

Abstract
T cell–mediated cancer immunotherapies, including anti–PD-1 and CAR-T cells, are becoming standard treatments for many cancer types. CAR-T therapy, in particular, has been successful in treating circulating, but not solid, tumors. One challenge limiting immunotherapy success is that tumors lacking T-cell infiltration do not respond to treatment. Therefore, one potential strategy to overcome resistance is to enhance the ability of T cells to traffic into tumors. Here, we describe an unbiased in vivo genetic screen approach utilizing the Sleeping Beauty (SB) mutagenesis system to identify candidate genes in T cells that might be modified to drive intratumoral T-cell accumulation. This screen identified over 400 candidate genes in three tumor models. These results indicated substantial variation in gene candidate selection, depending on the tumor model and whether or not mice were treated with anti–PD-1, yet some candidate genes were identified in all tumor models and with anti–PD-1 therapy. Inhibition of the most frequently mutated gene, Aak1, impacted chemokine receptor expression and enhanced T-cell trafficking in vitro and in vivo. Screen candidates should be further validated as therapeutic targets, with particular relevance to enhancing infiltration of adoptively transferred T cells into solid tumors.
Keywords: Sleeping Beauty, T-cell trafficking, CAR-T cells, immunotherapy, Aak1
Introduction
Cancer immunotherapy has become the standard-of-care in a variety of tumor types, but response rates remain suboptimal (1). In lymphoid malignancies, genetically redirected T cells expressing chimeric antigen receptors (CAR-T cells) have achieved clinical success, but this approach has had limited success in treating solid tumors, in part, due to poor trafficking of the CAR-T cells to the tumor site (2). Driving T cells into the tumor has been successful at enhancing efficacy in preclinical models (1,3,4). A better understanding of the mechanisms contributing to T-cell infiltration into tumors could enhance the success of T cell–based cancer immunotherapies.
Several high-impact forward genetic screens have been performed to increase our understanding of the underlying molecular mechanisms driving immunotherapy response (5–9). Most of these interrogate tumor cell–intrinsic mechanisms rather than immune mechanisms and utilize CRISPR or shRNA technologies that result in loss-of-expression of targeted genes after introduction of complex libraries of sgRNAs or shRNAs. These approaches are difficult to perform in endogenous immune cells in vivo and can produce off-target effects (10). The Sleeping Beauty (SB) approach offers several advantages over shRNA and CRISPR screens, including the potential to mutagenize the whole genome of endogenous T cells in vivo, the ability to induce overexpression or gain-of-function in addition to gene disruption, and no risk of undetectable “off-target” genomic modifications (11).
In this report, SB screens were performed in three distinct solid tumor models, as well as in two tumor models treated with anti–PD-1 therapy. A variety of candidate genes were identified. Many were unique to individual tumor models, whereas some were identified in all screens. These genes, including Aak1 (AP2-associated kinase 1), are not currently being explored as immunotherapy targets and represent potential new targets to enhance intratumoral T-cell accumulation, which may be particularly useful to the CAR-T cell and adoptive cell-transfer applications.
Methods
Animal information
All mice were housed in specific pathogen-free facility at the University of Iowa, and the University of Iowa Animal Care and Use Committee (IACUC) approved all uses in this study. T2Onc2/T2Onc3 double transgenic SB mice from two strains (6070/12740 and 6117/12775, maintained by the Dupuy Lab at the University of Iowa) were crossed with CD4-Cre mice from Jackson Laboratories (JAX stock #017336) to generate T-cell mutagenized mice (12). F1 offspring that inherited the Cre allele were used for genetic screening, and Cre allele presence was confirmed using PCR with primers TTATTCGGATCATCAGCTACAC (CreF) and ATCTGGCATTTCTGGGGATT (CreR). OT-1 TCR transgenic mice (Jackson Laboratories, JAX stock #003831) and wild-type C57BL/6N (Charles River, Strain Code 027) were obtained from the indicated vendor and directly used for T-cell migration (OT-1) and tumor growth studies (C57BL/6N).
Tumor cell lines
Cell lines were tested annually for mycoplasma and were negative at time of last test. A20 and EL4 were purchased from ATCC in 2010, and B16F0 were purchased from ATCC in 2016. Cell lines were authenticated by STR analysis in 2018 (IDEXX BioResearch). A20 and EL4 cell lines were cultured in R10 medium (Gibco), and LLC and B16F0 cells were cultured in DMEM (Gibco) supplemented with 1% penicillin/streptomycin (Gibco) and 10% heat-inactivated FBS (Hyclone, catalog #SH30070.03).
In vivo tumor growth
Tumor cell lines were resuspended in sterile 0.9% sodium chloride (Hospira) and injected subcutaneously, bilaterally, or unilaterally into the rear flank(s) of 6–12 week old T-cell mutagenized SB mice (genetic screen) or wild-type C57BL/6N mice (tumor growth experiments). The number of cells per injection site was as follows: 8,000 B16F0 cells, 1x106 LLC cells, 3x106 A20 cells, or 1x106 EL4 cells. Tumor growth was monitored by caliper measurement twice weekly, and mice were euthanized at experimental endpoint before tumors reached 2,000 mm at largest diameter. This corresponds to days 21 (B16F0), 20 (A20), and 16 (EL4) after tumor inoculation. Where indicated, treatment with anti–PD-1 (clone RMP1–14, BioXCell) or isotype control (clone 2A3, BioXCell) was administered twice weekly via intraperitoneal (i.p.) injection of 10 mg/kg, and treatment with a small molecule inhibitor of Aak1 (Aak1i, LP-935509 (13), Axon Medchem) was also administered twice weekly (concomitantly with anti–PD-1) via oral gavage of 10 mg/kg in sterile saline. Mice were randomized before starting treatment on day 3 of tumor growth (tumors not palpable) using a random number generator. Statistical methods were not used to determine cohort size and researchers were not blinded. For genetic screen data, tumors and spleens were removed at tumor growth endpoints indicated above. A portion of these tissues was dissociated using the GentleMACs tissue dissociator (Miltenyi) and used for flow cytometric quantification of T cells, and the remaining tissues were snap-frozen in liquid nitrogen and stored at −80°C until genomic DNA extractions could be performed. For tumor growth data, tumors and spleens were removed at tumor growth endpoints indicated above and dissociated using the gentleMACs tissue dissociator (Miltenyi Biotec) and used for flow cytometric quantification of T cells.
Candidate gene identification and additional notes regarding Supplementary Table S2
To identify transposon insertion sites in T cells, bulk genomic DNA was isolated from snap-frozen tumor and spleen tissues of T cell-mutagenized mice using GenElute™ Mammalian Genome DNA miniprep Kit (Sigma). DNA was sheared using the Covaris E220 sonicator, and DNA fragments containing transposon-genomic sequences were amplified via ligation-mediated PCR and submitted for sequencing at the Iowa Institute of Human Genetics on Illumina Hi-Seq2000/4000 as previously described (14–16). The detailed protocol for sequencing library generation and sequence data analysis is available in Additional File 2 from Feddersen, et al. (16).
Local transposition was previously handled by excluding insertion data from the local chromosome(17). However, we chose to include all chromosomes in the present analysis because these events would be present in both splenic and tumor signatures (rather than enriched in tumor-infiltrating T cells only). It is estimated that only ~10% of insertions are on the local chromosome in somatic screens (18), and two distinct transgenic lines were included in the screen presented here, such that local hopping events would be further diluted in the results. Transgenic strain data for each individual mouse can be found in Supplemental Table S1. Insertion sites were mapped to GRCm38 using a previously described Integration Analysis System (IAS) pipeline (16), which outputted the intermediate Source Data gff3 file containing all mapped insertion sites. Identification of gene-level transposon-induced driver mutations (gCIS) was performed using 5,000 bp as the input promoter region size (16). All tools and accompanying documentation, can be acquired through GitHub (https://github.com/addupuy/IAS.git)
Average enrichment scores were calculated by comparing normalized read abundance of individual insertion sites from individual mice in tumors compared to spleen. An enrichment score of 1 represented insertion sites observed only in tumor, 0.5 represented insertion sites equally represented in tumor and spleen, and 0 represented insertion sites observed only in spleen. Supplementary Table S2 contains the gene name (column A), the chromosomal address of the gene (columns B-E), the number of mice with any insertions in that gene (column F), the percentage of all mice with any insertions in that gene (column G), the p-value and false discovery rate to judge significance of gene mutation (columns H an d I), the number of mice whose insertions were in the gene itself or promoter region broken down by strand (columns J and K), the average enrichment score ranging from 0 [spleen only] to 1 [tumor only] (column L), and the animal IDs for each mouse where insertions in that gene were observed, formatted as ID=<tumor model>_<unique mouse number> (column M). Further, candidate genes that were previously identified as T-cell cancer-driver genes in a separate SB screen (column N) or in human cancer (column O) are annotated. Pathway analysis was performed using Database for Annotation, Visualization and Integrated Discovery (DAVID)(19).
Microarray analysis
Gene expression of Aak1 in intratumoral and splenic T cells from tumor bearing mice was taken from a publicly available microarray dataset (GSE53388), and details used to generate this dataset are published (7). Briefly, OT-1 T cells were adoptively transferred into B16 tumor-bearing mice, then purified from tumors or spleens such that gene expression from splenic T cells could be compared to gene expression from intratumoral T cells. For the present analysis, normalized gene expression values for intratumoral and splenic T cells for Aak1 probes (1420025_s_at, 1420026_at, 1434935_at, 1435038_s_at, 1441782_at, 1452632_at, 1452633_s_at) were selected from the publicly available data and plotted in Figure 4A. Samples used for analysis were GSM1290674, GSM1290675, GSM1290676, GSM1290692, GSM1290693, and GSM1290694.
Figure 4. Inhibition of Aak1 kinase activity enhances mouse T-cell migration.

A, Publicly available microarray data (7) was used to evaluate Aak1 gene expression of intratumoral T cells compared to splenic T cells from the same mice (n = 3 mice). Probe data for Aak1 was pulled and differential expression P values were determined by two-sided t-test for individual probes. All probes hybridized to exon 21 of Aak1. Bars indicate population mean, standard deviation, and min/max. Asterisk refers to probe location in panel B. B, Aak1 was the most frequently mutated gene in our screen. Transposon insertions were predominately clustered in intron 2, as indicated by the red triangles, with a few other insertion sites elsewhere in the gene (grey triangles). Though insertions were tightly clustered, transposon orientation was unbiased. Asterisk and blue triangle represent the region covered by microarray probes panel A. C, Cell surface expression of Cxcr3 on activated, primary mouse T cells treated with Aak1i or DMSO control for 1.5 hours was measured by flow cytometry. Each dot represents a technical replicate, and data are representative of three biological replicates (one-way ANOVA, error bars represent standard deviation, experiment was replicated three times). D, Migration of primary mouse T cells toward CXCL10 with Aak1i or DMSO vehicle control treatment (*P=0.03, ***P=0.0004, ****P<0.0001, two-way ANOVA with multiple comparisons, horizontal lines indicate means). E,F, B16F0 melanoma tumors treated with Aak1i or DMSO vehicle control were harvested at tumor growth day 21, and (E) CD4+ and (F) CD8+ T-cell infiltration was measured by flow cytometry. Each dot represents an individual mouse. Significance determined by one-way ANOVA. Data shown are representative of 3 replicates with 5 mice/group in each replicate.
Flow cytometry
B16F0, A20, and EL4 tumors or spleens were excised and fresh tissues were manually diced into small (~2mm) pieces and then further homogenized in RPMI (Gibco) without additives using a gentleMACS Dissociator (Miltenyi Biotec) on the mouse spleen setting. Cell suspensions were passed through a 70 µm mesh filter, and red blood cells were lysed with ACK buffer (made in-house with reagents purchased from Fisher Scientific according to the following recipe: 16.58g NH4Cl, 2g KHCO3, 74.4 mg Na2-EDTA, 1600ml ddH20, pH to 7.2 with 1N HCl) . Single-cell suspensions were labeled with Zombie viability dye (catalog # 423113, BioLegend), according to the manufacturer’s instructions, followed by extracellular labeling with FcR block (14–0161-86, eBioscience) and the following antibodies (BioLegend): CD45.2 (clone 104), CD3e (clone 17A2), CD4 (clone GK1.5), CD8a (clone 53–6.7), and CXCR3 (clone CXCR3–173) for 20 minutes. Labeled cells were washed twice and fixed in PBS containing 0.05% paraformaldehyde and counted on an LSR II Violet (Becton Dickinson) in the University of Iowa Flow Cytometry Facility. Flow data was analyzed using FlowJo software v.9. Cells were gated on live (Zombie violet negative) singlets (FSC-H vs. FSC-W) before proceeding with T-cell identification (CD45.2+CD3e+). T cell subsets were then gated (CD4+ versus CD8a+). CXCR3-positivity was determined based on FMO negative control lacking CXCR3 antibody.
Genetic modification of primary human T cells
Full length AAK1 or truncated AAK1 (dN80-AAK1, lacking the first 80 amino acids) cDNA was cloned into retroviral backbone pMIG (Addgene). Primary PBMCs from healthy donors were activated for 2 days before transduction with pMIG virus. Transductions were performed with polybrene and spinoculation at 1000 x g for 90 minutes at 32°C. Human peripheral blood was used for T-cell migration studies presented in Figure 5. All human peripheral blood was obtained from normal healthy donors (n = 12) with informed consent and in accordance with the protocol approved by the University of Iowa Institutional Review Board, and no additional human data was collected alongside specimens for migration studies.
Figure 5. Inhibition of Aak1 kinase activity enhances human T-cell migration toward CXCL10.
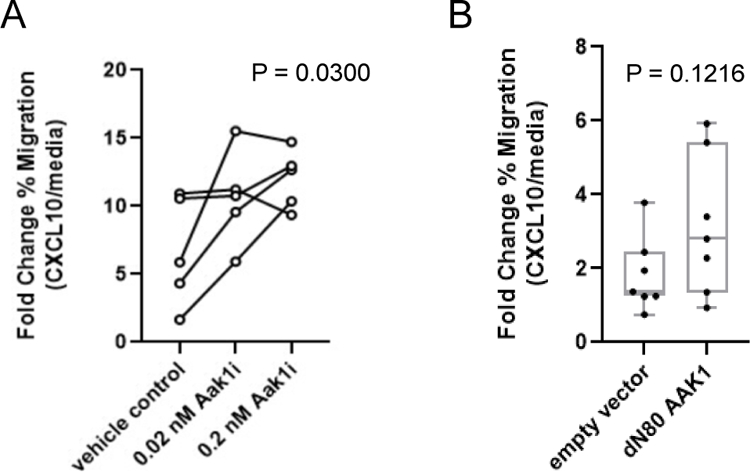
A, Primary human T cells (n=5) were isolated and cultured in vitro with Aak1i at the indicated concentrations or DMSO vehicle control. Migration of human T cells toward CXCL10 was assessed and presented as fold-change. P=0.0300, one-way ANOVA with multiple comparisons. Lines represent individual donors. B, Migration of primary human T cells genetically modified (or not) to overexpress mutant AAK1 (dN80 AAK1) toward CXCL10.P=0.1216, paired two-tailed t test, bars represent mean, min, and max with dots representing individual donors.
Transwell migration assays
Primary mouse splenocytes from OT-1 TCR transgenic mice were activated using 1 nM SIINFEKL peptide (Anaspec catalog # AS-60193-1) and cultured for 7–10 days. Activated T cells were then placed into migration media (1x Hank’s Buffered Saline Solution with 0.1% BSA) and 100 ul containing 5x105 cells plated on top transwell plates (Corning, catalog # 3387), with Aak1i or DMSO vehicle control. The bottom of the transwells contained migration media alone, or recombinant CXCL10 (Peprotech) and Aak1i in migration media. Cells were allowed to migrate for 1.5 hours as previously published (20). Similarly, primary human PBMCs were activated using anti-CD3/anti-CD28 beads (Invitrogen) for 7–10 days to allow upregulation of chemokine receptor expression. Cells at the same cell density as in mouse experiments were then plated in transwell plates (Corning) and allowed to migrate toward CXCL10 (20). For both mouse and human migration assays, cells in the top of the transwell and cells in the bottom well were harvested after migration and counted separately using a flow cytometer (LSR Violet, BD) and analyzed with FlowJo software. Mouse or human cells were gated on live (Zombie violet negative) singlets (FSC-H vs. FSC-W). Human cells were further gated on GFP positive (to count only cells successfully transduced with pMIG constructs). Percent migration was calculated (# bottom / # bottom + # top) and normalized to background migration (media-only) condition.
Statistical analysis
P values were calculated using the tests described in the individual figure legends using Graphpad Prism 7 (Graphpad Software), R, or SAS.
Data sharing statement
Sequence data are publicly available at the NCBI Sequence Read Archive (SRA) under BioProject Accession PRJNA641272.
Results
In vivo screen approach to identify T-cell genes that influence intratumoral accumulation
A common problem in the field of T cell–mediated immunotherapy is that many tumors lack adequate and productive intratumoral T-cell infiltrates. The factors governing this are diverse and can include a lack of immunogenic tumor antigens, systemic and local immune suppression, and tumor inaccessibility (21–23). Current efforts to improve cancer immunotherapy are largely focused on known immune checkpoint pathways, but novel alternative strategies may help advance the field. Thus, an unbiased, forward genetic screen was used to identify genes that contributed to T-cell infiltration into the tumor microenvironment. This screen was performed in vivo in endogenous T cells. T cells were randomly mutagenized by inducing tissue-specific expression of the SB machinery. This approach has a number of differences compared to more widely used CRISPR/Cas and shRNA screening tools (Table 1). The SB system is comprised of a DNA transposon and a transposase enzyme that cuts the transposon from the donor site and enables integration into random TA dinucleotides across the genome (11). In contrast to more widely used screening tools, insertional mutagenesis can both promote overexpression of downstream full-length or truncated genes through the transposon’s promoter sequence, and disrupt a gene by introducing a premature polyA signal (Figure 1A). The transposon itself serves as a sequence tag, allowing for specific identification of insertion sites via PCR with transposon-specific primers. Thus, identification of insertion sites can be done using bulk tissue without concern for contaminating genomic DNA from un-mutagenized cells (tumor cells and other non-T cells). The SB mice used in this experiment were engineered to carry a transgene with hundreds of copies of the DNA transposons (T2/Onc2 and T2/Onc3) and expressed the SB transposase upon Cre-induction, allowing endogenous mutagenesis in vivo without requiring additional gene delivery and eliminating the need for methods to normalize variability in library delivery.
Table 1.
Key differences between Sleeping Beauty and other popular screen tools.
| Sleeping Beauty (SB)(35,36) | CRISPR/Cas (37,38) | siRNA/shRNA (38,39) |
|---|---|---|
| Deliver transposon and transposase (co-transfected in vitro and transgenic mice available in vivo) | Deliver a library of sgRNAs (usually virally in vivo) | Deliver a library of shRNAs (usually virally in vivo) |
| All cells start with the same number of transposons | Cell-to-cell variability in sgRNA delivery efficiency can introduce bias | Cell-to-cell variability in shRNA delivery efficiency can introduce bias |
| No concern for off-target effects (mutations are self-labeling via transposon sequence) | Off-target effects still not well-characterized | Off-target effects are known to occur |
| Whole genome mutagenesis | Whole genome or targeted mutagenesis | Whole genome or targeted mutagenesis |
| Gain-of-function and loss-of-function capabilities | Loss-of-function only (knockout) | Loss-of-function only (knockdown) |
Figure 1. In vivo Sleeping Beauty (SB) screen to identify genes influencing intratumoral T-cell accumulation.

A, The SB transposon structure enables both gain- and loss-of-function mutagenesis upon insertion in or near a gene. The promoter sequence (MSCV or CAG) can overexpress full or partial transcripts, and the bi-directional polyA can disrupt expression. The transposon is flanked by inverted repeats (IRL and IRR) that facilitate transposon recognition by transposase. B, SB mice were bred to CD4-Cre mice, producing offspring with active transposition in CD4+ and CD8+ T cells. Mice were injected subcutaneously with syngeneic tumor cells lacking SB (two tumors per mouse) and treated twice weekly with anti-PD-1 or left untreated. At endpoint (day 21), both tumors and the spleen from each mouse were harvested and transposon insertion sites in the T-cell genome were identified by high-throughput sequencing. (n=154 untreated and n=53 anti-PD-1 treated mice) C, Insertion sites in tumor-infiltrating T cells (left and right) were compared to the insertion sites in splenic T cells from the same mouse to identify tumor-enriched insertion sites. Additional statistical analyses (gCIS) were performed on tumor-associated T-cell insertion sites to identify significantly mutated genes in tumor-infiltrating T cells across multiple individual mice. D, Intratumoral T-cell infiltration, as measured by flow cytometry (F139, 39=1.8267, P=0.03), in tumors with and without SB mutagenesis (n=85 independent mice combined into a single graph). Lines represent population mean and significance was determined by performing a two-sided F-test to compare variances on log-transformed values.
To induce transposition specifically in endogenous T cells, SB-transgenic mice were bred with CD4-Cre mice to induce transposition in CD4+ cells, thus mutagenizing both CD4+ and CD8+ T cells (Figure 1B). A similar cross was done previously with the goal of identifying genetic drivers of T-cell malignancy (24), and tumors developed with an average latency of 48.9 weeks. Thus, the present screen was designed to limit the age of mice included in the screen cohorts. Syngeneic tumor cells lacking SB (B16F0 melanoma, EL4 lymphoma, or LLC Lewis lung carcinoma) were injected subcutaneously on both flanks (single tumor model per animal). Additional information on cohort demographics, including SB strain and sex, is included in Supplementary Table S1. Tumors were allowed to grow either in untreated mice or mice treated with anti–PD-1, and then tissues were harvested to analyze T-cell infiltration and identify SB insertion sites. T-cell receptor (TCR) specificity was not defined in any screen, and intratumoral T cells were likely a polyclonal pool. We aimed to enrich for T cells specifically trafficking to the tumor microenvironment by requiring mutations to be identified in both tumors in each bilateral tumor model. Initial studies evaluated genetic signatures in CD4+ and CD8+ T cells separately by using live cell sorting prior to sequencing library preparation. Most of the insertion sites were identified in the CD8+ T cells, with very few identified in the CD4+ subpopulation (Supplementary Figure S1A). The T-cell selection process reduced the number of T cells available for analysis, possibly introducing bias (Supplementary Figure S1B). Subsequent sequence analysis was done on pooled T cells, and as such, identified mutations could have been in CD4+ or CD8+ T cells.
Insertion site analysis and candidate gene identification
Insertion sites in tumor-infiltrating T cells (observed in both the left tumor and right tumor) were compared to the insertion sites in splenic T cells from the same mouse to identify tumor-enriched insertion sites (Figure 1C). To eliminate abundant, non-tumor–associated T-cell insertion sites (e.g. genes, such as potential oncogenes which might be clonally expanded independent of an antitumor response), the splenic insertion site signature in each mouse was used as the background insertion pattern. An enrichment score for each insertion site within individual mice was calculated using normalized read abundances for left tumor versus spleen and right tumor versus spleen, and averaging this score. We then compared the present gene candidates to the tumor driver genes identified in a closely related CD4-Cre SB screen to identify any genes present in both lists (24). Of the top 856 significant gene candidates (FDR<0.01), only four were previously identified by SB mutagenesis as potential genetic drivers of T-cell malignancy (Cdkn2a, Oprm1, Myo16, and Rasgrf1). A list of genes known to be associated with human T-ALL was also compared to the present gene candidates (24). Similarly, only four of these genes associated with human T-cell malignancy were represented in the present candidate gene list (St6galnac3, Cdkn2a, Atxn1, and Oprm1). These were annotated as potential drivers of malignancy in the full gene candidate list (Supplementary Table S2). Therefore, we could be reasonably confident that the majority of candidate genes we identified were not strong drivers of T-cell malignancy.
Additional statistical analyses (gCIS) were performed on tumor-associated T-cell insertion sites to identify significantly mutated genes in tumor-infiltrating T cells across multiple individual mice (Figure 1C). Genes having insertions that were enriched in intratumoral T cells from multiple mice were identified as candidate genes that might functionally contribute to intratumoral T-cell infiltration. In support of this hypothesis, SB mutagenesis enhanced the magnitude of T-cell infiltration into B16 tumors in select mice (Figure 1D), whereas tumor size remained unchanged (Supplementary Figure S2). Combining all screen cohorts, gene common insertion site (gCIS) analysis identified 406 candidate genes that were significantly mutated in two or more mice at a higher frequency in intratumoral T cells compared to splenic T cells (enrichment score>0.5 and gCIS FDR<1x10–4)(Table 2, Supplementary Table S2).
Table 2.
Top 10 gene candidates hypothesized to influence T-cell functions important for intratumoral infiltration.
| Gene* | Function |
|---|---|
| Aak1 | Regulates AP2-mediated endocytosis, Notch and Wnt(29,40,41) |
| Ehhadh | Peroxisomal beta-oxidation of fatty acids(42) |
| Macrod2 | Haploinsufficient tumor suppressor in intestinal cancers(43) |
| Ckb | T cell differentiation and TCR signal enhancer(25) |
| Rnf214 | (Unknown function) |
| Sprr1b | Epidermal development, unknown T cell function(44) |
| Cpd | Secretory pathway and hormone processing(45) |
| Rpl48-ps1 | (Unknown function) |
| Son | Component of spliceosome, mediates alternative splicing(46) |
| Eif3b | Increased activity in activated T cells(26) |
Genes ranked in descending order of mutation frequency
Genetic selection of candidate genes varies with tumor model
The SB screening approach was designed to identify T-cell genes associated with increased intratumoral T-cell accumulation. We hypothesized that these candidate genes were likely to be involved in increasing T-cell proliferation, promoting prolonged T-cell viability, or enhancing trafficking to the tumor microenvironment. Two of the top ten most frequently mutated genes, Ckb and Eif3b, have been previously documented to be involved in T-cell activation, proliferation, and cytokine secretion downstream of TCR activation (25,26). A third, Son, is upregulated in CD8+ tumor-infiltrating lymphocytes (27). Not all tumor models sustained mutations in the top 10 genes, despite the top 10 genes having the lowest false discovery rates and the highest number of mice with insertions (Figure 2A). Aak1 was mutated in intratumoral T cells from multiple mice in all three tumor models. Note that the next most frequently mutated genes were not identified in all three tumor models. This could be a product of microenvironmental differences between tumor models influencing T-cell accumulation.
Figure 2. Gene candidate identification in tumor-infiltrating T cells obtained from untreated mice.

A, Percentage of tumor-bearing mice with insertions in the indicated gene across the three models (number of mice in each indicated). Top 10 gene candidates (all tumor models) were ranked by significance, then number of mice, and finally by enrichment score in three tumor models. B, DAVID pathway enrichment analysis was performed on the candidate genes with gCIS FDR<0.001 (544 genes). All functional pathways are listed, with the number of genes in candidate gene list in each functional category on the x-axis. Bars with asterisks are significantly enriched biological processes (P<0.002). C, Venn diagram of all gCIS identified in each tumor model. Numbers represent the total number of gCIS identified (no significance cutoff), illustrating the mutational overlap in intratumoral T cells between tumor models. Mutation frequency of the 24 genes shared by all three models is illustrated by the heatmap, with genes ranked in descending order of average mutation frequency. Cluster analysis (indicated above the heatmap) was performed by the R heatmap function and included mutation frequency for all genes.
Protein pathway analysis was performed on all candidate genes (gCIS FDR<0.001) using the Database for Annotation, Visualization and Integrated Discovery (DAVID)(19). Approximately 11% of the significant gene candidates identified were transcription factors (Supplementary Table S2). Enriched Gene ontology (GO) terms included signal transduction (41 genes), cell adhesion (30 genes), and adhesion molecules (21 genes). Mutations were significantly enriched in KEGG pathways, including those involved in axon guidance and glutamatergic synapse formation, providing further evidence that biological processes impacting cellular trafficking were represented in our screen results (Figure 2B, Supplementary Table S3).
Genes identified in more than one tumor model could impact intratumoral T-cell accumulation. Of the genes identified, only 24 were shared among all three tumor models (Figure 2C). Aak1 was the most frequently mutated gene in each of the three tumor models, making it an attractive gene candidate for further study.
Genetic selection of candidate genes varies with anti-PD-1 treatment
Anti-PD-1 therapies are now in wide use clinically, and there is great need to rationally identify promising combinatorial therapies to enhance the efficacy of immune checkpoint blockade. Thus, mice were treated twice weekly with anti-PD-1 to determine whether it would select for alternative gene candidates in intratumoral T cells. Consistent with published literature, anti-PD-1 therapy had little effect on B16 tumor growth and no impact on the percentage of intratumoral T cells in SB-mutagenized mice (Figure 3A, Supplementary Figure S3)(28). Anti-PD-1 treatment had no significant impact on EL4 tumor size at endpoint, but caused a reduction in the percentage of intratumoral CD4+ T cells and an increase in the percentage of intratumoral CD8+ T cells. Despite minimal control of tumor growth and mixed effects on T-cell infiltration, anti-PD-1 therapy significantly influenced genetic selection of insertion sites in intratumoral T cells in both tumor models. Comparison of candidate genes identified in T cells from tumors of mice treated with anti-PD-1 to candidate genes identified in T cells from untreated mice revealed greater overlap in candidate genes in B16 than in EL4, suggesting anti-PD-1 exerted stronger a more selective effect in EL4 than in B16 (Figure 3B). For the complete list of candidate genes identified in each tumor model and treatment group, see Supplementary Table S2 column M.
Figure 3. Impact of treatment with anti-PD-1 on genetic selection of candidate genes.

A, Percentage of tumor-infiltrating (left) CD4+ or (right) CD8+ T cells in SB-mutagenized mice with and without anti–PD-1 treatment (number of mice in each model and treatment cohort indicated in panel B), as analyzed by flow cytometry on whole tumor material at time of tumor harvest (day 21). Lines represent population means, and P values were calculated using a two-tailed t-test, and results were combined and analyzed as a single replicate. B, Venn diagram comparison of untreated (gray) versus treated (red) tumors in (left) B16F0 and (right) EL4 models. Numbers represent all genes identified by gCIS (no significance cutoff). C, Candidate intratumoral T-cell genes (gCIS FDR<1x10–4) that were shared between both treated and untreated tumors (Venn diagram intersect in panel B) and that were also significantly differentially mutated upon anti-PD-1 treatment (Fisher’s exact P<0.05). Mutation frequency in treated versus untreated (EL4, open circles; B16F0, closed circles) tumors is expressed as log2 fold change on the y-axis.
Anti-PD-1 therapy also influenced the mutation frequency of genes previously identified in T cells from untreated tumors. Of the hundreds of genes that were shared between treated and untreated cohorts, only 22 showed significant differential mutation frequencies upon treatment (Fisher’s Exact P<0.05, Figure 3C). Sprr1b (small proline-rich protein 1B) was mutated with ~5-times higher frequency in B16 tumors treated with anti-PD-1 compared to untreated B16 tumors (Figure 3C, log2 fold-change treated versus untreated), suggesting that Sprr1b mutation within T cells might synergize with anti-PD-1 to enhance their intratumoral infiltration. The most frequently mutated gene, Aak1, was also mutated to a higher degree in anti-PD-1–treated tumors.
Experimental validation of gene candidate Aak1 in T-cell migration and tumor infiltration
Aak1 was examined more closely as a potential immunotherapy target because it was the most frequently mutated gene in all three untreated tumor models, and its mutation frequency was increased by anti-PD-1 treatment. Analysis of previously published microarray data indicated differential expression of Aak1 in intratumoral T cells compared to splenic T cells (Figure 4A). Transposon insertion sites were clustered within intron 2 of Aak1, but transposon orientation was unbiased (Figure 4B). This region of the gene encodes the kinase domain, such that transposon insertions would likely truncate this domain. The tightly clustered pattern of transposon insertions within the Aak1 kinase domain suggested selective pressure for a gain-of-function or change-of-function (e.g. dominant-negative activity).
Aak1 phosphorylates Adapter Protein 2 (AP2), which regulates clathrin-mediated endocytosis (29). T-cell trafficking is controlled through chemokine signaling, and chemokine receptor expression on the cell surface is tightly regulated, with many chemokine receptors internalized via clathrin-mediated endocytosis following chemokine binding (30,31). Aak1 localizes to the leading edge of migrating HeLa cells (29), suggesting Aak1 may regulate endocytosis of chemokine receptors. Therefore, we hypothesized that mutation of Aak1 would impact T-cell trafficking, as well as receptor expression on the T-cell surface. To assess whether Aak1 kinase activity regulated chemokine receptor expression, primary splenocytes harvested from OT-1 transgenic mice were activated with cognate peptide and incubated for 7 days. T cells were then treated with a commercially available small molecule inhibitor of Aak1 (Aak1i, LP-935509)(13) for 1.5 hours before chemokine receptor Cxcr3 expression was measured by flow cytometry. This receptor was chosen because Cxcr3 signaling attracts T cells toward CXCL10 (IP-10), a chemokine frequently produced in the tumor microenvironment (32). Activated T cells treated with Aak1i had increased expression of Cxcr3 compared to controls (Figure 4C), suggesting Aak1 kinase activity impacted localization of Cxcr3. Treatment of activated T cells with Aak1i increased T-cell migration toward chemokine CXCL10 in an in vitro transwell migration assay (Figure 4D).
Because CXCL10 is not the only factor regulating T-cell infiltration into the tumor microenvironment in vivo, the impact of Aak1i on intratumoral T-cell infiltration was tested in vivo in the B16 melanoma model. Tumor-bearing mice were treated with Aak1i (13), anti–PD-1, combination of Aak1i and anti–PD-1, or vehicle control starting on day 3 of tumor growth. At the endpoint (day 21), T-cell infiltration was measured using flow cytometry. Aak1i increased intratumoral CD4+ T-cell infiltration in all mice treated with Aak1i (Figure 4E). However, Aak1i did not significantly impact CD8+ T-cell infiltration (Figure 4F), nor did it directly impact tumor cell proliferation in vitro (Supplementary Figure S4). Together, these results supported the hypothesis that therapeutically targeting Aak1 could enhance T-cell infiltration into tumors.
Similar to the mouse studies presented in Figure 4, primary human T cells from five healthy individuals were activated, and their ability to migrate toward CXCL10 was measured. Migration of T cells toward CXCL10 was enhanced by Aak1i treatment (Figure 5A), suggesting Aak1 played a role in human T-cell trafficking. Aak1i, although primarily targeting Aak1, also has off-target inhibitory effects on related BMP-2-inducible Protein Kinase (BIKE) and Cyclin G-Associated Kinase (GAK)(13). Therefore, we tested the role of Aak1 in T-cell migration using complimentary genetic approaches. Primary human T cells were engineered to overexpress the truncation mutant dN80 AAK1, which is designed to mimic the mutation produced by SB transposon insertion. In transwell assays, genetic modification of AAK1 enhanced T-cell migration toward CXCL10 in 5 of 7 individuals (Figure 5B). This supported the hypothesis that the dN80 truncation disrupted Aak1 kinase activity. Together with the clustered insertion site pattern (Figure 4B), which suggested a gain- or change-of-function, this suggests that the dN80 mutant functions as a dominant-negative mutation.
Discussion
T cell–mediated immunotherapy, including but not limited to CAR-T therapy, has the potential to be more effective if T-cell infiltration into the tumor microenvironment could be enhanced. The screen reported here was designed to identify genes that might be modified to enhance such infiltration. This screen has two advantages over previously reported screens. First, it allows for detection of both gain- and loss-of-function mutations. Second, it is an unbiased, whole genome screen performed in vivo. This approach allows for more comprehensive genomic interrogation to identify genes that might be modified to enhance intratumoral T-cell presence. This easy-to-use screen approach is flexible and can be readily adapted to other treatment types, temporal studies, or for screening additional immune cell types, including NK or B cells, by virtue of using alternative Cre transgenic mice (e.g. Granzyme B-CreERT2 (33)).
The tumor model or treatment impacted genetic selection, suggesting there were likely complex microenvironmental differences between tumor models that influenced T-cell trafficking. However, some genes were identified across all screen conditions, suggesting some commonalities existed that might allow for enhanced trafficking of T cells across tumor types and treatments. The imperfect overlap in gene candidates between screen cohorts may also be explained, in part, by the fact that screen saturation points had likely not been reached. Additional gene targets could be found that are common across tumor types and treatment strategies with additional screening.
Therapeutic strategies modulating these candidate genes or their products could be promising in combination with immune checkpoint blockade, as evidenced by the impact of anti-PD-1 therapy on gene selection and T-cell infiltration in preclinical tumor models. The effects of the Aak1 inhibitor were complex, and likely reflected the impact of the inhibitor on other molecular targets and cells other than T cells. This highlights the value of identifying ubiquitously expressed genes that can be genetically targeted in T cells, and have been overlooked as potential targets for cancer immunotherapy. Well-known immune checkpoint molecules including, Ctla4, Pdcd1 (PD-1), CD274 (PD-L1), Pdcd1lg2 (PD-L2), and a number of TNF family members (Tnfsf8, Tnfsf15, Tnfrs1b, Tnfrs8, Tnfrs11a, Tnfrs19, Tnfrs22, Tnfrs23) were identified as having more insertions in intratumoral T cells versus splenic T cells but did not reach the significance cutoff during gCIS analysis. The mutations that did reach statistical significance were, in large part, those impacting key signaling molecules that function downstream of receptors including immune checkpoints. It may be that changes in such downstream molecules are more efficient in enhancing intratumoral T-cell presence than changes in the receptors themselves. For example, ENPP1 is upregulated in T cells upon PD-1 receptor stimulation, and was identified during the screen (34). Thus, although most of the identified candidates have not previously been considered as immunotherapy targets, they may be capable of promoting T-cell infiltration if modified. Although many of these genes play vital roles in other tissue types, this does not prevent consideration of modifying them specifically in T cells that are being adoptively transferred as is done for CAR-T therapy.
Because many of the same biological processes required for an antitumor immune response are co-opted in T-cell leukemias and lymphomas (e.g. cell proliferation), it is also important to understand whether the gene candidates we identified might be pro-tumorigenic. Previously published work indicated that inducing SB mutagenesis using a CD4-Cre approach was successful in identifying genetic drivers of T-cell malignancies (24). Thus, the following precautions were taken to distinguish tumorigenic mutations from those that were predicted to enhance intratumoral T-cell accumulation. First, this screen was performed in young mice (6–9 weeks) before tumor development would be expected (average latency ~50 weeks). Second, clonal expansion in the spleen was used to establish a background mutation signature in each individual mouse. Third, little overlap was observed in the present screen candidates with genes previously identified as T-cell cancer drivers in SB models or human T-ALL. Combined, these data support the conclusion that these screen results have potential to be safely translated into adoptive transfer therapies.
In summary, we developed a screening strategy utilizing the SB transposon system to identify T-cell genes that could enhance T cell–mediated immunotherapies by increasing the number of intratumoral T cells. The advantages over other current screening platforms include the ability to easily mutagenize endogenous T cells, and the capacity to induce gain-of-function mutations that may be unique targets to enhance T cell-mediated immunotherapies. Once validated, gene candidates (including AAK1) could be genetically modified to benefit immune checkpoint blockade and adoptive T-cell therapies by enhancing T-cell infiltration into tumors.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the Iowa/Mayo Lymphoma SPORE (P50 CA97274) and the National Cancer Institute of the National Institutes of Health under Award Numbers K22CA225786 and P30CA086862. Sequencing data presented herein were obtained at the Genomics Division of the Iowa Institute of Human Genetics, which is supported, in part, by the University of Iowa Carver College of Medicine and the Holden Comprehensive Cancer Center. Flow cytometry data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine / Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center and the National Cancer Institute of the National Institutes of Health under Award Number 1 S10 OD016199-01A1.
Financial Support
Research reported in this publication was supported by the Iowa/Mayo Lymphoma SPORE (P50 CA97274) and the National Cancer Institute of the National Institutes of Health under Award Numbers K22CA225786 and P30CA086862.
Footnotes
Competing interests: The authors declare no conflicts of interest.
Conflict of Interest Statement
The authors declare no potential conflicts of interest.
References
- 1.Kim TK, Herbst RS, Chen L. Defining and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends in immunology 2018;39(8):624–31 doi 10.1016/j.it.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Frontiers in immunology 2018;9:1740 doi 10.3389/fimmu.2018.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trujillo JA, Sweis RF, Bao R, Luke JJ. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer immunology research 2018;6(9):990–1000 doi 10.1158/2326-6066.CIR-18-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71 doi 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature 2017;548(7669):537–42 doi 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017;547(7664):413–8 doi 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 2014;506(7486):52–7 doi 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018;359(6377):770–5 doi 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang L, Malu S, McKenzie JA, Andrews MC, Talukder AH, Tieu T, et al. The RNA-binding Protein MEX3B Mediates Resistance to Cancer Immunotherapy by Downregulating HLA-A Expression. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(14):3366–76 doi 10.1158/1078-0432.CCR-17-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 2018;561(7723):416–9 doi 10.1038/s41586-018-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeNicola GM, Karreth FA, Adams DJ, Wong CC. The utility of transposon mutagenesis for cancer studies in the era of genome editing. Genome biology 2015;16:229 doi 10.1186/s13059-015-0794-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001;15(5):763–74. [DOI] [PubMed] [Google Scholar]
- 13.Kostich W, Hamman BD, Li YW, Naidu S, Dandapani K, Feng J, et al. Inhibition of AAK1 Kinase as a Novel Therapeutic Approach to Treat Neuropathic Pain. The Journal of pharmacology and experimental therapeutics 2016;358(3):371–86 doi 10.1124/jpet.116.235333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, Liu P, et al. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer research 2009;69(20):8150–6 doi 10.1158/0008-5472.CAN-09-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riordan JD, Drury LJ, Smith RP, Brett BT, Rogers LM, Scheetz TE, et al. Sequencing methods and datasets to improve functional interpretation of sleeping beauty mutagenesis screens. BMC Genomics 2014;15:1150 doi 10.1186/1471-2164-15-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feddersen CR, Wadsworth LS, Zhu EY, Vaughn HR, Voigt AP, Riordan JD, et al. A simplified transposon mutagenesis method to perform phenotypic forward genetic screens in cultured cells. BMC Genomics 2019;20(1):497 doi 10.1186/s12864-019-5888-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brett BT, Berquam-Vrieze KE, Nannapaneni K, Huang J, Scheetz TE, Dupuy AJ. Novel molecular and computational methods improve the accuracy of insertion site analysis in Sleeping Beauty-induced tumors. PloS one 2011;6(9):e24668 doi 10.1371/journal.pone.0024668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 2005;436(7048):221–6 doi 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 19.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome biology 2007;8(9):R183 doi 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barreira da Silva R, Albert ML. Mouse CD8+ T Cell Migration in vitro and CXCR3 Internalization Assays. Bio-protocol 2017;7(6):e2185 doi 10.21769/BioProtoc.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan X, Zhang S, Deng Y, Wang P, Hou Q, Xu H. Prognostic Factors for Checkpoint Inhibitor Based Immunotherapy: An Update With New Evidences. Front Pharmacol 2018;9:1050 doi 10.3389/fphar.2018.01050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17(16):5343–52 doi 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Endres S, Kobold S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotherapy 2019;11(3):201–13 doi 10.2217/imt-2018-0111. [DOI] [PubMed] [Google Scholar]
- 24.Berquam-Vrieze KE, Nannapaneni K, Brett BT, Holmfeldt L, Ma J, Zagorodna O, et al. Cell of origin strongly influences genetic selection in a mouse model of T-ALL. Blood 2011;118(17):4646–56 doi 10.1182/blood-2011-03-343947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Li H, Wang X, Gao X, Liu X. Regulation of T cell development and activation by creatine kinase B. PloS one 2009;4(4):e5000 doi 10.1371/journal.pone.0005000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyamoto S, Patel P, Hershey JW. Changes in ribosomal binding activity of eIF3 correlate with increased translation rates during activation of T lymphocytes. The Journal of biological chemistry 2005;280(31):28251–64 doi 10.1074/jbc.M414129200. [DOI] [PubMed] [Google Scholar]
- 27.Giordano M, Henin C, Maurizio J, Imbratta C, Bourdely P, Buferne M, et al. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. The EMBO journal 2015;34(15):2042–58 doi 10.15252/embj.201490786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer immunology research 2016;4(10):845–57 doi 10.1158/2326-6066.CIR-16-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conner SD, Schmid SL. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J Cell Biol 2002;156(5):921–9 doi 10.1083/jcb.200108123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luther SA, Cyster JG. Chemokines as regulators of T cell differentiation. Nat Immunol 2001;2(2):102–7 doi 10.1038/84205. [DOI] [PubMed] [Google Scholar]
- 31.Neel NF, Schutyser E, Sai J, Fan GH, Richmond A. Chemokine receptor internalization and intracellular trafficking. Cytokine Growth Factor Rev 2005;16(6):637–58 doi 10.1016/j.cytogfr.2005.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 2017;17(9):559–72 doi 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bannard O, Kraman M, Fearon DT. Secondary replicative function of CD8+ T cells that had developed an effector phenotype. Science 2009;323(5913):505–9 doi 10.1126/science.1166831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nature medicine 2010;16(10):1147–51 doi 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mann MB, Jenkins NA, Copeland NG, Mann KM. Sleeping Beauty mutagenesis: exploiting forward genetic screens for cancer gene discovery. Curr Opin Genet Dev 2014;24:16–22 doi 10.1016/j.gde.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 36.O’Donnell KA. Advances in functional genetic screening with transposons and CRISPR/Cas9 to illuminate cancer biology. Curr Opin Genet Dev 2018;49:85–94 doi 10.1016/j.gde.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kweon J, Kim Y. High-throughput genetic screens using CRISPR-Cas9 system. Arch Pharm Res 2018;41(9):875–84 doi 10.1007/s12272-018-1029-z. [DOI] [PubMed] [Google Scholar]
- 38.Schuster A, Erasimus H, Fritah S, Nazarov PV, van Dyck E, Niclou SP, et al. RNAi/CRISPR Screens: from a Pool to a Valid Hit. Trends Biotechnol 2019;37(1):38–55 doi 10.1016/j.tibtech.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Schaefer C, Mallela N, Seggewiss J, Lechtape B, Omran H, Dirksen U, et al. Target discovery screens using pooled shRNA libraries and next-generation sequencing: A model workflow and analytical algorithm. PloS one 2018;13(1):e0191570 doi 10.1371/journal.pone.0191570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agajanian MJ, Walker MP, Axtman AD, Ruela-de-Sousa RR, Serafin DS, Rabinowitz AD, et al. WNT Activates the AAK1 Kinase to Promote Clathrin-Mediated Endocytosis of LRP6 and Establish a Negative Feedback Loop. Cell reports 2019;26(1):79–93 e8 doi 10.1016/j.celrep.2018.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorensen EB, Conner SD. AAK1 regulates Numb function at an early step in clathrin-mediated endocytosis. Traffic 2008;9(10):1791–800 doi 10.1111/j.1600-0854.2008.00790.x. [DOI] [PubMed] [Google Scholar]
- 42.Houten SM, Denis S, Argmann CA, Jia Y, Ferdinandusse S, Reddy JK, et al. Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids. J Lipid Res 2012;53(7):1296–303 doi 10.1194/jlr.M024463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakthianandeswaren A, Parsons MJ, Mouradov D, MacKinnon RN, Catimel B, Liu S, et al. MACROD2 Haploinsufficiency Impairs Catalytic Activity of PARP1 and Promotes Chromosome Instability and Growth of Intestinal Tumors. Cancer Discov 2018;8(8):988–1005 doi 10.1158/2159-8290.CD-17-0909. [DOI] [PubMed] [Google Scholar]
- 44.Koizumi H, Kartasova T, Tanaka H, Ohkawara A, Kuroki T. Differentiation-associated localization of small proline-rich protein in normal and diseased human skin. Br J Dermatol 1996;134(4):686–92 doi 10.1111/j.1365-2133.1996.tb06971.x. [DOI] [PubMed] [Google Scholar]
- 45.Varlamov O, Eng FJ, Novikova EG, Fricker LD. Localization of metallocarboxypeptidase D in AtT-20 cells. Potential role in prohormone processing. The Journal of biological chemistry 1999;274(21):14759–67 doi 10.1074/jbc.274.21.14759. [DOI] [PubMed] [Google Scholar]
- 46.Huen MS, Sy SM, Leung KM, Ching YP, Tipoe GL, Man C, et al. SON is a spliceosome-associated factor required for mitotic progression. Cell Cycle 2010;9(13):2679–85 doi 10.4161/cc.9.13.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.