

Synopsis
We report a new artificial hydrogenase made by covalent anchoring of the iron Knölker’s complex to a xylanase S212C variant. This artificial metalloenzyme was found to be able to catalyze efficiently the transfer hydrogenation of the benchmark substrate trifluoroacetophenone by sodium formate in water, yielding the corresponding secondary alcohol as a racemic. The reaction proceeded more than 3-fold faster with the XlnS212CK biohybrid than with the Knölker’s complex alone. In addition, efficient conversion of trifluoroacetophenone to its corresponding alcohol was reached within 60 h with XlnS212CK, whereas a ≈ 2.5 fold lower conversion was observed with Knölker’s complex alone as catalyst. Moreover, the data were rationalized with a computational strategy suggesting the key factors of the selectivity. These results suggested that the Knölker’s complex was most likely flexible and could experience free rotational reorientation within the active-site pocket of Xln A, allowing it to access the subsite pocket populated by trifluoroacetophenone.
Keywords: Artificial metalloenzymes, catalysis, hydrogenation, Knölker’s complex
Graphical Abstract
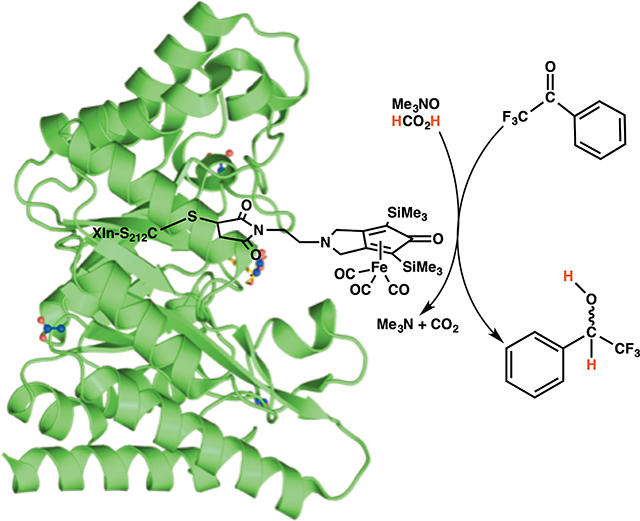
1. Introduction
The reduction of polarized multiple bonds (ketones, imines, etc.) plays a major role in modern organic synthesis. This reaction can be carried out using metal hydrides such as LiAlH4 (1), DIBALH (2), BH3 (3) or NaBH4 (4). However, these reducing agents are used in stoichiometric amounts and, as a result, generate numerous by-products. In addition, the control of chemo- and stereoselectivity can be difficult in the presence of these hydrides. Nonetheless, obtaining enantiomerically pure products resulting from such transformations (chiral alcohols or chiral amines) is a key step in the synthesis of pharmaceutical or agrochemical products. To this end, alternative synthesis routes are now commonly used, involving the metallo-catalyzed version of reduction reactions such as hydrogenation under high pressure of molecular hydrogen and hydrogen transfer reactions in the presence of a hydrogen atom donor (5). Depending on the nature of the metal and its ligands, the main advantage of this approach is the possibility to control regio-, chemo- and/or stereoselectivity of the reaction. Unfortunately, the most commonly used catalysts for this type of reaction are based on weakly available, expensive, and toxic noble metals such as ruthenium (5), iridium (6) or rhodium (7). The development of cheaper, more efficient, and eco-compatible catalysts has become a challenge of modern chemistry. In this context, iron-catalyzed hydrogenation has recently emerged as the most economical and environmentally preferred route of synthesis. It was recently demonstrated that Knölker type iron complexes could be efficient catalysts for the hydrogenation of aromatic ketones in water (8). However, this prior report has revealed that the reaction temperature was also decisive since no conversion was observed at 40 °C, and only 13% conversion was observed at 60 °C.
In addition to the enantioselective transformations catalyzed by metal complexes, catalysis by artificial metalloenzymes has become an interesting tool for the synthesis of enantiopure compounds in water (9). This technology combines the attractive features of enzymatic catalysis with those of chemical catalysis (10). Given the great potential of these biohybrid catalysts in asymmetric synthesis and, knowing that Knölker’s iron complexes are efficient catalysts in water, a recent report illustrated the synthesis of biotinylated Knölker’s complexes incorporated into streptavidin using the so-called “Trojan Horse” strategy (11). The artificial iron hydrogenases thus prepared catalyzed the reduction of ketones in aqueous medium, with the best conversion rates (26%) and enantiomeric excess (34%) obtained with para-methoxy-acetophenone as substrate (11).
Xylanases hydrolyze β−1,4 glycosidic bonds in the main chain of xylan, the major component of plant cell wall hemicellulose. In the pulp bleaching process, the enzymatic hydrolysis of xylan at high temperature has gained importance from an environmental standpoint. Consequently, growing interest in temperature-tolerant xylanases working under extreme pH and ionic strength conditions has led to the discovery, cloning, and characterization of numerous enzymes. This includes xylanase A (Xln A) from Streptomyces lividans, an enzyme exhibiting optimal activity at 60 °C and pH 6 (12). This robust biocatalyst can also be easily purified, and its sequence and three-dimensional structure are known (13), making it a good scaffold for the production of artificial metalloenzymes. It has been shown that non-covalently inserted Fe(III)- and Mn(III)-complexes of anionic porphyrins into Xln A leads to artificial metalloenzymes exhibiting good peroxidase activity (12). In addition, the Fe(III)-porphyrin-based hybrid biocatalysts Fe(III)(TpCPP)- and Fe(III)(TpSPP)-Xln A were found to catalyze the stereoselective oxidation of thioanisole in the presence of imidazole with roughly 40% enantiomeric excess (14,15). The most efficient biohybrid was undoubtedly observed for the stereoselective oxidation of styrene derivatives by oxone, catalyzed by the Mn(III)(TpCPP)-Xln A hemozyme, which led to an 80% ee in favor of the R isomer in the particular case of p-methoxystyrene (16).
In this paper, we report the preparation and characterization of a new artificial metalloenzyme that was obtained by the covalent attachment of a Knölker type iron complex into xylanase A. This artificial metalloenzyme can efficiently catalyze the transfer hydrogenation of the benchmark substrate trifluoroacetophenone by sodium formate in water, yielding the corresponding secondary alcohol as a racemic. The reaction proceeded more than 3-fold faster with the XlnS212CK biohybrid than with the Knölker’s complex alone. In addition, efficient conversion of trifluoroacetophenone to its corresponding alcohol was reached within 60 h with XlnS212CK, whereas a ≈ 2.5 fold lower conversion was observed with Knölker’s complex alone as catalyst.
2. Experimental section
2.1. Physical measurements
UV–visible spectroscopy studies were performed on double beam UVIKON 860XL and CARY 300BIO VARIAN spectrophotometers.
1H NMR and 13C NMR spectra were recorded on BRUKER AM 250, 300 and 360 spectrometers. Chemical shifts δ are given in ppm using the solvent as internal reference with respect to tetramethylsilane (TMS). Electrospray ionization mass spectrometry experiments were carried on a MicrOTOF-Q Bruker spectrometer.
2.2. Preparation of the Knölker’s complex 3-(trimethylsilyl)prop-2-yn-1-ol (S1) (11)
In a two-necked round bottom flask under argon atmosphere a 2.5 M solution of nBuLi in hexane (34.5 mL, 86.3 mmol) was added dropwise to a solution of propargylic alcohol (2.1 g, 37.5 mmol) in 100 mL dry THF at −78°C. The solution was slowly warmed up to room temperature and stirred for an additional 1h. The reaction mixture was cooled down to −78°C and trimethylsilyl chloride (11 mL, 86.3 mmol) was slowly added. The resulting mixture was stirred at room temperature overnight. The reaction was then cooled down to 0 °C and an aqueous solution of 2.6 mL of 95% sulfuric acid in 15 mL of water was added slowly. The resulting solution was stirred for an additional 3 h at room temperature. Water (20 mL) was added until dissolution of the inorganic salts. The aqueous layer was extracted with diethyl ether (3 × 50 mL). Organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by distillation (3.5 g, 73 %).
3-(trimethylsilyl)prop-2-ynyl bromide (S2) (11,17)
In a three necked round bottom flask under argon atmosphere, a 1 M solution of bromine in dry dichloromethane (38.4 mL, 38.4 mmol) was added dropwise to a solution of triphenylphosphine (10 g, 38.4 mmol) in 200 mL dry dichloromethane at 0°C. Pyridine (2.55 mL, 32 mmol) was then added, followed by the addition of a solution of S1 (4.1 g. 32 mmol) in 50 mL dry dichloromethane at 0°C. The resulting mixture was stirred at 0°C for 2 h and then at room temperature for 4 h. Pentane (500 mL) was then added, which resulted in the formation of a precipitate of triphenylphosphine oxide. The solution was then filtered through a pad of Celite® and silica and the solvents were removed under reduced pressure to afford compound S2 as a colorless oil (4.16 g, 68 %).
1H NMR (CDCl3, 300 MHz) δ(ppm/TMS): 3.91 (s, 2H), 0.18 (s, 9H).
Tert-butyl(2-aminoethyl)carbamate (S3) (11,17)
In a two necked round bottom flask under argon atmosphere, a solution of Boc2O (1 g, 4.6 mmol) in 25 mL dry dichloromethane was added dropwise to a solution of ethylenediamine (10 mL, 150 mmol) in 40 mL dry dichloromethane at 0°C. The reaction mixture was stirred 1 h at 0°C and then overnight at room temperature. Water (40 mL) was added and the organic layer was recovered, washed twice with 40 mL water and dried over Na2SO4. After filtration, the solvent was removed under reduced pressure and the residue was purified by chromatography on a neutral alumina column eluted with ethyl acetate/methanol (95/5) to afford S3 as a colorless oil (0.42 g, 57 %).
1H NMR (CDCl3, 300 MHz), δ(ppm/TMS): 4.86 (br s, 1H), 3.17 (q, J = 5.9 Hz, 2H), 2.80 (t, J = 5.9 Hz, 2H), 1.44 (s, 9H), 1.42 (br s, 2H).
ESI-MS m/z [M + H]+ : Calc. 161.1285, Found 161.1284
N-tert-Butoxycarbonyl,N’-Maleimido-Ethylenediamine (S4) (17)
In a round bottom flask, N-Methoxy-carbonyl maleimide (119 mg, 0.77 mmol) was dissolved in 10 mL of a saturated aqueous NaHCO3 solution on ice. The reaction mixture was stirred 40 min. on ice and then 1 h 30 min. at room temperature. The pH of the solution was adjusted to 3 with concentrated H2SO4 on ice. The aqueous layer was then extracted with ethyl acetate (3 × 150 mL). Organic layers were dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude product was then purified by chromatography on a silica gel column eluted with heptane/ethyl acetate (1/1) to afford S4 as a white solid (131 mg, 71 %).
1H NMR (CDCl3, 300 MHz), δ(ppm/TMS) : 4.86 (br s, 1H), 3.17 (q, J = 5.9 Hz, 2H), 2.80 (t, J = 5.9 Hz, 2H), 1.44 (s, 9H), 1.42 (br s, 2H).
N-(2-Aminoethyl)maleimide (S5) (17,18)
In a round bottom flask, compound S4 (130 mg, 0.54 mmol) was dissolved in 2 mL dichloromethane at 0° C. TFA (760 μL, 9.9 mmol) was added and the resulting solution was stirred for 2 h at room temperature. The mixture was concentrated to 1 mL and ice-cold diethyl ether (2 mL) was added. The obtained precipitate was filtered off and washed with ice-cold diethyl ether (3×10 mL).
1H NMR (D2O, 300 MHz), δ(ppm/TMS) : 6.88 (s, 2H), 3.81 (t, 2H), 3.20 (t, 2H).
ESI-MS m/z [M + H]+ : Calc. 141.0659, Found 141.0664
1-(2-(bis(3-(trimethylsilyl)prop-2-yn-1-yl)amino)ethyl)-1H-pyrrole-2,5-dione (S6) (11)
In a round bottom flask under argon atmosphere, S5 (90.5 mg, 0.36 mmol) and S3 (136 mg, 0.71 mmol) were added in 20 mL of dry acetonitrile. N,N-diisopropylethylamine (63 μL, 0.36 mmol) was added and the resulting mixture was stirred for 2h at room temperature, and then for an additional 16 h at 70° C. After cooling down, the solvent was removed under reduced pressure and 50 mL of water were added. The aqueous layer was then extracted with ethyl acetate (3 × 30 mL). Organic layers were dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude product was then purified with by chromatography on a silica gel column using a pentane/ ethyl acetate (95/5 to 90/10) gradient to obtain a white solid (70 mg, 54%)
1H NMR (CDCl3, 300 MHz), δ(ppm/TMS): 7 (s, 2H), 3.4 (s, 4H), 2.8 (t, 2H), 2.6 (t, 2H), 0.16 (s, 18H) ppm.
13C NMR (CDCl3, 300 MHz), δ(ppm/TMS): 170.6 (C q), 134 (CH), 100.8 (C q), 89.8 (C q), 49.4 (CH2), 43.4 (CH2), 35.36 (CH2), −0.07 (CH3).
ESI-MS m/z [M + H] +: calc. 361.1762, Found 361.1763
Knölker’s Complex (S7) (11)
In a round bottom flask under argon atmosphere, S6 (122 mg, 0.34 mmol) and Fe2(CO)9 (123 mg, 0.34 mmol) were introduced in 5 mL degassed and dry toluene. The reaction mixture was heated at 110°C for 14 h. The resulting brownish solution was cooled down to room temperature and purified by column chromatography on neutral alumina topped with a pad of Celite (eluent: dichloromethane/Cyclohexane, 6/4) to afford the desired product S7 as a yellow-brown solid (95 mg, 53%).
1H NMR (CHCl3, 300 MHz), δ(ppm/TMS) : 6.68 (s, 2H), 3.88 (d, 2H), 3.72 (t, 2H), 3.35 (d, 2H), 3.04 (t, 2H), 0.24 (s, 18H).
13C NMR (CHCl3, 300 MHz), δ(ppm/TMS) : 170.7 (C q), 134.1 (CH), 112.9 (C q), 69.2(C q), 53.3 (CH2), 52.3 (CH2), 36.5 (CH2), −0.8(CH3) ppm.
ESI-MS m/z [M + H]+ : Calc. 529.0908, Found. 529.0908
2.3. Protein expression and purification
Wild-type and mutant forms of Xln A were expressed and purified from the supernatant of S. lividans using a slightly modified version of a previously reported protocol (12). Genetic constructs for cysteine mutants at positions K48 and S212 were a generous gift from Prof. Claude Dupont (INRS). Briefly, S. lividans was first pre-cultured in 12 mL TSB medium for 2 days and further expressed for 72 h at 34°C by inoculation in M14 minimal medium using xylose as the sole carbon source. The culture supernatant was concentrated by ultrafiltration and dialyzed against 20 mM citric buffer (pH 4.5). The dialyzed enzyme solution was then loaded on a HiPrep CM FF 16/10 column (GE Healthcare) pre-equilibrated with 20 mM citric buffer (pH 4.5). The enzyme was eluted with a linear gradient of the same buffer containing 1 M NaCl. Fractions were collected and protein elution was monitored at 280 nm. Elution fractions were further analyzed by SDS-PAGE and fractions containing Xln A were collected, pooled, dialyzed against water, and lyophilized.
2.4. Monitoring of thiol accessibility
The accessibility of the C212 thiol group was monitored using Ellman’s test (19). In a standard assay, 5 μL of a 10 mM solution 5,5’-Dithio-bis-(2-nitrobenzoic acid) in water were added to 445 μL of reaction buffer (0.1 M sodium phosphate pH 8.0, 1 mM EDTA) in a 1 mL quartz cuvette. The absorbance at 412 nm was then monitored as a function of time, using a Jasco spectrophotometer, after the addition of 50 μL of a 100 μM protein solution until complete reaction. The final absorbance obtained at the plateau was noted Aeq and the initial absorbance measured before protein addition was noted A0. The reduced cysteine content was then calculated from the difference in absorbance Aeq - A0, corrected by the dilution factor, using an εM value of 14150 M−1.cm−1 at 412nm.
2.5. Molecular modeling
2.5.1. Construction of the Xln A-Knölker’s biohybrid complex
The crystal structure of Xln A from Streptomyces lividans (PDB entry 1E0W) was used as the starting template for construction of the Xln A-Knölker biohybrid complex (20). To predict backbone perturbations caused by introduction of the S212C substitution, a standard RosettaBackub protocol was used (21). The Universal Force Field (UFF) and Steepest Descent protocols available in the Avogadro 1.2.0 suite were applied to properly estimate conformation of the Knölker’s molecule (including the Fe ion as transition metal). The Xln A-Knölker’s complex was created using the Build-Structure tools available in UCSF Chimera 1.14. Amino acid residues in the vicinity of the Knölker’s molecule were then energy minimized using the force field provided by the Molegro Virtual Docker suite 6.0 (MVD). Through this energy minimization step, the thermodynamic barriers between the Xln A receptor protein and Knölker’s ligand were evaluated to extract the most realistic representations of this hybrid complex. During this particular analysis, both the Knölker’s complex ligand and Xln A were first considered as separated entities.
2.5.2. Conformational exchange experienced by Xln A
To simulate conformational exchange experienced by Xln A, we applied two coarse-grained methodologies that predict short- and long-range protein dynamics. To simulate local residue dynamics, the stochastic method based on a Monte Carlo analysis was applied using the CABSflex 2.0 server (22). Default server parameters were used for this analysis, which recently demonstrated results comparable to other all-atoms MD simulations protocols (i.e. similar to 10-ns simulations in aqueous solvent) (23). Long-range protein dynamics were predicted using a Normal Mode Analysis (NMA) methodology through the ElNémo server (24) NMA computed two trajectories with 10 models, corresponding to the lowest frequency normal modes. Parameters used a perturbation range of −100 and a cut-off of 8 Å. Collectivity scores were considered to manually select the best two elastic network models. All coarse-grained methodologies were performed using the S212C model obtained from the RosettaBackub server as starting PDB structure (25).
2.5.3. Molecular docking simulations
To simulate coordination of the Xln A-Knölker’s biohybrid complex with the trifluoroacetophenone (TFAC) substrate molecule, we performed virtual docking simulations using the MolDock scoring function implemented in the MVD suite 6.0 (26). The virtual docking procedure was performed in two sequential phases. We first performed blind (free) docking of the substrate on the whole biohybrid complex as receptor target, after which we used a reduced searching area acquired from the best docking conformers obtained from the first phase of the docking simulation. Simulations were performed without incorporation of water molecules and the best conformers arose from a pool of 1,000,000 docking combinations. Results from both sequential phases were compiled from 500 iterations per run and 20 runs per process. The ligand molecule was obtained from the PubChem database under accession CID number 9905.
2.6. Coupling of the Knölker’s complex to the S212C variant of Xln A
The concentration of accessible thiols calculated via Ellman’s test was used as the free cysteine concentration available for the coupling of the Knölker’s complex S7. Five equiv. of S7 diluted in DMSO were mixed with the Xylanase A S212C variant (XlnS212C) in the coupling buffer (HEPES 10 mM, 150 mM NaCl, pH 7.75), for a final DMSO concentration of 2% in the desired volume. The mixture was incubated overnight at 4°C. The mixture was loaded on a gel filtration column (HiLoad 16/600 Superdex™ 75, GE Healthcare) equilibrated in buffer A (HEPES 10 mM, 150 mM NaCl, pH 8) that was previously incubated with Chelex® 100 resin and filtered. The coupling efficiency was measured by using Ellman’s test and MALDI-TOF MS analysis. The molar extinction coefficient of the Knölker’s complex was determined in HEPES 10 mM, NaCl 150 mM, pH 7 containing 5 % ethanol and was found to be similar to that measured in ethanol, εΜ = 3100 M−1.cm−1 at 280 nm. The molar extinction coefficient of the biohybrid at 280 nm was calculated by adding the computed molar extinction coefficient of the protein variant to that measured for the Knölker’s complex moiety and was found to be εΜ = 67,500 M−1.cm−1.
2.7. Protein mass spectrometry analysis
Protein solutions were desalted on a Zeba Spin column (Thermo) against 10 mM HEPES buffer pH 8 and then diluted in a sinapinic acid matrix for MALDI-ToF MS analysis in linear mode by using a MALDI-ToF/ToF UltrafleXtreme spectrometer (Bruker Daltonics).
2.8. Catalysis of the hydrogen transfer reaction
Reactions were carried out in 1 mL sealed vials for a 200 μL reaction volume. In each vial, 86.2 μL of a 69.6 μM XlnS212CK solution in 10 mM HEPES 150 mM NaCl pH 8 (final concentration: 30 μM), 5 μL of a 3.6 mM Me3NO solution in water (final concentration: 90 μM), 40.8 mg of sodium formate (final concentration: 3 M) and 10 μL of a 500 mM TFAC solution in ethanol (final concentration: 25 mM) and 10 mM HEPES 150 mM NaCl pH 8 were added for a final volume of 200 μL with 7.5% of EtOH. The vials were tightly closed and set in a dry block (Reacti-therm heating and stirring module) heated at 65°C with a vigorous stirring. The reaction was stopped after various times and 20 μL of a 31 mM acetophenone solution in acetonitrile were then added. The organic products were then extracted twice with 500 μL of ethyl acetate, dried with Na2SO4 and filtered. The samples were then analyzed by GC.
GC conditions used to determine the conversion: Zebron ZB Semi-Volatiles column (25 m, 0.25 mm, 0.25 μm) and helium vector gaz. Injector temperature: 300°C, oven temperature : 50 – 54°C, 1°C.min−1, then 54 – 150°C, 15°C.min−1, and 150 – 220°C, 50°C.min−1, then hold at 220°C for 1 min. Detection by FID at 300°C
GC conditions used to determine the ee of the product: Hydrodex y-DiMOM column (25 m, 0.25 mm, 0.25 μm) and helium vector gaz. Injector temperature: 240°C, oven temperature :100–118°C, 5°C.min−1 hold 15 min, then 118–150°C, 10°C.min−1 and hold for 1 min. Detection by FID at 300°C.
3. Results and discussion
3.1. Xln A mutant selection and modeled biohybrid formation
Covalently attaching an artificial complex to the active-site cavity of an enzyme to yield a newly designed, stable, and active biocatalyst remains a challenging task to this day, especially in light of issues pertaining to protein and biohybrid stability. To help with proper positioning and orientation of the Knölker’s complex in the solvent-exposed active site of Xln A, active-site residues K48 and S212 were selected as potential targets for cysteine mutagenesis, in addition to being subjected to molecular modeling investigation. Residues K48 and S212 are located on opposite sides of the active-site cavity, allowing subsequent formation of a covalent adduct between the enzyme and the Knölker’s complex using the free thiol group of the cysteine side chain. Our experimental results showed that biohybrid formation was unsuccessful at residue 48, most likely due unfavorable steric clashes between the enzyme and the Knölker’s complex upon covalent anchoring at this position (Figure 1). However, coupling of the Knölker’s complex to Xln A mutant S212C was successfully achieved, yielding the XlnS212CK enzyme variant (Figure 1).
Figure 1.

Coupling of the S7 Knölker’s complex to XlnS212C and XlnK48C enzyme variants : synthesis of the xylanase A-derived biohybrid XlnS212CK.
In accordance with experimental results, molecular modeling confirmed that the Knölker’s complex can be accommodated in the active-site of Xln A upon covalent anchoring at this position (Figure 2A).
Figure 2.

Biohybrid model of the Knölker’s complex covalently anchored to Xln A. A) Energy-minimized structure of Xln A covalently bound to the Knölker’s complex at mutated position C212. The left panel shows the surface of the Xln A active-site cavity (green) bound to the Knölker’s complex (ball-and-stick model). The right panel shows residue side-chains that stabilize the Knölker’s complex in the active-site cavity of Xln A. Surface transparency was applied to illustrate the active-site loop harboring residue C212. Residue side chains are labeled and shown before (blue) and after (gray) energy minimization. The Knölker’s complex is shown in sandy brown and the C212 substitution is labeled in green. B) Molecular dynamics simulations illustrating short-range (left panel) and long-range (right panel) conformational exchange experienced by Xln A. The C212 loop is colored red (left) and highlighted by a dashed circle (right). For long-range trajectory simulations, the starting Xln A conformer is shown as a blue ribbon.
However, the MolDock scoring function of the MVD force field revealed that this biohybrid remains energetically unfavorable when subjected to side-chain energy stabilization in the vicinity of the active-site cavity. Even after energy minimization, residues delineating the catalytic pocket of Xln A remain highly unstable (≈ 80 kcal/mol for the biohybrid complex) (Figure 2A, right panel). These results illustrate that an energy contribution is required to stabilize the Knölker’s complex and to establish proper thermodynamic conditions essential for enzyme-ligand stabilization. This also strongly suggests that formation of the Xln A-Knölker’s biohybrid requires conformational rearrangements within the protein scaffold.
To provide a more realistic model representation of the experimentally observed biohybrid complex, we performed Monte Carlo and Normal Mode Aanalysis (NMA) simulations to estimate short- and long-range conformational exchange experienced by the enzyme in solution. Coarse-grained simulations strongly suggest that the active-site cavity of Xln A experiences short- and long-range conformational rearrangements to allow Knölker’s complex stabilization within the active-site cavity (Figure 2B). Upon structural reorganization, energetically favorable protein conformers that stabilize biohybrid complex formation were readily observed, specifically for short-range flexibility models, which displayed favorable biohybrid complex energy values (ΔG up to −20 kcal/mol).
3.2. Synthesis of the Knölker’s complex S7
The Knölker’s Complex (S7) bearing a maleimido group that enables its covalent anchoring into the XlnS212C variant by reaction with the thiol function of C212 was prepared in seven steps, as described in Scheme 1.
Scheme 1.

Tert-butyl(2-aminoethyl)carbamate S3 was first prepared in an overall 50 % yield in two steps starting by the trimethyl silylation of propargylic alcohol by trimethylsilyl chloride, followed by a bromination by Br2 in the presence of triphenyl phosphine and pyridine in dichloromethane, as described in the literature (Scheme 1) (11,17). N-(2-Aminoethyl)maleimide S5 was then prepared in an overall 40 % yield in 3 steps involving the monoprotection of one amino group of ethylenediamine by Boc2O, followed by the introduction of the maleimido group by peptide coupling with N-Methoxy-carbonyl maleimide in the presence of sodium hydrogenocarbonate and, finally, deprotection of the amino group by trifluoroacetic acid (Scheme 1) (17,18). The 1-(2-(bis(3-(trimethylsilyl)prop-2-yn-1-yl)amino)ethyl)-1H-pyrrole-2,5-dione ligand S6 was then obtained in a 54 % yield by reaction of S3 with S5 in dry acetonitrile in the presence of N,N-diisopropylethylamine, as described in the experimental section (11). Final introduction of the iron(III) ion was performed by reaction of S7 with Fe2(CO)9 in dry toluene under inert atmosphere, producing the S7 Knölker’s complex at 53 % yield, which was then fully characterized by 1H and 13C NMR and ESI MS.
3.3. Synthesis and characterization of the XlnS212CK biohybrid
The reactivity and accessibility of the thiol function of cysteine 212 in the XlnS212C enzyme variant was first assessed by the time-resolved Ellman’s assay using sulfhydryl reagent 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB) that reacts with sulfhydryl groups and releases 2-nitro-5-thiobenzoate (TNB), which displays a maximum absorption at 412 nm (19). The final labeled cysteine concentration was equal to protein concentration, indicating full accessibility, reactivity, and lack of disulfide bridge formation at cysteine 212, even after several weeks of storage without any reducing agent. The XlnS212C variant was then functionalized by incubation with 5 equivalents of the S7 Knölker’s complex dissolved in DMSO overnight at 4°C, in coupling buffer (HEPES 10 mM, 150 mM NaCl, pH 7.75) (Figure. 1). This resulted in the formation of the XlnS212CK biohybrid, bearing Knölker’s complex S7 covalently linked to cysteine 212, which was further characterized by MALDI-ToF mass spectrometry (Figure 3). Peaks at 33158 ± 5 and 33598 ± 5 Da were respectively observed for the XlnS212C variant and for the xylanase A-derived biohybrid XlnS212CK, in agreement with the covalent anchoring of one Knölker’s complex S7 per protein after loss of three CO ligands (Figure 3).
Figure 3.

Maldi-ToF spectra of the XlnS212C variant measured before (blue) and after (red) functionalization with the Knölker’s complex compound.
3.4. Aqueous transfer hydrogenation of trifluoroacetophenone by metal complexes
Trifluoroacetophenone (TFAC) S8 was chosen as a benchmark substrate to delineate the catalytic activity of the newly synthesized complexes in aqueous transfer hydrogenation reactions using formate as a hydrogen source (Fig. 4). First, this substrate may be readily reduced into the corresponding α-(trifluoromethyl)benzyl alcohol S9 thanks to the electron-attracting character of the CF3 substituent (27). Second, TFAC is well known to be hydrogenated into asymmetric trifluoromethyl benzyl alcohol by naturally occurring hydrogenases like carbonyl reductase (28), which makes it a perfect candidate for studying the enantioselectivity of the catalysis.
Figure 4.

Hydrogenation of trifluoroacetophenone TFAC S8 by sodium formate catalyzed by Knölker’s complex S7 or by the XlnS212CK biohybrid.
The hydrogenation of TFAC was carried out in aqueous medium, under the following reaction conditions: a 25 mM solution of the carbonyl substrate in HEPES buffer (pH 8) was allowed to react at 65° C with 3 M sodium formate in the presence of 90 μM Me3NO and 30 μM catalyst (29).
The same conditions were then chosen to explore the catalytic activity of the XlnS212CK artificial hydrogenase, and the influence of Xln A on the kinetics and selectivity of the hydrogenation reaction catalyzed by Knölker’s complex cofactor S7 was investigated.
3.5. Hydrogenation of trifluoroacetophenone by the XlnS212CK biohybrid
The reaction was followed by gas chromatrography using acetophenone as internal standard. Whereas the reaction was negligible in the absence of catalyst (data not shown), it was catalyzed by both the Knölker’s iron complex S7 and the Xln A-Knölker’s iron biohybrid complex XlnS212CK. XlnS212CK catalyzed the hydrogenation of TFAC into alcohol S9 with an initial rate of 8.8 μM.h−1, whereas Knölker’s complex S7 alone was poorly soluble in water thus producing alcohol S9 with an initial rate of only 2 μM.h−1 (data not shown). Under those conditions, the reaction thus proceeded about 4.4-fold faster with the XlnS212CK biohybrid than with the Knölker’s complex S7 alone as catalyst. It is noteworthy that, in ethanol the Knökler’s complex S7 was soluble and produced alcohol S9 with an initial rate of 2.6 μM.h−1. Over time, the production of alcohol S9 slowed down to reach a plateau after about 60 hours. After reaching the plateau, approximately 2.5-fold more alcohol S9 was produced in the presence of the biohybrid XlnS212CK than in the presence of the Knölker’s complex S7 alone as catalyst (Fig. 5).
Figure 5.

Time course of hydrogenation of TFAC by XlnS212CK (ρ) in aqueous buffer in comparison with CK alone in EtOH (π).
This confirmed that the XlnS212CK biohybrid was a better catalyst for the hydrogenation of TFAC by sodium formate in water, which could potentially be explained by the fact that the protein scaffold may facilitate solubilization of the hydrophobic Knölker’s complex and the TFAC substrate in water.
Finally, the enantiomeric excess was measured using a chiral GC column, as described in the experimental section. Trifluoromethyl benzyl alcohol was formed as a racemic mixture, exhibiting poor control of the substrate orientation and/or stabilization inside the hydrophobic pocket of the XlnS212CK biohybrid.
3.6. Virtual docking of trifluoroacetophenone to the XlnS212CK biohybrid
To rationalize these experimental results and to simulate a pre-coordination complex that could favor hydrogenation of the TFAC molecule, we performed virtual docking of this substrate to XlnS212C and XlnS212CK. Blind virtual docking was first performed on free XlnS212C to uncover pockets and/or preferred hot-spots that favor TFAC binding to the enzyme. After testing over 1,000,000 blind docking combinations, we found that the most favorable TFAC ligand conformers are always preferentially positioned within a given subsite of the Xln A binding pocket (ΔG = −62 kcal/mol, Figure 6A). Even upon anchoring of the Knölker’s complex to the enzyme scaffold, docking simulations illustrate that the most favorable TFAC binding site remains unchanged, targeting the same active-site pocket within Xln A, even after conformational re-arrangements of the active-site cavity (ΔG values up to - 81 kcal/mol, Figure 6B–D). Interestingly, none of our docking models show catalytically productive atomic coordination between the Knölker’s complex and the TFAC substrate molecule, further supporting the lack of enantioselectivity conferred by the XlnS212CK biohybrid scaffold. Indeed, this observation suggests that the Knölker’s complex is most likely flexible and can experience free rotational reorientation within the active-site pocket of Xln A, allowing it to access the subsite pocket populated by TFAC.
Figure 6.

Virtual docking of the trifluoroacetophenone (TFAC) substrate to the XlnS212C and XlnS212CK biohybrid complexes used as receptor targets. A) Blind virtual docking of TFAC to the XlnS212C variant obtained from the RosettaBackrub server. B) Docking of TFAC to the energetically minimized cavity of the XlnS212CK biohybrid complex. C) Docking of TFAC to an energetically favorable XlnS212CK conformer extracted from Monte Carlo simulations. D) Virtual docking of TFAC to the open state conformation of XlnS212CK, obtained from Normal Mode Analysis. In all structures, TFAC and the Knölker’s complex are colored red and sandy brown, respectively. Red and blue protein surfaces highlight hydrophobic and hydrophilic surface areas, respectively.
3.7. Reaction mechanism
Based on our results and previously reported work (30), a three-step mechanism can be proposed for the reduction of TFAC by sodium formate catalyzed by the XlnS212CK biohybrid (Scheme 1). First, in agreement with the necessity to add Me3NO in the medium for the reaction to occur, it is reasonable to assume that oxidation by Me3NO of one CO ligand stabilized by the iron atom in the starting Knölker’s complex S7 provides the activated 16-electron iron species S7b. The subsequent hydrogen activation should lead to the hydride iron complex S7c (30).
The C=O bond may then be reduced either through a one-step process (as shown in Scheme 1) or through a stepwise process. Finally, the reduced products are released and the Knölker’s complex S7 regenerated.
4. Conclusions
In conclusion, we have demonstrated that a new artificial hydrogenase made by covalent grafting of the iron Knölker’s complex S7 bearing a maleimido reactive function anchored to a xylanase S212C variant can efficiently catalyze the transfer hydrogenation of the benchmark substrate TFAC by sodium formate in water, yielding the corresponding secondary alcohol as a racemic. The reaction proceeded more than 3-fold faster with the XlnS212CK biohybrid than with the Knölker’s complex S7 alone. In addition, efficient conversion of TFAC to its corresponding alcohol were reached within 60 h with XlnS212CK, whereas ≈ 2.5 fold lower conversion was observed with Knölker complex S7 alone as catalyst. As previously reported for metal complexes such as metalloporphyrins (13,14), this confirmed that incorporation of iron Knölker’s complex S7 into the hydrophobic pocket of a protein such as Xln A is productive and favorable, most likely due to increased solubility provided by the protein scaffold for such hydrophobic substrate and Knölker moiety. In contrast, incorporation of the Knölker’s complex in the chiral cavity of XlnS212C did not induce the expected reaction enantioselectivity, suggesting poor control of substrate and/or Knölker’s complex orientation with respect to the iron center within the active-site pocket of Xln A.
On the basis of these results, we aim to build new artificial metalloenzymes based on the same scaffold. To improve activity of the biohybrid catalyst, water-soluble metalloporphyrins incorporating different metal centers such as ruthenium will be synthesized. Improving enantioselectivity may be achieved either by chemical modifications such as using spacers of variable lengths between the xylanase protein and the Knölker’s complex, or by performing additional mutagenesis within the active-site pocket of Xln A to better accommodate and improve complex stabilization.
Scheme 2.

Postulated mechanism of the catalytic hydrogenation by iron artificial metalloenzyme in water.
5. Acknowledgements
The authors thank Nhung Nguyen-Thi and Roger Dubuc (INRS) for helpful technical assistance with protein expression and purification. This work was supported in part by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) under award number RGPIN-2016-05557 (to N.D.), in addition to a grant from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award R01GM105978 (to N.D.). N.D. is the recipient of a Fonds de Recherche Québec – Santé (FRQS) Research Scholar Junior 2 Career Award (number 32743). This work was supported in part by an ANR grant Carb2zyme (ANR-17-CE11-0014-02) and the LabEx CHARM3AT (ANR-11-LABEX-0039)
6. References
- [1].Ashby EC, Boone JR (1976) J. Am. Chem. Soc. 98, 5524–5531. [Google Scholar]
- [2].Dickson HD, Smith SC, Hinkle KW (2004) Tetrahedron Lett. 45, 5597–5599. [Google Scholar]
- [3].Du D-M, Fang T, Xu J, Zhang S-W (2006) Org. Lett. 8, 1327–1330. [DOI] [PubMed] [Google Scholar]
- [4].Johnson RM, Rickborn B (1970) J. Org. Chem. 35, 1041–1045. [Google Scholar]
- [5].Naota T, Takaya H. Murahashi, (1998) Chem. Rev. 98, 2599–2660. [DOI] [PubMed] [Google Scholar]
- [6].Vivancos Á, Beller M, Albrecht M (2018) ACS Catal. 8, 17–21. [Google Scholar]
- [7].Schrock RR & Osborn JA (1976) J. Am. Chem. Soc. 98, 4450–4455. [Google Scholar]
- [8].Mérel DS, Elie M, Lohier J-F, Gaillard S, Renaud J-L (2013) ChemCatChem 5, 2939–2945. [Google Scholar]
- [9].Letondor C, Humbert N, Ward TR (2005) Proc. Natl. Acad. Sci. 102, 4683–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, Lebrun V, Reuter R, Köhler V, Lewis J,C, Ward T,R, (2018) Chem. Rev. 118, 142–231. [DOI] [PubMed] [Google Scholar]
- [11].Mérel DS, Gaillard S, Ward TR, Renaud J-L (2016) Catal. Lett. 146, 564–569. [Google Scholar]
- [12].Ricoux R, Dubuc R, Dupont C, Marechal J-D, Martin A, Sellier M, Mahy J-P, (2008) Bioconjug. Chem. 19, 899–910. [DOI] [PubMed] [Google Scholar]
- [13].Ducros V, Charnock SJ, Derewenda U, Derewenda ZS, Dauter Z, Dupont C, Shareck F, Morosoli R, Kluepfel D, and Davies JG (2000) J. Biol. Chem. 275, 23020–23026. [DOI] [PubMed] [Google Scholar]
- [14].Ricoux R, Allard M, Dubuc R, Dupont C, Maréchal J-D, Mahy J-P, (2009) Org. Biomol. Chem. 7, 3208–3011. [DOI] [PubMed] [Google Scholar]
- [15].Mahy J-P, Raffy Q, Allard M, Ricoux R (2009) Biochimie 91, 1321–1323. [DOI] [PubMed] [Google Scholar]
- [16].Allard M, Dupont C, Muñoz Robles V, Doucet N, Lledós A, Maréchal J-D, Urvoas A, Mahy J-P, Ricoux R (2012) ChemBioChem 13, 240–251. [DOI] [PubMed] [Google Scholar]
- [17].Uehara T, Rokugawa T, Kinoshita M, Nemoto S, Fransisco Lazaro GG, Hanaoka H, Arano Y, (2014) Bioconjug. Chem. 25, 2038–2045. [DOI] [PubMed] [Google Scholar]
- [18].Richter M, Chakrabarti A, Ruttekolk IR, Wiesner B, Beyermann M, Brock R, Rademann J, (2012) Chem. - Eur. J 18, 16708–16715. [DOI] [PubMed] [Google Scholar]
- [19].Ellman GL (1959) Arch. Biochem. Biophys. 82, 70–77. [DOI] [PubMed] [Google Scholar]
- [20].Ducros V, Charnock SJ, Derewenda U, Derewenda ZS, Dauter Z, Dupont C, Shareck F, Morosoli R, Kluepfel D, Davies GJ (2016) J Biol Chem. 275, 23020–23026. [DOI] [PubMed] [Google Scholar]
- [21].Lauck F, Smith CA, Friedland GF, Humphris EL, Kortemme T (2010) Nucleic Acids Res. 38, W569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kuriata A, Gierut AM, Oleniecki T, Ciemny MP, Kolinski A, Kurcinski M, Kmiecik S (2018) Nucleic Acids Res. 46, W338–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jamroz M, Orozco M, Kolinski A, Kmiecik S (2013) J Chem Theory Comput. 9,119–125. [DOI] [PubMed] [Google Scholar]
- [24].Suhre K, Sanejouand YH (2004) Nucleic Acids Res. 32, W610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lauck F, Smith CA, Friedland GF, Humphris EL, Kortemme T (2010) Nucleic Acids Res. 38, W569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Thomsen R, Christensen MH (2006) J Med Chem. 49, 3315–21. [DOI] [PubMed] [Google Scholar]
- [27].Madern N, Talbi B, Salmain M (2013) Appl. Organomet. Chem. 27, 6–12 (2013). [Google Scholar]
- [28].Honda K, Ono T, Okano K, Miyake R, Dekishima Y, Kawabata H, (2019) J. Biosci. Bioeng. 127, 145–149. [DOI] [PubMed] [Google Scholar]
- [29].Pagnoux-Ozherelyeva A, Pannetier N, Mbaye MD, Gaillard S, Renaud J-L (2012) Angew. Chem. 124, 5060–5064. [DOI] [PubMed] [Google Scholar]
- [30].Lator A, Gaillard S, Poater A, & Renaud JL (2018) Chemistry. 24, 5770–5774. [DOI] [PubMed] [Google Scholar]