

Abstract
Toxoplasma gondii is a protozoan parasite of great importance in human and veterinary health. The frontline treatment of antifolates suffers a variety of drawbacks, including toxicity and allergic reactions, underscoring the need to identify novel drug targets for new therapeutics to be developed. We previously showed that the Toxoplasma lysine acetyltransferase (KAT) GCN5b is an essential chromatin remodeling enzyme in the parasite linked to the regulation of gene expression. We have previously established that the KAT domain is a liability that can be targeted in the parasite by compounds like garcinol; here, we investigate the potential of the bromodomain as a targetable element of GCN5b. Bromodomains bind acetylated lysine residues on histones, which helps stabilize the KAT complex at gene promoters. Using an inducible dominant-negative strategy, we found that the GCN5b bromodomain is critical for Toxoplasma viability. We also found that the GCN5-family bromodomain inhibitor, L-Moses, interferes with the ability of the GCN5b bromodomain to associate with acetylated histone residues using an in vitro binding assay. Moreover, L-Moses displays potent activity against Toxoplasma tachyzoites in vitro, which can be overcome if parasites are engineered to over-express GCN5b. Collectively, our data support the GCN5b bromodomain as an attractive target for the development of new therapeutics.
Keywords: Toxoplasma, epigenetics, chromatin, histone acetylation, bromodomain, drug development
Graphical Abstract
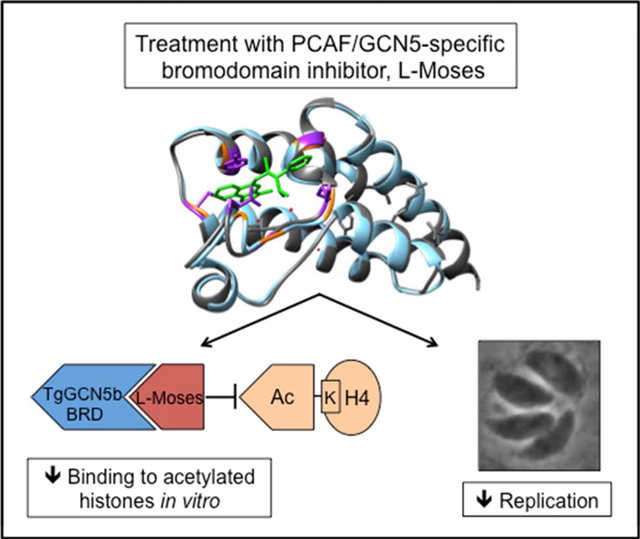
1. Introduction
The obligate intracellular protozoan parasite Toxoplasma gondii poses a significant health risk to humans and livestock, necessitating the development of safe and effective drugs (1). Toxoplasma belongs to phylum Apicomplexa, which is comprised of other medically relevant pathogens including Plasmodium (malaria), Cryptosporidium (cryptosporidiosis), and Eimeria (poultry coccidiosis). Toxoplasma resides in nucleated cells of warm-blooded animals and has been estimated to have infected about one-third of the world’s population (2).
Acute infection is characterized by rapidly proliferating parasites (tachyzoites) but is often asymptomatic in immunocompetent hosts. A healthy immune system controls the infection within weeks, at which point Toxoplasma establishes a chronic, latent infection characterized by its conversion into tissue cysts (bradyzoites) (3). As the parasite is not eradicated from the host, it can reactivate to cause acute opportunistic disease in immunocompromised individuals, such as those infected with HIV. Extensive tissue damage can occur during reactivated toxoplasmosis that results in severe complications including Toxoplasma encephalitis and ocular toxoplasmosis (4).
The first line of therapy for toxoplasmosis is a combination of pyrimethamine and sulfadiazine, often administered with leucovorin to reduce cytotoxicity (5). Pyrimethamine and sulfadiazine inhibit parasite replication by blocking the folate metabolic pathway, but the treatment is not effective against latent tissue cysts. The treatment is also toxic, putting patients at risk of bone marrow suppression and dermatologic and gastrointestinal events; discontinuation or change in this treatment regimen due to adverse events occurred in over 55% of studies analyzed in a systematic review (6). As a result of these shortcomings, there is an urgent need for the identification of novel drug targets to facilitate the development of better-tolerated treatments for toxoplasmosis.
A promising area of investigation for potential drug targets in Toxoplasma includes protein complexes that “read” and “write” posttranslational modifications (PTM) such as lysine acetylation (7). The addition and removal of acetyl marks from lysine residues is mediated by lysine acetyltransferases (KATs) and lysine deacetylases (KDACs), respectively. Lysine acetylation has emerged as a key PTM taking place on hundreds of proteins throughout various compartments in the cell. Extensive lysine acetylation occurs in both Toxoplasma and the fellow apicomplexan parasite Plasmodium on proteins that function in a variety of areas including metabolism, translation, chromatin biology, and stress responses (8, 9). In addition, lysine acetylation of histones is correlated with stage-specific gene expression and stage conversion in Toxoplasma (10).
Two GCN5 family KATs have been identified in Toxoplasma and designated GCN5a and GCN5b (11). Each has a lengthy N-terminal extension, a KAT domain, and a single C-terminal bromodomain, which is a ~110 amino acid protein motif established to bind acetylated lysines (12). Knockout studies indicate that GCN5a is dispensable for tachyzoite proliferation, but required for proper stress responses (13). Conversely, multiple attempts to knockout GCN5b have failed, suggesting it is essential for tachyzoite viability (13, 14). In support of this idea, we previously observed that when a catalytically inactive form of GCN5b (GCN5bE703G) was ectopically expressed, it exerted a dominant-negative effect that resulted in arrest of parasite proliferation (14). Additionally, ChIP-chip analysis demonstrated that GCN5b is enriched at genes involved in a wide spectrum of critical functions, including transcription, translation, and metabolism (14). We have also shown that the KAT inhibitor garcinol targets the KAT domain of GCN5b; treatment of Toxoplasma with garcinol in vitro decreases GCN5b auto-acetylation, alters expression of genes linked to GCN5b control, and inhibits parasite replication (15). These data, along with a recent genome-wide fitness screen reporting a score of −3.0, strongly support GCN5b as an essential gene (16).
In addition to the inhibition of the KAT domain, the bromodomain of GCN5b may also serve as a targetable element. A variety of small molecules have been developed to interfere with binding of the bromodomain to acetylated lysines, and a number of these have already been reported to have inhibitory activity against Toxoplasma and other protozoan pathogens, such as Plasmodium falciparum, Trypanosoma brucei and Trypanosoma cruzi (17–21). L-Moses (also known as L-45) is a triazolopthalazine-based chemical probe that binds specifically to PCAF/GCN5 family bromodomains, capable of displacing their association with histone H3.3 in cultured HEK-293 cells (22).
In this study, we analyzed the GCN5b bromodomain as a potential new drug target against Toxoplasma tachyzoites. Modeling of the GCN5b bromodomain showed considerable homology at key residues linked to acetyl-lysine binding. We mutated these residues to express an inducible dominant-negative form of GCN5b that lacked a fully functional bromodomain. Parasites expressing GCN5b with a mutated bromodomain exhibited a significant fitness defect. We also found that L-Moses interferes with GCN5b bromodomain binding to acetyl-lysines and has potent activity against tachyzoites in vitro. Parasites engineered to overexpress wild-type GCN5b showed decreased sensitivity to L-Moses compared to the parental line, consistent with the conclusion that the GCN5b bromodomain is a major target. Together, these findings highlight the importance of the GCN5b bromodomain as an attractive target for future therapeutics.
2. Materials and methods
2.1. Chemicals
Shield (CheminPharma) was prepared in absolute ethanol and stored at −20°C. L-Moses dihydrochloride was purchased from To cris (catalog no 6251) and prepared in DMSO.
2.2. Host cell and parasite culture
Primary human foreskin fibroblast (HFF) cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Corning), without antibiotic/antimycotic, supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco/Invitrogen). Toxoplasma gondii Type I RH strain parasites were propagated in confluent monolayers of HFF cells and cultured in DMEM, without antibiotic/antimycotic, supplemented with 1% heat-inactivated FBS as previously described (23). Host cell and parasite cultures were maintained at 37°C with 5% CO2 in a humidified incubator.
Plasmid DNA was transfected into RHΔHX parasites by electroporation and selected in 20 μM chloramphenicol; individual clones were isolated by limiting dilution in 96-well plates as previously described (23). For assays involving Shield-based regulation, the designated parasites were cultured with vehicle (100% EtOH) or Shield for 48 hours.
2.3. Plasmid construction
To construct an expression plasmid to generate recombinant GCN5b bromodomain (BRD) protein in E. coli, a region of TgGCN5b (amino acids 877–986 of TGGT1_243440 from ToxoDB) encoding the BRD (amino acids 888–985) was amplified by PCR from a previously reported plasmid (14) using the following primers which include 15bp ends that are complementary to the target site in the pGEX-4T1 plasmid: sense primer (5’-TGGATCCCCGGAATTCCAGATTCCAGGTCTTCTGCAGTG) and anti-sense primer (5’- GTCGACCCGGGAATTCCAGTTGCTTCTGCTGCTG). The TgGCN5b BRD sequence was inserted into the pGEX-4T1 vector at the EcoRI site (939) using the InFusion cloning system (Takara), for expression of TgGCN5b BRD protein fused to an N-terminal GST tag. To generate a fusion protein that expresses a mutant TgGCN5b BRD, tandem alanine substitutions (Y963A/N964A/Y970A) were created using the Q5 site-directed mutagenesis kit (NEB; E0554S) with the following primers: sense primer (5’-CAAACGATTGCTTACAAATACGCGAACGAG) and anti-sense primer (5’-CTGGTGGGCGGCCTGCCGACAGTTCTTGAAC).
To generate a plasmid for inducible ectopic expression of GCN5b with a mutated bromodomain (Y963A/N964A/Y970A) in T. gondii, the ptubXFLAG::CAT plasmid encoding full-length GCN5b with N-terminal destabilization (dd) and HA tags previously described in (14) was subjected to Q5 site-directed mutagenesis using the same Q5 primers as listed above. The wild-type and mutant ptubXFLAG::CAT plasmids were linearized with NotI before transfection into parasites.
2.4. Purification of GCN5b bromodomain protein
GCN5b BRD wild-type and mutant pGEX-4T1 plasmids were transformed into BL21 (DE3) competent E. coli cells, which were grown to an OD600 of 0.6–0.8 and incubated with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 16°C to induce protein expression. IPT G-induced bacteria were centrifuged at 20,000 rpm for 25 minutes at 4°C and bacterial pellets were incubated with 20 mL of lysis buffer (50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5% glycerol (v/v), 1% Triton X-100 (v/v), 1 mM DTT, lysozyme from chicken egg white (300 ug/mL) (Sigma Aldrich; L6876), and an EDTA-free protease inhibitor cocktail tablet (Sigma Aldrich; 11873580001)) for 1 hr at 4°C, following which 250 units of benzonase nuclease (Novagen; 2894174) and 1 mM MgCl2 were added and bacteria were incubated for an additional 30 minutes at 4°C. The lysed bacteria were centrifuged at 20,000 rpm for 25 minutes at 4°C and the soluble protein was purified and concentrated (with a 30 kDa MWCO concentrator) using the Amicon Pro Affinity Concentration Kit for GST fusion proteins (Millipore, ACK5030GS).
2.5. In vitro binding assays
In vitro binding assays were conducted to assess recombinant GST-tagged GCN5b BRD (amino acids 888–985) binding to acetylated histone H4 peptide in a similar manner as described in (24) with minor modifications. First, biotinylated histone peptides H4 (corresponding to residues 1–21 of human histone H4) (Epigentek, R-1007) and tetra-acetylated H4 (K5/8/12/16) (corresponding to residues 1–18 of human histone H4) (Epigentek, R-1008) were linked to streptavidin magnetic beads (NEB). To link histone peptides and streptavidin beads, 625 μL of streptavidin bead slurry was washed twice in 150 mM salt solution (150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 0.1% Triton X-100, 10% glycerol, 1 mM DTT) using a magnetic stand, and then resuspended in 625 μL of 150 mM salt solution. 100 μL of histone peptide (100 μM) was added to the bead slurry and incubated overnight at 4°C. Histone peptide-linked streptavidin beads were washed three times in 150 mM salt solution to remove unbound peptide and resuspended in 625 μL of 150 mM salt solution with 0.8% sodium azide. Bead samples were stored at 4°C.
Beads linked to unmodified H4 peptide, as well as streptavidin beads alone, were used as negative controls in binding/inhibition assays. 10 μL samples of histone-linked beads and control bead samples were washed once with 150 mM salt solution and resuspended in 250 μL150 mM salt solution for binding assays or 150 mM salt solution supplemented with vehicle or inhibitor for inhibitor assays. 1 μg BRD protein was added to the beads and samples were incubated overnight at 4°C. Beads were then washed five times with a 500 mM salt solution (500 mM NaCl, 50 mM Tris-HCl (pH 7.5), 0.1% Triton X-100, 10% glycerol, 1 mM DTT) for a total of 1 hour at 4°C. Magnetic beads were boiled in 4X SDS loading buffer (40% glycerol, 240 mM Tris/HCl pH 6.8, 8% SDS, 0.04% bromophenol blue, 5% beta-mercaptoethanol) for 10 minutes at 95°C to elute bound bromodomain protein. Beads were magnetically separated from the boiled samples and the supernatant was run on an SDS page gel. GST-tagged BRD protein was detected by western blot analysis.
2.6. Western blot analysis
Protein samples were run on denaturing 4–12% Tris-acetate polyacrylamide gradient gels (Invitrogen). GST-tagged fusion proteins were probed with anti-GST rabbit polyclonal antibody (1:2,000; Sigma, G7781). HA-tagged fusion proteins were probed with anti-HA rat monoclonal antibody (1:2,000; Roche #11867423001). The major surface antigen p30 was probed with Toxoplasma P30 mouse monoclonal antibody (1:1,000; Genway GWB-44A329). Anti-rabbit, anti-rat, or anti-mouse antibodies conjugated with horseradish peroxidase (1:5,000; GE Healthcare) were used as secondary antibodies. Blots were visualized by electrochemiluminescence using the Chemiluminescence Western Blot Substrate (Pierce) and imaged by FluorChem E machine (ProteinSimple).
2.7. Immunofluorescence assays
Localization of proteins in Toxoplasma was determined by immunofluorescence assay (IFA) as previously described (14). HFFs were grown to confluence on coverslips in a 12-well plate and inoculated with freshly lysed parasites. 18 hours post-infection, medium was aspirated and coverslips were fixed with 4% paraformaldehyde in PBS for 10 minutes, washed with PBS, and then permeabilized with 0.2% Triton X-100 in 3% BSA-PBS for 15 minutes. Plates were incubated with anti-HA rat monoclonal antibody (1:2,000; Roche, #11867423001) for 2 hours at room temperature, washed with PBS 3×5 minutes, and incubated with anti-rat Alexa Fluor 488 antibody (1:2,000; Invitrogen, A-11006) and 4′,6-diamidino-2-phenylindole (DAPI) (1:1,000) for 1 hour at room temperature. Samples were washed once more with PBS three times for 5 minutes each and visualized using a Nikon Eclipse 80i microscope.
2.8. Modeling of TgGCN5b BRD
The predicted 3D structure of the Toxoplasma GCN5b BRD (amino acids 877–986) was obtained via I-Tasser using unbiased settings (25–27) and visualized with UCSF Chimera (28). The UCSF matchmaker function was used to superimpose BRD structures.
2.9. Parasite growth assays
For all growth assays, confluent HFF monolayers were inoculated with freshly harvested intracellular parasites that were scraped, syringe-lysed with a 25-gauge needle, and counted by hemocytometer. Plaque assays were used to measure parasite viability (23). 500 parasites were used to inoculate HFF monolayers in a 12-well plate. Parasite lines were added in technical triplicate within each plate. After two hours of invasion, uninvaded parasites were aspirated, wells were washed twice with medium, and fresh medium supplemented with vehicle or drug was added. Plates were left to incubate undisturbed for five to seven days and then fixed with 100% ice-cold methanol and stained with crystal violet for visualization of plaques. Plates were imaged by FluorChem E machine (ProteinSimple) and host cell lysis was calculated using ImageJ analysis (29). Average percentages of host cell area lysed were compared between parasite lines by One-way ANOVA with Dunnett’s multiple comparisons post-hoc.
The IC50 of L-Moses was determined by reporter assay established by (30) with the modifications reported in (31). 100 RHβ1 parasites stably expressing bacterial β-galactosidase were added to each well of a black 96-well plate. After 2 hours of invasion, wells were washed twice with DMEM lacking phenol red (Thermofisher Scientific), and phenol red-free DMEM supplemented with vehicle or drug was added. Drug was serially diluted across the plate by multichannel pipette. The IC50 of pyrimethamine has been previously established by β-galactosidase assay and thus was used as a control (30). After four days of incubation, 100 μM of the β-galactosidase substrate chlorophenolred-β-D-galactopyranoside (CPRG) (Sigma, #10884308001) was added to each well. 24 hours later, β-galactosidase activity was measured by reading absorbance at 570 nm (excitation) and 630 nm (emission) with the Synergy H1 Plate Reader (Biotek). Assays were conducted in technical triplicate and average absorbance values obtained from three biological replicates were normalized to wells containing uninfected host cells (maximum inhibition) and infected host cells treated with vehicle (DMSO) (minimum inhibition). Absorbance values were plotted and the IC50 was calculated by non-linear regression-variable slope (4 parameters).
Parasite replication was measured by doubling assay (32). Briefly, 1×105 parasites were added to HFF monolayers in a 6-well plate. After two hours of invasion, wells were washed twice with medium and fresh medium supplemented with vehicle or drug was added. Plates were fixed with 100% ice-cold methanol and stained with Hema 3 solutions I and II (Fisher, 23–122937/23–122952) at 24 and 36 hours post-infection to visualize parasites and vacuoles. The number of parasites was counted in 150 random vacuoles for three biological replicates and the average percent of vacuoles containing indicated numbers of parasites between parasite lines at designated drug concentrations were compared by two-way ANOVA with Tukey’s multiple comparisons post-hoc.
2.10. Host cell cytotoxicity assay
To determine the effect of L-Moses on HFF cell metabolism, a CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega; G3582) was conducted (33). This colorimetric assay measures the reduction of the MTS tetrazolium compound to a formazan dye mediated by NAD(P)H-dependent dehydrogenase enzymes. Phenol red-free medium supplemented with vehicle (DMSO) or drug was added to confluent HFF cells in a black 96-well plate and serially diluted across the plate by multichannel pipette. Cells treated with 1% SDS were used as a positive control for inhibition of cell metabolic activity. CellTiter 96® AQueous One Solution Reagent was added to each well and incubated for four hours. The formation of formazan dye was quantified by measuring absorbance at 490 nm using the Synergy H1 Plate Reader (Biotek). Assays were performed in technical triplicate and the absorbance values from four biological replicates were averaged, the absorbance of the media alone (no cells) was subtracted, and absorbance from treated cells were normalized to absorbance from cells treated with vehicle.
2.11. Statistical analyses
All statistical analyses were conducted using GraphPad Prism version 8.3.0 for Windows, GraphPad Software, La Jolla California USA, http://www.graphpad.com.
3. Results and Discussion
3.1. The bromodomain of GCN5b is important for parasite fitness
We previously employed a dominant-negative strategy to determine that the lysine acetyltransferase (KAT) domain of GCN5b was critical for parasite growth and viability (14). Expression of a recombinant version of GCN5b containing an inactive KAT domain induced parasite growth arrest
We sought to employ a similar dominant-negative strategy here to determine if the GCN5b bromodomain is also important for normal parasite growth in vitro. Protein sequence alignments of GCN5 bromodomains from other representative species to that of Toxoplasma GCN5b revealed conservation of key residues implicated in the interaction with acetylated lysine residues (Fig. 1A) (12). These amino acids include two conserved tyrosines (Y) and an internal asparagine (N): Y963/N964/Y970, which line the acetyl-lysine binding pocket formed between the ZA and BC loops (Fig. 1A–B). To ensure that these conserved residues would ablate the ability to bind to acetyl-lysine, we expressed a wild-type (WT) and mutant form of the GCN5b bromodomain (amino acids 877–986) in E. coli, fused to an N-terminal GST tag for purification purposes (Fig. 1C). To create the mutant bromodomain, each of the three conserved residues (Y963, N964, and Y970) were replaced with alanines. We tested the ability of the WT and mutant GCN5b bromodomains to interact with acetylated histone H4 peptide (tetra-acetylated at lysines 5/8/12/16) using an in vitro binding assay (24). Results show that WT GCN5b bromodomain binds to acetylated H4 while the mutant GCN5b bromodomain protein does not (Fig. 1D).
Fig. 1. Alanine mutations render the TgGCN5b bromodomain non-functional in vitro.
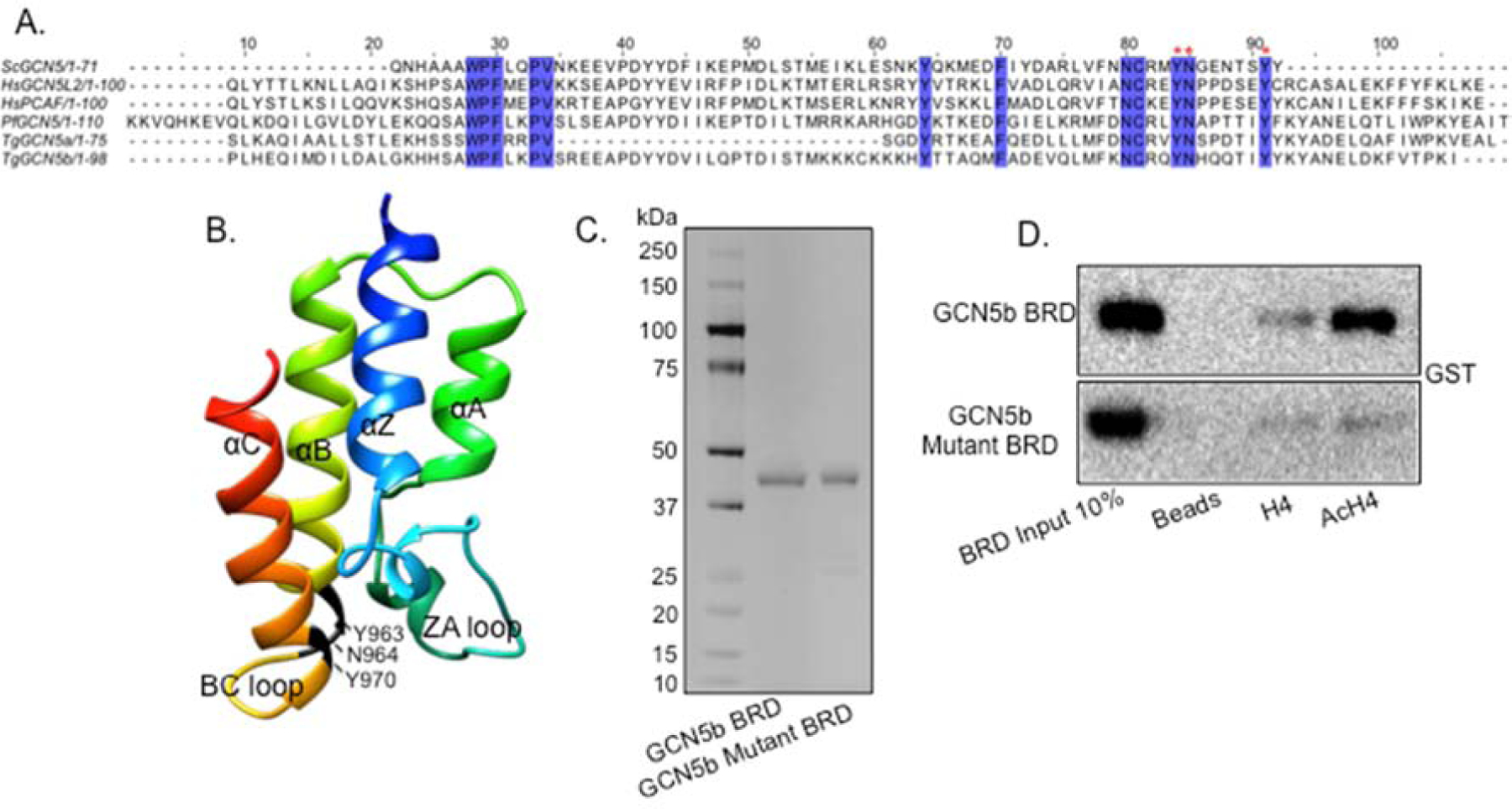
(A) Alignment of amino acid sequences from GCN5 bromodomains in Saccharomyces cerevisiae (Sc), Homo sapiens (Hs), Plasmodium falciparum (Pf), and Toxoplasma gondii (Tg). Highlighted amino acids are conserved among species. Red asterisks denote amino acids that were mutated to alanine in this study. (B) Predicted structure of Toxoplasma GCN5b bromodomain using I-Tasser. Amino acids that underwent alanine substitution are indicated. (C) Silver stain of recombinant, GST-tagged wild-type and mutant recombinant GCN5b bromodomain (BRD) proteins purified from E. coli. (D) BRD binding assay to monitor wild-type and mutant GCN5b BRD association with histone H4 (unmodified or acetylated, Ac), probed with anti-GST.
We then proceeded to generate clonal transgenic parasites in type I RH strain that express full-length GCN5b with or without these mutations (Y963A/N964A/Y970A) in the bromodomain. Both WT and mutant GCN5b were fused to an N-terminal destabilization domain (dd) and HA epitope tag, denoted asddHAGCN5b orddHAGCN5bMutBRD. The dd tag sends the fusion protein to the proteasome for degradation unless it is blocked by including the ligand Shield in the culture media (34). Expression of ectopicddHAGCN5b orddHAGCN5bMutBRD was controllable by Shield, as assessed by immunofluorescence assay (IFA) and western blotting for the HA tag (Fig. 2A–B). Notably, the IFA reveals that the bromodomain mutations did not disrupt nuclear localization of GCN5b (Fig. 2B).
Fig 2. Ectopic expression of a mutated GCN5b bromodomain arrests parasite proliferation.
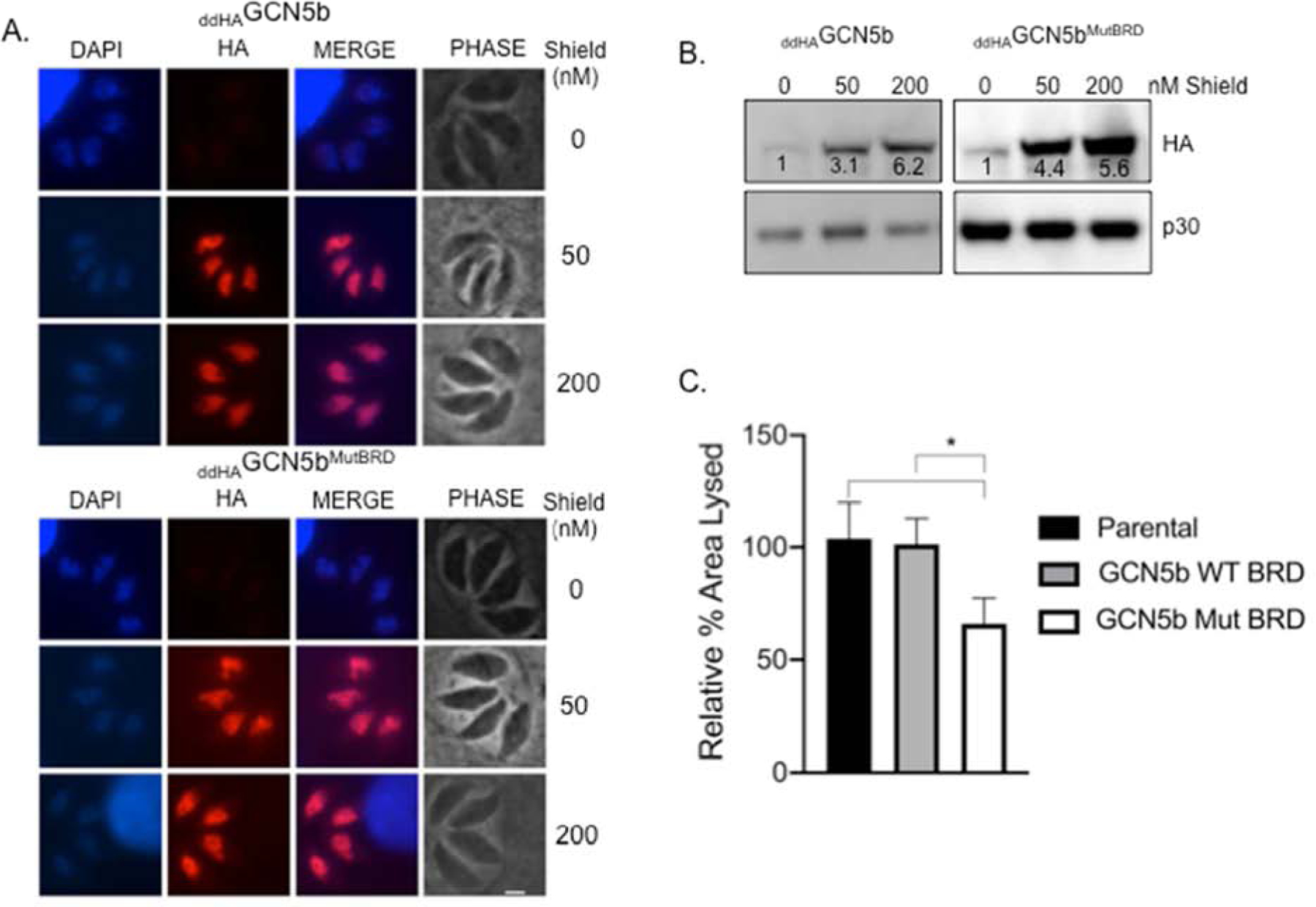
(A and B)ddHAGCN5b andddHAGCN5bMutBRD parasites after 48 hours of incubation with vehicle or Shield. (A) Immunofluorescence assays to monitor expression ofddHAGCN5b andddHAGCN5bMutBRD by Shield. Blue indicates DAPI stain and red anti-HA stain. Scale bar represents 2 μm. (B) Western blot to monitor expression ofddHAGCN5b andddHAGCN5bMutBRD by Shield. Numeric values represent the band densities of Shield-treated parasites relative to vehicle-treated parasites, normalized to the loading control (Toxoplasma p30). (C) Quantification of host cell area lysed inddHAGCN5b andddHAGCN5bMutBRD parasites treated with 200 nM Shield relative to parental (RHΔHX) parasites treated with vehicle from three independent plaque assays performed in technical triplicate. Error bars represent standard deviation. *, P<0.05 by one-way ANOVA with Dunnett’s multiple comparisons post-hoc.
To determine if the mutant bromodomain had an impact on parasite viability, we performed standard plaque assays and assayed the relative area of host cell lysis. As we have seen before (14), expression of ectopicddHAGCN5b had no effect on parasite proliferation compared to the parental line (RHΔHX). In contrast, expression ofddHAGCN5bMutBRD resulted in ~35% decreased lysis of the host cell monolayer (Fig. 2C), suggesting that a functional GCN5b bromodomain is important for parasite viability.
3.2. L-Moses inhibits GCN5b bromodomain binding activity in vitro
To further investigate the role of the GCN5b bromodomain in parasite viability, we tested whether the bromodomain chemical probe, L-Moses, could interfere with the ability of the GCN5b bromodomain to bind to acetyl-lysine. A previous study describing the co-crystallization structure (PDB identification 5TPX) of the Plasmodium falciparum GCN5 bromodomain complexed with L-Moses illustrated that L-Moses binds within the acetyl-lysine binding pocket (22). Superimposition of this co-crystal structure using UCSF Chimera software (28) with the predicted model of the Toxoplasma GCN5b bromodomain generated by I-Tasser shows that the acetyl-lysine binding sites between these two apicomplexan KATs are similar (Fig. 3A). It is noted that the amino acids within the PfGCN5 bromodomain that are involved in the interaction with L-Moses are conserved in the Toxoplasma GCN5b bromodomain, making it likely that L-Moses targets Toxoplasma GCN5b in a similar manner (Fig. 3A). Two of the amino acids in the PfGCN5 bromodomain that interact with L-Moses (Y1442 and N1436, which correspond to Y970 and N964 in Toxoplasma, respectively) include residues we mutated to alanine in this study, further supporting the critical role of these residues in the function of the Toxoplasma GCN5b bromodomain.
Fig. 3. Treatment with L-Moses inhibits TgGCN5b bromodomain binding activity in vitro.
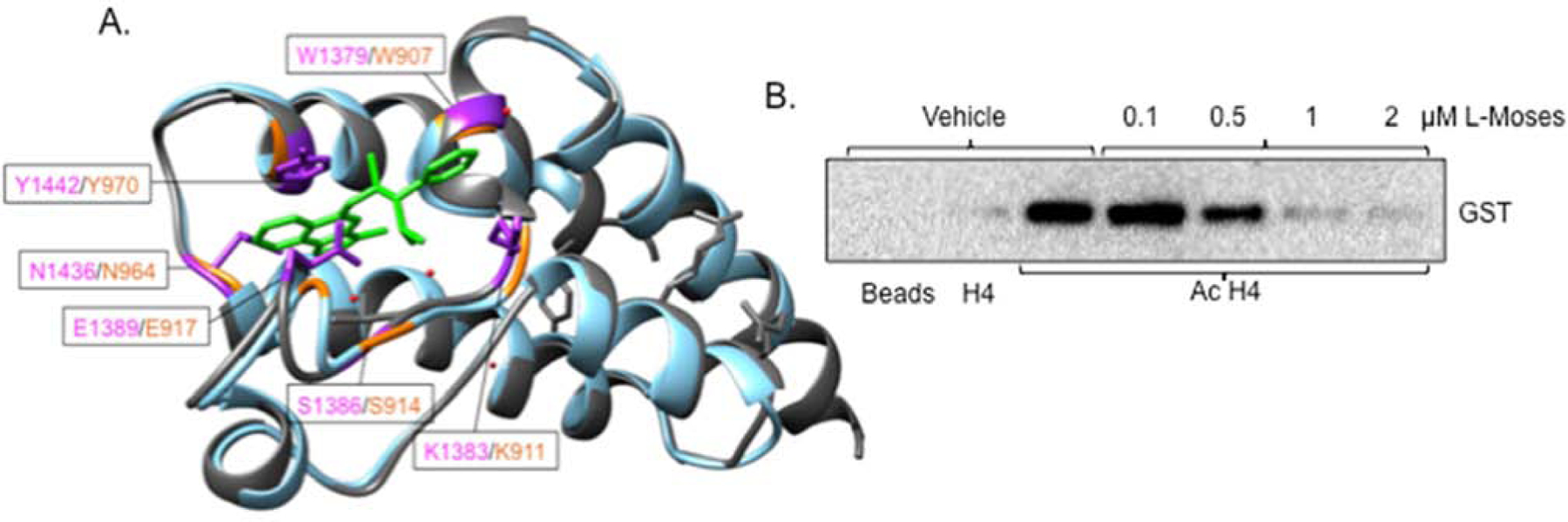
(A) Predicted model of TgGCN5b bromodomain (blue) superimposed with the PfGCN5 bromodomain (grey) bound to L-Moses (green) (PDB identification of PfGCN5 complexed to L-Moses is 5tpx) using Chimera Matchmaker. Purple residues are those in P. falciparum that interact with L-Moses. Orange text represents the corresponding residues in Toxoplasma. Red dots represent water molecules. (B) BRD binding assay. Western blot was probed with anti-GST to display recombinant, GST-tagged GCN5b bromodomain binding to H4 or acetylated H4 (AcH4) in the presence of vehicle or L-Moses.
To assess the activity of L-Moses against the Toxoplasma GCN5b bromodomain in vitro, we tested the ability of L-Moses to inhibit GCN5b bromodomain binding to acetylated histone H4. While the vehicle control (DMSO) has no effect on binding of the GCN5b bromodomain to acetylated H4, L-Moses inhibits GCN5b bromodomain binding to acetylated H4 in a dose-dependent manner (Fig. 3B). Together, these data suggest that the function of GCN5 bromodomains in apicomplexan parasites can be disrupted by small molecule inhibitors such as L-Moses.
3.3. L-Moses inhibits Toxoplasma replication
Given that L-Moses inhibits GCN5b bromodomain binding activity in vitro, we examined the effect of L-Moses on tachyzoite proliferation in HFF host cells. To quantitate parasite growth, we cultured RH strain parasites engineered to express a β-galactosidase reporter (30) in increasing concentrations of L-Moses. Results show that L-Moses inhibits parasite proliferation at an IC50 of ~0.6 μM (Fig. 4A). L-Moses did not exhibit overt effects on the morphology of the HFF host cells, nor was it cytotoxic at concentrations up to 100 μM according to an MTS viability assay (Fig. 4B) (33). To determine whether the GCN5b bromodomain is a target of L-Moses in the parasite, we utilized a previously generated clonal parasite line that constitutively overexpresses full-length GCN5b (HAGCN5b). If GCN5b is a target of L-Moses, overexpression of GCN5b would be expected to confer some resistance to the compound. Both a parasite plaque assay and doubling assay revealed that L-Moses decreases viability and replication of parental (RHΔHX) parasites (Fig. 4C–D). In contrast, parasites constitutively overexpressing GCN5b showed decreased susceptibility to L-Moses compared to the parental line. Parental parasites exhibited significantly fewer and smaller plaques thanHAGCN5b overexpressing parasites in response to L-Moses treatment (Fig. 4C). Similarly, at 36 hours post-infection, parental parasites showed a significant reduction in parasite doubling with L-Moses treatment compared toHAGCN5b overexpressing parasites (Fig. 4D).
Fig.4. Treatment with L-Moses inhibits Toxoplasma replication.
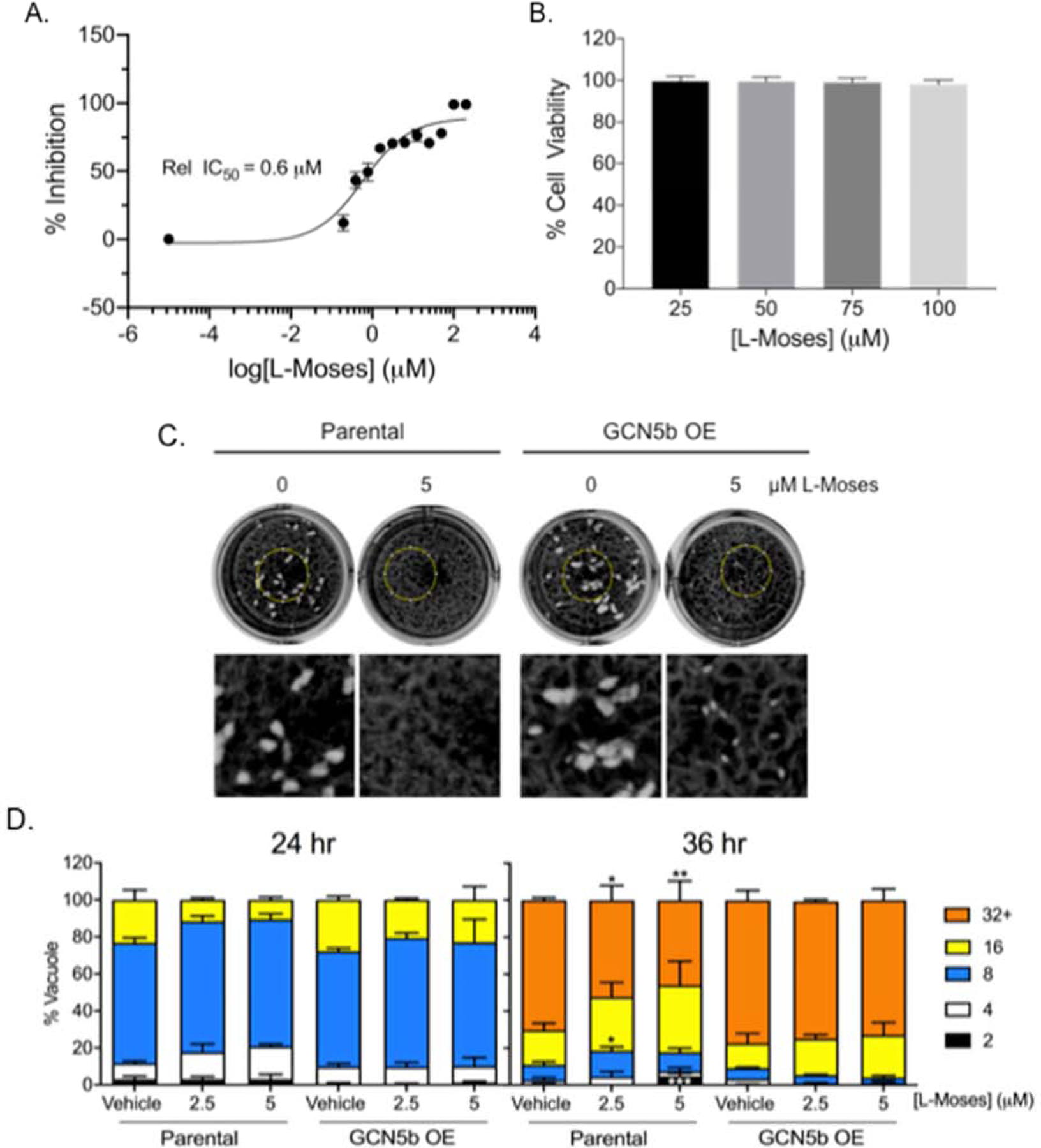
(A) Proliferation assay showing average percent inhibition of the reporter activity of RH parasites constitutively expressing β-galactosidase in the presence of increasing concentrations of L-Moses. Shown are data from three independent experiments performed in technical triplicate. (B) MTS assays showing average percentage of host cell viability in L-Moses-treated cells relative to vehicle-treated cells. Data from four independent experiments performed in technical triplicate are shown. (C) Plaque assay showing fitness of parental parasites versus parasites over-expressing GCN5b, treated with vehicle or L-Moses. Images under the wells are magnified areas of the regions outlined by the yellow circle. This is representative of three independent trials showing similar results. (D) Doubling assay showing the average percent of vacuoles containing the indicated number of parasites per vacuole.150 random vacuoles were counted from three independent experiments. *, P<0.05, **, P<0.01, and ***, P<0.001 between parasite lines at the given concentration of drug by two-way ANOVA with Tukey’s multiple comparisons post-hoc. For all graphs, error bars represent standard deviation.
These data do not rule out additional mechanisms of action for L-Moses, but lend support to the idea that the GCN5b bromodomain is a major target.
4. Conclusions
There is an urgent need to identify new drug targets to treat infections caused by apicomplexan parasites. We previously showed that GCN5b is an essential gene in Toxoplasma, and that the KAT domain is critical for protein function (14). Here, we present evidence that the bromodomain of GCN5b is a novel and attractive drug target important for the viability of tachyzoites. Based on our findings, we propose that the rationally designed bromodomain inhibitor L-Moses is a promising lead compound against Toxoplasma. The effects of L-Moses against tissue cysts remains to be examined; this is an important question to resolve in future studies as there is evidence that both bromodomain-containing KATs, GCN5a and GCN5b, play roles in bradyzoite development (10, 13).
Given that the Toxoplasma GCN5b BRD shares a high degree of sequence identity (55%) (Fig. 1A) and structural similarity (Fig. 3A) with the PfGCN5b BRD, it is likely that L-Moses will also show activity against Plasmodium and possibly other apicomplexan parasites.
Further structure-activity relationship testing will be necessary to identify an even more selective inhibitor for the parasite BRD, with minimal toxicity to the host. Our established in vitro binding assay employing the recombinant GCN5b bromodomain and acetylated H4 peptide substrate could be scaled up to identify new small molecules that interfere with this critical domain. Furthermore, virtual screening approaches, such as in (35), can be utilized to identify and design inhibitors with specificity for the apicomplexan target over the human target.
Highlights.
GCN5b is an essential lysine acetyltransferase in the parasite Toxoplasma gondii.
The GCN5b bromodomain binds to acetylated histones via conserved residues.
Mutating the GCN5b bromodomain decreases parasite viability.
L-Moses blocks GCN5b bromodomain binding to acetylated histones.
L-Moses has anti-Toxoplasma activity in vitro.
Acknowledgments
This work was supported by research grants from the National Institutes of Health (NIH AI116496 to WJS) and a Junior Investigator Pilot Grant from the IU Center for AIDS Research (VJ). The authors wish to thank members of the Sullivan laboratory and Drs. Gustavo Arrizabalaga and Stacey Gilk for helpful discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill D, Dubey JP. Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect. 2002;8(10):634–40. [DOI] [PubMed] [Google Scholar]
- 2.Hill DE, Chirukandoth S, Dubey JP. Biology and epidemiology of Toxoplasma gondii in man and animals. Anim Health Res Rev. 2005;6(1):41–61. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan WJ Jr. , Jeffers V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS microbiology reviews. 2012;36(3):725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeffers V, Tampaki Z, Kim K, Sullivan WJ, Jr. A latent ability to persist: differentiation in Toxoplasma gondii. Cellular and molecular life sciences : CMLS. 2018. doi: 10.1007/s00018-018-2808-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss LM, Dubey JP. Toxoplasmosis: A history of clinical observations. International journal for parasitology. 2009;39(8):895–901. doi: 10.1016/j.ijpara.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Harari RR, Goodwin E, Casoy J. Adverse Event Profile of Pyrimethamine-Based Therapy in Toxoplasmosis: A Systematic Review. Drugs in R&D. 2017;17(4):523–44. doi: 10.1007/s40268-017-0206-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marmorstein R, Zhou MM. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harbor perspectives in biology. 2014;6(7):a018762. doi: 10.1101/cshperspect.a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeffers V, Sullivan WJ Jr. Lysine Acetylation Is Widespread on Proteins of Diverse Function and Localization in the Protozoan Parasite Toxoplasma gondii. Eukaryot Cell. 2012;11(6):735–42. doi: 10.1128/EC.00088-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miao J, Lawrence M, Jeffers V, Zhao F, Parker D, Ge Y, et al. Extensive lysine acetylation occurs in evolutionarily conserved metabolic pathways and parasite-specific functions during Plasmodium falciparum intraerythrocytic development. Mol Microbiol. 2013;89(4):660–75. doi: 10.1111/mmi.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saksouk N, Bhatti MM, Kieffer S, Smith AT, Musset K, Garin J, et al. Histone-modifying complexes regulate gene expression pertinent to the differentiation of the protozoan parasite Toxoplasma gondii. Mol Cell Biol. 2005;25(23):10301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatti MM, Livingston M, Mullapudi N, Sullivan WJ Jr. Pair of unusual GCN5 histone acetyltransferases and ADA2 homologues in the protozoan parasite Toxoplasma gondii. Eukaryot Cell. 2006;5(1):62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735):491–6. [DOI] [PubMed] [Google Scholar]
- 13.Naguleswaran A, Elias EV, McClintick J, Edenberg HJ, Sullivan WJ. Toxoplasma gondii Lysine Acetyltransferase GCN5-A Functions in the Cellular Response to Alkaline Stress and Expression of Cyst Genes. PLoS Pathog. 2010;6(12):e1001232. doi: 10.1371/journal.ppat.1001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Dixon SE, Ting LM, Liu TK, Jeffers V, Croken MM, et al. Lysine acetyltransferase GCN5b interacts with AP2 factors and is required for Toxoplasma gondii proliferation. PLoS Pathog. 2014;10(1):e1003830. doi: 10.1371/journal.ppat.1003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeffers V, Gao H, Checkley LA, Liu Y, Ferdig MT, Sullivan WJ, Jr. Garcinol Inhibits GCN5-Mediated Lysine Acetyltransferase Activity and Prevents Replication of the Parasite Toxoplasma gondii. Antimicrob Agents Chemother. 2016;60(4):2164–70. doi: 10.1128/AAC.03059-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, et al. A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell. 2016;166(6):1423–35 e12. doi: 10.1016/j.cell.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulz D, Mugnier MR, Paulsen EM, Kim HS, Chung CW, Tough DF, et al. Bromodomain Proteins Contribute to Maintenance of Bloodstream Form Stage Identity in the African Trypanosome. PLoS Biol. 2015;13(12):e1002316. doi: 10.1371/journal.pbio.1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeffers V, Yang C, Huang S, Sullivan WJ Jr. Bromodomains in Protozoan Parasites: Evolution, Function, and Opportunities for Drug Development. Microbiology and molecular biology reviews : MMBR. 2017;81 (1). doi: 10.1128/MMBR.00047-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia P, Alonso VL, Serra E, Escalante AM, Furlan RLE. Discovery of a Biologically Active Bromodomain Inhibitor by Target-Directed Dynamic Combinatorial Chemistry. ACS medicinal chemistry letters. 2018;9(10):1002–6. doi: 10.1021/acsmedchemlett.8b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chua MJ, Robaa D, Skinner-Adams TS, Sippl W, Andrews KT. Activity of bromodomain protein inhibitors/binders against asexual-stage Plasmodium falciparum parasites. International journal for parasitology Drugs and drug resistance. 2018;8(2):189–93. doi: 10.1016/j.ijpddr.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramallo IA, Alonso VL, Rua F, Serra E, Furlan RLE. A Bioactive Trypanosoma cruzi Bromodomain Inhibitor from Chemically Engineered Extracts. ACS combinatorial science. 2018;20(4):220–8. doi: 10.1021/acscombsci.7b00172. [DOI] [PubMed] [Google Scholar]
- 22.Moustakim M, Clark PG, Trulli L, Fuentes de Arriba AL, Ehebauer MT, Chaikuad A, et al. Discovery of a PCAF Bromodomain Chemical Probe. Angewandte Chemie. 2017;56(3):827–31. doi: 10.1002/anie.201610816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roos DS, Donald RG, Morrissette NS, Moulton AL. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 1994;45:27–63. [DOI] [PubMed] [Google Scholar]
- 24.Xu L, Cheng A, Huang M, Zhang J, Jiang Y, Wang C, et al. Structural insight into the recognition of acetylated histone H3K56ac mediated by the bromodomain of CREB-binding protein. FEBS J. 2017;284(20):3422–36. doi: 10.1111/febs.14198. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5(4):725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 2017;18(1):529. doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McFadden DC, Seeber F, Boothroyd JC. Use of Toxoplasma gondii expressing beta-galactosidase for colorimetric assessment of drug activity in vitro. Antimicrob Agents Chemother. 1997;41(9):1849–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varberg JM, LaFavers KA, Arrizabalaga G, Sullivan WJ, Jr. Characterization of Plasmodium Atg3-Atg8 Interaction Inhibitors Identifies Novel Alternative Mechanisms of Action in Toxoplasma gondii. Antimicrob Agents Chemother. 2018;62 (2). doi: 10.1128/AAC.01489-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fichera ME, Bhopale MK, Roos DS. In vitro assays elucidate peculiar kinetics of clindamycin action against Toxoplasma gondii. Antimicrob Agents Chemother. 1995;39(7):1530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doggett JS, Nilsen A, Forquer I, Wegmann KW, Jones-Brando L, Yolken RH, et al. Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc Natl Acad Sci U S A. 2012;109(39):15936–41. doi: 10.1073/pnas.1208069109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herm-Gotz A, Agop-Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, et al. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat Methods. 2007;4(12):1003−−5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar A, Bhowmick K, Vikramdeo KS, Mondal N, Subbarao N, Dhar SK. Designing novel inhibitors against histone acetyltransferase (HAT: GCN5) of Plasmodium falciparum. European journal of medicinal chemistry. 2017;138:26–37. doi: 10.1016/j.ejmech.2017.06.009. [DOI] [PubMed] [Google Scholar]