanisms that protect from Xi instability remain unknown. Here, we show that macroH2A1.2—an RS-protective histone variant enriched on the Xi—is required for Xi integrity and female survival. Mechanistically, macroH2A1.2 counteracts its structurally distinct and equally Xi-enriched alternative splice variant, macroH2A1.1. Comparative proteomics identified a role for macroH2A1.1 in alternative end-joining (alt-EJ), which accounts for Xi anaphase defects in the absence of macroH2A1.2. Genomic instability was rescued by simultaneous depletion of macroH2A1.1 or alt-EJ factors, and mice deficient for both macroH2A1 variants harbor no overt female defects. Notably, macroH2A1 splice variant imbalance affected alt-EJ capacity also in tumor cells. Together, these findings identify macroH2A1 splicing as a modulator of genome maintenance that ensures Xi integrity and may, more broadly, predict DNA repair outcome in malignant cells.
Graphical Abstract
One Sentence Summary:
Splicing-regulated macroH2A1 histone variants control genome maintenance and ensure inactive X chromosome stability and female viability.
Blurb
Sebastian et al. demonstrate that balanced expression of macroH2A1 splice variants is essential to ensure genome integrity. Deletion of macroH2A1.2 in mice results in X chromosome instability through aberrant alternative end-joining mediated by macroH2A1.1. MacroH2A1 splice variant imbalance is also predictive of DNA repair outcome in cancer cells.
Introduction
Susceptibility to genome instability is intimately linked to the surrounding chromatin environment (Dabin et al., 2016). A prominent example is the highly condensed, inactive X chromosome (Xi), which acts as an epigenetic hotspot for genomic aberrations. Compared to autosomes and its active counterpart (Xa), the Xi exhibits elevated risk for genome instability in the form of chromosome loss, micronuclei formation and mutation during ageing and malignancy (Jager et al., 2013; Machiela et al., 2016; Russell et al., 2007). The Xi is characterized by a repressive chromatin environment, which ensures dosage compensation of X-linked gene expression in female mammals but interferes with DNA replication initiation. As a result, the Xi is among the latest replicating chromosomes and its unique replication dynamics have been associated with increased susceptibility to replication stress (RS) (Koren and McCarroll, 2014). Accumulation of under-replicated DNA can cause DNA fragility and DNA double-strand strand breaks (DSBs) in late S/G2, pointing to RS as a likely contributor to Xi-specific genome instability (Bakhoum et al., 2017; Minocherhomji et al., 2015). The mechanisms that protect female cells from this inherent Xi instability remain to be investigated.
RS is epigenetically modulated by the homologous recombination (HR)-promoting macroH2A1.2 histone variant, which forms protective chromatin domains across fragile DNA (Khurana et al., 2014; Kim et al., 2018). Both macroH2A1.2 and its alternative splice variant macroH2A1.1 are enriched on the mammalian Xi to form the Xi macro-body (Costanzi and Pehrson, 1998) (Fig. S1A). Of note, macro-histones are dispensable for initiation and maintenance of X chromosome inactivation (XCI) (Changolkar et al., 2007), and the functional relevance of Xi-enriched macro-histones remains elusive. Here, we investigate macroH2A1.2 function using a splice variant-specific knockout mouse model. We identify a novel, splicing-modulated role for macroH2A1 variants in genome maintenance, which provides a rationale for macroH2A1.2 accumulation on the Xi and has implications not only for Xi integrity but also more broadly for DNA repair pathway control in malignant cells.
Results
MacroH2A1.2 ensures anaphase integrity and female viability.
To selectively inactivate macroH2A1.2, we generated splice variant-specific knockout mice via targeted inactivation of the macroH2A1.2-encoding exon 6a in ES cells. MacroH2A1.2 expression was undetectable in macroH2A1.2−/− mice, while expression of the macroH2A1.1 variant was increased by ~ two-fold both at the overall RNA / protein level and on the Xi, consistent with exclusive and compensatory use of the macroH2A1.1-encoding exon 6b (Fig. 1A, B, Fig. S1B–D). MacroH2A1.2-deficient male mice were born at the expected rate. However, female macroH2A1.2−/− mice were 40 – 50 % less frequent than males (Fig. 1C, D. Fig. S1E). Loss of females was observed as early as embryonic day E 9.5 with a male-to-female ratio of 1.75 at E 9.5 and 1.9 at E 12.5 (Fig. 1D), indicative of a female-biased, developmental defect. Similar to previous findings in mice lacking both macroH2A1 splice variants (Changolkar et al., 2007), ablation of macroH2A1.2 did not interfere with XCI: (i) expression and spreading of the XCI-initiating Xist non-coding RNA occurred normally in the absence of macroH2A1.2 in two distinct ES cell-based XCI models (Fig. 1E, F, Fig. S1F–N) (Engreitz et al., 2013; Jonkers et al., 2008), and (ii) repression of Xi-linked genes in differentiating, macroH2A1.2-deficient ES cells was comparable to WT cells (Fig. S1F). Female-biased embryonic lethality in the absence of XCI defects has been observed previously as a result of aberrant replication and concomitant genome instability (McNairn et al., 2019). Supporting a role for macroH2A1.2 in the latter, the majority of surviving macroH2A1.2−/− females showed varying levels of X aneuploidy in peripheral blood-derived mono-nuclear cells (PBMCs) (Fig. 1C, G, Fig. S1E). No X aneuploidy was detected in PBMCs from KO males or WT females (Fig. S1O). Since numerical chromosomal aberrations have been linked to RS in both yeast and humans (Ait Saada et al., 2017; Burrell et al., 2013), we asked if macroH2A1.2 loss promotes RS at the Xi. Using co-Immuno-fluorescence (IF) analysis for phosphorylated single-stranded DNA binding protein RPA32 (pRPA), a marker of RS (Marechal and Zou, 2015) and trimethylated histone H3K27, a marker for the Xi (Plath et al., 2003), we observed a significant increase in Xi-associated RS in macroH2A1.2-deficient MEFs but not WT cells that were arrested at G2/M following treatment with a low dose of the DNA polymerase inhibitor aphidicolin (Fig. 1H). Together, these findings uncover a role for macroH2A1.2 in protecting from Xi-associated RS, Xi aneuploidy and female-biased lethality.
Fig. 1. MacroH2A1.2 loss impairs female survival and X ploidy.
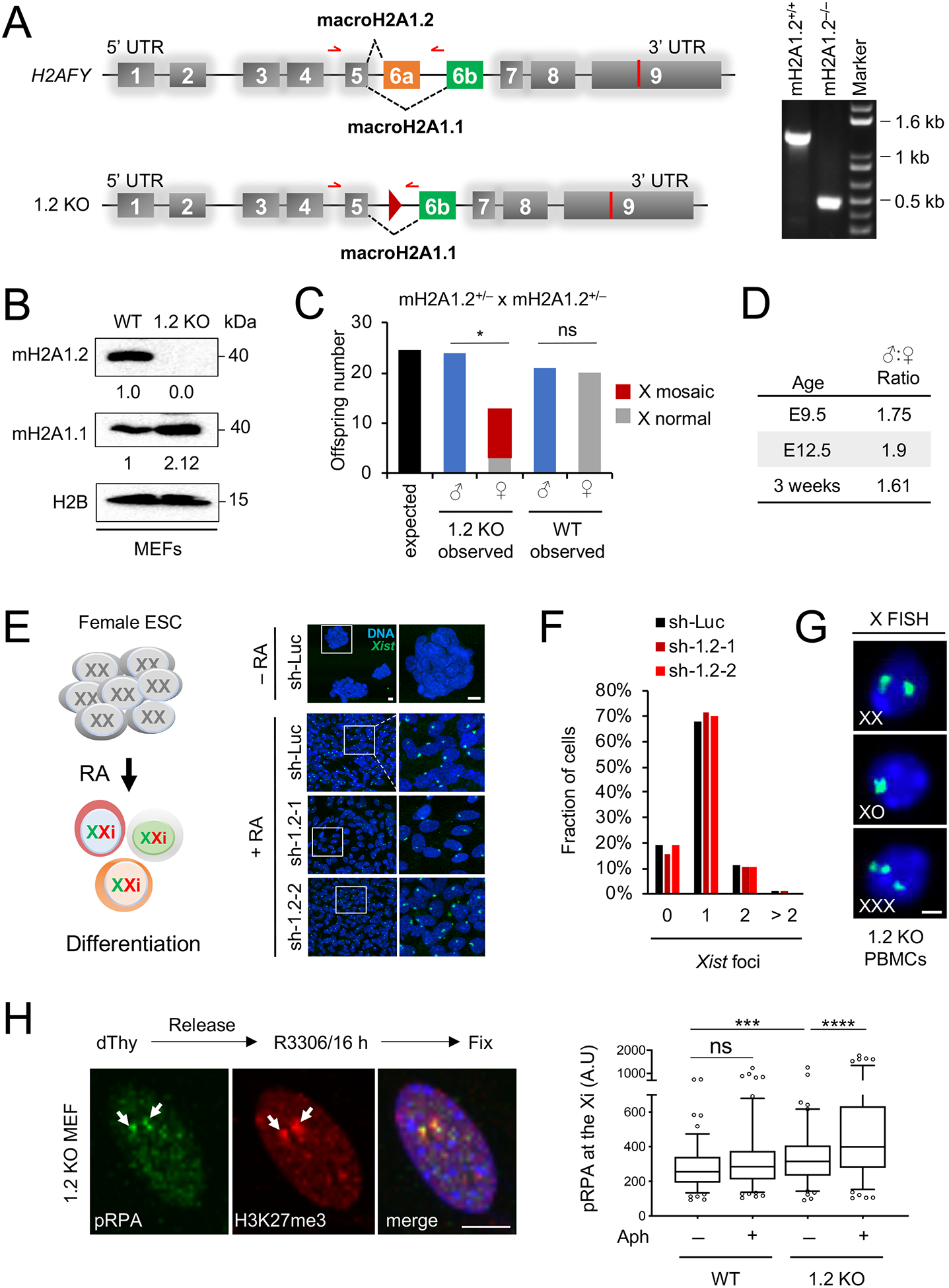
(A) Schematic of the macroH2A1-encoding H2AFY gene and the macroH2A1.2 knockout allele (1.2 KO), not to scale. Exon 6a was deleted by Cre/loxP-mediated recombination, resulting in a residual loxP site (triangle). Alternative splicing resulting in macroH2A1.1 or macroH2A1.2 is indicated. Red arrows depict primers used for PCR genotyping, resulting in a 1.3 kb fragment for the WT allele and 0.5 kb for the 1.2 KO allele. A representative PCR for the indicated genotypes is shown. (B) Western blot for macroH2A1 variants in MEFs from WT and 1.2 KO littermates. (C) Number of WT and 1.2 KO males and females at time of weaning, based on 28 litters from macroH2A1.2+/− parents. X-mosaicism was determined as in (G) for all female KO offspring, and for a subset of male KO and female WT offspring, see Fig S1O. * p = 0.01, based on a two-tailed binomial test, ns: not significant. (D) Male:Female ratio in 1.2 KO mice analyzed at the indicated ages, twoway comparisons between each of the three age groups revealed no significant differences based on Fisher’s exact test. (E) Xist induction in differentiating female mouse ES cells expressing a control shRNA (sh-Luc, n = 218 cells) or one of two distinct shRNAs against macroH2A1.2, sh-1.2–1 (n = 260 cells) and sh-1.2–2 (n = 379 cells), see Table S2 for shRNA sequences. A schematic for retinoic-acid induced ES cell differentiation is shown. Xist was detected by RNA FISH, nuclei were stained with Hoechst 33342, representative images and enlarged inlays are shown, scale bars: 10 μm. (F) Fraction of cells with the indicated number of Xist foci, based on (E). (G) X-chromosome FISH in PBMCs from 1.2 KO mice. Female cells with X counts other than XX were considered X-mosaic, scale bar: 10 μm. (H) Representative IF images and quantification showing pRPA localization to the Xi. H3K27me3 is used to mark the Xi (white arrows). A minimum of 100 cells were analyzed per group. scale bar: 10 μm. *** p < 0.001, **** p < 0.0001, based on Mann-Whitney U test. See also Fig. S1, Table S2.
MacroH2A1.2 loss promotes Xi-associated anaphase instability
Aneuploidy is the result of defective anaphase progression. To determine if macroH2A1.2 helps ensure anaphase integrity, we first measured the overall frequency of mitotic aberrations including DNA bridges and lagging chromosomes in mouse embryonic fibroblasts (MEFs) from both WT and macroH2A1.2−/− mice. MacroH2A1.2 deficiency caused a pronounced increase in anaphase defects (Fig. 2A, B), which were further aggravated by treatment with etoposide, an inhibitor of type II topoisomerases (TOP2) that interferes with decatenation of under-replicated genomic DNA to promote DNA fragility (Downes et al., 1994) (Fig. 2A, B). Treatment with etoposide was restricted to 1 h prior to cell harvest, pointing to DSB formation in G2/M as the main cause for etoposide-induced anaphase defects. Anaphase defects further coincided with increased accumulation of cells in G2 and a reduction in mitotic index, supporting a defect in mitotic entry or progression (Fig. S2A, B). The majority of anaphase bridges and lagging chromosomes did not contain centromeres, ruling out kinetochore-related segregation defects (Fig. S2C). Together, these observations identify a role for macroH2A1.2 in the suppression of anaphase bridge formation.
Fig. 2. MacroH2A1.2 protects from Xi-specific anaphase defects and chromosomal instability.
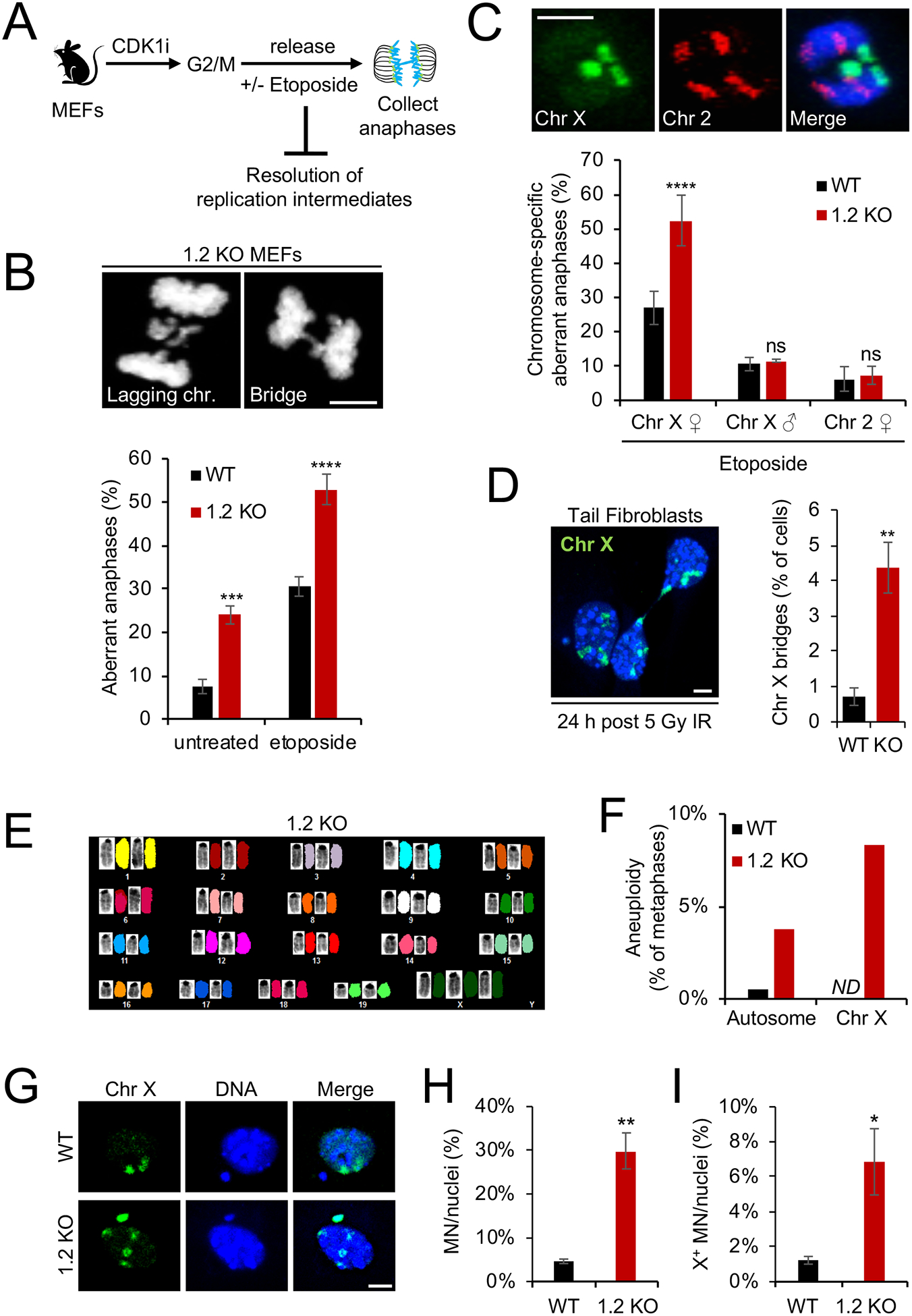
(A) Schematic for anaphase collection. MEFs from WT or macroH2A1.2 KO (1.2 KO) mice were arrested at G2/M via inhibition of CDK1 (CDK1i), followed by release in the presence or absence of etoposide and analysis 50 min thereafter. (B) Frequency of anaphase aberrations in WT or 1.2 KO MEFs in the presence or absence of etoposide. At least 70 metaphases were analyzed per sample, representative images for lagging chromosomes and DNA bridges are shown, scale bar: 10 μm. (C) Frequency of X or chromosome 2 specific anaphase aberrations expressed as percent of total aberrant anaphases in male or females MEFs treated with etoposide. Representative FISH images are shown, scale bar: 10 μm. At least 50 metaphases were analyzed per sample. (D) Frequency of X-chromosome bridges in asynchronous WT and 1.2 KO mouse tail fibroblasts 24 h after 5 Gy of IR. At least 100 metaphases were analyzed per sample, a representative FISH image is shown. DNA was counterstained with Hoechst 33342. Scale bar: 10 μm. (E) Representative spectral karyotype (SKY) analysis of a 1.2 KO metaphase with X aneuploidy. Chromosome-specific FISH paints unambiguously identify all 22 mouse chromosomes. (F) Frequency of autosomal or X chromosomal aneuploidy in WT (n = 34 metaphases) and 1.2 KO MEFs (n = 36 metaphases), based on SKY analysis in (E). One of two representative experiments is shown. ND: not detected. Autosomes with one or more aneuploidy events across 34 metaphases include chromosomes 1, 3, 5, 6, 8, 9, 10, 12, 13, 14, 16, 17, 19. (G) X FISH counterstained with DAPI identifies X+ micronucleus and X aneuploidy in 1.2 KO MEF nucleus. A WT nucleus with X− micronucleus is shown as a control. Scale bar: 10 μm. (H, I) Fraction of total (H) and X+ micronuclei (I) in WT and 1.2 KO MEFs following etoposide treatment, normalized to total cell number and mitotic index (see Fig. S2B). Values are expressed as mean and SD from three independent fibroblast preparations, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, based on Student’s two-tailed t-test.. See also Fig. S2.
To determine whether macroH2A1.2 loss-associated anaphase defects are biased towards the Xi, we performed selective spectral karyotyping using fluorescent in situ hybridization (FISH) probe sets specific for either the X chromosome or a comparably sized autosome, chromosome 2. Chromosome X accounted for ~ 60% of all DNA bridges or lagging chromosomes in anaphases from etoposide-treated macroH2A1.2−/− MEFs, compared to ~ 30% in WT cells (Fig. 2C). In contrast, anaphase defects involving chromosome 2 were comparable between WT and macroH2A1.2−/− MEFs and represented ~ 5% of all DNA bridges, as expected based on chromosome size. Moreover, the frequency of X-specific anaphase aberrations in male MEFs was unaffected by macroH2A1.2 deletion and was overall significantly lower than in females, consistent with the Xi being less stable than the Xa (Fig. 2C) (Jager et al., 2013; Machiela et al., 2016; Russell et al., 2007). Increased Xi-anaphase defects were further observed upon ionizing radiation (IR) and persisted into the subsequent G1 phase in the form of macro-body-associated, nucleoplasmic bridges (Fig. 2D, Fig. S2D).
Aberrant formation and / or resolution of anaphase bridges can cause chromosome missegregation, aneuploidy and micronuclei accumulation (Bakhoum et al., 2017). Spectral karyotyping of macroH2A1.2-deficient metaphase MEFs revealed an increase in aneuploidy that was most pronounced for X chromosomes (Fig. 2E, F), consistent with our findings in PBMCs from surviving macroH2A1.2−/− female mice. Little to no aneuploidy was observed in WT cells. Moreover, macroH2A1.2 loss resulted in a five-fold increase in micronuclei formation in response to etoposide-induced DSB formation in G2/M, compared to WT cells (Fig. 2G, H). X-FISH analysis of the same cells revealed both increased X chromosome-positive micronuclei and X aneuploidy (Fig. 2G, I). Micronuclei from macroH2A1.2−/− MEFs were also significantly enriched for the Xi chromatin mark H3 trimethyl-K27 (Fig. S2E) (Jager et al., 2013). Together, these observations support the notion that anaphase aberrations involving the late-replicating Xi promote enhanced genome instability, which may be at least in part responsible for female-biased lethality of macroH2A1.2-deficient mice.
Variant-specific proteomes reveal a role for macroH2A1.1 in alternative end joining
The above findings stand in sharp contrast to previous reports in mice deficient in both macroH2A1 splice variants, which show no overt defects in female survival or genome integrity (Changolkar et al., 2007). We thus considered the possibility that the phenotypes observed in macroH2A1.2−/− MEFs and mice may be a consequence of unbalanced macroH2A1.1 function. Consistent with this notion, macroH2A1 splice variants have opposing roles in various cellular processes, including the control of HR and cell growth (Chen et al., 2015; Khurana et al., 2014; Kim et al., 2019). MacroH2A1 splice variants differ in 33 aa within the macrodomain, which allows macroH2A1.1 but not macroH2A1.2 to bind ADP-ribose derivatives (Kustatscher et al., 2005).
To understand the molecular basis for differential macroH2A1 splice variant functions, we sought to comprehensively interrogate variant-specific protein interactomes. Flag-tagged macroH2A1 cDNA (F-mH2A1) was targeted into a single genomic locus in HEK293 T-REx cells, ensuring expression levels comparable to WT macroH2A1.2. Note that endogenous macroH2A1.1 is poorly expressed in immortalized cells including HEK293 cells (Fig. 3A, Fig. S3A). Nuclear protein lysates from F-mH2A1.1- and F-mH2A1.2-expressing cell lines were subjected to Flag-co-immunoprecipitation (IP), followed by differential dimethyl-labeling using CH2O (light) and CD2O (medium), respectively (Fig. 3A). Co-IP specificity was validated using H2B as positive control and PCNA as negative control (Fig. 3B). Subsequent comparative mass spectrometry (MS) analysis of two independent co-IP experiments identified 314 common interactors, including known macroH2A1 binding proteins such as core histones and the FACT chaperone complex (Table S1, Fig. 3C) (Kim et al., 2018). Only four macroH2A1 interactors exhibited a reproducible, greater than two-fold preference for macroH2A1.1: poly(ADP)-ribose polymerase 1 (PARP1), arginine and serine rich coiled-coil 2 (RSRC2), tyrosyl-DNA phosphodiesterase 1 (TDP1) and DNA ligase 3 (LIG3). With an approximately four-fold enrichment over macroH2A1.2, LIG3 showed the strongest macroH2A1.1 preference, which was independently confirmed by Western blot (Fig. 3D). LIG3 interaction with macroH2A1.1 was sensitive to PARP inhibition (Fig. 3D, Fig. S3B), and a similar effect was observed for PARP1, in agreement with earlier reports (Ouararhni et al., 2006; Timinszky et al., 2009) (Fig. 3D).
Fig. 3: Splice variant interactomes identify macroH2A1.1 as a mediator of alt-EJ.
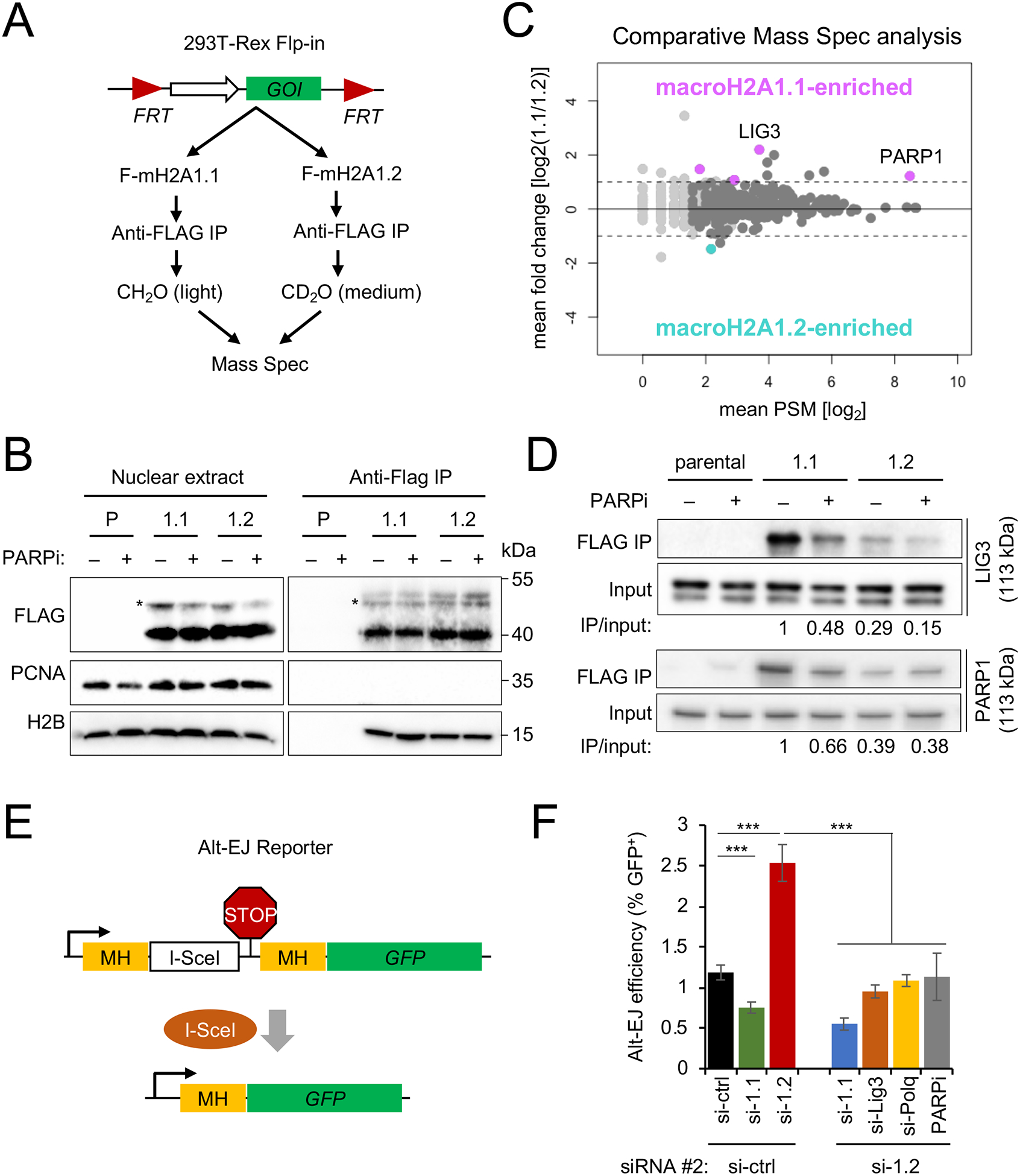
(A) Schematic for macroH2A1 splice variant interactome isolation and comparative MS. HEK293 TREx cells carrying a single-copy cDNA encoding of Flag-macroH2A1.1 (F-mH2A1.1) or Flag-macroH2A1.2 (F-mH2A1.2) inserted via Flp recombination were subjected to Flag-IP, followed by stable isotope dimethyl-labeling and combined MS. GOI: gene of interest. (B) Western blot for the indicated proteins in nuclear lysates or Flag-IP lysates from parental (P) and macroH2A1 splice variant-transgenic HEK293 T-REx cells (1.1, 1.2), in the presence or absence of PARPi. Asterisks indicate monoubiquitinated F-mH2A1 isoforms (Kim et al., 2017) (C) Analysis of macroH2A1 splice variant interactomes from (A). Data points reflect mean PSM and log2 fold changes from two independent MS experiments. Proteins with a PSM > 0 are shown, proteins with PSM > 2 are depicted in dark gray, proteins with a log2 fold change ≥ 1 and a PSM > 2 in both replicates are shown in pink (macroH2A1.1-enriched) or teal (macroH2A1.2-enriched). (D) Western blot analysis for LIG3 (top panels) and PARP1 (bottom panels) following Flag-IP as in (B) in the presence or absence of PARPi. Signal intensities were normalized to input and are expressed relative to the untreated macroH2A1.1 IP sample. One of two independent experiments is shown. (E) Schematic for alt-EJ reporter assay. A Stop codon (STOP) flanked by microhomologies (MH) prevents GFP expression. DSB induction by the endonuclease I-SceI and subsequent repair by alt-EJ deletes the stop codon and activates GFP expression. (F) Alt-EJ efficiency in reporter cells expressing a control siRNA (si-ctrl) or an siRNA against macroH2A1.2 (si-1.2), combined with PARP inhibitor (PARPi) or a second siRNA, against macroH2A1.1 (si-1.1), LIG3 (si-Lig3) or POLQ (si-Polq). Alt-EJ efficiency is expressed as the fraction of GFP+ cells. Values represent mean and SD (n = 3 replicates). *** p < 0.001, based on Student’s two-tailed t-test. See also Fig. S3 and Table S1.
Moreover, all four macroH2A1.1 interactors were previously shown to be poly-ADP-ribosylated (Gibson et al., 2016; Jungmichel et al., 2013), suggesting that macroH2A1.1-specific binding involves its poly(ADP-ribose)-binding domain (Kustatscher et al., 2005).
Two of the four macroH2A1.1 interactors (LIG3 and PARP1) are centrally involved in alternative non-homologous end joining (alt-EJ) – a DNA end resection-dependent, microhomology-mediated DNA repair pathway that frequently results in chromosomal aberrations and mutations (Gostissa et al., 2011). Consistent with a role for macroH2A1.1 in this repair pathway, siRNA-mediated macroH2A1.1 depletion caused a significant reduction in alt-EJ frequency in a FACS-based reporter assay (Bennardo et al., 2008) (Fig. 3E, F, Fig. S3C). In contrast, depletion of the HR-permissive macroH2A1.2 variant increased alt-EJ efficiency more than two-fold in a manner that was entirely dependent on the presence of macroH2A1.1 and the alt-EJ effectors LIG3 and POLQ (Ceccaldi et al., 2015; Gostissa et al., 2011; Mateos-Gomez et al., 2015) (Fig. 3F, Fig. S3C). Depletion of either variant had only a minor, and comparable effect on canonical non-homologous end joining (cNHEJ, Fig. S3D, E) (Bennardo et al., 2008). Together with our previous work (Khurana et al., 2014; Kim et al., 2018; Kim et al., 2019), these findings thus identify macroH2A1 as a splicing-modulated, epigenetic regulator of repair outcome at resected DNA ends.
Alt-EJ and macroH2A1.1 account for macroH2A1.2 loss-induced anaphase defects
Next, we asked whether macroH2A1.1 function in alt-EJ may be responsible for the anaphase defects observed in the absence of macroH2A1.2. While neither HR nor classical NHEJ are active in mitosis (Lee et al., 2014; Orthwein et al., 2014), a recent report has identified a role for alt-EJ in the micro-homology-mediated joining of DNA ends after replication fork breakage in mitotic extracts (Deng et al., 2019). Aberrant activation of alt-EJ may thereby account for chromosome fusions of mitotic DSBs, resulting in increased anaphase aberrations. To test this hypothesis, we assessed anaphase defects in WT and macroH2A1.2−/− MEFs in the presence or absence of siRNA-mediated depletion of macroH2A1.1, Lig3 or Polq, or when Parp1 was transiently inhibited upon release into mitosis. Inactivation of either of these factors was sufficient to restore the frequency of macroH2A1.2 loss-associated anaphase defects back to wild-type levels (Fig. 4A, B, Fig. S4A). A similar rescue was observed when using etoposide to increase DSB formation during mitotic progression (Fig. S4B). Moreover, the frequency of anaphase aberrations in MEFs from mice deficient in both macroH2A1 splice variants (Changolkar et al., 2007) was comparable to that in WT MEFs (Fig. 4C).
Figure 4: MacroH2A1.1 drives anaphase defects via alt-EJ upon macroH2A1.2 loss.
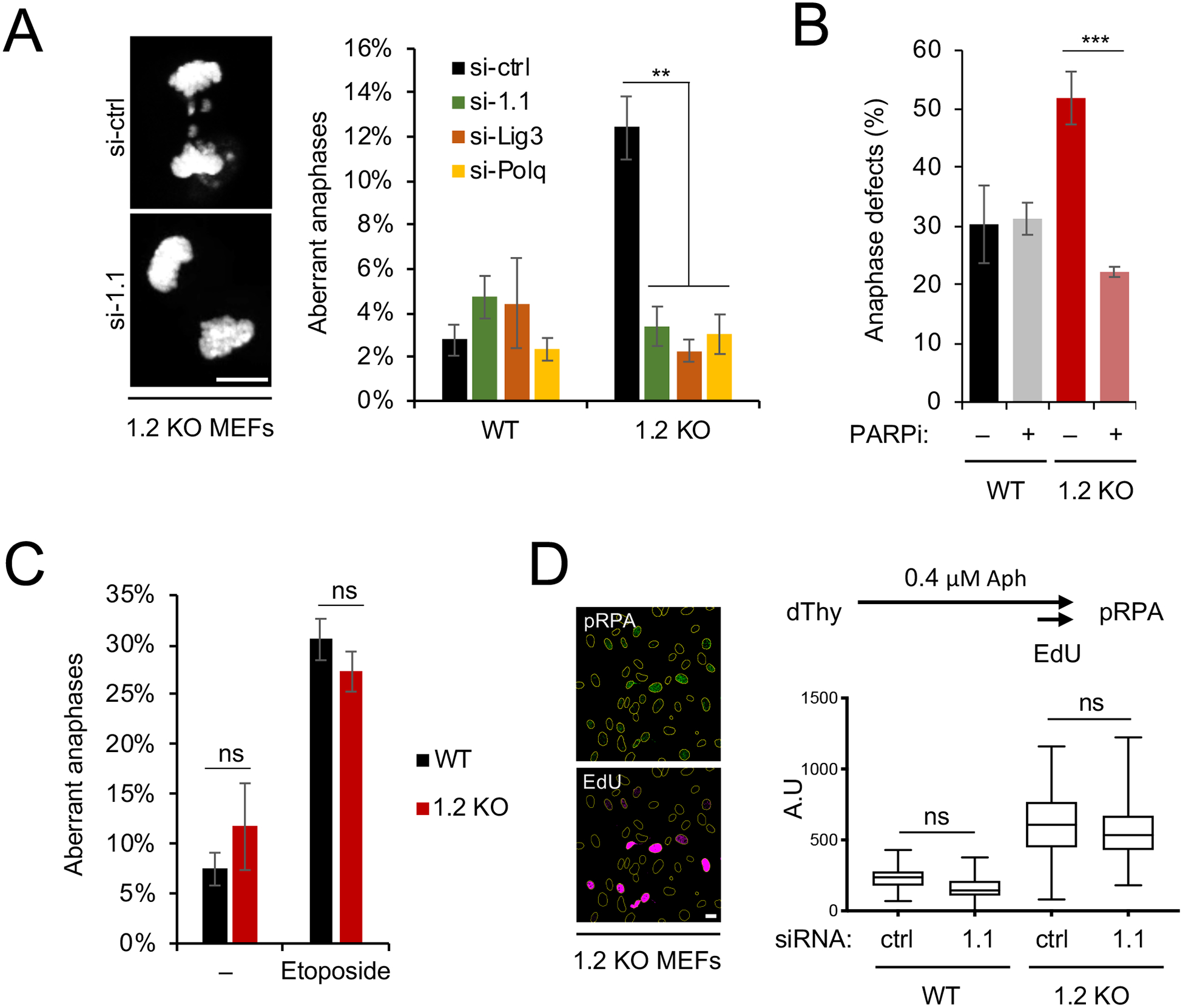
(A) Anaphase aberrations in WT or macroH2A1.2 KO (1.2 KO) MEFs expressing the indicated siRNAs. Anaphases were collected as described in Fig. 2A in the absence of etoposide. Values represent mean and SD (n = 3 replicates), at least 40 anaphases were analyzed per replicate and sample. Scale bar: 10 μm. (B) Fraction of anaphase aberrations in etoposide-treated WT and macroH2A1.2 KO MEFs in the presence or absence of PARP inhibitor (PARPi). Values represent mean and SD (n = 3 replicates, > 60 metaphases per replicate). (A, B) ** p < 0.01, *** p < 0.001 based on Student’s two-tailed t-test. (C) Fraction of anaphase aberrations in MEFs from macroH2A1 KO mice (Changolkar et al., 2007) in the presence or absence of etoposide. Values are expressed as mean and SD (n = 3 replicates, >50 metaphases per replicate), ns: not significant. (D) pRPA intensity in EdU+ MEFs. Representative images are shown, scale bar: 20 μm. ns: not significant based on ANOVA with multiple comparison assuming non-Gaussian distribution. See also Fig. S4.
To rule out that macroH2A1.1 depletion altered the macroH2A1.2 loss-associated increase in RS, and hence the source of the G2/M lesions that underlie mitotic aberrations, we assessed RS levels in the presence or absence of each macroH2A1 variant. RNA interference-mediated macroH2A1.1 depletion was performed in WT and macroH2A1.2 KO MEFs in the presence of aphidicolin (Fig. 4D). S phase cells were identified based on EdU incorporation and RS was measured using RPA32 phosphorylation as a readout (Marechal and Zou, 2015). Consistent with our previous work (Kim et al., 2018), macroH2A1.2 loss resulted in a pronounced increase in RS. MacroH2A1.1 depletion, on the other hand, did not induce RPA phosphorylation in WT cells, and did not further increase RS in macroH2A1.2 KO cells. Taken together, these data point to a dual role of macroH2A1.2 in preventing chromosome segregation defects: (i) the suppression of excessive RS (Kim et al., 2018) (Fig. 4D); and (ii) the prevention of aberrant alt-EJ of mitotic DSBs (Fig. 4A–C). While RS is unaffected by the presence or absence of macroH2A1.1, alt-EJ depends on expression of the alternative splice variant.
MacroH2A1 splice-variants modulate alt-EJ efficiency in tumor cells
Providing a physiological context for macroH2A1 splice variant imbalance, previous reports demonstrated aberrant macroH2A1 alternative splicing in tumor tissue, often resulting in predominant macroH2A1.2 variant expression (Cantarino et al., 2013; Novikov et al., 2011). More recently, we reported an RS-protective role for macroH2A1.2 across fragile regions genome-wide that was particularly apparent in a rapidly dividing tumor cell line (Kim et al., 2018). In agreement with the latter, increased genome instability triggered by the absence of macroH2A1.2 was not strictly limited to the X chromosome (Fig. 2F). Together, these findings point to a broader role for macroH2A1 alternative splicing as a modulator of genome maintenance, particularly in cancer cells. In agreement with reports in tumor tissue (Cantarino et al., 2013; Novikov et al., 2011), the NCI-60 panel of cancer cell lines exhibits predominant macroH2A1.2 variant expression with a mean abundance of 78% of total macroH2A1, ranging from 30% to 98% (Fig. 5A). The NCI-60 panel, thus, allowed us to examine if macroH2A1 splice variant expression has predictive value for DSB repair pathway choice in a cancer setting.
Figure 5: MacroH2A1 variants predict DSB repair pathway choice in tumor cells.
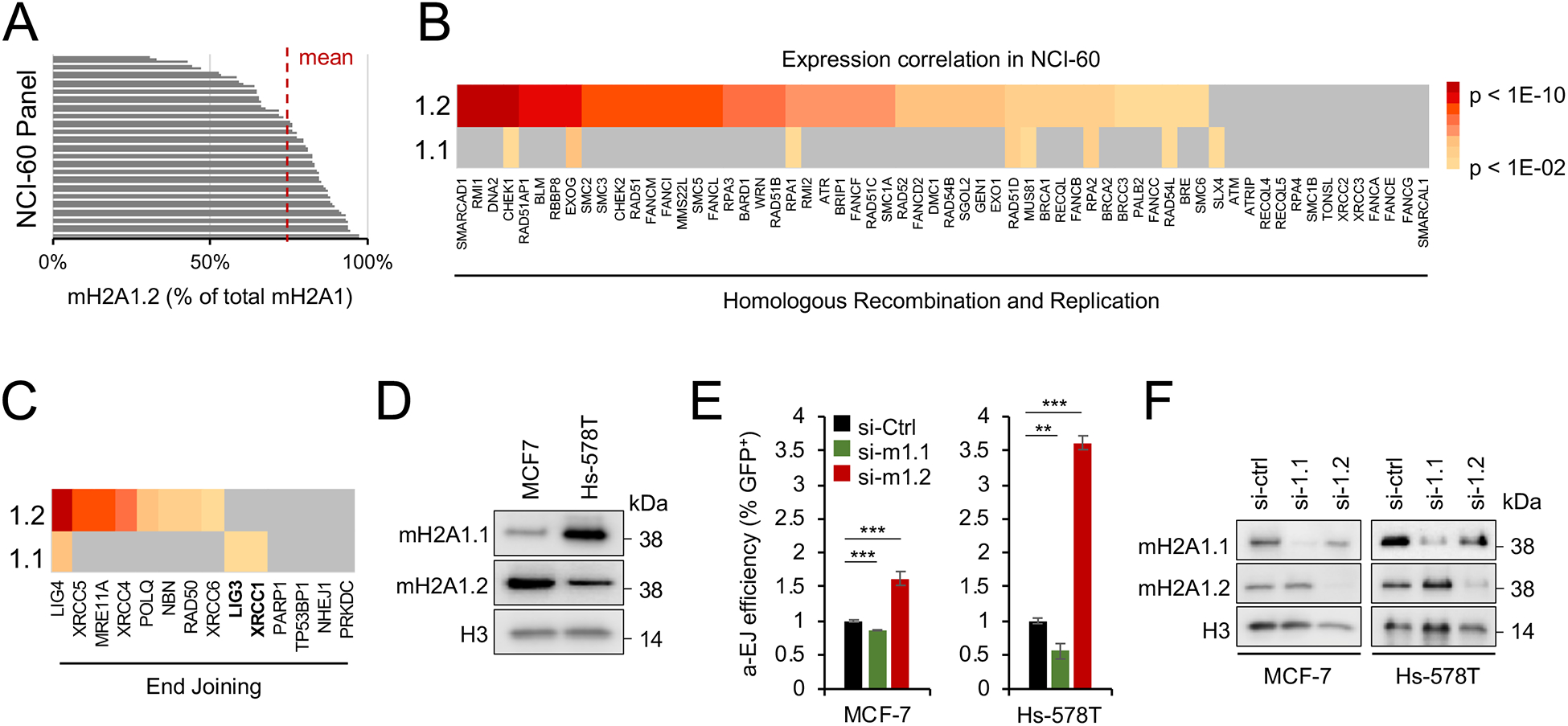
(A) Relative abundance of macroH2A1.2 splice variant expression in NCI60 tumor cell lines based on RNA-Seq expression analysis in CellMinerCDB. (B, C) Correlation of macroH2A1 splice variant expression with genes involved in genome maintenance pathways across NCI-60 tumor cell lines. Genes associated with HR and replication pathways (B) or end joining (EJ) pathways (C) are shown. Heat maps represent p values of the expression correlation, significance was assessed by two-tailed Z test. (D) Western blot using two NCI60 cell lines with high or low macroH2A1.1 or macroH2A1.2 levels. (E) Relative efficiency of alt-EJ in MCF-7 and Hs-578T cells with KD of macroH2A1.1 or macroH2A1.2. ** p < 0.01, *** p < 0.001 based on Student’s two-tailed t-test. (F) Western blot showing siRNA mediated knockdown of macroH2A1.1 and macroH2A1.2 cells from (E). See also Fig. S4.
Gene expression correlation analysis of well-defined HR / RS- or EJ-associated repair factors with either macroH2A1 splice variant was performed using the CellMinerCDB tool (http://discover.nci.nih.gov/cellminercdb) (Rajapakse et al., 2018). Consistent with the role of macroH2A1.2 in homology-directed DSB repair (Khurana et al., 2014; Kim et al., 2018; Kim et al., 2019), we uncovered a strong correlation between macroH2A1.2 and HR / RS-protective repair factors (Fig. 5B, Fig. S4C, D). In contrast, macroH2A1.1 expression was neither indicative of HR / RS nor most canonical EJ components across NCI-60 cell lines. Instead, macroH2A1.1 positively correlated with two alt-EJ components, LIG3 and XRCC1, indicative of a link between macroH2A1.1 expression and alt-EJ in cancer cells (Fig. 5B, C, Fig. S4D). To experimentally test the latter, we measured alt-EJ efficiency in two NCI-60 breast cancer cell lines that express either high (Hs-578T) or low macroH2A1.1 levels (MCF7) and reciprocal macroH2A1.2 levels (Fig. 5D). Confirming our findings in U2OS cells, macroH2A1.1 depletion by siRNA resulted in a reduction in alt-EJ reporter activity in both NCI-60 cell lines, whereas macroH2A1.2 knockdown significantly increased alt-EJ (Fig. 5E, F). Of note, the extent to which alt-EJ changed upon macroH2A1 variant manipulation correlated with the overall abundance of macroH2A1.1. Together, these findings are in agreement with our mechanistic studies in MEFs and point to a broader role for macroH2A1 splice variants as predictive markers for DNA repair pathway choice.
Discussion
Providing a long elusive rationale for macroH2A1.2 enrichment on the Xi, we demonstrate here that macroH2A1.2 is essential for Xi genome maintenance and female survival. We further define opposing roles for macroH2A1 splice variants in the regulation of DSB repair and show that genomic integrity of the Xi depends on a balanced expression of both variants (Fig. S4E). In contrast with the function of macroH2A1.2 during HR and RS, our data demonstrate that macroH2A1.1 acts as a chromatin effector of alt-EJ, which mediates aberrant DSB joining and anaphase aberrations when macroH2A1.2 is absent. Underlining broader physiological relevance, we find that differential modulation of DSB repair by macroH2A1 splice variants extends from healthy to cancer cells.
The female-biased lethality observed in macroH2A1.2-deficient mice is in agreement with a recent report demonstrating that RS can trigger an excessive inflammatory response, which is preferentially lethal to female embryos. Inflammation was linked to the accumulation of micronuclei and was in part facilitated by the absence of the anti-inflammatory properties of testosterone (McNairn et al., 2019). Together with our findings, this suggests that female viability in macroH2A1.2−/− mice is adversely affected by a combination of increased genome instability and micronuclei formation at the Xi, and an overall exacerbated inflammatory response of female mice to genomic damage.
Underlining the central role for aberrant genome maintenance as a driver for the adverse physiological consequences of macroH2A1.2 loss, inactivation of alt-EJ is sufficient to rescue macroH2A1.2 loss-induced anaphase defects. Similarly, co-depletion of macroH2A1.1 restored Xi genome instability and female-biased lethality. While increased RS persists in the absence of both macroH2A1 variants (Fig 4D), alt-EJ of the resulting DNA lesions cannot efficiently occur without macroH2A1.1, preventing chromosome fusions and subsequent anaphase bridges. How DNA breaks are repaired when alt-EJ is impaired remains to be fully determined, but cellular mechanisms exist that tether mitotic DNA breaks until more faithful repair pathways are reactivated in the subsequent cell cycle (Bakhoum et al., 2017; Leimbacher et al., 2019; Spies et al., 2019). It is, however, noteworthy that MEFs deficient in both macroH2A1 variants showed a trend towards increased chromosomal aberrations (Fig. 4C), which raises the possibility that a lack of both variants may result in measurable and potentially harmful chromosomal defects over time. Future studies are needed to address the impact of macroH2A1 variant loss on tumorigenesis and age-associated genome instability.
While our observations explain Xi enrichment of the RS-protective macroH2A1.2 variant, the physiological relevance of macroH2A1.1 on the Xi remains to be further investigated. It is tempting to speculate that macroH2A1.1 may help to modulate PARP1 function beyond alt-EJ, given that PARP enzymes have Xi-specific roles in both genome maintenance and XCI (Menissier de Murcia et al., 2003; Nusinow et al., 2007). Independent of its genomic location, macroH2A1.1-rich chromatin is furthermore an important regulator of mitochondrial respiration via its ability to modulate nuclear NAD+ consumption (Posavec Marjanovic et al., 2017). In addition, the macroH2A1.1 interactors LIG3, TDP1 and PARP1 are central mediators of DNA base excision repair (BER) of single-stranded DNA lesions (Plo et al., 2003; Tomkinson et al., 2001), raising the possibility that macroH2A1.1 may also play a role in BER. However, more work is needed to investigate this possibility. Importantly, given that BER has not been implicated in the repair of etoposide-induced DNA lesions (Pommier et al., 2016), and that we are able to rescue macroH2A1.2 loss-associated mitotic defects by depleting the alt-EJ effector Polq, our findings strongly support the notion that the adverse effect of macroH2A1.1 on anaphase integrity involves alt-EJ rather than the BER pathway. It is worth noting that Polq depletion was recently found to sensitize cells to RS through a mechanism that remains to be fully determined (Wang et al., 2019). As such, Polq depletion may aggravate the impact of macroH2A1.2 loss with regard to S phase-associated DNA damage, while nevertheless protecting from subsequent alt-EJ-associated anaphase aberrations.
Although macroH2A1.1 accumulation may come at the cost of an alt-EJ permissive chromatin environment, the latter is generally counterbalanced by the presence of macroH2A1.2. Moreover, macroH2A1.1 expression correlates with cellular differentiation and proliferative arrest (Chen et al., 2015; Gaspar-Maia et al., 2013; Wan et al., 2017). As a result, cells with high macroH2A1.1 levels are less likely to undergo mitosis and concomitant alt-EJ-associated anaphase defects, while alt-EJ proficiency may provide an important backup DSB repair pathway in G1, where HR is unavailable.
Supporting the physiological relevance of our findings, macroH2A1 alternative splicing outcome changes during cellular or developmental transitions, which is particularly well documented for malignant transformation and progression (Dardenne et al., 2012; Novikov et al., 2011). Consistent with its RS-protective role, macroH2A1.2 supports proliferation of both primary and tumor cells (Kim et al., 2018; Kim et al., 2019), whereas macroH2A1.1 expression negatively correlates with tumor growth (Chen et al., 2015; Pazienza et al., 2016; Sporn et al., 2009; Wan et al., 2017). Our findings in a panel of 60 tumor cell lines now point to macroH2A1 splice variant abundance as a pathologically relevant indicator of repair outcome and pathway choice in cancer cells. We anticipate that the assessment and/or manipulation of macroH2A1 splice variant abundance, as well as a better understanding of what determines their unique protein interactomes, may provide novel strategies to manipulate genome maintenance and therapeutic outcome during malignant growth.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philipp Oberdoerffer (Philipp.Oberdoerffer@nih.gov).
Materials Availability
MacroH2A1.2 KO mice generated in this study have been deposited to the NCI Mouse Repository, Cat#01BQO, strain name B6.Cg-Macroh2a1<tm1.2Pobe>/Nci; synonym: B6.Cg-macroH2A1.2 KO.
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Original/source data for figures 1–5 in the paper is available [Mendeley Data, doi:10.17632/xh2z6m4t75.1].
Mass spectrometry data have been deposited in MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) under the identifier MassIVE: MSV000085351.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
macroH2A1.2−/− mice.
Gene-targeted macroH2A1.2fl-neo/+ ES cells (see below) were injected into C57BL/6 albino (cBRD/cBRD) blastocysts and chimeric males were crossed to C57BL/6 females. macroH2A1.2fl-neo/+ mice were bred to β-actin Cre transgenic mice (Lewandoski et al., 1997) (back-crossed to C57Bl/6 for more than 20 generations) for germline deletion of the neoR-E6a cassette, resulting in mice heterozygous for the macroH2A1.2 KO allele (macroH2A1.2+/−). The latter were interbred to generate macroH2A1.2−/− mice. All animal breeding and experimentation followed guidelines approved by the National Institutes of Health Institutional Animal Care and Use Committee.
Cell Lines.
Human female lung IMR90 (Coriell) and male foreskin BJ fibroblasts (ATCC) were cultured in MEM (Gibco), supplemented with 10% (v/v) FBS (Gemini), 2 mM L-Glutamine (Gibco), 1 mM sodium pyruvate (Sigma) and 0.1 (1X) mM Non-Essential Amino Acids (NEAA, Gibco). Alt-EJ U2OS cells (gift from J. Stark, City of Hope) were cultured in DMEM (Gibco) with 10% FBS. HEK293 T-REx cells (gift from The Broad Institute, Cambridge) were grown in DMEM with 10% FBS containing Blasticidin (Invivogen, 15 μg/ml) and hygromycin B (Thermo Fisher, 100 μg/ml). MEFs were grown in DMEM (Gibco) containing β-mercaptoethanol and Pen/Strep (Gibco), supplemented with 10% FBS. mTFs were gown in mTF media (1:1 DMEM and Ham’s F10, 20% FBS, Non-Essential Amino Acids, Pen/Strep, β-mercaptoethanol, Sodium pyruvate, HEPES and L-Glutamine, all from Gibco). pSM33 male ES cells (Engreitz et al., 2013) and MS2-Xist female ES cells (Jonkers et al., 2008) were a kind gift from Kathrin Plath, UCLA. ES cells were grown on mitomycin C-treated mouse embryonic fibroblasts (feeder cells) in ES cell media (Knockout DMEM (Invitrogen), 15% FBS, 2 mM L-glutamine, 100 μM Non-Essential Amino Acids, 1% Pen/Strep, 0.1 % β-mercaptoethanol, 1000 U/mL mLIF (Invitrogen)). All cells were maintained at 37 °C in a humidified incubator containing 5% CO2. Primary cells and ES cell lines were not tested for mycoplasm, all other cell lines were mycoplasm-free.
METHODS DETAILS
macroH2A1.2 gene targeting.
5’ and 3’ arms of homology were PCR-amplified and inserted into pEZ-Frt-lox-DT (a gift from K. Rajewsky, Addgene, plasmid #11736). A 840 bp H2AFY exon 6a (E6a)-containing DNA fragment was PCR-amplified and inserted downstream of an Frt- and LoxP flanked neoR gene resulting in a loxP-flanked FRT-neoR-E6a cassette. See Table S3 for primer sequences. Gene targeting was performed in C57BL/6 Bruce4 ES cells as described previously (Lewandoski et al., 1997). Neomycin-resistant ES cells were analyzed for correct transgene integration by Southern Blot analysis, using PCR-generated 5’ and 3’ external probes and a 5’ internal probe in combination with HindIII or BamHI digests of genomic ES cell DNA, see Table S3 for primer sequences. The targeted allele is referred to as macroH2A1.2fl-neo.
Isolation of tail fibroblasts, embryonic fibroblasts and PBMCs.
Mouse tail fibroblasts (mTF) were prepared from 6 week-old mice. Briefly, animals were euthanized, and the tails were de-skinned. The skin was wiped in 70% ethanol and cut into small pieces (2 X 2 mm) in mTF media (see above) containing 1.6 mg/ml collagenase II (Thermo Fisher). After 24 h, a cell strainer was used to remove hair and debris. One million cells were plated onto a 10 cm dish in fresh mTF medium and passaged when confluent. For mouse embryonic fibroblasts (MEFs), embryos from WT and KO littermates were isolated on E12.5 and processed as described previously (Durkin et al., 2013). Individual MEF strains were genotyped for sex using SRY PCR (see Table S3 for primer sequences) and stored at passage 2. For experiments, WT and KO MEFs from identical passage numbers were used, passage numbers ranged from 3–6. For PBMCs, blood samples from animals were drawn by retro-orbital bleeding and cultured in phytohaemagglutinin stimulated RPMI media (Gibco) with 10% FBS and Pen/Strep for 48 h.
Cell line generation and treatments.
Targeted, Flp-recombinase-mediated insertion of a single copy of the Flag-macroH2A1.1- or Flag-macroH2A1.2-encoding transgenes into HEK293 T-REx cells was performed as described previously (Kim et al., 2018). SiRNAs were transfected using DF-1 reagent following the manufacturer’s instructions (Dharmacon) and analyzed at 72 – 96 h post transfection. Lentiviral infection of LKO.1 shRNA-expression vectors was carried out by spin infection (2500 rpm, 90 min, Beckman-Coulter Allegra X-12R centrifuge) with 8 μg/ml polybrene (Sigma), Cells were incubated overnight prior to virus removal and selection with puromycin (1 μg/ml, Invitrogen). See Table S2 for siRNA and shRNA sequences. To induce Xist expression, pSM33 cells were plated as single cells without feeder cells on 0.1% gelatin (Millipore) coated cover slips overnight. Doxycycline (2 μg/ml) was added subsequently for 8 h and the coverslips were processed for RNA FISH as described below. For differentiation experiments in MS2-Xist ES cells, cells were depleted of feeder cells and plated onto acid-etched, gelatin coated coverslip for 24 h in ES cell media without mLIF. Subsequently, all-trans retinoic acid (R2625, Sigma) was added and cells were allowed to differentiate for 6 days, followed by RNA FISH or RNA collection. For undifferentiated controls, feeder cell-depleted ES cells were grown on coverslips for 24 hours with mLIF containing ES media and fixed for RNA FISH or RNA collection. For G2/M arrest, cells were treated with 10 μM CDK1 inhibitor (CDK1i) RO3306 for 16 h. For PARP inhibition, cells were treated with 10 μM veliparib (Selleckchem), for topoisomerase II inhibition, cells were treated with 5 μM etoposide for the indicated times. Irradiation experiments were performed using an X-ray irradiator (X-rad 320, Precision X-Ray).
Alternative end-joining (alt-EJ) and canonical NHEJ (cNHEJ) assays.
Detection of alt-EJ was performed using a previously described alt-EJ U2OS reporter cell line (Bennardo et al., 2008). Briefly, cells were first transfected siRNAs on day 1. On day 2, cells were transfected with 1 μg of I-SceI expression vector pCBASceI (Addgene #26477) or 1 μg of pcDNA3-EGFP (Addgene #13031) using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s instructions. Where indicated, PARPi treatment was performed 24 h prior to analysis. The fraction of GFP-positive cells was assayed 72 h post siRNA transfection using a FACSCalibur (BD, USA). Values were normalized to transfection efficiency based on pcDNA3-EGFP. For experiments involving transient transfection, alt-EJ reporter plasmid (EJ2) was transfected along with I-SceI expression vector. Detection of cNHEJ was performed using a previously described cNHEJ U2OS reporter cell line (Bennardo et al., 2008).
Cell cycle analyses.
For DNA content analyses, cells were fixed in 70% ethanol at −20 °C overnight. Fixed cells were resuspended in propidium-iodide (PI) / RNase A staining buffer (BD Pharmingen) at room temperature for 20 min. DNA content was determined by flow cytometry (FACS Calibur, BD Pharmingen). For mitotic index analyses, chromatin extracts were collected 30 min after release from G2/M arrest and analyzed for H3-pS10 content using the phospho-S10 detection kit (Abcam, ab115127) following the manufacturer’s instructions.
RNA isolation and RT-PCR.
Total RNA was extracted using the RNeasy Mini Kit according to the manufacturer’s instructions (QIAGEN). cDNA was synthesized from 0.5–1 μg of total RNA using the Superscript III reverse transcriptase RT-PCR system (Thermo Fisher), and expression of the indicated genes was analyzed by quantitative RT-PCR using a LightCycler 480 II (Roche) (see Table S3 for primer sequences). The standard comparative cycle threshold method was used for cDNA quantification.
Antibodies.
The following antibodies were used for western blot: α-macroH2A1.2 (Millipore, MABE61), α-macroH2A1.1 (CST, 12455), α-Lig3 (GeneTex, GTX103172), α-FLAG (Agilent, 200472), α-PCNA (Santa Cruz, sc-56), α-H2B (Abcam, ab1790), α-GAPDH (Santa Cruz, sc-32233), α-PARP1 (CST, 9542S), α-H3 (CST, 9715). Secondary antibodies were goat anti-mouse IgG-HRP (Santa Cruz, sc-2005) and goat anti-rabbit IgG-HRP (Santa Cruz, sc-2004). Primary antibodies for IF were: α-macroH2A1.1 (CST, 12455), α-macroH2A1.2 (Millipore, MABE61), α-centromere (Antibodies incorporated, 15-234-0001), α-PAR (Trevigen, 4335-MC-100), H3K27me3 (Abcam, ab6002), phosphor RPA32 (Ser4 / Ser8) (Bethyl, A300–245A). Secondary antibodies were from Life Technologies (goat-α-mouse, goat-α-rabbit or goat-α-human conjugated to Alexa Fluor 488 or 568).
Immunostaining and imaging.
Cells were fixed and in PTEMF buffer (20 mM PIPES pH 6.8, 10 mM EGTA, 0.2% Triton X-100, 1 mM MgCl2 and 4% paraformaldehyde) at room temperature and then permeabilized in 0.5% Triton X-100. Cells were washed and blocked for 1 h at room temperature (3% BSA, 0.5% Triton X-100 in PBS). Primary and secondary antibody stainings were carried out in 3% BSA and PBS-T. For S phase discrimination, cells were pulsed with 10 μM EdU (Life Technologies) at 37°C, prior to fixation. Incorporated EdU was Click-labeled using azide-linked Alexa Fluor-647 (Life Technologies). Confocal z-stacks were acquired using a Zeiss LSM 780 microscope. Images were displayed and analyzed as maximum intensity projections.
Anaphase analyses and micronuclei detection.
For anaphases, MEFs were plated onto gelatin coated coverslips for 6 h followed by treatment with CDK1i. Cells were washed 3 times with pre-warmed PBS and allowed to progress into anaphase in the presence or absence of etoposide or veliparib. Cells were fixed and processed for IF or DNA-FISH 50 min after CDK1i washout. Unbiased imaging was performed using the field-tiling method followed by manual, non-blinded scoring of all detectable anaphases. For micronuclei analyses, MEFs were seeded in 96-well plates, treated with CDK1i as above and released into etoposide containing media. Cells were fixed 2 h post release to allow for completion of mitosis. Cells were then processed for IF and subjected to high-throughput imaging (CV7000, Yokogawa). An unbiased ImageJ detection program was used to measure the number of micronuclei and nuclei, based on size, intensity and circularity. H3K27me3 signal intensity was recorded for each micronucleus and H3K27me3 positive micronuclei were defined using a cutoff value established based on the distribution across all samples. For X-micronuclei detection using DNA-FISH, cells were grown on coverslips and treated as above. Images were blinded for unbiased, manual detection of X-micronuclei.
DNA FISH.
Flow-sorted chromosome X or chromosome 2 was labeled with fluorescence tagged dUTP (Enzo Life Sciences) by DOP-PCR (degenerate oligomer-primed polymerase chain reaction) (Telenius et al., 1992). For mTFs or MEFs, cells were grown on coverslips, fixed in freshly prepared methanol:acetic acid (3:1) for 15 min, washed and stored at −20 in 70% ethanol until use. For PBMCs, cells were swollen in 75 mM KCl, fixed as above and spread on coverslips. Samples in 70% ethanol were serially dehydrated in 80, 90 and 100% ethanol, air dried and transferred to 50% de-ionized formamide in SSC. Samples were then denatured with probes at 75 °C for 2 min, incubated overnight in a humidified chamber at 37 °C, washed in 0.4X SSC for 2 min at 72 °C, counterstained with Hoechst 33342 and mounted using antifade. Anaphase FISH images were acquired on a Zeiss LSM780 confocal microscope. PBMC FISH images were acquired using an epi-fluorescence microscope (Imager Z2, Zeiss) and HiFish acquisition software (GenASIs, Applied Spectral Imaging,Inc., CA).
RNA-FISH and analyses.
RNA FISH probes for Xist RNA FISH were prepared from full-length mouse Xist plasmid using CGH Bioprime array kit (Life Technologies, #18095011) following the manufacturer’s instructions. Alexa Fluor-488 labelled dUTPs were used to label probes. Cells grown on coverslips were fixed for 15 min in 4% PFA, washed two times in PBS and stored in 70% ethanol in −20 until use. Cells were washed in PBS and in 10% formamide prior to incubating with the RNA FISH probe at 37 °C overnight. Coverslips were then washed in 10% formamide and 2X SSC followed by counterstaining with Hoechst 33342 and mounting in antifade. Images were acquired using LSM 780 (Zeiss) confocal microscope and unbiased Xist detection was performed using KNIME software as described below.
Xist segmentation analyses.
For Xist foci detection in male ES cells, we adapted a previously developed image analysis workflow (Jowhar et al., 2018) or detecting Xist foci in the full field of view (FOV). Briefly, the workflow uses undecimated wavelet-based spot detection algorithm (Olivo-Marin, 2002) for segmenting the Xist foci in the FOV. We used up to four wavelet scales for segmenting the Xist foci. The per-scale threshold factors were set to lower values to enable detection of all Xist signal to ensure reliable foci detection with the same set of values. Bona fide Xist foci were visually distinguished from background foci and used for quantitative assessment and comparison. Nuclear segmentation tools could not be employed in undifferentiated, colony forming cells. For Xist foci detection in differentiated female ES cells, we used the three workflows described in (Jowhar et al., 2018). Briefly, the first workflow includes a nuclear segmentation module and is followed by an interactive module which allows the end-user to annotate segmented objects into good (visually accurate segmentation) and bad (over/under segmentation) classes, calculates their morphological features, and trains a binary random forest (RF) classifier for rejecting mis-segmented nuclei (bad class). The second workflow segments nuclei in 2D, applies the trained RF classifier from previous workflow to reject mis-segmented 2D nuclei, and crops Xist foci from corresponding spectral channel(s) using the binary 2D nucleus mask. The third workflow segments Xist foci using the un-decimated wavelet-based spot detection algorithm described earlier and extracts quantitative parameters of the detected Xist foci, namely, integrated intensity (a.u.), mean intensity (a.u.), and area (pixels). The image processing steps and workflows described above were implemented into bespoke pipelines in the KNIME (Berthold et al., 2008). Analytics Platform (Version 3.2.1, 64-bit) using compatible KNIME Image Processing Nodes (Dietz and Berthold, 2016), Python and R Scripting Nodes. All KNIME workflows were either executed on a dedicated workstation running Microsoft Windows 2012 Server R2 (64-bit) with 16-cores of AMD Opteron 6212 processor (2.7 GHz) and 256 GB RAM or on a dedicated high-performance batch cluster compute node (Biowulf, CIT, NIH) running RedHat Enterprise Linux 7.4 with 28 cores of Intel X2680, 240 GB RAM, 4 K80 Nvidia GPUs and 400 GB of solid state local storage.
Spectral Karyotyping.
Metaphases were arrested by incubation with Colcemid (15210–040, KaryoMax ® Colcemid Solution, Invitrogen) (10 μg/ml) 3 hours prior to harvest. Cells were collected and treated with hypotonic solution (KCL 0.075 M) for 15 minutes at 37°C and fixed with methanol:acetic acid 3:1. Slides were prepared and aged overnight prior to SKY analysis. Metaphases were hybridized with the 21-color mouse SKY paint kit (FPRPR0030, ASI) according to the manufacturer’s protocol. Hybridization was carried out in a humidity chamber at 37°C for 16 hours. The post-hybridization rapid wash procedure was used with 0.4X SSC at 72°C for 4 minutes. Detection was carried out following ASI manufacturer’s protocol. Spectral images of the hybridized metaphases were acquired using Hyper Spectral Imaging system (Applied Spectral Imaging Inc., CA) mounted on top of an epi-fluorescence microscope (Imager Z2, Zeiss). Images were analyzed using HiSKY 7.2 acquisition software (Genasis, Applied Spectral Imaging Inc., CA). G-banding was simulated by electronic inversion of DAPI counterstaining. An average of 20 mitoses of comparable staining intensity and quality was examined per cell line and compared for chromosomal abnormality. The karyotype was determined by comparison to the standard ideogram of banding patterns for mouse chromosomes (Nesbitt and Francke, 1973). Abnormalities were then separated as either aneuploidy or other aberrations (deletions, translocations, etc.). Events were summated between autosomes and X chromosomes and the number of events for autosomes was normalized to the total number of autosomes and the number of nuclei analyzed.
Cellular extract preparation and immunoblotting.
Cells were lysed in RIPA lysis buffer (150 mM NaCl, 1% NP-40, 1% Na-deoxycholate, 0.1% SDS, 25 mM Tris-HCl pH 7.5) supplemented with protease inhibitors (Sigma) and phosphatase inhibitors (Calbiochem). Lysates were sonicated, centrifuged, and heated with reducing sample buffer (375 mM Tris-HCl pH 6.8, 9% SDS, 50% glycerol, 9% β-mercaptoethanol, 0.03% bromophenol blue). Lysates of equal protein amount based on BCA assay (Thermo Fisher) were separated by SDS-PAGE and subjected to western blotting using the indicated primary antibodies. HRP-conjugated secondary antibodies were used for signal detection by enhanced chemiluminescence (Amersham).
Co-immunoprecipitation.
HEK293 T-REx cells expressing Flag-tagged macroH2A1.1 or macroH2A1.2 were used for co-IP experiment along with parental cell line as control. Where indicated, PARPi was added at a concentration of 10 μM / 6 million cells for 24 h. Equal numbers of cells were used for IP experiment. Briefly, cells were washed with PBS and resuspended in hypotonic buffer (20 mM HEPES, 10 mM KCl, 2mM MgCl2, 10% glucose, 0.5% NP-40) followed by MNase digestion (0.2 U/μl, 4°C, 60 min) in digestion buffer (20 mM HEPES, 150 mM KCl, 10% Glycerol, 3 mM CaCl2). 20 mM EGTA was used to terminate the reaction. Using a 27G needle, each sample was aspirated 10 times followed by centrifugation at 1000 g, retrieval of the supernatant and IP with M2 magnetic beads (50% slurry) at 4 °C for 60min. Post IP, beads were washed 4 times using wash buffer (20 mM HEPES, 150 mM KCl, 0.1% NP-40) followed by 50 mM HEPES. Immunoprecipitated proteins were solubilized by incubating the beads with SDS sample buffer containing 10% β-mercaptoethanol for 5 min at 95 °C, followed by western blotting. LIG3 signal intensities were quantified using Image Lab software (BIO-RAD), IP intensity was normalized to input.
Digestion and dimethyl labeling of affinity purified protein complex.
Affinity purified protein complexes were resuspended in 30 μL of 25 mM HEPES (pH 8.4) and subjected to overnight digestion with 2 μg trypsin (Promega) reconstituted in 100 mM triethylammonium bicarbonate (TEAB) (Sigma) at 37°C. Digested samples were recovered, dried and resuspended in 100 μl of 100 mM TEAB. In-solution dimethylation of the tryptic digest was performed using CH2O (Sigma) for light labeling or CD2O (Sigma) for medium labeling. Labeling reagent comprising 4 μl of 4% CH2O/CD2O mixed with equal amount of 0.6 M NaBH3CN, was added to reconstituted tryptic digest and incubated for 1 hr at room temperature. The labeling reaction was quenched by addition of 16 μl of 1% ammonia. Differentially labeled sample pairs were mixed, dried and de-salted using C18 spin columns (Thermo Fisher) according to the manufacturer’s protocol. The peptide mixture was reconstituted in 25% ACN/0.1% FA (100 μl) and fractionated using strong cation exchange (SCX) liquid chromatography (LC) as described in (Das et al., 2010). Thirty-six SCX fractions were pooled in 12 fractions based on the intensity profile and vacuum dried. Dried samples were reconstituted in 0.1% trifluoroacetic acid prior to MS analysis.
Mass Spectrometry acquisition and data analysis.
Protein samples were subjected to nanoflow liquid chromatography (Thermo Easy nLC 1000, Thermo Scientific) coupled to high resolution tandem MS (Fusion, Thermo Scientific). MS scans were performed in the Orbitrap analyzer at a resolution of 60,000 with an ion accumulation target set at 2e6 over a mass range of 350–1500 m/z, followed by MS/MS analysis in an iontrap with an ion accumulation target set at 1e5. MS2 precursor isolation width was setup at 1.6 m/z, normalized collision energy was 29, and charge state 1 and unassigned charge states were excluded. Acquired MS/MS spectra were searched against a human uniprot protein database, using a SEQUEST and the resulting peptides were filtered at a maximum of 1% FDR using the percolator validator algorithms in the Proteome Discoverer 1.4 software (Thermo Scientific, CA). The precursor ion tolerance was set at 10 ppm and the fragment ions tolerance was set at 0.6 Da along with methionine oxidation included as a dynamic modification. For dimethyl labeling on the N-terminus and lysine, 28.0313 and 32.0564 Da were filled in for light and medium labels respectively. More than 2 non-redundant peptides from each protein group was used for protein quantification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Splice variant correlation analysis in NCI-60 cells.
Gene expression correlation analyses across the NCI-60 panel were determined using CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/), a web-based resource for elucidating gene expression determinants across various cancer cell line data sets (Rajapakse et al., 2018). In brief, RNA-seq transcript expression levels for a well-annotated set of repair genes were obtained from CellMiner\Query Genomic Data (https://discover.nci.nih.gov/cellminer/queryLoad.do). Values for macroH2A1 isoform 1.1 are based on refseq NM_138609, values for macroH2A1.2 represent the summation of NM_001040158, NM_138610, and NM_004893. The patterns for the composite gene transcript versus isoforms transcript levels were compared using Pearson’s correlations, and p-values were determined by two-tailed Z test using R computing (http://www.r-project.org).
Statistical analyses.
All statistical tests and values / meaning of n are listed in the Figure legends. The following statistical tests were used as appropriate: Student’s two-tailed t-test, Fisher’s exact test, Mann-Whitney U test, two-tailed binomial test, two-tailed Z test, ANOVA with multiple comparison assuming non-Gaussian distribution.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-macroH2A1.2 | Millipore | Cat#MABE61, RRID:AB_10807977 |
| α-macroH2A1.1 | Cell Signaling Technologies | Cat#12455, RRID:AB_2797923 |
| α-Lig3 | GeneTex | Cat#GTX103172, RRID:AB_2036773 |
| α-FLAG | Agilent | Cat#200472, RRID:AB_10596649 |
| α-PCNA | Santa Cruz | Cat#sc-56, RRID:AB_628110 |
| α-H2B | Abcam | Cat#ab1790, RRID:AB_302612 |
| α-GAPDH | Santa Cruz | Cat#sc-32233, RRID:AB_627679 |
| α-PARP1 | Cell Signaling Technologies | Cat#9542, RRID:AB_2160739 |
| α-H3 | Cell Signaling Technologies | Cat#9715, RRID:AB_331563 |
| α-centromere | Antibodies incorporated | Cat#15-234-0001, RRID:AB_2687472 |
| α-PAR | T revigen | Cat#4335-MC-100, RRID :AB_2572318 |
| α-H3K27me3 | Abcam | Cat#ab6002, RRID:AB_305237 |
| α-phosphor RPA32 (Ser4 / Ser8) | Bethyl | Cat#A300–245A, RRID:AB_210547 |
| Goat anti-mouse IgG-HRP | Santa Cruz | Cat#sc-2005, RRID:AB_631736 |
| Goat anti-rabbit IgG-HRP | Santa Cruz | Cat#sc-2004, RRID:AB_631746 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Aphidicolin | Sigma | Cat#A0781 |
| EdU (5-ethynyl-2’-deoxyuridine) | ThermoFisher Scientific | Cat#E10187 |
| Alexa Fluor 647-azide | ThermoFisher Scientific | Cat#A10277 |
| Hoechst 33342 | ThermoFisher Scientific | Cat#H3570 |
| RO3306 | Selleckchem | Cat#S7747 |
| Alexa Fluor 488–5-dUTP | ThermoFisher Scientific | Cat#C11397 |
| Anti-FLAG M2 Magnetic beads | Sigma | Cat#M8823 |
| Veliparib (PARPi) | Selleckchem | Cat#S1004 |
| Puromycin | Sigma | Cat#P8833 |
| Collagenase II | ThermoFisher Scientific | Cat#17101015 |
| Phytohaemagglutinin | Sigma | Cat#L1668 |
| PI/RNase staining buffer | BD PharMingen | Cat#550825 |
| cOmplete Mini EDTA-free protease inhibitor | Roche | Cat#11836170001 |
| KaryoMAX Colcemid | ThermoFisher Scientific | Cat#15210–040 |
| mLIF | ThermoFisher Scientific | Cat#PMC9484 |
| Doxycycline | Sigma | Cat#D9891 |
| All-trans retinoic acid | Sigma | Cat#R2625 |
| Critical Commercial Assays | ||
| Click-iT EdU Alexa Fluor 647 Imaging Kit | ThermoFisher Scientific | Cat#C10340 |
| Phosphatase inhibitor cocktail set II | Millipore | Cat#524625 |
| SuperscriptIII | ThermoFisher Scientific | Cat#18080044 |
| TURBO DNase | ThermoFisher Scientific | Cat#AM1907 |
| Lipofectamine 2000 | ThermoFisher Scientific | Cat#11668 |
| CGH Bioprime array kit | ThermoFisher Scientific | Cat#18095011 |
| Histone H3 (phospho S10) assay kit | Abcam | Cat#ab115127 |
| 21-color mouse SKY paint kit | Applied spectral imaging | Cat#FPRPR0030 |
| Deposited Data | ||
| Mass spectrometry analyses, see Table S1 | This study | MassIVE: MSV000085351 |
| Experimental Models: Cell Lines | ||
| BJ | ATCC | Cat#CRL-2522, RRID:CVCL_3653 |
| IMR-90 | Coriell | Cat#I90–15, RRID:CVCL_0347 |
| T-REx-293 Cell Line | Thermo Fisher Scientific | Cat#R78007, RRID:CVCL_U427 |
| FLAG-macroH2A1.1 (in HEK293 T-REx) | This study | N/A |
| FLAG-macroH2A1.2 (in HEK293 T-REx) | This study | N/A |
| MCF-7 | ATCC | Cat#HTB-22, RRID:CVCL_0031 |
| Hs 578T | ATCC | Cat#HTB-126, RRID:CVCL_0332 |
| pEJ5 U2OS (cNHEJ) | Gift from Jeremy Stark | N/A |
| pEJ2 U2OS (alt-EJ) | Gift from Jeremy Stark | N/A |
| C57BL/6 Bruce4 ES cells | Lewandoski et al., 1997 | N/A |
| MS2-Xist female ES cells | Jonkers et al., 2008 | N/A |
| pSM33 male ES cells | Engreitz et al., 2013 | N/A |
| macroH2A1.2 WT and KO MEFs | This study | N/A |
| Tail fibroblasts from WT and KO mice | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| MacroH2A1.2 KO mice | This study | NCI Mouse Repository, Cat#01BQO |
| MacroH2A1 KO mice | Changolkar et al., 2007 | N/A |
| β-actin Cre transgenic mice | Lewandoski et al., 1997 | The Jackson Laboratory, Cat#B6N. FVB-Tmem 163Tg(ACTB-cre)2Mrt/CjDswJ |
| Oligonucleotides | ||
| shRNA targeting sequences, see Table S2 | This study | N/A |
| siRNA targeting sequences, see Table S2 | This study | N/A |
| Primers for gene targeting, see Table S3 | This study | N/A |
| Primers for RT-PCR, see Table S3 | This study | N/A |
| Primers for genotyping, see Table S3 | This study | N/A |
| Recombinant DNA | ||
| EJ2 | Gift from Jeremy Stark | RRID :Addgene_44025 |
| pCBASceI | Gift from Maria Jasin | RRID :Addgene_26477 |
| pcDNA3-EGFP | Gift from Doug Golenbock | RRID:Addgene_13031 |
| pEZ-Frt-lox-DT | Gift from Klaus Rajewsky | RRID:Addgene_11736 |
| Full-length mouse Xist plasmid | Gift from Kathrin Plath | N/A |
| pLKO.1 | Gift from David Root | RRID:Addgene_10878 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| FlowJo | BD Biosciences | https://www.flowjo.com |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Proteome Discoverer 1.4 | ThermoFisher Scientific | https://www.thermofisher.com/us/en/home.html |
| R | R Core Team | http://www.r-project.org |
| KNIME | Berthold et al., 2008 | https://www.knime.com/ |
| HiFish acquisition software | GenASIs, Applied Spectral Imaging | https://spectral-imaging.com/ |
| HiSKY 7.2 | Genasis, Applied Spectral Imaging | https://spectral-imaging.com/ |
| Spot detection algorithm | Olivo-Marin, 2002 | N/A |
| Other | ||
| CellMinerCDB | NIH | https://discover.nci.nih.gov/cellminercdb/ |
Highlights.
The macroH2A1.2 histone variant promotes Xi stability and female survival in mice
Balanced macroH2A1 splice variant expression ensures genome integrity
macroH2A1.1 promotes alt-EJ and genome instability in macroH2A1.2 knockout mice
MacroH2A1 splice variants affect DNA repair pathway choice in cancer cells
Acknowledgments
We thank A. Nussenzweig for critical reading of the manuscript, S. Kozlov, K. Plath, K. Rajewsky and J. Stark for reagents, T. Karpova for imaging support, S. Das and T. Andresson for Mass Spectrometry analyses, E. Southon and the Mouse Cancer Genetics Program at NCI-Frederick for gene targeting and ES cell injection, and the Biowulf, High-Performance Computing Group, Center for Information Technology (CIT), NIH, for their computational support. High-throughput imaging work was performed at the High-Throughput Imaging Facility (HiTIF) / Center for Cancer Research / National Cancer Institute / NIH. This work was supported by a grant from the Daiichi Sankyo Foundation of Life Science (Japan) to E.H. and the Intramural Research Program of the NIH, NCI, Center for Cancer Research (Z01 BC006150 to Y.P. and ZIA BC010411 to M.I.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Data and materials availability
All data is available in the main text or the supplementary materials.
Supplementary Materials:
Supplementary Information includes Supplementary Figures S1 – S4 and Supplementary Tables S2, S3.
Supplementary Excel file:
Supplementary Table S1, related to Figure 3 and Supplementary Figure S3. MacroH2A1 splice variant interactomes.
References
- Ait Saada A, Teixeira-Silva A, Iraqui I, Costes A, Hardy J, Paoletti G, Freon K, and Lambert SAE (2017). Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol Cell 66, 398–410 e394. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Kabeche L, Compton DA, Powell SN, and Bastians H (2017). Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer 3, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, and Stark JM (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold MR, Cebron N, Dill F, Gabriel TR, Kotter T, Meinl T, Ohl P, Sieb C, Thiel K, and Wiswedel B (2008). KNIME: The Konstanz Information Miner. Stud Class Data Anal, 319–326. [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarino N, Douet J, and Buschbeck M (2013). MacroH2A--an epigenetic regulator of cancer. Cancer Lett 336, 247–252. [DOI] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. (2015). Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changolkar LN, Costanzi C, Leu NA, Chen D, McLaughlin KJ, and Pehrson JR (2007). Developmental changes in histone macroH2A1-mediated gene regulation. Mol Cell Biol 27, 2758–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, and Gamble MJ (2015). MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol Cell 59, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi C, and Pehrson JR (1998). Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393, 599–601. [DOI] [PubMed] [Google Scholar]
- Dabin J, Fortuny A, and Polo SE (2016). Epigenome Maintenance in Response to DNA Damage. Mol Cell 62, 712–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardenne E, Pierredon S, Driouch K, Gratadou L, Lacroix-Triki M, Espinoza MP, Zonta E, Germann S, Mortada H, Villemin JP, et al. (2012). Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat Struct Mol Biol 19, 1139–1146. [DOI] [PubMed] [Google Scholar]
- Das S, Bosley AD, Ye X, Chan KC, Chu I, Green JE, Issaq HJ, Veenstra TD, and Andresson T (2010). Comparison of strong cation exchange and SDS-PAGE fractionation for analysis of multiprotein complexes. J Proteome Res 9, 6696–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wu RA, Sonneville R, Kochenova OV, Labib K, Pellman D, and Walter JC (2019). Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol Cell 73, 915–929 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz C, and Berthold MR (2016). KNIME for Open-Source Bioimage Analysis A Tutorial. Adv Anat Embryol Cell Biol 219, 179–197. [DOI] [PubMed] [Google Scholar]
- Downes CS, Clarke DJ, Mullinger AM, Gimenez-Abian JF, Creighton AM, and Johnson RT (1994). A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature 372, 467–470. [DOI] [PubMed] [Google Scholar]
- Durkin ME, Qian X, Popescu NC, and Lowy DR (2013). Isolation of Mouse Embryo Fibroblasts. Bio Protoc 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, et al. (2013). The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341, 1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar-Maia A, Qadeer ZA, Hasson D, Ratnakumar K, Leu NA, Leroy G, Liu S, Costanzi C, Valle-Garcia D, Schaniel C, et al. (2013). MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat Commun 4, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, and Kraus WL (2016). Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 353, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Alt FW, and Chiarle R (2011). Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol 29, 319–350. [DOI] [PubMed] [Google Scholar]
- Jager N, Schlesner M, Jones DT, Raffel S, Mallm JP, Junge KM, Weichenhan D, Bauer T, Ishaque N, Kool M, et al. (2013). Hypermutation of the inactive X chromosome is a frequent event in cancer. Cell 155, 567–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Monkhorst K, Rentmeester E, Grootegoed JA, Grosveld F, and Gribnau J (2008). Xist RNA is confined to the nuclear territory of the silenced X chromosome throughout the cell cycle. Mol Cell Biol 28, 5583–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowhar Z, Shachar S, Gudla PR, Wangsa D, Torres E, Russ JL, Pegoraro G, Ried T, Raznahan A, and Misteli T (2018). Effects of human sex chromosome dosage on spatial chromosome organization. Mol Biol Cell 29, 2458–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, and Nielsen ML (2013). Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol Cell 52, 272–285. [DOI] [PubMed] [Google Scholar]
- Khurana S, Kruhlak MJ, Kim J, Tran AD, Liu J, Nyswaner K, Shi L, Jailwala P, Sung MH, Hakim O, et al. (2014). A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep 8, 1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Chan DW, Jung SY, Chen Y, Qin J, and Wang Y (2017). The Histone Variant MacroH2A1 Is a BRCA1 Ubiquitin Ligase Substrate. Cell Rep 19, 1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Sturgill D, Sebastian R, Khurana S, Tran AD, Edwards GB, Kruswick A, Burkett S, Hosogane EK, Hannon WW, et al. (2018). Replication Stress Shapes a Protective Chromatin Environment across Fragile Genomic Regions. Mol Cell 69, 36–47 e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Sun C, Tran AD, Chin PJ, Ruiz PD, Wang K, Gibbons RJ, Gamble MJ, Liu Y, and Oberdoerffer P (2019). The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening. Nat Struct Mol Biol 26, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, and McCarroll SA (2014). Random replication of the inactive X chromosome. Genome Res 24, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, and Ladurner AG (2005). Splicing regulates NAD metabolite binding to histone macroH2A. Nat Struct Mol Biol 12, 624–625. [DOI] [PubMed] [Google Scholar]
- Lee DH, Acharya SS, Kwon M, Drane P, Guan Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto JA, et al. (2014). Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell 54, 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimbacher PA, Jones SE, Shorrocks AK, de Marco Zompit M, Day M, Blaauwendraad J, Bundschuh D, Bonham S, Fischer R, Fink D, et al. (2019). MDC1 Interacts with TOPBP1 to Maintain Chromosomal Stability during Mitosis. Mol Cell 74, 571–583 e578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M, Meyers EN, and Martin GR (1997). Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol 62, 159–168. [PubMed] [Google Scholar]
- Machiela MJ, Zhou W, Karlins E, Sampson JN, Freedman ND, Yang Q, Hicks B, Dagnall C, Hautman C, Jacobs KB, et al. (2016). Female chromosome X mosaicism is age-related and preferentially affects the inactivated X chromosome. Nat Commun 7, 11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2015). RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res 25, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, and Sfeir A (2015). Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNairn AJ, Chuang CH, Bloom JC, Wallace MD, and Schimenti JC (2019). Female-biased embryonic death from inflammation induced by genomic instability. Nature 567, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, et al. (2003). Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J 22, 2255–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, and Hickson ID (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. [DOI] [PubMed] [Google Scholar]
- Nesbitt MN, and Francke U (1973). A system of nomenclature for band patterns of mouse chromosomes. Chromosoma 41, 145–158. [DOI] [PubMed] [Google Scholar]
- Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, and Gamble MJ (2011). QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol Cell Biol 31, 4244–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DA, Hernandez-Munoz I, Fazzio TG, Shah GM, Kraus WL, and Panning B (2007). Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J Biol Chem 282, 12851–12859. [DOI] [PubMed] [Google Scholar]
- Olivo-Marin JC (2002). Extraction of spots in biological images using multiscale products. Pattern Recogn 35, 1989–1996. [Google Scholar]
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, and Durocher D (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193. [DOI] [PubMed] [Google Scholar]
- Ouararhni K, Hadj-Slimane R, Ait-Si-Ali S, Robin P, Mietton F, Harel-Bellan A, Dimitrov S, and Hamiche A (2006). The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev 20, 3324–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazienza V, Panebianco C, Rappa F, Memoli D, Borghesan M, Cannito S, Oji A, Mazza G, Tamburrino D, Fusai G, et al. (2016). Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis. Epigenetics Chromatin 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, and Zhang Y (2003). Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135. [DOI] [PubMed] [Google Scholar]
- Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, and Pommier Y (2003). Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2, 1087–1100. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, and Nitiss JL (2016). Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nature reviews. Molecular cell biology 17, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posavec Marjanovic M, Hurtado-Bages S, Lassi M, Valero V, Malinverni R, Delage H, Navarro M, Corujo D, Guberovic I, Douet J, et al. (2017). MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD(+) consumption. Nat Struct Mol Biol 24, 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, Iorio F, Sousa FG, Elloumi F, Aladjem MI, et al. (2018). CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience 10, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell LM, Strike P, Browne CE, and Jacobs PA (2007). X chromosome loss and ageing. Cytogenet Genome Res 116, 181–185. [DOI] [PubMed] [Google Scholar]
- Spies J, Lukas C, Somyajit K, Rask MB, Lukas J, and Neelsen KJ (2019). 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat Cell Biol 21, 487–497. [DOI] [PubMed] [Google Scholar]
- Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T, Schnabel P, and Ladurner AG (2009). Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 28, 3423–3428. [DOI] [PubMed] [Google Scholar]
- Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, and Tunnacliffe A (1992). Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13, 718–725. [DOI] [PubMed] [Google Scholar]
- Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, et al. (2009). A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat Struct Mol Biol 16, 923–929. [DOI] [PubMed] [Google Scholar]
- Tomkinson AE, Chen L, Dong Z, Leppard JB, Levin DS, Mackey ZB, and Motycka TA (2001). Completion of base excision repair by mammalian DNA ligases. Prog Nucleic Acid Res Mol Biol 68, 151–164. [DOI] [PubMed] [Google Scholar]
- Wan D, Liu C, Sun Y, Wang W, Huang K, and Zheng L (2017). MacroH2A1.1 cooperates with EZH2 to promote adipogenesis by regulating Wnt signaling. J Mol Cell Biol 9, 325–337. [DOI] [PubMed] [Google Scholar]
- Wang Z, Song Y, Li S, Kurian S, Xiang R, Chiba T, and Wu X (2019). DNA polymerase theta (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J Biol Chem 294, 3909–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original/source data for figures 1–5 in the paper is available [Mendeley Data, doi:10.17632/xh2z6m4t75.1].
Mass spectrometry data have been deposited in MassIVE (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) under the identifier MassIVE: MSV000085351.